

Abstract
A large number of investigations have demonstrated the participation of the immune system in the pathogenesis of hypertension. Studies focusing on macrophages and Toll-like receptors have documented involvement of the innate immunity. The requirements of antigen presentation and co-stimulation, the critical importance of T cell–driven inflammation, and the demonstration, in specific conditions, of agonistic antibodies directed to angiotensin II type 1 receptors and adrenergic receptors support the role of acquired immunity. Experimental findings support the concept that the balance between T cell–induced inflammation and T cell suppressor responses is critical for the regulation of blood pressure levels. Expression of neoantigens in response to inflammation, as well as surfacing of intracellular immunogenic proteins, such as heat shock proteins, could be responsible for autoimmune reactivity in the kidney, arteries, and central nervous system. Persisting, low-grade inflammation in these target organs may lead to impaired pressure natriuresis, an increase in sympathetic activity, and vascular endothelial dysfunction that may be the cause of chronic elevation of blood pressure in essential hypertension.
Keywords: autoimmunity, blood pressure, hypertension, immunity, lymphocytes, macrophages.
“Immunity maybe inborn or acquired. The former is always natural, that is to say, independent of direct intervention of human art. Acquired immunity (may be) spontaneous or maybe the result of human intervention.” 1
Immune reactivity in experimental and human hypertension has been reported in occasional investigations for more than half a century, but in most cases it has been considered epiphenomena associated with the increase in blood pressure. In early studies, the observation that most strongly suggested a pathogenic role for the immune system in hypertension was the demonstration that a functional thymus was a requirement for the maintenance of hypertension in various animal models.2–4 Renewed interest in the immune pathogenesis of hypertension has followed the findings that patients with essential hypertension have markers of systemic inflammation,5,6 the discovery of agonistic antibodies directed to angiotensin II and adrenergic receptors in specific conditions associated with high blood pressure,7 and the demonstration that the suppression of T cell–driven inflammation in target organs corrects, ameliorates or prevents experimental hypertension.8,9
The immune system is activated as an innate response to injury and as an antigen-specific acquired immune response. Both types of immunological activation are present in hypertension. In this review we shall discuss the evidence that causally associates innate and acquired immune reactivity to hypertension and the role played by inflammation in the kidney, arteries, and central nervous system (CNS) in the pathogenesis of high blood pressure.
INNATE IMMUNITY IN HYPERTENSION
The innate immune system is responsible for the immediate inflammatory response recruited as a defense mechanism against infections or in response to tissue injury. Dendritic cells, macrophages, natural killer (NK) T cells, and Toll-like receptors (TLRs) of the inflammasomes represent components of the innate immune system that have been investigated in hypertension. Evidence for the participation of other components of innate immunity remains limited at the present time.10
Dendritic cells
Dendritic cells are increased, infiltrating the kidney and arterial walls in hypertension models. Dendritic cells promote a differentiation of T cells toward a CD4+ IL-17 phenotype in response to aldosterone.11 In addition, they likely play a role as antigen-presenting cells because their number is increased in experimental models in which the hypertensive response is ameliorated by inhibition of co-stimulatory pathways (discussed in the section of adaptive immunity).
Macrophages
A consistent finding in experimental models of hypertension is the infiltration of macrophages in the kidney and periadventitial areas in the aorta and medium-sized arteries. A reduction in macrophage infiltration is associated with improvement of hypertension in spontaneously hypertensive rats (SHRs),12 Dahl salt-sensitive rats,13 hypertension induced by angiotensin II14 and aldosterone,15 salt-dependent hypertension,16–18 and autoimmune hypertensive renal disease.19 Wenzel et al. 20 studied the role of myelomonocytic cells in the pathogenesis of hypertension and vascular dysfunction by the daily administration of low doses of diphtheria toxin in the LysMiDTR mice with induced expression of the diphtheria toxin receptor. This strategy depleted circulating monocytes and protected the mice from developing angiotensin II–induced hypertension. This effect was reversed by the adoptive transfer of monocytes. Concordant with these results, osteopetrotic mice that have a mutation in the colony-stimulating factor gene and a generalized deficiency in macrophage populations are protected against hypertension induced with angiotensin II and aldosterone.21 In another study, treatment with a CCR2 antagonist, which blocks a chemokine receptor responsible for macrophage infiltration in the arterial wall, completely reversed the influx of macrophages and significantly reduced hypertension.22 Similar improvement was found blocking the monocyte chemoattractant protein (MCP-1) receptor in angiotensin II and desoxycorticosterone acetate (DOCA)-salt–induced hypertension.23 However, in other investigations the depletion of macrophages did not result in the improvement of blood pressure.24,25 The disagreement in these experiment may be due to the lack of discrimination between proinflammatory and protective (antihypertensive) effects of macrophages. For example, Titze and colleagues26,27 have shown that skin macrophages respond to the interstitial tonicity-responsive enhancer binding protein (TONEBP) with the production of VEGF-C, which stimulates the development of lymphatic capillaries and sequesters Na+ and Cl− in the interstitium, ameliorating hypertension.
NK cells
Interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α), interleukin 2 (IL-2), and interleukin 4 (IL-4) are all rapidly released by NK cells, and 2 important studies have suggested they may play a role in hypertension-related inflammation. Koomans et al. 28 have shown that monocytes and NK cells present a program of reciprocal activation in hypertension. Their studies have demonstrated that the inflammation and vascular dysfunction induced by angiotensin II are associated with the accumulation of NK cells and macrophages in the aortic wall. Angiotensin-induced cellular infiltration in vascular walls is drastically reduced in the Tbx21−/− mice. TBx2 drives IFN-γ production, and the response to angiotensin may be restored in the Tbx21−/− mice by the combined administration of IFN-γ–competent NK cells and monocytes, but not by the adoptive transfer of either one of them alone. The role of NK cells was also shown by the studies of Taherzadeh et al.29 who were able to induce susceptibility to L-NAME hypertension and vascular remodeling in the constitutively resistant Th2-biased BALB-C mice by the introduction of an NK gene complex of the Th1-biased C57BL/6 mice.
Inflammasomes and Toll-like receptors
The innate immune response depends on the recognition of molecular patterns that activate the multimeric protein complexes that constitute the inflammasomes and result in the activation of caspase 1, which induces the secretion of proinflammatory cytokines and a form of cell death called pyroptosis.30 In addition, the inflammasome induces an effective antigen presentation to naive T cells and thereby plays a crucial role of shaping the subsequent adaptive immune response. Molecular patterns recognized by the infammasome are pathogen-associated and danger-associated molecular patterns. In the pathogenesis of essential hypertension, pathogen-associated molecular patterns appear to be less relevant; nevertheless, 1 study31 reported that periodontitis is associated with a higher risk of hypertension in postmenopausal women.
Among the pattern recognition receptors, the TLRs are the best studied and, up to the present time, the only group that has been shown to play a role in inflammation related to hypertension. TLRs are expressed by T and B lymphocytes, antigen-presenting cells, and somatic cells, including endothelial and vascular smooth muscle cells. In adult SHRs there is an increase in TLR4 expression, and administration of anti-TLR4 antibody reduced serum interleukin 6 levels, improved endothelial-induced vasodilatation, and corrected hypertension.32 TLR4 knockout mice fail to develop L-NAME–induced hypertension.33 Increased TLR7/8- and TLR9-mediated interleukin 6 release was observed in splenic cell cultures of SHRs (but not Wistar rats) subjected to cholinergic (nicotinic) stimulation.33
Peripheral blood monocytes of patients with essential hypertension show increased TLR4 mRNA levels that become downregulated after 12 weeks of intense antihypertensive treatment.34 In a subgroup of patients with essential hypertension, the activation of the inflammasome has been suggested by the finding of a mutation in the gene of the sensor molecule, NPLR3.35
A large number of danger-associated molecular patterns related to vascular inflammation and dysfunction are capable of activating TLR4 and TLR2, including angiotensin II,36 C-reactive protein,37 uric acid,38 and Heat shock proteins (HSPs) 6039 and 70.40 HSP60,41 HSP70,42,43 C-reactive protein,44 and uric acid45 have also been associated with essential hypertension. Recently, it has been theorized that fetal programming of hypertension could result from activation of the innate immune system driven by danger-associated molecular patterns originating from the placenta or fetal tissues in complicated pregnancies.46
ADAPTIVE IMMUNITY IN HYPERTENSION
The adaptive immune response is the reaction of the immune system to specific antigens that results in immunological memory. Evidence of its role in hypertension includes studies of antigen presentation, lymphocyte activation, and antibody production.
Antigen presentation
The activation of T cells by antigens is facilitated by T-cell co-stimulation via B7 (CD80) ligands. Recently Vinh et al.47 showed an increased number of activated dendritic cells in angiotensin II and DOCA-salt hypertension and found that the hypertensive response could be prevented by pharmacological inhibition of the CD28/B7 co-stimulatory pathway. Amelioration of angiotensin II–induced hypertension was also observed in the B7 knockout mice that lack co-stimulatory proteins CD80 and CD86. Furthermore, the hypertensive response to angiotensin was restored by engrafting wild-type bone marrow in B7 knockout mice.47
Proinflammatory T-cell activation
More than a decade ago, it was shown that angiotensin II infusions induced a Th1-type cell-mediated immune response characterized by increase in IFN-γ and a decrease in IL-4 in the spleen and kidney.48 Subsequently, a large number of studies have shown T lymphocytes in target organs are driving inflammation and hypertension. These studies will be discussed in the next section. In humans, indirect evidence of the role of T lymphocytes was reported by Seaberg et al.,49 who found that, after controlling for age, race, body mass index, and smoking, untreated HIV-infected patients with chronic low lymphocyte counts had a lower prevalence of systolic hypertension than HIV patients treated with antiretroviral drugs in whom the prevalence of hypertension increased over time and was similar to uninfected control subjects. Additional evidence was given in the studies of Herrera et al.,50 who examined the effects of immunosuppression in 8 patients with essential hypertension who were prescribed mycophenolate mofetil (MMF) for the treatment of rheumatoid arthritis or psoriasis. During treatment with MMF, blood pressure was significantly reduced without changes in diet or antihypertensive treatment.
TNF-α and IL-6 may be increased51,52 or unchanged53 in hypertensive patients. One cytokine of potential importance is IL-17, which is elevated in the circulation in hypertensive patients.54 Madhur et al.54 also observed an increase in IL-17 production in the angiotensin II infusion model and found that the IL-17 knockout mice had less arterial immune cell infiltration and lower blood pressure during the chronic phase of this model.
Recently, Youn et al.55 found that hypertensive patients have increased numbers of CD8+ cytotoxic T cells with immunosenescence markers: loss of CD28 and acquisition of CD57. The loss of CD28 in hypertensive patients appears contrary to the role played by the co-stimulatory CD28/B7 molecules in hypertension demonstrated in the studies of Vinh et al.47 An explanation for this apparent paradox was suggested in an editorial commentary56 that hypothesized that inflammation in target organs could be responsible for the formation of increasing numbers of neoantigens and repeated bouts of antigen stimulation would lead to long-term CD28 loss in hypertensive patients.
Regulatory T cells
T regulatory lymphocytes (Tregs) are a subpopulation of T cells that limit immune responses and suppress inflammatory reactivity. Tregs are generated in the thymus (natural Tregs) and in the periphery in response to stimuli that induce immune tolerance. In mice, forkhead box p3 (Foxp3) is a specific marker for Tregs. In humans, Foxp3 expression is not always associated with regulatory function, and an additional subtype of CD4 Tregs (Foxp3–IL-10+), induce suppression of the immune response. Demonstration of a role for Tregs in experimental models of hypertension suggests indirectly the involvement of the immune system in the pathogenesis of hypertension.
Barhoumi et al.57 demonstrated that the administration of CD4+CD25+ Foxp3 Tregs reduces vascular immune cell infiltration, improves endothelial vasodilatation, and reduces blood pressure in mice receiving angiotensin II infusions. Similar benefits were observed in aldosterone-induced hypertension.58 In contrast, Kavan et al.59 did not find changes in blood pressure despite improvement of cardiac hypertrophy and fibrosis. Different experimental designs may be the reason for the contrasting results in these studies. Although a direct demonstration of the participation of Tregs in the modulation of genetic models of hypertension has not been investigated, the aortic expression of Foxp3, transforming growth factor beta, and IL-10, markers of immune suppression associated with Tregs, were all increased in consomic rats in which chromosome 2, a site of genes responsible for inflammation and hypertension, was transferred from normotensive Brown Norway to Dahl salt-sensitive rats. As a result, vascular inflammation and salt-dependent hypertension were suppressed in the consomic rats.60
Antibodies in hypertension
The generation of antibodies is an indication of B cell–mediated adaptive immune response. Table 1 summarizes recent investigations that have reported associations between specific antibodies and hypertension. Most of these investigations have focused on antibodies that bind to receptors mimicking the natural ligands and induce a stimulatory effect (agonistic antibodies). The most comprehensively studied is the agonistic antibody directed to the second extracellular loop of the angiotensin II type 1 receptor (AA-AT1r), first described in women with preeclampsia and also found in patients with essential hypertension, transplant rejection, systemic sclerosis, aldosterone-producing adenoma, and 8%–14% of normal individuals.7,61–67 Hypertensive patients with circulating AA-AT1r have an enhanced response to treatment with AT1r blockers.65 Other autoantibodies have also been identified: agonistic antibodies against alpha-1 adrenergic receptor (AA-α1Ar) are directed against the first or the second receptor loop and are expected to enhance peripheral vasoconstriction. They have been found in patients with essential refractory hypertension.68–70 Agonistic antibodies against the beta-1 adrenergic receptors (AA-β1AR) bind to the second receptor loop in cardiomyocytes71 and are capable of increasing cardiac output and blood pressure and of inducing experimental cardiomyopathy. Zhou et al.72 have found an antibody directed against the L-type voltage-gated calcium channels in 30% of patients with essential hypertension and 6.7% of healthy control subjects. It is assumed that this antibody may be agonistic and, by increasing the calcium concentration in vascular smooth muscle cells, would cause vasoconstriction and increase peripheral resistance. The hypertension resulting from the agonistic antibodies appears to be primarily due to increased peripheral vascular resistance by vasoconstriction and increased cardiac output. There is also the possibility, unexplored at the present time, that sympathetic and angiotensin overactivity induced by agonistic antibodies could have collateral immune-mediated reactivity.
Table 1.
Serum antibodies in hypertension
| Autoantibodies | Molecular targets | Clinical conditions |
|---|---|---|
| Anti–angiotensin II type 1 receptors (agonistic) | Second extracellular loop | Eclampsia and pre-eclampsia, 7,61,62 essential hypertension, 64,65 transplant rejection, 66 systemic sclerosis 63 adrenal adenoma 67 |
| Anti–alpha adrenergic receptors (agonistic) | First or second extracellular loop | Refractory hypertension 68–70 |
| Anti–beta-1 adrenergic receptors (agonistic) | Second extracellular loop (cardiomyocytes) | Dilated cardiomyopathy 71 |
| Anti–L-type voltage-gated calcium channels (agonistic) | Alpha1c-subunit of L-type Ca2+ channel used as antigen for detection | Essential hypertension 72 |
| Anti–heat shock proteins | HSP70 HSP65 |
Increased titers in essential hypertension and atherosclerosis 41,43,73 |
| Anti–endothelial cell | Endothelial cells | Borderline hypertension
74,75
Eclampsia 76 |
The agonistic characteristic was demonstrated in anti–angiotensin type I receptors, anti– alpha adrenergic receptors, and anti–beta adrenergic receptors and assumed in the anti–L-type voltage-gated calcium channels.
Abbreviation: HSP, heat shock protein.
Antibodies against HSPs have also been found in hypertensive patients,41,43,73 and, rather than being pro-hypertensive, the antibodies are likely part of an adaptive immune response against HSP70 in which the immune cell–driven inflammation in target organs is the pivotal element for the development of salt-sensitive hypertension. Finally, circulating immunoglobulin G and immunoglobulin M antibodies with affinity for endothelial cells have been found in patients with borderline hypertension74,75 and eclampsia.76
INFLAMMATION IN TARGET ORGANS: PATHOPHYSIOLOGY OF HYPERTENSION
Inflammation in the kidney, arteries, and CNS is central in the pathogenesis of hypertension.
Renal inflammation
Renal immune cell infiltration is a constant feature in experimental and clinical studies of hypertension. Renal biopsies taken during sympathectomies done for the treatment of hypertension more than half a century ago showed clusters of T lymphocytes in tubulointerstitial areas.8 Increased numbers of immune cells were also shown in autopsy studies of white and black hypertensive patients,77 and a recent study also reported in renal biopsies of 7 patients with hypertensive nephrosclerosis the tubulointerstital infiltration of CD4 and CD8 lymphocytes and increased expression of the T-cell chemokine, IFN-inducible T-cell α chemoattractant.78
In all experimental studies shown in Table 2, 2–4,12–17,43,79–92 a reduction in immune cell infiltration with immunosuppressive therapy or by genetic manipulations resulted in amelioration or correction of hypertension. In the SHRs,12 in salt-sensitive hypertension,17 and in hypertensive patients,77 the severity of hypertension is correlated with the intensity of immune cell renal infiltration. In salt-sensitive hypertension, the severity of hypertension is directly correlated with the renal angiotensin II concentration and inversely correlated with the plasma angiotensin II concentration, which, as expected, is suppressed by the high-salt diet.17
Table 2.
Studies on the role of renal inflammation in the pathogenesis of hypertension
| Treatment | Experimental models | Findings |
|---|---|---|
| Athymic (nude) mice/ neonatal thymectomy | Renal infarct, 2 DOCA-salt, 3 NZB mice 4 | Acute hypertension unchanged; late (chronic) phase hypertension corrected. Reduction in renal inflammation |
| Adrenal steroids/6 MP | Renal infarct 79 | Improvement in hypertension. Reduction in antikidney and antiartery antibodies |
| Transfer of splenic cells from DOCA-salt hypertensive rats | Normotensive rats 80 | Induction of renal inflammation and hypertension (by immune reactivity against arterial walls) |
| Cyclophosphamide | NZB mice, 81 SHRs 82 | Reduction in perivascular immune cell infiltration in the kidneys. Amelioration of hypertension |
| Cyclosporin A | dTGF rats 83 | Reduced renal injury, proteinuria, and immune cell infiltration. Amelioration of hypertension |
| Mycofenolate mofetil | SHRs, 11 Dahl SS, 13 post all SSHBP, 16,17 post L-NAME SSHBP, 18 DOCA-salt, 15 chronic lead toxicity, 84 overload proteinuria, 85 cellophane wrapped kidney, 86 prenatally programmed hypertension, 87 dTGF rats 14 | Reduction of immune cell infiltration, angiotensin II, and oxidative stress. Amelioration of hypertension and prevention of SSHBP |
| Ehanercept/dexamethasone | dTGF rats 14 | Reducton in renal inflammation and renal injury. No effect on blood pressure |
| Inhibition of NFkB | SHRs, 88 dTGF rats 89 | Reduced renal injury, proteinuria, and immune cell infiltration. Amelioration of hypertension in SHRs, not in the dTGF rats |
| Mutation of Rag 1 gene | Dahl salt-sensitive rat 90 | Reduction in renal immune cell infiltration and albuminuria and amelioration of salt-induced hypertension |
| CD247 knockout | Dahl salt-sensitive rat 91 | Reduction in renal T-cell infiltration and correction of hypertension |
| Induction of immune tolerance to HSP70 | L-NAME–induced SSHBP 43 | Reduction in renal immune cell infiltration, prevention of SSHBP |
Studies demonstrating the role of renal inflammation in the pathogenesis of hypertension.
Abbreviations: 6MP, 6 mercaptopurine; DOCA, deoxycorticosterone acetate; dTGF, double transgenic rat harboring human renin and angiotensinogen genes; NfKB, nuclear factor kappa B; NZB, New Zealand black; SHR, spontaneously hypertensive rat; SS, salt sensitive; SSHBP, salt-sensitive hypertension.
How is hypertension induced by renal inflammation? The complex pathophysiology of pressure natriuresis93 is outside the limits of this review, but impairment in this relationship is central in the development of hypertension that represents an adaptive response to maintain sodium balance.94 Franco et al.95 found that in salt-sensitive hypertension, accumulation of tubulointerstitial immune cells is directly associated with blunting of pressure-dependent natriuresis (Figure 1). Crowley et al. 96 gave insight to the relationship between lymphocyte activity and urinary sodium excretion, demonstrating that scid mice, which have deficient lymphocyte activity, were protected against angiotensin II–dependent hypertension by a pressure-induced natriuresis that was a consequence of renal overexpression of eNOS and COX2 and higher generation of nitric oxide and prostaglandin E2.
Figure 1.
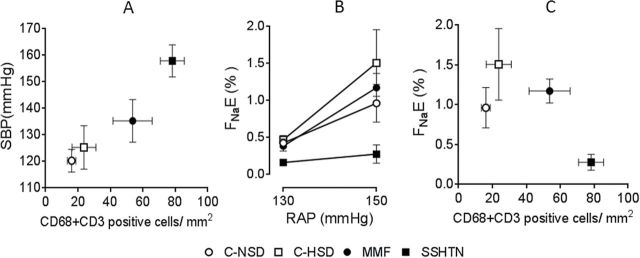
Pressure natriuresis studies in salt-sensitive hypertension induced by transient inhibition of nitric oxide synthase. Control groups received normal (C-NSD; open circles) and high (C-HSD; open squares) salt diet. Experimental groups were studied after 4 weeks of a high salt diet, started after discontinuation of 3 weeks of oral administration of L-NAME given alone (SSHTN; closed squares) or in association with mycophenolate mofetil (MMF; 20mg/kg/day; closed circles). (a) Salt-sensitive hypertension is directly correlated with the intensity tubulointerstitial immune cell infiltration. (b and c) Pressure natriuresis studies are done adjusting renal artery pressure (RAP) by an aortic clamp at 90, 110, 130 and 150mm Hg. (b) Fractional sodium excretion (FNaE) at 150mm Hg of RAP is suppressed in the SSHTN group and restored to normal in the MMF group. (c) Immune cell infiltration (CD3+ cells = lymphocytes; CD68 cells = macrophages) is directly correlated with natriuresis at 150mm Hg RAP. Abbreviation: SBP, systolic blood pressure. Figures made with data from Franco et al.95
Renal inflammation, immune cell infiltration, and augmented angiotensin II activity17,86 may be generated in renal tubular cells97 and in infiltrating cells16,18 because T cells have a functional renin-angiotensin system.98 In the kidney, angiotensin II impairs pressure natriuresis, and this effect is counteracted by L-arginine,99 but the relationship between angiotensin activity and hypertension is complex and dependent on the type of cells expressing AT1rs. Elegant studies by Crowley et al.100 demonstrated that bone marrow chimeras lacking AT1r have similar baseline blood pressure as wild-type controls and, surprisingly, presented an augmented hypertensive response to angiotensin II infusions, indicating a protective role of AT1r in the bone marrow–derived cells against the hypertensive actions of angiotensin II.
Vascular inflammation
Vascular inflammation is a characteristic of hypertension. In experimental models of hypertension, there is infiltration of CD4 and CD8 T cells, macrophages, and dendritic cells in perivascular tissue and adventitia in large (aorta) and medium-sized (mesenteric arteries) vessels.57–59,101 In the kidney, immune cells are preferentially found surrounding renal arteries.13 The reasons for the perivascular accumulation of immune cells are not defined, but there are sympathetic nerve endings in these areas, and perivascular inflammation is critically dependent on the CNS.102
Suppression of vascular inflammation has been associated with the correction of hypertension in various experimental models (Table 3).21–23,57–60,101–106 The experiments of Guzic et al.101 showed, for the first time, that adoptive transfer of T cells restored the full hypertensive response to angiotensin II in mice genetically devoid of T and B lymphocytes (rag−/− mice) that were resistant to angiotensin II. Interestingly, hypertension related to life stress is also associated with vascular inflammation; maternal separation, a recognized animal model for behavioral stress in early life, results in exaggerated sensibility to angiotensin and vascular inflammation in adult life. These findings are not observed in the rag−/− mice and restored by adoptive T lymphocyte transfer.105 Because oxidative stress is generated by inflammation and angiotensin II, it is somewhat surprising that deletion of extracellular superoxide dismutase (SOD3) in vascular tissue does not modify inflammation or angiotensin II–induced hypertension.107
Table 3.
Studies showing the role of vascular inflammation in the pathogenesis of hypertension
| Hyperetension | Experimental model | Results |
|---|---|---|
| Angiotensin II–induced hypertension | Rag−/− mice 101 | Resistance to angiotensin II hypertension. Adoptive transfer of T cells induces vascular inflammation and restores sensitivity to angiotensin II |
| IL-17−/− mice 54 | IL-17 needed for vascular inflammation and hypertension | |
| Adoptive transfer of T regulatory cells 57 | Improvement of inflammation and amelioration of hypertension | |
| Deletion of vascular superoxide dismutase 3 103 | Reduction in nitric oxide. Inflammation and hypertension unaffected | |
| MK2−/− mice 104 | Suppression of ICAM-1, MCP1, and vascular inflammation. Amelioration of hypertension | |
| Osteopetrotic mice (Op/Op) deficient in m-CSF 21 | Reduced vascular inflammation, increased AC-induced vascular relaxation, no increase in media thickness | |
| Selective ablation of myelomonocytic cells 20 | Reduced number of monocytes in blood and in vessels. Reduced inflammation, increased vascular relaxation. Transfer of monocytes reversed effects | |
| Aldosterone-induced hypertension | Adoptive transfer of T regulatory cells 58 | Suppression of vascular inflammation and correction of hypertension |
| DOCA-salt | CCR2 antagonist 22 | Suppressed vascular macrophage, correction of hypertension |
| Stress-induced hypertension | Effects of early life stress on the response to angiotensin II 105 | Increased vascular inflammation, increased hypertension in response to angiotensin II |
| Cold-induced hypertension | IL-6−/− 106 | Reduction in inflammation in arteries, heart and kidney. Amelioration of hypertension |
Studies showing the relevance of vascular inflammation in the pathogenesis of experimental hypertension.
Abbreviations: AC, acetylcholine; CCR2, chemokine (C-C motif) receptor 2; CSF, colony stimulating factor; DOCA, deoxycorticosterone acetate; ICAM-1, intercellular adhesion molecule 1; IL-6, interleukin 6; IL-17, interleukin 17; MCP1, monocyte chemoattractan protein 1; MK2, mitogen activated protein kinase 2; Rag−/−, recombination activating gene knockout; SOD, superoxide dismutase.
The mechanisms by which vascular inflammation favors the development of hypertension are related to increased vascular tone and impairment in arterial relaxation. The latter is a critical physiological response mediated by nitric oxide activity in normotensive humans to counteract increased sympathetic vasoconstriction.108 A number of experimental studies have shown that, when vascular inflammation is present, the endothelial (acetylcholine-induced) relaxation in contracted aortic rings is incomplete and the norepinephrine-induced vasoconstriction is enhanced.57,58,60,101 A role of inflammation in the impairment of the vascular physiology is demonstrated by the restoration of the vasodilatation capacity as a result of the adoptive transfer of Tregs.57,58
CNS inflammation
The CNS, the sympathetic nervous system (SNS), and the immune system are interconnected in the physiological modulation of hemodynamic and immune responses.109 In general, epinephrine and norepinephrine inhibit selectively Th1 and favor Th2 immune responses, but the preexisting state of T cells determines the ultimate responses of sympathetic SNS activation. For example, CD4 cells cultured under Th1-promoting conditions respond to norepinephrine with a robust production of IFN-α.109 The increases in peripheral vascular resistance, cardiac output, and sodium reabsorption resulting from activation of the SNS are well recognized,110 and, in addition, adrenergic stimulation increases the TLR-mediated production of proinflammatory cytokines by macrophages.111 In contrast with the abundance of data on the participation of the SNS in immune responses, scarce information exists on the inhibitory effects of parasympathic stimulation on immunity. It has been found that loss of parasympathetic downregulation of innate immune responses may favor hypertension in SHRs.112 Recent studies have focused on the antihypertensive potential of cholinergic activity mediated by α-7 nicotinic acetylcholine receptors. In SHRs, the α-7 nicotinic receptors in the spleen are reduced, and it has been suggested that, as a result, there is overexpression of inflammatory cytokines in several organs, including the kidney.113 However, further studies are needed to define the role of this pathway in the pathogenesis of hypertension.
Many of the studies examining the role inflammation within the brain in hypertension have focused on circumventricular regions because these areas lack a blood–brain barrier and have an abundance of AT1r. The importance of the integrity of this region in hypertension was shown by Marvar et al.,102 who found that lesions in the anteroventral third ventricle impair activation of lymphocytes and their homing to the arterial walls and, thereby, ameliorate angiotensin II–induced hypertension. Induction of oxidative stress in the subfornical organ in the hypothalamus by the knockdown of superoxide dismutase increases vascular infiltration of activated lymphocytes and worsens hypertension114 and, conversely, reducing the superoxide generation by the knockdown of the p22 subunit of the NADPH oxidase ameliorates angiotensin II–dependent hypertension.115 In the same model, suppressing inflammation in the subfornical organ with minocycline or by the induction of IL-10 overexpression ameliorates hypertension.115 The participation of brain receptors of angiotensin II is also critical in other models of hypertension; for example, DOCA-salt hypertension is ameliorated if the AT1rs in the subfornical organ are knocked down.116 Recent studies by Siramula et al.117 have demonstrated the role of TNF in the CNS in the development of hypertension. These investigators showed that inhibition of brain TNF by intracerebroventricular infusion of etanercep protects rats against angiotensin II–induced hypertension by restoring the balance between prohypertensive (ACE and AT1R) and antihypertensive (ACE2, Mas, and AT2 receptors) axes of the renin-angiotensin system, in association with inhibition of inflammatory cytokines and oxidative stress in the paraventricular nucleus. Because hypertensive patients have circulating markers of inflammation,5,6 it is relevant that systemic inflammation may cause inflammation in the CNS: proinflmmatory cytokines are produced in the rostral ventrolateral medulla in response to 2 weeks of infusion of lipopolysacharide.118
Other recent investigations have centered on the nucleus tractus solitarii in the brain stem, which is a pivotal region for the regulation of arterial pressure. In SHRs, the proinflammatory molecule junctional adhesion molecule 1 (JAM 1) is highly overexpressed in the vascular endothelial cells in this area in association with dysregulation of the expression of several genes associated with inflammation. These findings were associated with accumulation of leukocytes within the capillaries in this region, a finding considered important in the development of hypertension.119
In summary, the mechanisms involved in the development of hypertension resulting from inflammation in the CNS include SNS stimulation and the activation and homing of T cells for the induction of arterial inflammation.
RECENT RESEARCH ON AUTOANTIGENS IN HYPERTENSION
A critical area of study in the immune pathogenesis of hypertension is the identification of antigen(s) responsible for driving the immune response in essential hypertension. Because the agonistic antibodies are present in only a fraction of the hypertensive patients, other antigens may be playing a role in the vast majority of patients with essential hypertension. The possibility that multiple antigens play a role in the pathogenesis of hypertension is suggested by the demonstration of T-cell senescence markers compatible with repeated bouts of neoantigens generation.56 We suggested9 that autoimmune reactivity to HSPs could play a role in hypertension because these molecules, which function as chaperones of nascent proteins, are very immunogenic when accessing the extracellular compartment. We centered our studies on HSP70 because it is expressed in the kidney, arteries, and CNS by stimuli associated with hypertension, such as angiotensin II, oxidative stress, sympathetic stimulation, and stress.8 In addition, HSP70 is increased in the kidney in animal models of hypertension,42 and patients with essential hypertension have anti-HSP70 serum antibodies.42,43,73 In salt-sensitive hypertension induced by transient L-NAME administration, we found that induction of immune tolerance to HSP70 prevented renal inflammation and hypertension. Furthermore, the adoptive transfer of IL-10–producing Tregs from tolerized rats corrected the salt-dependent hypertension. Finally, salt-induced increments in blood pressure resulted from genetically induced overexpression of HSP70 in the kidney of rats previously sensitized to HSP70.43 Taken together these experiments suggest that HSP70 should be investigated as one of the potential antigens in human essential hypertension.
CONCLUDING REMARKS
Investigations in the last decade have provided compelling evidence that innate and adaptive immunity play a central role in hypertension (Figure 2). The role of B cells is evidenced by the generation of agonistic antibodies directed to angiotensin II and adrenergic receptors. The most definite data supporting the role of agonistic antireceptor antibodies correspond to the AA-AT1r in eclampsia and preeclampsia. However, pregnancy-associated hypertension is an acute, self-limited condition. In salt-sensitive primary hypertension, studies to date suggest a role for T cell–mediated inflammation and T cell–suppressor responses. Inflammation-derived neoantigens and surfacing of intracellular immunogenic proteins, such as HSP70, could be central in the development of autoimmunity that would drive a low-grade inflammation in target organs that underpins the increase in blood pressure. Therapeutic strategies of potential clinical use may result from the insights gained in studies on the role of immunity in hypertension.
Figure 2.

Innate and adaptive immune responses are closely interrelated, and prohypertensive stress signals drive immune involvement in the pathogenesis of hypertension. Immune reactivity in target organs induces local inflammation that is responsible for impairment in pressure diuresis, suppression of endothelial vasodilation capacity, and increased sympathetic activity.
DISCLOSURES
R.J.J. is on the Scientific Board of Amway, XORT Therapeutics, and Rivermend Health and has patent and patent applications related to lowering uric acid or blocking fructose metabolism in the treatment of hypertension and metabolic disorders. B.R.-I., H.P., and Y.Q. declared no conflict of interest.
ACKNOWLEDGMENTS
The work in the authors’ laboratories is supported by funding from grants from FONACYT, Venezuela (FC-2005000283 to B.R.-I.) and the National Heart Lung and Blood Institute, National Institutes of Health, United States (HL-68607 to R.J.J.).
REFERENCES
- 1. Metchnikoff E. Immunity in Infective Diseases. Translated from the French by Binnie Francis G. Cambridge University Press: London, 1905, p. 10. [Google Scholar]
- 2. Svendsen UG. The importance of thymus in the pathogenesis of the chronic phase of hypertension in mice following partial infarction of the kidney. Acta Pathol Microbiol Scand (A) 1977; 85:539–547. [DOI] [PubMed] [Google Scholar]
- 3. Svendsen UG. Evidence for an initial, thymus independent and a chronic, thymus dependent phase of DOCA and salt hypertension in mice. Acta Pathol Microbiol Scand 1976; 84:523–528. [DOI] [PubMed] [Google Scholar]
- 4. Bataillard A, Freiche JC, Vincent M, Touraine JL, Sassard J. Effects of neonatal thymectomy on blood pressure and immunological characteristics of genetically hypertensive rats of the Lyon strain. J Hypertens 1986; 4:5445–5447. [Google Scholar]
- 5. Chae CU, Lee RT, Rifai N, Ridker PM. Blood pressure and inflammation in apparently healthy men. Hypertension 2001; 38:399–403. [DOI] [PubMed] [Google Scholar]
- 6. Engström G, Janzon L, Berglund G, Lind P, Stavenow L, Hedblad B, Lindgärde F. Blood pressure increase and incidence of hypertension in relation to inflammation-sensitive plasma proteins. Arterioscler Thromb Vasc Biol 2002; 22:2054–2058. [DOI] [PubMed] [Google Scholar]
- 7. Xia Y, Kellems RE. Angiotensin receptor agonistic autoantibodies and hypertension: preeclampsia and beyond. Circ Res 2013; 113:78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rodriguez-Iturbe B, Pons H, Quiroz Y, Lanaspa M, Johnson RJ. Autoimmunity in the pathogenesis of hypertension. Nature Rev Nephrol 2014; 10: 56–62. [DOI] [PubMed] [Google Scholar]
- 9. Rodriguez-Iturbe B, Vaziri ND, Herrera-Acosta J, Johnson RJ. Oxidative Stress, Renal infiltration of immune cells and salt-sensitive hypertension: all for one and one for all. Am J Physiol Renal Physiol 2004; 286: F606–F616. [DOI] [PubMed] [Google Scholar]
- 10. Ryan MJ. Immune mechanisms in hypertension. In Granger ND, Granger J, eds, Colloquium Series on Integrated System Physiology. Morgan & Claypool Life Sciences: San Rafael, CA, 2013, p. 33–44. [Google Scholar]
- 11. Herrada AA, Contreras FJ, Marini NP, Amador CA, Gonzalez PA, Cortes CM, Riedel CA, Carvajal CA, Figueroa F, Michea LF, Fardella CE, Kalergis AM. Aldosterone promotes autoimmune damage by enhancing Th17-mediated immunity. J Immunol 2010; 184:191–202. [DOI] [PubMed] [Google Scholar]
- 12. Rodriguez-Iturbe B, Quiroz Y, Nava M, Bonet L, Chavez M, Herrera-Acosta J, Johnson RJ, Pons HA. Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. Am J Physiol Renal Physiol 2002; 282:F191–F201. [DOI] [PubMed] [Google Scholar]
- 13. De Miguel C, Das S, Lund H, Mattson DL. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 2010; 298:R1136–R1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F, Fiebeler A, Ju X, Finckenberg P, Theuer J, Viedt C, Kreuzer J, Heidecke H, Haller H, Zenke M, Luft FC. Immunosuppressive treatment protects against angiotensin ii-induced renal damage. Am J Pathol 2002; 161:1679–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boesen EI, Williams DL, Pollock JS, Pollock DM. Immunosuppression with mycophenolate mofetil attenuates the development of hypertension and albuminuria in deoxycorticosterone acetate-salt hypertensive rats. Clin Exp Pharmacol Physiol 2010; 37:1016–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rodríguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincón J, Chávez M, Parra G, Herrera-Acosta J, Gomez-Garre D, Largo R, Egido J, Johnson RJ. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int 2001; 59:2222–2232. [DOI] [PubMed] [Google Scholar]
- 17. Franco M, Martínez F, Quiroz Y, Galicia O, Bautista R, Johnson RJ, Rodríguez-Iturbe B. Renal angiotensin II concentration and interstitial infiltration of immune cells are correlated with blood pressure levels in salt-sensitive hypertension. Am J Physiol Reg Integr Comp Physiol 2007; 293:R251–R256. [DOI] [PubMed] [Google Scholar]
- 18. Quiroz Y, Pons H, Gordon KL, Rincón J, Chávez M, Parra G, Herrera-Acosta J, Gómez-Garre D, Largo R, Egido J, Johnson RJ, Rodríguez-Iturbe B. Mycophenolate mofetil prevents the salt-sensitive hypertension resulting from short-term nitric oxide synthesis inhibition. Am J Physiol Renal Physiol 2001; 281:F38–F47. [DOI] [PubMed] [Google Scholar]
- 19. Venegas-Pont M, Sartori-Valinotti JC, Maric C, Racusen LC, Glover PH, McLemore GR, Jr, Jones AV, Reckelhoff JF, Ryan MJ. Rosiglitazone decreases blood pressure and renal injury in a female mouse model of systemic lupus erythematosus. Am J Physiol Regul Integr Comp Physiol 2009; 296:R1282–R1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, Karbach SH, Schwenk M, Yogev N, Schulz E, Oelze M, Grabbe S, Jonuleit H, Becker C, Daiber A, Waisman A, Münzel T. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 2011; 124:1370–1381. [DOI] [PubMed] [Google Scholar]
- 21. De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler Thromb Vasc Biol 2005; 25:2106–2113. [DOI] [PubMed] [Google Scholar]
- 22. Chan CT, Moore JP, Budzyn K, Guida E, Diep H, Vinh A, Jones ES, Widdop RE, Armitage JA, Sakkal S, Ricardo SD, Sobey CG, Drummond GR. Reversal of vascular macrophage accumulation and hypertension by a CCR2 antagonist in deoxycorticosterone/salt-treated mice. Hypertension 2012; 60:1207–1212. [DOI] [PubMed] [Google Scholar]
- 23. Elmarakby AA, Quigley JE, Olearczyk JJ, Sridhar A, Cook AK, Inscho EW, Pollock DM, Imig JD. Chemokine receptor 2b inhibition provides renal protection in angiotensin II–salt hypertension. Hypertension 2007; 50:1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pires PW, Girgla SS, McClain JL, Kaminski NE, van Rooijen N, Dorrance AM. Improvement in middle cerebral artery structure and endothelial function in stroke-prone spontaneously hypertensive rats after macrophage depletion. Microcirculation 2013; 20:650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zandbergen HR, Sharma UC, Gupta S, Verjans JW, van den Borne S, Pokharel S, van Brakel T, Duijvestijn A, van Rooijen N, Maessen JG, Reutelingsperger C, Pinto YM, Narula J, Hofstra L. Macrophage depletion in hypertensive rats accelerates development of cardiomyopathy. J Cardiovasc Pharmacol Ther 2009; 14:68–75. [DOI] [PubMed] [Google Scholar]
- 26. Titze J, Matchnik A. Sodium sensing in the interstitium and relationship to hypertension. Curr Opin Nephrol Hypertens 2010; 19:385–392. [DOI] [PubMed] [Google Scholar]
- 27. Wiig H, Schröder A, Neuhofer W, Jantsch J, Kopp C, Karlsen TV, Boschmann M, Goss J, Bry M, Rakova N, Dahlmann A, Brenner S, Tenstad O, Nurmi H, Mervaala E, Wagner H, Beck FX, Müller DN, Kerjaschki D, Luft FC, Harrison DG, Alitalo K, Titze J. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J Clin Invest 2013; 23:2803–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kossmann S, Schwenk M, Hausding M, Karbach SH, Schmidgen MI, Brandt M, Knorr M, Hu H, Kröller-Schön S, Schönfelder T, Grabbe S, Oelze M, Daiber A, Münzel T, Becker C, Wenzel P. Angiotensin II-induced vascular dysfunction depends on interferon-γ-driven immune cell recruitment and mutual activation of monocytes and NK-cells. Arterioscler Thromb Vasc Biol 2013; 33:1313–1319. [DOI] [PubMed] [Google Scholar]
- 29. Taherzadeh Z1, VanBavel E, de Vos J, Matlung HL, van Montfrans G, Brewster LM, Seghers L, Quax PH, Bakker EN. Strain-dependent susceptibility for hypertension in mice resides in the natural killer gene complex. Am J Physiol Heart Circ Physiol 2010; 298:H1273–H1282. [DOI] [PubMed] [Google Scholar]
- 30. Schroder K, Tschopp J. The inflammasomes. Cell 2010; 140:821–832. [DOI] [PubMed] [Google Scholar]
- 31. Taguchi A, Sanada M, Suei Y, Ohtsuka M, Lee K, Tanimoto K, Tsuda M, Ohama K, Yoshizumi M, Higashi Y. Tooth loss is associated with an increased risk of hypertension in postmenopausal women. Hypertension. 2004; 43:1297–1300. [DOI] [PubMed] [Google Scholar]
- 32. Bomfim GF, Dos Santos RA, Oliveira MA, Giachini FR, Akamine EH, Tostes RC, Fortes ZB, Webb RC, Carvalho MH. Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin Sci (Lond) 2012; 122:535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sollinger D, Eißler R, Lorenz S, Strand S, Chmielewski S, Aoqui C, Schmaderer C, Bluyssen H, Zicha J, Witzke O, Scherer E, Lutz J, Heemann U, Baumann M. Damage-associated molecular pattern activated Toll-like receptor 4 signalling modulates blood pressure in L-NAME-induced hypertension. Cardiovasc Res 2014; 101:464–472. [DOI] [PubMed] [Google Scholar]
- 34. Marketou ME, Kontaraki JE, Zacharis EA, Kochiadakis GE, Giaouzaki A, Chlouverakis G, Vardas PE. TLR2 and TLR4 gene expression in peripheral monocytes in nondiabetic hypertensive patients: the effect of intensive blood pressure-lowering. J Clin Hypertens (Greenwich) 2012; 14:330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Omi T, Kumada M, Kamesaki T, Okuda H, Munkhtulga L, Yanagisawa Y, Utsumi N, Gotoh T, Hata A, Soma M, Umemura S, Ogihara T, Takahashi N, Tabara Y, Shimada K, Mano H, Kajii E, Miki T, Iwamoto S. An intronic variable number of tandem repeat polymorphisms of the cold-induced autoinflammatory syndrome 1 (CIAS1) gene modifies gene expression and is associated with essential hypertension. Eur J Hum Genet 2006; 14:1295–1305. [DOI] [PubMed] [Google Scholar]
- 36. Ji Y, Liu J, Wang Z, Liu N. Angiotensin II induces inflammatory response partly via toll-like receptor 4-dependent signaling pathway in vascular smooth muscle cells. Cell Physiol Biochem 2009; 23:265–276. [DOI] [PubMed] [Google Scholar]
- 37. Liu N, Liu J, Ji Y, Liu P. Toll-like receptor 4 signaling mediates inflammatory activation induced by C-reactive protein in vascular smooth muscle cells. Cell Physiol Biochem 2010; 25:467–476. [DOI] [PubMed] [Google Scholar]
- 38. Liu-Bryan R, Scott P, Sydlaske A, Rose DM, Terkeltaub R. Innate Immunity Conferred By Toll-Like Receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum 2005; 52:2936–2946. [DOI] [PubMed] [Google Scholar]
- 39. deGraaf R, Kloppenburg G, Titslaard PJ, Bruggerman CA, Stassen F. Human heat shock protein 60 stimulates smooth muscle cell proliferation through Toll-like receptors 2 and 4. Microbes Infect 2006; 8:1859–1865. [DOI] [PubMed] [Google Scholar]
- 40. Asea A, Rehli M, Kabingu F, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70. Role for toll-like receptor (TLR) 2 and TLR 4. J Biol Chem 2002:277: 15028–15034. [DOI] [PubMed] [Google Scholar]
- 41. Pockley AG, De Faire U, Kiessling R, Lemne C, Thulin T, Frostegård J. Circulating heat shock protein and heat shock protein antibody levels in established hypertension. J Hypertens 2002; 20:1815–1820. [DOI] [PubMed] [Google Scholar]
- 42. Parra G, Quiroz Y, Salazar J, Bravo Y, Pons H, Chavez M, Johnson Rj, Rodriguez-Iturbe B. Experimental induction of salt-sensitive hypertension is associated with lymphocyte proliferative response to HSP70. Kidney Int 2008; 74:S55–S59. [DOI] [PubMed] [Google Scholar]
- 43. Pons H, Ferrebuz A, Quiroz Y, Romero-Vasquez F, Parra G, Johnson RJ, Rodriguez-Iturbe B. Immune reactivity to heat shock protein 70 expressed in the kidney is cause of salt sensitive hypertension. Am. J. Physiol Renal Physiol 2013; 304:F289–F299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Davey Smith G, Lawlor DA, Harbord R, Timpson N, Rumley A, Lowe GD, Day IN, Ebrahim S. Association of C-reactive protein with blood pressure and hypertension. Life course confounding and Mendelian randomization tests of causality. Arterioscler Thromb Vasc Biol 2005; 25:1051–1056. [DOI] [PubMed] [Google Scholar]
- 45. Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. J Am Med Assoc 2008; 300:924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thomson JA, Webb RC. Potential role of Toll-like receptors in programming of vascular dysfunction. Clin Sci (Lond) 2013; 125:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, Weyand CM, Harrison DG, Guzik TJ. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation 2010; 122:2529–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shao J, Nangaku M, Miyata T, Inagi R, Yamada K, Kurokawa K, Fujita T. Imbalance of T-cell subsets in angiotensin II-infused hypertensive rats with kidney injury. Hypertension 2003; 42:31–38. [DOI] [PubMed] [Google Scholar]
- 49. Seaberg EC, Munoz A, Lu M, Detels R, Margolick JB, Riddler SA, Williams CM, Phair JP. Association between highly active antiretroviral therapy and hypertension in a large cohort of men followed from 1984 to 2003. AIDS 2005; 19:953–960. [DOI] [PubMed] [Google Scholar]
- 50. Herrera J, Ferrebuz A, MacGregor EG, Rodriguez-Iturbe B. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol 2006; 17:S218–S225. [DOI] [PubMed] [Google Scholar]
- 51. Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens 2005; 19:149–154. [DOI] [PubMed] [Google Scholar]
- 52. Chamarthi B, Williams GH, Ricchiuti V, Srikumar N, Hopkins PN, Luther JM, Jeunemaitre X, Thomas A. Inflammation and hypertension: the interplay of interleukin-6, dietary sodium, and the renin-angiotensin system in humans. Am J Hypertens 2011; 24:1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sheu WHH, Lee WJ, Chang RL, Chen YT. Plasma tumor necrosis factor alpha levels and insulin sensitivity in hypertensive subjects. Clin Exp Hypertens 2000; 22:595–606. [DOI] [PubMed] [Google Scholar]
- 54. Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 2010; 55:500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Youn J-C, Yu HT, Lim BJ, Koh MJ, Lee J, Chang D-Y, Choi YS, Lee S-H, Kang S-M, Jang Y, Yoo OJ, Shin E-C, Park S. Immunosensescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension 2013; 62:126–133. [DOI] [PubMed] [Google Scholar]
- 56. Madhur MS, Harrison DJ. Senescent T cells and hypertension: new ideas about old cells. Hypertension 2013; 126:13–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, Paradis P, Schiffrin EL. T Regulatory Lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension 2011; 57:469–476. [DOI] [PubMed] [Google Scholar]
- 58. Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A, Neves MF, Laurant P, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension 2012; 59:324–330. [DOI] [PubMed] [Google Scholar]
- 59. Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, Rahn HP, Plehm R, Wellner M, Elitok S, Gratze P, Dechend R, Luft FC, Muller DN. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation 2009; 119:2904–2912. [DOI] [PubMed] [Google Scholar]
- 60. Viel EC, Lemarié CA, Benkirane K, Paradis P, Schiffrin EL. Immune regulation and vascular inflammation in genetic hypertension. Am J Physiol Heart Circ Physiol 2010; 298:H938–H944. [DOI] [PubMed] [Google Scholar]
- 61. Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jüpner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest 1999; 103:945–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Siddiqui AH, Irani RA, Blackwell SC, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibody is highly prevalent in preeclampsia: correlation with disease severity. Hypertension 2010; 55:386–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Riemekasten G, Philippe A, Näther M, Slowinski T, Müller DN, Heidecke H, Matucci-Cerinic M, Czirják L, Lukitsch I, Becker M, Kill A, van Laar JM, Catar R, Luft FC, Burmester GR, Hegner B, Dragun D. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann Rheum Dis 2011; 70:530–536 [DOI] [PubMed] [Google Scholar]
- 64. Fu ML, Herlitz H, Schulze W, Wallukat G, Micke P, Eftekhari P, Sjögren KG, Hjalmarson A, Müller-Esterl W, Hoebeke J. Autoantibodies against the angiotensin receptor (AT1) in patients with hypertension. J Hypertens 2000; 18:945–953. [DOI] [PubMed] [Google Scholar]
- 65. Wei F, Jia XJ, Yu SQ, Gu Y, Wang L, Guo XM, Wang M, Zhu F, Cheng X, Wei YM, Zhou ZH, Fu M, Liao YH; SOT-AT1 Study Group. Candesartan versus imidapril in hypertension: a randomised study to assess effects of anti-AT1 receptor autoantibodies. Heart 2011; 97:479–484. [DOI] [PubMed] [Google Scholar]
- 66. Dragun D, Muller DN, Brasen JH, Fritsche L, Nieminen-Kelha M, Dechend R, Kintscher U, Rudolph B, Hoebeke J, Eckert D, Mazak I, Plehm R, Schonemann C, Unger T, Budde K, Neumayer HH, Luft FC, Wallukat G. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N Engl J Med 2005; 352:558–569. [DOI] [PubMed] [Google Scholar]
- 67. Rossitto G, Regolisti G, Rossi E, Negro A, Nicoli D, Casali B, Toniato A, Caroccia B, Seccia TM, Walther T, Rossi GP. Elevation of angiotensin-II type-1-receptor autoantibodiestiter in primary aldosteronism as a result of aldosterone-producing adenoma. Hypertension 2013; 61:526–533. [DOI] [PubMed] [Google Scholar]
- 68. Fu ML, Herlitz H, Wallukat G, Hilme E, Hedner T, Hoebeke J, Hjalmarson A. Functional autoimmune epitope on alpha 1-adrenergic receptors in patients with malignant hypertension. Lancet 1994; 344:1660–1663. [DOI] [PubMed] [Google Scholar]
- 69. Liao YH, Wei YM, Wang M, Wang ZH, Yuan HT, Cheng LX. Autoantibodies against AT1-receptor and alpha1-adrenergic receptor in patients with hypertension. Hypertens Res 2002; 25:641–646. [DOI] [PubMed] [Google Scholar]
- 70. Wenzel K, Haase H, Wallukat G, Derer W, Bartel S, Homuth V, Herse F, Hubner N, Schulz H, Janczikowski M, Lindschau C, Schroeder C, Verlohren S, Morano I, Muller DN, Luft FC, Dietz R, Dechend R, Karczewski P. Potential relevance of alpha(1)-adrenergic receptor autoantibodies in refractory hypertension. PLoS One 2008; 3:e3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jahns R, Boivin V, Hein L, Triebel S, Angermann CE, Ertl G, Lohse MJ. Direct evidence for a beta 1-adrenergic receptor-directed autoimmune attack as a cause of idiopathic dilated cardiomyopathy. J Clin Invest 2004; 113:1419–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhou ZH, Wang J, Xiao H, Chen ZJ, Wang M, Cheng X, Liao YH. A novel autoantibody in patients with primary hypertension: antibody against L-type Ca2+ channel. Chin Med J (Engl) 2008; 121:1513–1517. [PubMed] [Google Scholar]
- 73. Wu T, Ma J, Chen S, Sun Y, Xiao C, Gao Y, Wang R, Poudrier J, Dargis M, Currie RW, Tanguay RM. Association of plasma antibodies against the inducible Hsp70 with hypertension and harsh working conditions. Cell Stress Chaperones 2001; 6:394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Papadopoulos DP, Makris TK, Krespi P, Papazachou U, Stavroulakis G, Hatzizacharias A, Votteas V. Antiendothelial cell antibody levels in healthy normotensives with high normal blood pressure. Clin Exp Hypertens 2006; 28:663–667. [DOI] [PubMed] [Google Scholar]
- 75. Frostegård J, Wu R, Gillis-Haegerstrand C, Lemne C, de Faire U. Antibodies to endothelial cells in borderline hypertension. Circulation 1998; 98:1092–1098. [DOI] [PubMed] [Google Scholar]
- 76. Rappaport VJ, Hirata G, Yap HK, Jordan SC. Anti-vascular endothelial cell antibodies in severe preeclampsia. Am J Obstet Gynecol 1990; 162:138–146. [DOI] [PubMed] [Google Scholar]
- 77. Hughson MD, Gobe GC, Hoy WE, Manning RD, Jr, Douglas-Denton R, Bertram JF. Associations of glomerular number and birth weight with clinicopathological features of African Americans and whites. Am J Kidney Dis 2008; 52:18–28. [DOI] [PubMed] [Google Scholar]
- 78. Youn JC, Yu HT, Lim BJ, Koh MJ, Lee J, Chang DY, Choi YS, Lee SH, Kang SM, Jang Y, Yoo OJ, Shin EC, Park S. Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension 2013; 62:126–133. [DOI] [PubMed] [Google Scholar]
- 79. White FN, Grollman A. Autoimmune factors associated with infarction of the kidney. Nephron 1964; 1:93–102. [DOI] [PubMed] [Google Scholar]
- 80. Olsen F. Transfer of arterial hypertension by splenic cells from DOCA-salt hypertensive and renal hypertensive rats to normotensive recipients. Acta Pathol Microbiol Immunol Scand 1980; 88:1–5. [DOI] [PubMed] [Google Scholar]
- 81. Svendsen UG. Spontaneous hypertension and hypertensive vascular disease in the NZB strain of mice. Acta Pathol Microbiol Scand (A) 1977; 85:548–554. [DOI] [PubMed] [Google Scholar]
- 82. Khraibi AA, Normal RA, Dzielak DJ. Chronic immunosuppression attenuates hypertension in Okamoto spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 1984; 247:H722––H726. [DOI] [PubMed] [Google Scholar]
- 83. Mervaala E, Müller DN, Park JK, Dechend R, Schmidt F, Fiebeler A, Bieringer M, Breu V, Ganten D, Haller H, Luft FC. Cyclosporin A protects against angiotensin II-induced end-organ damage in double transgenic rats harboring human renin and angiotensinogen genes. Hypertension 2000; 35:360–366. [DOI] [PubMed] [Google Scholar]
- 84. Bravo Y, Quiroz Y, Ferrebuz A, Vaziri ND, Rodriguez-Iturbe B. Mycophenolate mofetil administration reduces renal inflammation, oxidative stress and arterial pressure in rats with lead-induced hypertension. Am J Physiol Renal Physiol 2007; 293:F616–F623. [DOI] [PubMed] [Google Scholar]
- 85. Alvarez V, Quiroz Y, Nava M, Pons H, Rodríguez-Iturbe B. Overload proteinuria is followed by salt-sensitive hypertension caused by renal infiltration of immune cells. Am J Physiol Renal Physiol 2002; 283:F1132–F1141. [DOI] [PubMed] [Google Scholar]
- 86. Vanegas V, Ferrebuz A, Quiroz Y, Rodríguez-Iturbe B. Hypertension in page (cellophane wrapped) kidney is due to interstitial nephritis. Kidney Int 2005; 68:1161–1170. [DOI] [PubMed] [Google Scholar]
- 87. Stewart T, Jung FF, Manning J, Vehaskari VM. Kidney immune cell infiltration and oxidative stress contribute to prenatally programmed hypertension. Kidney Int 2005; 68:2180–2188. [DOI] [PubMed] [Google Scholar]
- 88. Rodríguez-Iturbe B, Ferrebuz A, Vanegas V, Quiroz Y, Mezzano S, Vaziri ND. Early and sustained inhibition of nuclear factor kappa B prevents hypertension in spontaneously hypertensive rats. J Pharmacol Exp Ther 2005; 315:51–57. [DOI] [PubMed] [Google Scholar]
- 89. Muller DN, Dechend R, Mervaala EM, Park JK, Schmidt F, Fiebeler A, Theuer J, Breu V, Ganten D, Haller H, Luft FC. NF-kB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension 2000; 35:193–201. [DOI] [PubMed] [Google Scholar]
- 90. Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H. Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol 2013; 304:R407–R414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Rudemiller N, Lund H, Jacob HJ, Geurts AM, Mattson DL; PhysGen Knockout Program. CD247 modulates blood pressure by altering T-lymphocyte infiltration in the kidney. Hypertension 2014; 63:559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Rodriguez-Iturbe B, Quiroz Y, Ferrebuz A, Parra G, Vaziri ND. Evolution of renal interstitial inflammation and NF-kB activation in spontaneously hypertensive rats. Am J Nephrol 2004; 24:587–594. [DOI] [PubMed] [Google Scholar]
- 93. Rodriguez-Iturbe B, Franco M, Johnson RJ. Impaired pressure natriuresis is associated with interstitial inflammation in salt-sensitive hypertension. Curr Opin Nephrol Hypertens 2013; 22:37–44. [DOI] [PubMed] [Google Scholar]
- 94. Guyton AC, Coleman TG, Cowley AW, Jr, Scheel KW, Manning RD, Jr, Norman RA., Jr Arterial pressure regulation: overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med 1972; 52:584–594. [DOI] [PubMed] [Google Scholar]
- 95. Franco M, Tapia E, Bautista R, Pacheco U, Santamaria J, Quiroz Y, Johnson RJ, Rodriguez-Iturbe B. Impaired pressure natriuresis resulting in salt-sensitive hypertension is caused by tubulointerstitial immune cell infiltration in the kidney. Am J Physiol Renal Physiol 2013; 304:F982–F990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol 2010; 298:R1089–R1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Navar LG, Satou R, Gonzalez-Villalobos RA. The increasing complexity of the intratubular renin-angiotensin system. J Am Soc Nephrol 2012; 23:1130–1132. [DOI] [PubMed] [Google Scholar]
- 98. Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, Weyand C, Harrison DG. Regulation of T-cell function by endogenously produced angiotensin II. Am J Physiol Regul Integr Comp Physiol 2009; 296:R208–R216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Das S, Mattson DL. Exogenous L-arginine attenuates the effects of angiotensin II on renal hemodynamics and the pressure natriuresis-diuresis relationship. Clin Exp Pharmacol Physiol 2014; 41:270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Crowley SD, Song YS, Sprung G, Griffiths R, Sparks M, Yan M, Burchette JL, Howell DN, Lin EE, Okeiyi B, Stegbauer J, Yang Y, Tharaux PL, Ruiz P. A role for angiotensin II type 1 receptors on bone marrow-derived cells in the pathogenesis of angiotensin II dependent hypertension. Hypertension 2010; 55:99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of T cell in the genesis of angiotensin II-induced hypertension and vascular dysfunction. J Exp Med 2007; 204:2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ, Harrison DG. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ Res 2010; 107:263–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lob HE, Vinh A, Li L, Blinder Y, Offermanns S, Harrison DG. Role of vascular extracellular superoxide dismutase in hypertension. Hypertension 2011; 58:232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ebrahimian T, Li MW, Lemarié CA, Simeone SM, Pagano PJ, Gaestel M, Paradis P, Wassmann S, Schiffrin EL. Mitogen-activated protein kinase-activated protein kinase 2 in angiotensin II-induced inflammation and hypertension: regulation of oxidative stress. Hypertension 2011; 57:245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Loria AS, Pollock DM, Pollock JS. Early life stress sensitizes rats to angiotensin II-induced hypertension and vascular inflammation in adult life. Hypertension 2010; 55:494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Crosswhite P, Sun Z. Ribonucleic acid interference knockdown of interleukin 6 attenuates cold-induced hypertension. Hypertension 2010; 55:1484–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lob HE, Vinh A, Li L, Blinder Y, Offermanns S, Harrison DG. Role of vascular extracellular superoxide dismutase in hypertension. Hypertension 2011; 58:232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Skarphendinsson JO, Elam M, Jungersten L, Wallin BG. Sympathetic nerve traffic correlates with the release of nitric oxide in humans: implications for blood pressure control. J Physiol 1997; 501:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Elenkov IJ, Wilder RlI, Chrousos GP, Vizi S. The sympathetic nerve—an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev 2000; 52:595–638. [PubMed] [Google Scholar]
- 110. Malpas SC. Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiol Rev 2010; 90:513–557. [DOI] [PubMed] [Google Scholar]
- 111. Grisanti LA, Woster AP, Dahlman J, Sauter ER, Combs CK, Porter JE. α1-Adrenergic receptors positively regulate Toll-like receptor cytokine production from human monocytes and macrophages. J Pharmacol Exp Ther 2011; 338:648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Harwani SC, Chapleau MW, Legge KL, Ballas ZK, Abboud FM. Neurohormonal modulation of the innate immune system is proinflammatory in the prehypertensive spontaneously hypertensive rat, a genetic model of essential hypertension. Circ Res 2012; 111:1190–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Li DJ, Evans RG, Yang ZW, Song SW, Wang P, Ma XJ, Liu C, Xi T, Su DF, Shen FM. Dysfunction of the cholinergic anti-inflammatory pathway mediates organ damage in hypertension. Hypertension 2011; 57:298–307. [DOI] [PubMed] [Google Scholar]
- 114. Lob HE, Marvar PJ, Guzik TJ, Sharma S, McCann LA, Weyand C, Gordon FJ, Harrison DG. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension 2010; 55:277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Lob HE, Schultz D, Marvar PJ, Davisson RL, Harrison DG. Role of the NADPH oxidases in the subfornical organ in angiotensin II-induced hypertension. Hypertension 2013; 61:382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Hilzendeger AM, Cassell MD, Davis DR, Stauss HM, Mark AL, Grobe JL, Sigmund CD. Angiotensin type 1a receptors in the subfornical organ are required for deoxycorticosterone acetate-salt hypertension. Hypertension 2013; 61:716–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Sriramula S, Cardinale JP, Francis J. Inhibition of TNF in the brain reverses alterations in RAS components and attenuates angiotensin II-induced hypertension. PLoS One 2013; 8:e63847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wu KLH, Chan SHH, Chan JYH. Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. J Neuroinflamm 2012; 9:212–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Gouraud SS, Waki H, Bhuiyan ME, Takagishi M, Cui H, Kohsaka A, Paton JF, Maeda M. Down-regulation of chemokine Ccl5 gene expression in the NTS of SHR may be pro-hypertensive. J Hypertens 2011; 29:732–740. [DOI] [PubMed] [Google Scholar]