

Summary
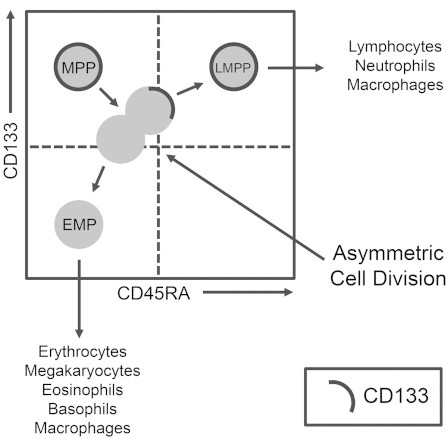
Hematopoietic stem and progenitor cells (HSPCs) can self-renew and create committed progenitors, a process supposed to involve asymmetric cell divisions (ACDs). Previously, we had linked the kinetics of CD133 expression with ACDs but failed to detect asymmetric segregation of classical CD133 epitopes on fixed, mitotic HSPCs. Now, by using a novel anti-CD133 antibody (HC7), we confirmed the occurrence of asymmetric CD133 segregation on paraformaldehyde-fixed and living HSPCs. After showing that HC7 binding does not recognizably affect biological features of human HSPCs, we studied ACDs in different HSPC subtypes and determined the developmental potential of arising daughter cells at the single-cell level. Approximately 70% of the HSPCs of the multipotent progenitor (MPP) fraction studied performed ACDs, and about 25% generated lymphoid-primed multipotent progenitor (LMPP) as wells as erythromyeloid progenitor (EMP) daughter cells. Since MPPs hardly created daughter cells maintaining MPP characteristics, our data suggest that under conventional culture conditions, ACDs are lineage instructive rather than self-renewing.
Graphical Abstract
Highlights
-
•
The HC7 anti-CD133 antibody allows analyses of ACDs on fixed human HSPCs
-
•
HC7 and AC133 anti-CD133 antibodies allow tracking of CD133 in living HSPCs
-
•
Cells of the MPP fraction divide asymmetrically to create LMPP- and EMP-like cells
-
•
ACDs of MPPs are lineage instructive rather than self-renewing
In this article, Giebel, Görgens, and colleagues demonstrate at the single-cell level that most human hematopoietic progenitor cells with multipotent potential (MPPs) perform non-self-renewing cell divisions in vitro. With their first in vitro cell division, MPPs lose their multipotency and create pairs of differently specified daughter cells, one a lymphomyeloid-committed CD133+ progenitor and the other an erythromyeloid-committed CD133− progenitor.
Introduction
Hematopoietic stem cells (HSCs) are defined as clonogenic cells that are able to self-renew and generate hematopoietic progenitor cells (HPCs) of all hematopoietic lineages. Triggered by the discovery of HSC niches (Calvi et al., 2003; Schofield, 1978; Zhang et al., 2003), the understanding of the mechanisms and molecules involved in cell-fate decisions of HSCs has increased considerably (Lévesque et al., 2010; Lymperi et al., 2010). Recently, experimental evidence has been provided that HSCs and distinct HPCs occupy different cellular niches: while lymphoid progenitors inhabit endosteal niches, murine HSCs reside in perivascular niches that specifically depend on mesenchymal stromal cells (MSCs) and endothelial cells (Ding and Morrison, 2013; Greenbaum et al., 2013). In addition to extrinsic factors provided by the environments of the different hematopoietic niches, hematopoietic stem and progenitor cells (HSPCs) contain the capability to divide asymmetrically, demonstrating that intrinsically controlled programs also participate in cell-fate specification processes (Giebel, 2008; Görgens and Giebel, 2010).
Evidence for the occurrence of asymmetric cell divisions (ACDs) during human early hematopoiesis was initially provided by the observation that ∼30% of dividing CD34+ or CD34+CD38low/− cells created daughter cells that followed different proliferation kinetics and adopted different cell fates (Brummendorf et al., 1998; Huang et al., 1999; Punzel et al., 2002). At a similar proportion, dividing CD133+CD34+ HSPCs were found to create CD133lowCD34+ cells (Beckmann et al., 2007). By studying the subcellular distribution of cell-surface antigens that are differentially expressed on CD133+CD34+ and CD133lowCD34+ cells, we previously identified four cell-surface antigens that segregate asymmetrically in 20%–30% of dividing HSPCs and confirmed the hypothesis that human HSPCs can divide asymmetrically (Beckmann et al., 2007). Recently, we comprehensively compared the developmental potential of human umbilical cord blood (UCB)-derived CD34+ cells that expressed either high CD133 (CD133+) or low/no CD133 (CD133−) levels on their cell surface. We demonstrated that CD133+CD34+ HSPCs can be subdivided by means of their CD45RA, CD38, and CD10 expression into different cell fractions, being enriched for multipotent progenitors (MPPs; CD133+CD34+CD38−CD45RA−CD10−), lymphoid-primed multipotent progenitors (LMPPs; CD133+CD34+CD38−CD45RA+CD10−), multilymphoid progenitors (MLPs; CD133+CD34+CD38−CD45RA+CD10+), or granulocyte-macrophage progenitors (GMPs; CD133+CD34+CD38+CD45RA+CD10−). The vast majority of CD133−CD34+ progenitors were found to belong to the erythromyeloid lineage whose common progenitors were determined to be erythromyeloid progenitors (EMPs; CD133−CD34+ CD38+CD45RA−CD10−) (Görgens et al., 2013b).
By studying the relationships of these subpopulations to each other, it was found that GMPs are able to create neutrophils but unexpectedly lack the potential to form eosinophils and basophils. Furthermore, and against the prevailing assumption, the GMPs were found to be derivatives of the same branch of hematopoiesis as the lymphocytes, pointing toward altered lineage relationships in human hematopoiesis (Görgens et al., 2013b). Accordingly, we recently proposed a revised model of human hematopoiesis (Görgens et al., 2013a, 2013b). Another outcome of this study was the observation that under the conditions used, MPPs cannot self-renew in vitro; following their first in vitro cell division, they apparently create CD133-positive LMPPs and CD133-negative EMPs, maybe by means of ACD (Görgens et al., 2013a, 2013b). Enforcing assumed roles of ACDs in this lineage-separation process, asymmetric segregation of CD133 molecules was observed in a proportion of dividing CD34+ cells at the intracellular level (Fonseca et al., 2008). In contrast, and independent of its intracellular distribution, the extracellular component of CD133 appeared to be symmetrically distributed on all dividing CD34+ cells (Beckmann et al., 2007; Fonseca et al., 2008).
In addition to the cell-fate analyses and ACD studies, we compared the distribution of CD133 at the subcellular level on freshly isolated and cultured HSPCs. Upon cultivation, HSPCs adopt a polarized morphology, forming a leading edge at the front and a leukocyte-specific structure, the uropod, at the rear (Giebel et al., 2004; Rajendran et al., 2009). While CD133 showed a rather random appearance on freshly isolated HSPCs, it redistributes to the uropod tips in cultured HSPCs (Giebel et al., 2004; Görgens et al., 2012). In our studies, we learned that the CD133 epitopes that are recognized by commonly used anti-CD133 antibodies (AC133 and AC141) are sensitive to paraformaldehyde fixation (Giebel et al., 2004). Both of these antibodies have been reported to recognize spatially distinct, glycosylation-dependent residues on the extracellular CD133 loops (Bidlingmaier et al., 2008; Miraglia et al., 1997). Recently, a novel monoclonal anti-CD133 antibody, the HC7 antibody, has been described, which specifically recognizes glycosylation-independent protein residues of both extracellular loops of CD133 (Swaminathan et al., 2010). This antibody had been successfully used in a variety of applications including western blot, immunofluorescence, flow cytometry, and immunohistochemistry using cancer cell lines (Swaminathan et al., 2010).
To test for the usefulness of the HC7 antibody in hematopoietic stem cell research, we comprehensively compared the presence and subcellular distribution of HC7 and glycosylation-dependent CD133 epitopes on freshly isolated and cultured UCB-derived CD133+CD34+ HSPCs. Furthermore, we tested whether binding of the HC7 or the AC133 antibody exerts any functional impact on the biology of human HSPCs, including their potential to home and engraft into immunodeficient nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice. After showing that neither AC133 nor HC7 antibodies recognizably alter biological features of human CD133+CD34+ cells and that both antibodies allow detection of living, asymmetrically dividing HSPCs, we compared the ACD rates in MPP-, LMPP-, and GMP-enriched fractions of HC7-stained cells and analyzed MPP daughter cells at a single-cell level, both phenotypically and functionally.
Results
Subcellular Distribution of HC7 and AC133 Epitopes on Polarized and Mitotic Human HSPCs
Recently, a novel anti-CD133 antibody has been described whose epitopes, unlike those of classical AC133 and AC141 anti-CD133 antibodies, were claimed to be insensitive to fixation (Swaminathan et al., 2010). Because of our interest in analyzing the subcellular distribution of CD133 on polarized and mitotic HSPCs, we decided to test for the usability of the HC7 antibody in human hematopoietic stem cell research.
At first, to analyze the specificity of the HC7 antibody, we compared the ability of the HC7 antibody and that of the commercially available AC133, AC141, and W6B3C1 anti-CD133 antibodies to bind to the ectopically expressed CD133 splice variants. To this end, we transduced cells of the human CD133− erythroleukemic cell line K562 with lentiviral vectors, encoding the human CD133 splice variant s1 or s2. Flow cytometric analyses revealed that none of the antibodies bound to nonmanipulated or enhanced GFP (EGFP)-expressing control K562 cells. In contrast, all antibodies bound to CD133-transduced K562 cells, regardless whether they expressed the s1 or the s2 EGFP variants of CD133 (Figure S1 available online). Noteworthy, all antibodies showed comparable staining intensities, which expectedly increased with higher EGFP intensities. Since we did not recognize differences in the staining intensity among the commercially available antibodies, we decided to focus on the comparison of HC7 and AC133 and did not include AC141 and W6B3C1 in our further analyses.
Next, we studied the subcellular distribution of HC7 and AC133 antigens on polarized as well as mitotic human HSPCs that were fixed in paraformaldehyde before staining. According to our previous finding that upon cultivation CD133+CD34+ cells adopt a migration phenotype and redistribute CD133 to the tip of the uropod (Giebel et al., 2004), we performed these analyses with CD34+ cells that had been cultured for 50–60 hr in the presence of early-acting cytokines. Regardless of whether we counterstained the HC7 or AC133 with Cy3- or Alexa 488-conjugated secondary antibodies, respectively, double-stained CD34+ cells revealed a high overlap among the HC7 and AC133 antigens, with the highest concentration of both antigens at the tips of the uropods (Figure 1A). Confirming our previous data, asymmetric distributions of AC133 epitopes were not observed in any of the dividing HSPCs studied. As described before, the AC133 epitopes were found to be concentrated in the region of the cleavage furrow or in later mitotic stages on midbodies, respectively (Beckmann et al., 2007). Remarkably, on mitotic HSPCs, the HC7 epitopes revealed a different distribution than the AC133 epitopes. On the vast majority of the mitotic HSPCs studied, HC7 epitopes covered areas of the cell surface of arising daughter cells that were negative for the AC133 staining. Strikingly, in all of five independent experiments, mitotic HSPCs were identified (27.5% ± 4.8%) displaying HC7 expression mainly in one of the arising daughter cells and thus revealing asymmetric HC7 epitope distributions (Figure 1B; Table S2). These findings implied that on mitotic HSPCs, extracellular HC7 epitopes either segregate differently than AC133 epitopes or the fixation conditions used cause artifacts, either affecting the distribution of AC133 or of HC7 epitopes.
Figure 1.
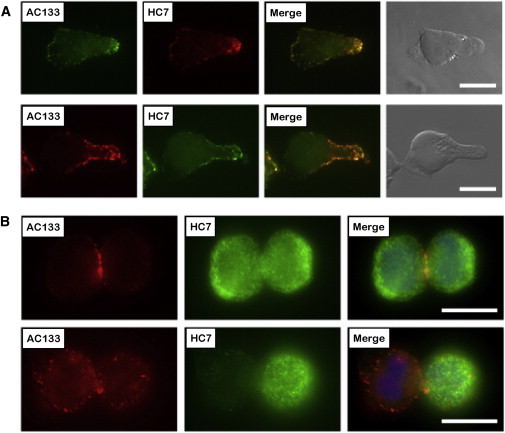
Subcellular Distribution Analyses of HC7, but Not AC133, Epitopes Allow Detection of Asymmetrically Dividing Cells within Paraformaldehyde-Fixed HSPC Samples
(A) Subcellular localization of AC133 and HC7 epitopes on 50–60 hr cultured, morphologically polarized CD133+CD34+ cells.
(B) Epitope localization on mitotic cells with symmetric (upper row) or asymmetric distribution (lower row) of the HC7 epitopes.
Scale bars, 10 μm. See also Figure S1 and Table S2 for a summary of observed frequencies of asymmetric versus symmetric HC7 epitope distributions.
HC7 and AC133 Label Identical Subpopulations of Human HSPCs
To exclude fixation artifacts, we compared the expression of AC133 and HC7 antigens on both freshly isolated human CD34+ cells and CD34+ cells that were cultured for 50–60 hr by flow cytometry. To discriminate distinct subpopulations, we costained the CD34+-enriched cells with anti-CD34, anti-CD38, and anti-CD45RA antibodies as well as with phycoerythrin (PE)-conjugated HC7 and allophycocyanin (APC)-conjugated AC133 antibodies.
The CD34+ cells showed slightly higher contrasts with the conjugated HC7-PE antibody than with the AC133-APC antibody. However, the general pattern and the percentages of all identifiable subpopulations are comparable to each other, and the direct comparison plots show that the more AC133 epitopes were expressed on given cells, the higher was their HC7 intensity, independently of whether freshly isolated or cultured CD34+ cells were analyzed (Figure 2A). To this end, we could not detect any differences in HC7 and AC133 epitope expression. Thus, the flow cytometric analyses did not support the notion that HC7 and AC133 epitopes might be expressed independently from each other.
Figure 2.
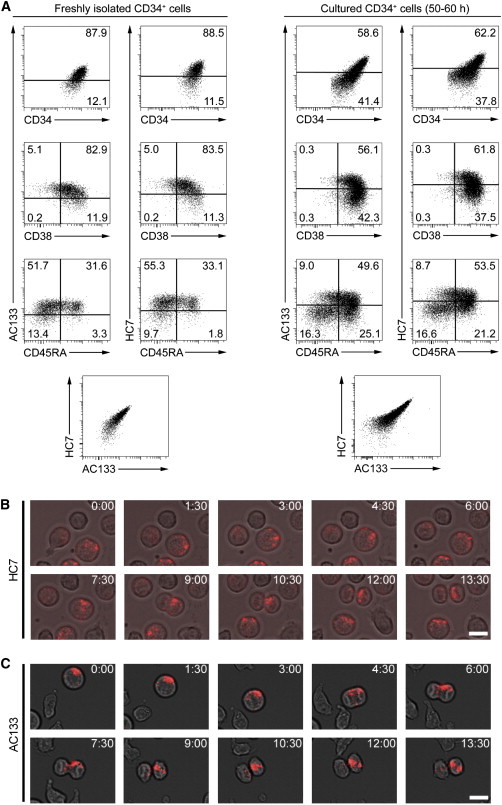
AC133 and HC7 Antibodies Allow Tracking of ACDs in Living HSPCs over Time
(A) AC133 and HC7 epitope expression on either freshly isolated CD34+ cells (left panel) or CD34+ cells that had been cultured for 50–60 hr in the presence of early-acting cytokines before (right panel). Cells were stained simultaneously with anti-AC133, anti-HC7, anti-CD34, anti-CD38, and anti-CD45RA antibodies. To analyze the expression of AC133 and HC7 epitopes on different HSPC subpopulations, obtained expression levels were plotted to those of CD34, CD38, and CD45RA and compared to each other.
(B) Time-lapse images (extracted from Movie S1) showing asymmetric segregation of HC7 epitopes of a mitotic CD133+CD34+ cell.
(C) Time-lapse images showing asymmetric segregation of AC133 epitopes of a mitotic CD133+CD34+ cell. Scale bars, 10 μm.
Both HC7 and AC133 Allow Detection of Living, Asymmetrically Dividing Human HSPCs
To test for the subcellular distribution of HC7 and AC133 epitopes on living HSPCs, we performed live-cell analyses of CD133+CD34+ cells. To this end, we stained cultured CD34+ cells with PE-conjugated HC7 or PE-conjugated AC133 antibodies and reseeded them. Stained cells were tracked for 5 hr by time-lapse microscopy, taking photos every 90 s. Related to the subcellular distribution on fixed HSPCs, both epitope types were found to be highly enriched at uropod tips of polarized cells (Figure S2). To analyze mitotic cells, 105 HC7- and 96 AC133-stained HSPCs were tracked over time. Similar to the fixed and HC7-stained mitotic HSPCs, 30.5% of the living, HC7-stained cells revealed asymmetric segregation or CD133 epitopes. Remarkably, and in contrast to the fixed-cell analyses, a similar amount (29.1%) of AC133-stained HSPCs showed asymmetric CD133 segregation as well (Figures 2B and 2C; Movie S1). Thus, we conclude that both HC7 and AC133 epitopes segregate asymmetrically on a proportion of dividing HSPCs and that in mitotic stages, fixation interferes with the detection of AC133 epitopes, but not with that of HC7 epitopes. Furthermore, and provided binding of the HC7 or the AC133 antibody does not affect any relevant biological process in HSPCs, both anti-CD133 antibodies can be considered as valuable tools for elaborated living-cell analyses within the early human hematopoietic compartment.
Binding of HC7 or AC133 Antibodies Does Not Affect the Cell Polarization, Migration, or Cell Adhesion Capabilities of Human HSPCs
A number of antibodies directed against cell-surface molecules (e.g., the cell adhesion molecules VLA-4, VLA-5, LFA-1, and CD44 or the phosphatase CD45) have been found to interfere with biological features of human HSPCs and leukemia-initiating cells, including their homing and engraftment and/or differentiation capabilities (Jin et al., 2006; Kollet et al., 2001; Shivtiel et al., 2011; Zanjani et al., 1999). Since homing is a multistep process that depends on cellular polarization, migration, and cell adhesion (Görgens et al., 2012; Lapidot et al., 2005), we investigated whether binding of HC7 or AC133 antibodies to HSPCs interferes with any of these processes.
To this end, we assessed the potential impact of HC7 and AC133 binding on the ability of HSPCs to adopt and maintain polarized morphologies. Following the experimental strategy we had used previously to dissect the polarization process of UCB-derived HSPCs (Görgens et al., 2012), we cultured freshly isolated CD34+ cells in the presence or absence of HC7, AC133, or isotype control antibodies and quantified the amount of polarized cells after 16 hr (Figure 3A) and 50–60 hr (Figure 3B). In a second setting, we added these antibodies to CD34+ cells that had been cultured for 50–60 hr before and assessed the amount of polarized cells 2 hr later (Figure 3C). In none of the experimental settings did we detect any functional impact of the HC7 or AC133 antibodies on the establishment or maintenance of the cell polarization process of HSPCs (Figures 3A–3C). Hence, neither HC7 nor AC133 antibodies affect the cellular polarization of human HSPCs.
Figure 3.
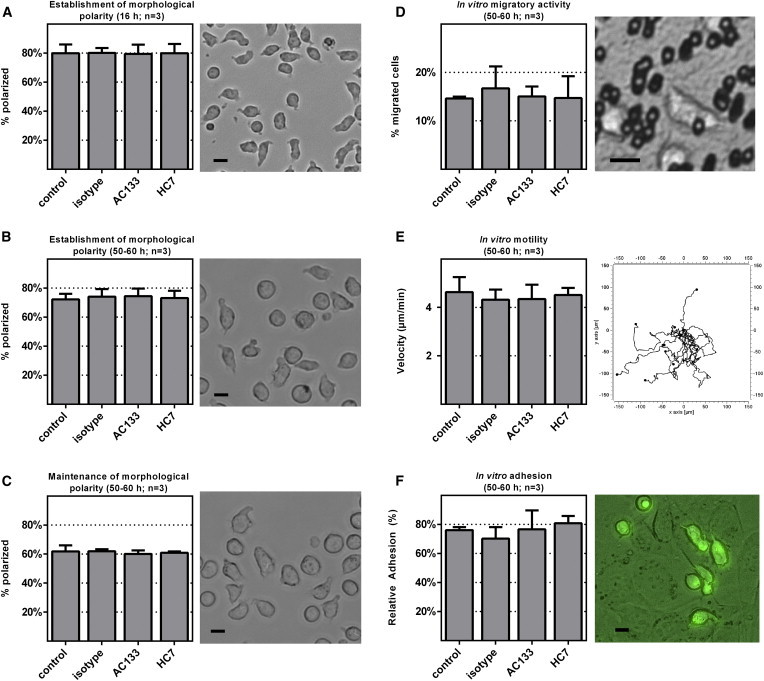
Binding of Neither AC133 nor HC7 Antibodies Recognizably Affects Cell Biological Properties of CD133+CD34+ Cells
The impact of AC133 and HC7 antibody binding on cell biological properties of cultured CD133+CD34+ cells was analyzed in different settings: on the establishment of morphological cell polarity (antibodies were added before cultivation) (A and B), on the maintenance of morphological cell polarity (antibodies were added following cultivation) (C), on their in vitro migratory activity measured in the transwell assay (D), on their in vitro motility measured by tracking their movements via time-lapse microscopy (E), and on their cell adhesion capabilities measured as the frequency of cells that tightly adhered to AFT024 stromal cells (F). In all settings, nontreated (control) or isotype-treated CD34+ cells (isotype) were used as controls. Pictures to the right of the bar graphs show images of cultured HSPCs at specified time points (A–C), on the microporous membrane (3 μm pore size) of a transwell insert (D), a center-corrected plot of the migration patterns of individually tracked HSPCs (E), and of PKH2-stained CD34+ cells that adhered to AFT024 stromal cells (F). n, number of independent experiments; arrow bars are given as SD of the mean; scale bars, 10 μm.
Next, the potential impact of HC7 and AC133 antibody binding on the in vitro migration capabilities of CD34+ cells was studied. In a first approach, we quantified HSPC migratory activity in transwell assays. In a second approach, the motility and velocity of individual HSPCs were determined by time-lapse microscopy. For the transwell experiments, CD34+ cells that had been cultured for 50–60 hr were transferred onto 3-μm-pore-sized transwell inserts, either nonlabeled or labeled with HC7, AC133, or isotype control antibodies. After, 12–14 hr, transmigration rates were recorded. We did not observe any difference in the ratio of transmigrated CD34+ cells in any of the settings (Figure 3D). For the quantification of the migratory velocity, individual CD34+ cells that had been cultured for 50–60 hr in the presence or absence of the corresponding antibodies were tracked by time-lapse imaging for 2 hr. Again, we did not detect any significant difference between the individual samples (Figure 3E). Thus, binding of HC7 and AC133 antibodies does not interfere with the in vitro migratory capabilities of UCB-derived CD34+ cells.
Finally, we compared the adhesion capabilities of non-anti-CD133-antibody-labeled and antibody-labeled CD34+ cells. To this end, we transferred nonlabeled and labeled PKH2-stained CD34+ cells that had been cultured for 50–60 hr into a cell adhesion assay performed on press-to-seal slides (Wagner et al., 2007). CD34+ cells were allowed to attach to preseeded AFT024 stromal cells for 60 min. After sealing and turning the slide upside down, and following incubation for 60 min, the percentage of CD34+ cells that were still attached to the stromal cells was quantified. Similar to the previous experiments, we did not detect any difference among the antibody-treated or control cells. Accordingly, the data indicate that binding of HC7 or AC133 antibodies does not interfere with the cell adhesion capabilities of CD34+ cells (Figure 3F).
The Binding of HC7 and AC133 Antibodies to Human HSPCs Does Not Affect Their CFC Capabilities
Next, we compared whether the HC7 or AC133 antibodies affect the development of HSPCs within the colony-forming cell (CFC) assay. We labeled 50–60 hr cultured CD34+ cells with the HC7 or AC133 anti-CD133 antibodies. Subsequently, fluorescent cell sorting was performed to separate CD133+CD34+ and CD133−CD34+ cells (Figure 4A) and transfer selected numbers of purified cells into the CFC assay (Figure 4B). Regardless of whether the CD34+ cells were labeled with the HC7 or AC133 antibody, the sorted cell fractions formed comparable colony numbers in all settings (Figure 4C). In good agreement with our previous studies (Görgens et al., 2013b), progenitors with colony-forming unit granulocyte-macrophage (CFU-GM) potentials were almost exclusively recovered within the CD133+CD34+ cell fractions, while progenitors revealing erythroid and colony-forming unit mixed (CFU-MIX) potentials were highly enriched within the CD133−CD34+ cell fractions (Figure 4D). Since no differences in the quality and quantity of colonies were observed in CFC assays of corresponding HC7- and AC133-labeled CD34+ cell fractions, we conclude that binding of these antibodies does not affect the colony-formation potential of UCB-derived HSPCs.
Figure 4.
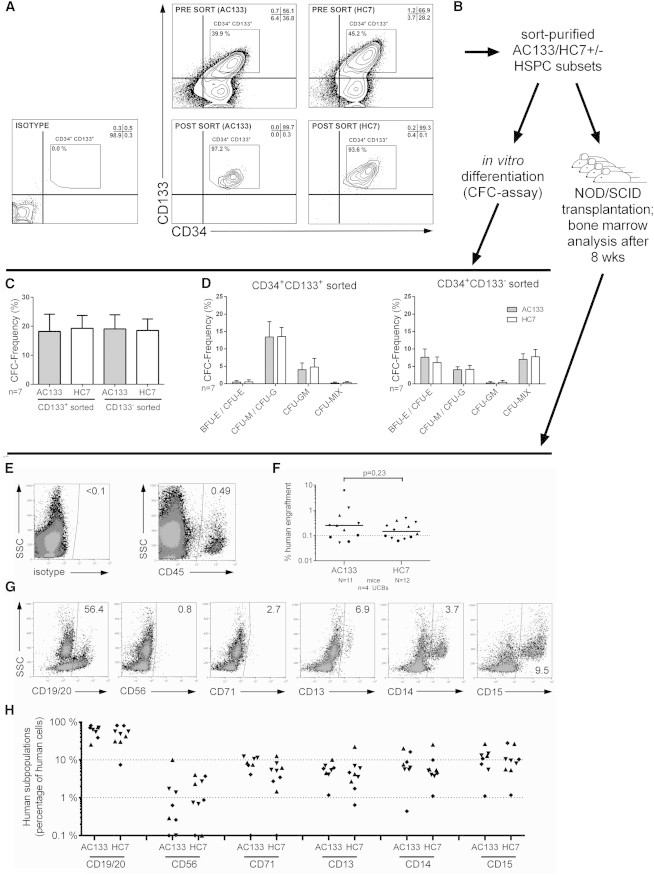
AC133- and HC7-Stained CD34+ Cells Reveal Comparable In Vitro and In Vivo Differentiation Potentials
(A) CD34+ cells cultured for 50–60 hr were stained with either AC133 or HC7 and anti-CD34 antibodies and flow cytometrically analyzed before (presort) or after fluorescent cell sorting of CD133+CD34+ cells (postsort).
(B) Experimental strategy for in vitro and in vivo analysis of sorted cell fractions using either AC133 or HC7 antibodies.
(C and D) Total CFC frequencies of AC133- and HC7-stained CD133+CD34+ and CD133−CD34+ cells (C) as well as the total frequencies of the obtained colony subtypes (D).
(E and F) CD34+ cells cultured for 50–60 hr were purified by fluorescent cell sorting according to the gating strategy shown in (A). Aliquots of 50,000 CD133+CD34+ cells were transplanted in four independent experiments to a total of 23 NOD/SCID mice. The total human cell engraftment was determined upon measuring the content of human CD45+ cells in the mice’s bone marrow 8 weeks after transplantation (E) and plotted in comparison to each other (F).
(G) The content of hematopoietic subpopulations within the human CD45+ cell fractions was determined by using markers specific for the detection of B cells (CD19/20), natural killer cells (CD56), erythroid cells (CD71), myeloid cells (CD13), monocytes/macrophages (CD14), and granulocytes (CD15).
(H) Obtained results were plotted in comparison to each other.
n, number of independent experiments; N, number of mice; arrow bars are given as SD of the mean.
HC7- and AC133-Stained HSPCs Reveal Comparable Engraftment Potentials
With the example of CD38, it had been demonstrated that antibody labeling might have profound inhibitory effects on the engraftment of human HSPCs in NOD/SCID mice (Taussig et al., 2008). To test whether binding of the HC7 antibody has any impact on the capacity of human HSPCs to home and engraft into the bone marrow of NOD/SCID mice, we compared multilineage engraftment potentials of HC7- and AC133-labeled human HSPCs. According to our previous findings that SCID-repopulating cells are exclusively contained within the CD133+CD34+ subpopulations (Görgens et al., 2013b), we purified CD133+CD34+ cells from UCB-derived CD34+ cells that had been cultured for 50–60 hr and were labeled with either the HC7 or AC133 antibody (Figures 4A and 4B).
NOD/SCID mice were transplanted with 5 × 104 cells of either sorted CD133+CD34+ cell population. After 8 weeks, human cell engraftment in the bone marrow of transplanted mice was quantified using anti-human CD45 antibodies (Figure 4E). We detected comparable engraftment rates in mice transplanted with HC7-labeled CD34+ cells and those with AC133-labeled CD34+ cells (Figure 4F). Multilineage analyses of the bone marrow and quantification of the number of human cells expressing lymphoid (CD19/20 and CD56) or erythromyeloid (CD71, CD13, CD14, and CD15) antigens did not show any quantitative differences between both experimental groups (Figures 4G and 4H). Thus, our data did not reveal any specific impact of the binding of HC7 or AC133 antibodies on the transplantation outcomes. In summary, our in vitro and in vivo data provide evidence that binding of HC7 and AC133 antibodies to human HSPCs does not alter any relevant biological features of labeled human HSPCs.
The Vast Majority of HSPCs of the MPP, but Not LMPP or GMP, Fraction Perform Asymmetric Cell Divisions In Vitro
Previously, we analyzed the developmental capacity of separated offspring of primitive human hematopoietic cells at a single-cell level. We showed that more than 80% of very primitive cells, those with myeloid-lymphoid initiating cell (ML-IC) potential, gave rise to daughter cells adopting different cell fates. In contrast, a higher proportion of committed progenitors created identically developing daughter cells (Giebel et al., 2006). The observation that only 20%–30% of CD133+CD34+ cells showed asymmetric segregation of CD53, CD62L, CD63, or CD71 (Beckmann et al., 2007) or of the CD133 epitopes (Table S2), respectively, implies different ACD rates among different HSPC subpopulations. To this end, we decided to determine the ACD rates of CD133+CD34+ cells belonging to the MPP, LMPP, or GMP subpopulation (Figure 5A). Due to the complexity of the envisioned experiments and the fact that the HC7, but not AC133, antibody recognizes asymmetrically segregating CD133 epitopes on fixed HSPCs, we decided to perform the following experiments with the HC7 antibody only.
Figure 5.
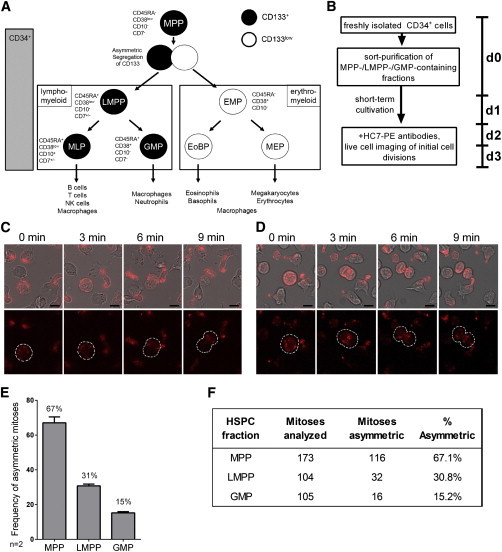
Most Mitotic HSPCs of the MPP-Enriched, but Not LMPP- and GMP-Enriched, Cell Fractions Reveal Asymmetric Segregation of HC7 Epitopes
(A) Recently, we proposed a revised version of the hematopoietic tree predicting different lineage relationships (Görgens et al., 2013b). Accordingly, MPPs can be enriched as CD133+CD34+CD45RA−CD38lowCD10−CD7−, LMPPs as CD133+CD34+CD45RA+CD38lowCD10−CD7+/−, and GMPs as CD133+CD34+CD45RA+CD38+CD10−CD7− cells.
(B) Experimentally, MPP-, LMPP-, and GMP-enriched fractions were sort purified from freshly isolated CD34+ cells (Figure S3) and cultured for 50–60 hr in the presence of early acting cytokines. After staining with PE-conjugated HC7 antibodies, cells were tracked via time-lapse microscopy (90 s time intervals) and discriminated regarding their HC7 epitope segregation behavior, either as asymmetrically or symmetrically dividing cells.
(C and D) Example of an asymmetrically dividing cell of the MPP fraction (C) and a symmetrically dividing cell of the GMP fraction (D).
(E) Comparison of the ACD rates within the MPP-, LMPP-, and GMP-enriched fractions obtained in two independent experiments.
(F) The embedded table summarizes the data.
n, number of independent experiments; arrow bars are given as SD of the mean. See also Figure S3.
To study ACD rates in MPPs, LMPPs, and EMPs, cells of all fractions were purified by fluorescent cell sorting (Figure S3) and cultured for 50–60 hr before they were labeled with anti-HC7-PE antibodies and reseeded for time-lapse microscopy (Figure 5B). Labeled cells were tracked for 5 hr, taking photos every 90 s (Figures 5C and 5D). A total of 67.1% ± 3.4% of the mitotic cells of the MPP fraction, 30.8% ± 1.1% of the LMPP fraction, and 15.2% ± 0.7% of the GMP fraction revealed asymmetric segregation patterns of HC7 epitopes. The remaining mitotic cells of the analyzed cell fractions revealed rather symmetrical segregation of HC7 epitopes (Figures 5E and 5F). Together, these findings confirm the long-lasting hypothesis that more primitive human hematopoietic cells display higher ACD rates in vitro than committed progenitors (Giebel et al., 2006).
Cells of the MPP Fraction Create LMPP- and EMP-like Daughter Cells
The observation that most cells of the MPP fraction divide asymmetrically, coupled to the finding that MPPs create LMPPs and EMPs in bulk cultures (Görgens et al., 2013a, 2013b), implies that ACDs control segregation of the lymphomyeloid and erythromyeloid lineages from MPPs by creating a pair of daughter cells, one inheriting LMPP and the other EMP potential (Figure 6A). To test this hypothesis, we decided to perform single-cell analysis of individual cells of the MPP fraction, i.e., of CD133+CD34+CD45RA−CD38low/− cells (Figure 6A). For setting up and validating the experimental strategy, we studied the population kinetics of bulk MPP cultures first. To this end, two independent experiments were performed with five repetitions each. A total of 100 or 10 cells of the MPP fractions were purified by fluorescent cell sorting and deposited into wells of a 96-well plate, respectively, and cultured for 6 days. After confirming the occurrence of cell proliferation in all given wells by light microscopy, the resulting progeny fractions were analyzed by flow cytometry. In good agreement with our previous data (Görgens et al., 2013b), cells reflecting the LMPP (CD133+CD34+CD45RA+) and EMP phenotype (CD133−CD34+CD45RA−) were found in all progeny fractions (Figure 6B), implying that reduction of the cell numbers does not affect the appearance of LMPP- and EMP-like daughter cells. Next, in two independent experiments, a total of 339 individual cells of the MPP fraction were deposited into individual wells of a 96-well plate, raised for 6 days, and analyzed by flow cytometry and, in selected cases, in CFC assays (Figure 6C). Following cultivation, 292 of them performed at least one cell division, creating up to 36 sibling cells (Figure 6D). A total of 95 randomly chosen wells with progeny fractions were analyzed by flow cytometry. As summarized in Figure 6E and exemplarily shown in Figure 6F, 23 (24.2%) fractions contained cells revealing phenotypic characteristics of both lymphomyeloid and erythromyeloid cells, 44 (46.3%) of lymphomyeloid cells, and 23 (24.2%) of erythromyeloid cells. None of the cells of the remaining 5 (5.3%) fractions showed lymphomyeloid or erythromyeloid characteristics.
Figure 6.
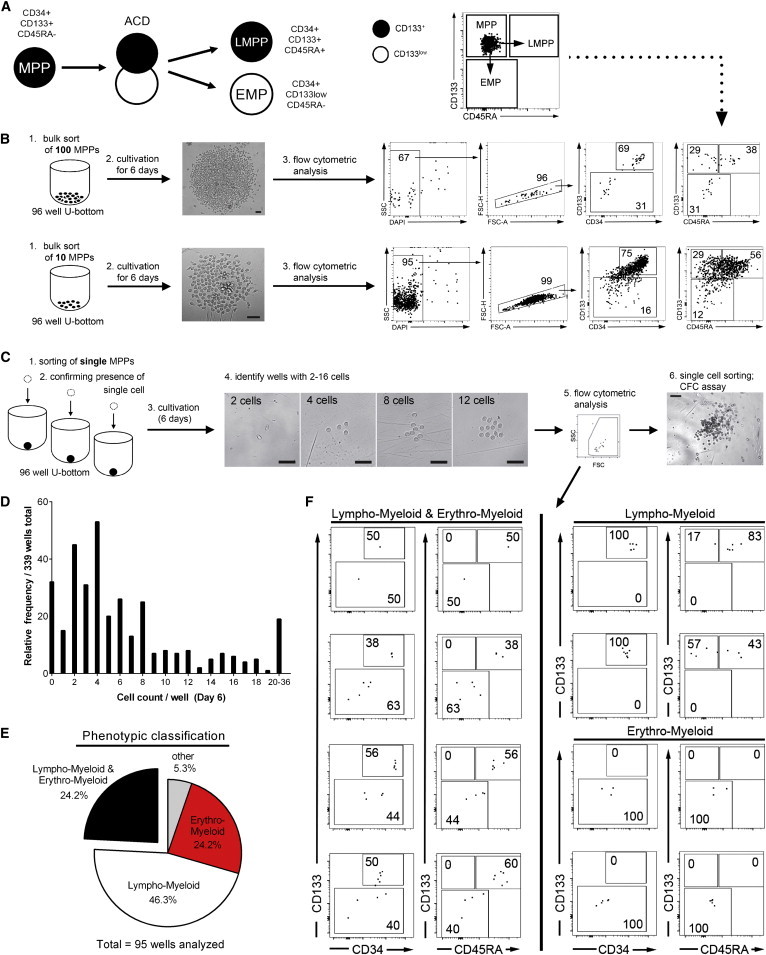
Cells of the MPP Fraction Create CD133+Lymphomyeloid as well as CD133low Erythromyeloid Daughter Cells
(A) Model proposing that MPPs (CD133+CD45RA−) divide asymmetrically to create lymphomyeloid (LMPP; CD133+CD45RA+) and erythromyeloid (EMP; CD133low) daughter cells. The dot plot of purified cells of the MPP fraction indicates the classification of MPPs, LMPPs, and EMPs according to their expression levels of CD133 and CD45RA, and arrows indicate proposed shifts of MPP daughter cells into either the LMPP or EMP fraction, respectively.
(B) Experimental strategy to analyze progeny fractions received from 100 (upper row) or 10 (lower row) sorted cells of MPP fractions, respectively. In two independent experiments, cells of the MPP fraction (Figure S3) were deposited by fluorescent cell sorting into individual wells of U-bottom 96-well plates, cultured for 6 days, and stained with anti-CD34, anti-CD133, and anti-CD45RA antibodies. Dead cells were excluded from the analyses via DAPI staining; doublets were discriminated by forward scatter area versus forward scatter height gating. Scale bars, 50 μm.
(C) Experimental strategy to analyze progeny fractions received from individually sorted cells of MPP fractions. Following deposition of single cells into individual wells of a 96 well U-bottom plates, the plating efficiency was determined by bright-field microscopy. Cells were cultured for 6 days. Arising progeny were enumerated by bright-field microscopy and analyzed like the progeny of the bulk cultures described in (C). Scale bars represent 50 μm (4) and 200 μm (6).
(D) Distribution reflecting the enumeration of the progeny fraction sizes of 339 individually deposited cells of MPP fractions.
(E) Distribution of the immunophenotypes of progeny fractions obtained from 95 individually deposited cells of MPP fractions.
(F) Representative examples of flow cytometric analyses of progeny fractions obtained from individually deposited cells of MPP fractions.
See also Figure S4.
To confirm the LMPP and EMP characteristics at a functional level, offspring of 12 MPP candidates creating lymphomyeloid and erythromyeloid siblings were sorted in parallel to their analysis as single cells in CFC assays. In 8 series out of 12, at least 1 lymphomyeloid and 1 erythromyeloid sibling formed a colony, either of the CFU-GM or the CFU-MIX/CFU-erythrocyte type, respectively (Figure S4). This demonstrates the existence of cells within the MPP fraction that indeed contain the dual capability to create LMPP- and EMP-like cells in vitro.
Cells of the MPP Fraction Create LMPP- and EMP-like Daughter Cells by Means of ACDs
To test whether ACDs control separation of LMPP and EMP lineages, we established a method to track initial mitoses of single cells of the MPP fraction, aiming to subsequently separate arising daughter cells and to analyze their differentiation potential in CFC assays (Figure 7A). In three independent experiments, a total of 288 single cells of the MPP fractions were deposited into individual wells of a 96-well plate. After confirming the single-cell deposition in 269 cases by light microscopy, cells were tracked for 16 hr (Figure 7A). In total, 100 mitoses were observed, which could clearly be scored on the bases of HC7 antibody reactivity as either symmetric (37%) or asymmetric (67%) (Figures 7B and S5; Movies S2 and S3). To separate the daughter cells for subsequent cell-fate analyses, the content of each of these wells were distributed in equal proportions to three wells of a 96-well plate. Light microscopic analyses confirmed that we managed to separate the daughter cells of 39 initially deposited cells that in terms of their HC7 epitope divided asymmetrically and of 25 cells that apparently performed symmetric cell divisions (SCDs; Table S3). Before CFC medium was added to the wells, fluorescent microscopy allowed us to distinguish wells containing CD133+ or CD133− siblings of ACDs, respectively. As summarized in Figures 7D and 7E, in 29 of the cases resulting from ACDs and in 15 resulting from SCDs, both daughter cells formed colonies. According to their appearance and objectivized by flow cytometric cell analyses (Figures 7C and S6), the prevailing colony combination following ACD was with 18 cases in which the CD133− sibling created a CFU-MIX colony and the CD133+ sibling a CFU-GM colony. In contrast, with 13 cases, the prevailing combination of siblings that were created by SCDs was one in which both siblings formed CFU-GM colonies (Figures 7D and 7E; Table S4). Of note, pairs of daughter cell with a CD133+ sibling creating a CFU-MIX colony and a CD133− sibling a CFU-GM colony were not observed in any of the experiments. These results support the initial hypothesis that under the conditions used, most MPPs divide asymmetrically to create LMPPs and EMPs. Thus, within the MPP compartment, ACDs promote lineage instruction rather than self-renewal of multipotent HSPCs.
Figure 7.
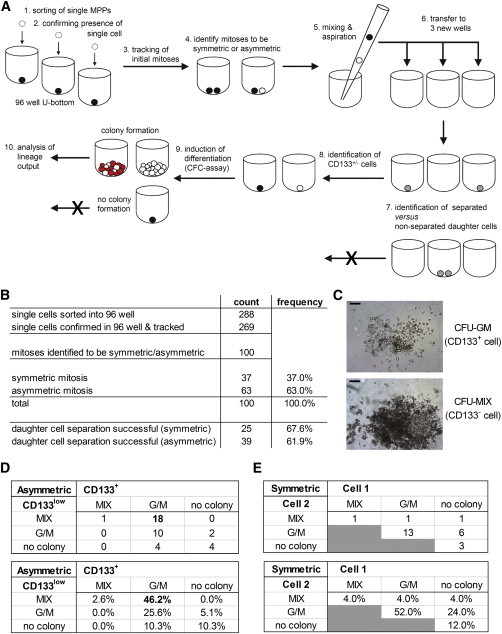
Large Proportions of Asymmetrically Dividing Cells of the MPP Fraction Create LMPP- and EMP-like Daughter Cells
(A) Experimental strategy for the separation and functional analysis of individual daughter cells arising from cells of the MPP fraction that were deposited as single, HC7-stained cells and tracked for 16 hr via time-lapse microscopy with 8–12 min time intervals. The content of the wells was analyzed by bright-field microscopy. Movies of the wells containing two daughter cells were analyzed for the occurrence of ACDs or SCDs. The whole media content of wells whose daughter cells clearly arose from an ACD or SCD were mixed and aspirated with a standard pipette to separate the daughter cells and distributed to three new wells. Successful separations were confirmed by bright-field microscopy. Siblings of ACD were discriminated as CD133+ and CD133− by fluorescence microscopy. Wells with successfully separated daughter cells were overlaid with CFC assay medium and cultured for 14 days. Arising CFC colonies were scored according to their phenotype and analyzed by flow cytometry (Figure S5) according to our previous description (Görgens et al., 2013b).
(B) Cumulative statistical overview of three independent separation experiments. In total, 39 or 25 cells of the MPP fraction performed confirmed ACDs or SCDs, respectively, and created daughter cells, both of which formed colonies (details are given in Table S3).
(C) Example of the prevailing colonies types that were formed (either a CD133+ daughter cell-derived CFU-GM colony or a CD133− daughter cell-derived CFU-MIX colony). Scale bars, 200 μm.
(D and E) Combined output of the colony assays of the siblings resulting from ACDs (D) or SCDs (E), respectively (details are given in Table S4).
See also Figures S5 and S6, Tables S3 and S4, and Movies S2 and S3.
Discussion
By using the HC7 anti-CD133 antibody, we demonstrate that almost all human HSPCs of the MPP fraction with confirmed lymphomyeloid and erythromyeloid developmental potential perform ACDs in vitro to create a lymphomyeloid CD34+CD133+CD45RA+ and a erythromyeloid CD34+CD133−CD45RA− daughter cell. In this context, we first comprehensively tested for the usability of the HC7 anti-CD133 antibody in human hematopoietic stem cell research. Side-by-side comparison of HC7 and the classical AC133 anti-CD133 antibody revealed that on fixed mitotic HSPCs, HC7, in contrast to AC133, allows detection of asymmetric CD133 distribution. Otherwise, no difference was detected regarding either their epitope expression on human HSPC subsets or their subcellular distribution on polarized or living mitotic HSPCs. Therefore, we conclude that the different appearance on fixed mitotic cells is an artifact caused by paraformaldehyde fixation rather than a difference in the HC7 and AC133 epitope distribution on HSPCs. Side-by-side comparison of HC7 or AC133 binding to human CD133+ HSPCs did not reveal a recognizable impact of any of these antibodies on the cell biological features of HSPCs, including their homing and repopulation capabilities in NOD/SCID mice. Thus, we consider the binding of both antibodies as functional neutral and conclude that they provide valuable tools for live-cell analyses of human CD133+ HSPCs.
Accordingly, we used the HC7 antibody to analyze the ACD behavior of defined CD133+ HSPC subpopulations. By tracking cells of the MPP fraction, almost 70% of them were found to perform ACDs in vitro, independent of whether they were tracked in bulk or single-cell cultures. In contrast, only 30% or 15% of the cells of the LMPP or GMP fractions performed ACDs, respectively. The high frequency of ACDs within the MPP fraction is in line with our previous observation that more than 80% of human HSPCs with long-term myeloid (LTC-IC) and long-term lymphoid potential (NK-IC), the ML-ICs, created daughter cells realizing different cell fates: while one daughter cell inherited the ML-IC capability, the other daughter cell appeared to be more restricted (Giebel et al., 2006). The functional data of the ML-IC assay were in good agreement with the assumption that a hallmark of many somatic stem cells is their ability to divide asymmetrically and that dysregulation of ACDs can result in tumor formation, as has been observed at the example larval neural stem cells of Drosophila, the neuroblasts, which regularly divide asymmetrically (Chang et al., 2012; Homem and Knoblich, 2012), and in different mammalian tumors, including oligodendrogliomas, mammary tumors, and leukemia (Cicalese et al., 2009; Sugiarto et al., 2011; Wu et al., 2007).
However, the observation of our functional and phenotypic single-cell analyses that most cells of the MPP fractions divide asymmetrically to create LMPP- and EMP-like cells, coupled to our previous observation that MPPs are lost over time (Görgens et al., 2013a, 2013b), suggests that ACDs within the MPP fraction instruct lineage diversification rather than control the self-renewal of MPPs. Even though this prediction might appear contradictory to our previous finding that HSPCs with ML-IC potential always create at least one daughter cell inheriting the ML-IC potential (Giebel et al., 2006), one has to consider that LMPPs, like MPPs, also contain LTC-IC and NK-IC potential (Görgens et al., 2013b). Consequently, a reduction of the MPP to the LMPP potential cannot be resolved within the ML-IC assay in its current form (Görgens et al., 2013a). Thus, it remains an open question whether human HSCs indeed contain the potential to perform self-renewing ACDs, as proposed before (Giebel et al., 2006), or if they exclusively control lineage diversification.
A very well-studied process in which ACDs exclusively control lineage diversification is the development of the external sensory organs of Drosophila, the fly’s bristles. Here, so-called sensory organ precursors repetitively divide asymmetrically to form the four different cells of a given external sensory organ (Gho et al., 1999). Unlike the situation in neuroblasts, mutations disturbing the asymmetry of occurring cell divisions result not in the expansion of the more primitive precursor cells but rather in cell-fate switches of one of the daughter cells into the other (Gho et al., 1999; Giebel and Wodarz, 2012).
Future studies might indicate whether ACDs within the early human hematopoietic compartment are exclusively related to those observed in the development of the external sensory organs of Drosophila or whether at least some HSPCs contain capabilities to perform neuroblast-like, self-renewing ACDs.
Experimental Procedures
Cell Preparation and Cell Culture
Human UCB was obtained from donors after informed consent according to the Declaration of Helsinki. The experimental usage of UCB samples was approved by the local ethics commission. Mononuclear cells (MNCs) were isolated from individual sources by Ficoll (Biocoll Separating Solution, Biochrom AG) density gradient centrifugation and highly enriched by magnetic cell separation as described previously (Giebel et al., 2004). If not used immediately, cells were cultured in a humidified atmosphere at 37°C and 5% CO2 at densities of 0.5–1 × 105 cells/ml in Iscove’s modified Dulbecco’s medium (Lonza) supplemented with 20% fetal bovine serum (Biochrom), 100 U/ml penicillin, and 100 U/ml streptomycin (Life Technologies) and with early-acting cytokines (FLT3L, stem cell factor, and thrombopoietin, each at 10 ng/ml final concentration [all PeproTech]).
Flow Cytometric Analyses and Cell Sorting
Antibodies used for flow cytometric analysis and sorting are listed in Table S1. For analyses of human graft compositions in mouse bone marrow, sample cells were labeled with antibodies against CD13, CD14, CD15, CD19, CD20, CD45, CD56, and CD71. Prior to the labeling, Fc receptors were blocked using purified rat anti-mouse CD16/CD32 monoclonal antibodies (Becton Dickinson [BD]).
For flow cytometric sorting, cultured CD34+ cells were labeled with PE-conjugated AC133 and anti-CD34 antibodies or with unconjugated HC7 antibody, counterstained with PE-conjugated goat-anti-mouse secondary antibodies (Dianova), saturated with mouse serum (Dianova), and subsequently stained with anti-CD34 antibodies. For flow cytometric sorting of MPPs, LMPPs, and GMPs, freshly isolated CD34+ cells were labeled with antibodies against CD34, CD133, CD38, CD45RA, CD10, and CD7 as described before (Görgens et al., 2013b).
Flow cytometric analyzes were performed on a FC500 flow cytometer (Beckman Coulter), a LSRII (BD), and a FACSAria I (BD). Dead cells were excluded by propidium iodide or DAPI staining. Isotype controls were used to set up gating strategies.
Cells were sort purified using a FACSAria I cell sorter (BD). The sort purity was routinely assessed by recovery of sorted cells and was >99.5%.
Functional In Vitro Differentiation Assays
For colony-forming cell (CFC) assays, 200 sorted cells were seeded into 1 ml MethoCult GF H4434 (STEMCELL Technologies); individual cells were immediately deposited into or for the individual daughter cell analyses overlaid with 200 μl MethoCult GF H4434, respectively. Hematopoietic colonies were scored after 12–14 days.
Statistics
Experimental results from independent experiments are reported as SD. Significance analyses and data assembly were performed with the paired, two-sided Student’s t test using Microsoft Excel and GraphPad Prism.
Author Contributions
B.G., A.G., and A-K.L. conceived the study and designed the experiments; B.G. and A.G. wrote the manuscript; A.G., A-K.L., and M.M. performed the experiments; B.G., A.G., and A-K.L. analyzed the data; A.G. and B.G. designed and assembled all figures; A.K., J.D., H.H., and P.A.H. contributed reagents, materials, and analysis tools; and all authors discussed the data and the manuscript.
Acknowledgments
The authors thank Stefan Radtke for general discussion and experimental support; Joachim Göthert, Philip Dammann, and Gero Hilken for their expertise and support regarding animal studies; and Ali Sak and Michael Gronenberg for their support in mouse irradiation. UCB samples were kindly provided by Angela Köninger and Rainer Kimmig (Department of Gynecology and Obstetrics, University Hospital Essen). The cDNA of the Prominin-1 splice variant s1 was kindly provided by Christiane Knobbe (Heinrich-Heine University Düsseldorf). The hybridoma clone 7 developed by Swaminathan et al. was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by Department of Biology, The University of Iowa (Iowa City, IA). This study was supported by grants from the Deutsche Forschungsgemeinschaft (GI 336/4-1 to B.G.) and the Stem Cell Network North Rhine Westphalia (to A.G.).
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Contributor Information
André Görgens, Email: andre.goergens@uk-essen.de.
Bernd Giebel, Email: bernd.giebel@uk-essen.de.
Supplemental Information
References
- Beckmann J., Scheitza S., Wernet P., Fischer J.C., Giebel B. Asymmetric cell division within the human hematopoietic stem and progenitor cell compartment: identification of asymmetrically segregating proteins. Blood. 2007;109:5494–5501. doi: 10.1182/blood-2006-11-055921. [DOI] [PubMed] [Google Scholar]
- Bidlingmaier S., Zhu X., Liu B. The utility and limitations of glycosylated human CD133 epitopes in defining cancer stem cells. J. Mol. Med. 2008;86:1025–1032. doi: 10.1007/s00109-008-0357-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummendorf T.H., Dragowska W., Zijlmans J.M.J.M., Thornbury G., Lansdorp P.M., Zijlmans JMJM Asymmetric cell divisions sustain long-term hematopoiesis from single-sorted human fetal liver cells. J. Exp. Med. 1998;188:1117–1124. doi: 10.1084/jem.188.6.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvi L.M., Adams G.B., Weibrecht K.W., Weber J.M., Olson D.P., Knight M.C., Martin R.P., Schipani E., Divieti P., Bringhurst F.R. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- Chang K.C., Wang C., Wang H. Balancing self-renewal and differentiation by asymmetric division: insights from brain tumor suppressors in Drosophila neural stem cells. Bioessays. 2012;34:301–310. doi: 10.1002/bies.201100090. [DOI] [PubMed] [Google Scholar]
- Cicalese A., Bonizzi G., Pasi C.E., Faretta M., Ronzoni S., Giulini B., Brisken C., Minucci S., Di Fiore P.P., Pelicci P.G. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138:1083–1095. doi: 10.1016/j.cell.2009.06.048. [DOI] [PubMed] [Google Scholar]
- Ding L., Morrison S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495:231–235. doi: 10.1038/nature11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca A.V., Bauer N., Corbeil D. The stem cell marker CD133 meets the endosomal compartment—new insights into the cell division of hematopoietic stem cells. Blood Cells Mol. Dis. 2008;41:194–195. doi: 10.1016/j.bcmd.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Gho M., Bellaïche Y., Schweisguth F. Revisiting the Drosophila microchaete lineage: a novel intrinsically asymmetric cell division generates a glial cell. Development. 1999;126:3573–3584. doi: 10.1242/dev.126.16.3573. [DOI] [PubMed] [Google Scholar]
- Giebel B. Cell polarity and asymmetric cell division within human hematopoietic stem and progenitor cells. Cells Tissues Organs (Print) 2008;188:116–126. doi: 10.1159/000112842. [DOI] [PubMed] [Google Scholar]
- Giebel B., Wodarz A. Notch signaling: numb makes the difference. Curr. Biol. 2012;22:R133–R135. doi: 10.1016/j.cub.2012.01.006. [DOI] [PubMed] [Google Scholar]
- Giebel B., Corbeil D., Beckmann J., Höhn J., Freund D., Giesen K., Fischer J., Kögler G., Wernet P. Segregation of lipid raft markers including CD133 in polarized human hematopoietic stem and progenitor cells. Blood. 2004;104:2332–2338. doi: 10.1182/blood-2004-02-0511. [DOI] [PubMed] [Google Scholar]
- Giebel B., Zhang T., Beckmann J., Spanholtz J., Wernet P., Ho A.D., Punzel M. Primitive human hematopoietic cells give rise to differentially specified daughter cells upon their initial cell division. Blood. 2006;107:2146–2152. doi: 10.1182/blood-2005-08-3139. [DOI] [PubMed] [Google Scholar]
- Görgens A., Giebel B. Self-renewal of primitive hematopoietic cells: a focus on asymmetric cell division. In: Kadereit S., Udolph G., editors. Umbilical Cord Blood: A Future for Regenerative Medicine. World Scientific Publishing Company; Singapore: 2010. pp. 51–75. [Google Scholar]
- Görgens A., Beckmann J., Ludwig A.K., Möllmann M., Dürig J., Horn P.A., Rajendran L., Giebel B. Lipid raft redistribution and morphological cell polarization are separable processes providing a basis for hematopoietic stem and progenitor cell migration. Int. J. Biochem. Cell Biol. 2012;44:1121–1132. doi: 10.1016/j.biocel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- Görgens A., Radtke S., Horn P.A., Giebel B. New relationships of human hematopoietic lineages facilitate detection of multipotent hematopoietic stem and progenitor cells. Cell Cycle. 2013;12:3478–3482. doi: 10.4161/cc.26900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görgens A., Radtke S., Möllmann M., Cross M., Dürig J., Horn P.A., Giebel B. Revision of the human hematopoietic tree: granulocyte subtypes derive from distinct hematopoietic lineages. Cell Reports. 2013;3:1539–1552. doi: 10.1016/j.celrep.2013.04.025. [DOI] [PubMed] [Google Scholar]
- Greenbaum A., Hsu Y.M., Day R.B., Schuettpelz L.G., Christopher M.J., Borgerding J.N., Nagasawa T., Link D.C. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495:227–230. doi: 10.1038/nature11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homem C.C., Knoblich J.A. Drosophila neuroblasts: a model for stem cell biology. Development. 2012;139:4297–4310. doi: 10.1242/dev.080515. [DOI] [PubMed] [Google Scholar]
- Huang S., Law P., Francis K., Palsson B.O., Ho A.D. Symmetry of initial cell divisions among primitive hematopoietic progenitors is independent of ontogenic age and regulatory molecules. Blood. 1999;94:2595–2604. [PubMed] [Google Scholar]
- Jin L., Hope K.J., Zhai Q., Smadja-Joffe F., Dick J.E. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 2006;12:1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- Kollet O., Spiegel A., Peled A., Petit I., Byk T., Hershkoviz R., Guetta E., Barkai G., Nagler A., Lapidot T. Rapid and efficient homing of human CD34(+)CD38(-/low)CXCR4(+) stem and progenitor cells to the bone marrow and spleen of NOD/SCID and NOD/SCID/B2m(null) mice. Blood. 2001;97:3283–3291. doi: 10.1182/blood.v97.10.3283. [DOI] [PubMed] [Google Scholar]
- Lapidot T., Dar A., Kollet O. How do stem cells find their way home? Blood. 2005;106:1901–1910. doi: 10.1182/blood-2005-04-1417. [DOI] [PubMed] [Google Scholar]
- Lévesque J.P., Helwani F.M., Winkler I.G. The endosteal ‘osteoblastic’ niche and its role in hematopoietic stem cell homing and mobilization. Leukemia. 2010;24:1979–1992. doi: 10.1038/leu.2010.214. [DOI] [PubMed] [Google Scholar]
- Lymperi S., Ferraro F., Scadden D.T. The HSC niche concept has turned 31. Has our knowledge matured? Ann. N Y Acad. Sci. 2010;1192:12–18. doi: 10.1111/j.1749-6632.2009.05223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miraglia S., Godfrey W., Yin A.H., Atkins K., Warnke R., Holden J.T., Bray R.A., Waller E.K., Buck D.W. A novel five-transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood. 1997;90:5013–5021. [PubMed] [Google Scholar]
- Punzel M., Zhang T., Liu D., Eckstein V., Ho A.D. Functional analysis of initial cell divisions defines the subsequent fate of individual human CD34(+)CD38(-) cells. Exp. Hematol. 2002;30:464–472. doi: 10.1016/s0301-472x(02)00781-6. [DOI] [PubMed] [Google Scholar]
- Rajendran L., Beckmann J., Magenau A., Boneberg E.M., Gaus K., Viola A., Giebel B., Illges H. Flotillins are involved in the polarization of primitive and mature hematopoietic cells. PLoS ONE. 2009;4:e8290. doi: 10.1371/journal.pone.0008290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells. 1978;4:7–25. [PubMed] [Google Scholar]
- Shivtiel S., Lapid K., Kalchenko V., Avigdor A., Goichberg P., Kalinkovich A., Nagler A., Kollet O., Lapidot T. CD45 regulates homing and engraftment of immature normal and leukemic human cells in transplanted immunodeficient mice. Exp. Hematol. 2011;39:1161–1170. doi: 10.1016/j.exphem.2011.08.012. [DOI] [PubMed] [Google Scholar]
- Sugiarto S., Persson A.I., Munoz E.G., Waldhuber M., Lamagna C., Andor N., Hanecker P., Ayers-Ringler J., Phillips J., Siu J. Asymmetry-defective oligodendrocyte progenitors are glioma precursors. Cancer Cell. 2011;20:328–340. doi: 10.1016/j.ccr.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan S.K., Olin M.R., Forster C.L., Cruz K.S., Panyam J., Ohlfest J.R. Identification of a novel monoclonal antibody recognizing CD133. J. Immunol. Methods. 2010;361:110–115. doi: 10.1016/j.jim.2010.07.007. [DOI] [PubMed] [Google Scholar]
- Taussig D.C., Miraki-Moud F., Anjos-Afonso F., Pearce D.J., Allen K., Ridler C., Lillington D., Oakervee H., Cavenagh J., Agrawal S.G. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112:568–575. doi: 10.1182/blood-2007-10-118331. [DOI] [PubMed] [Google Scholar]
- Wagner W., Wein F., Roderburg C., Saffrich R., Faber A., Krause U., Schubert M., Benes V., Eckstein V., Maul H., Ho A.D. Adhesion of hematopoietic progenitor cells to human mesenchymal stem cells as a model for cell-cell interaction. Exp. Hematol. 2007;35:314–325. doi: 10.1016/j.exphem.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Wu M., Kwon H.Y., Rattis F., Blum J., Zhao C., Ashkenazi R., Jackson T.L., Gaiano N., Oliver T., Reya T. Imaging hematopoietic precursor division in real time. Cell Stem Cell. 2007;1:541–554. doi: 10.1016/j.stem.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanjani E.D., Flake A.W., Almeida-Porada G., Tran N., Papayannopoulou T. Homing of human cells in the fetal sheep model: modulation by antibodies activating or inhibiting very late activation antigen-4-dependent function. Blood. 1999;94:2515–2522. [PubMed] [Google Scholar]
- Zhang J., Niu C., Ye L., Huang H., He X., Tong W.G., Ross J., Haug J., Johnson T., Feng J.Q. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.