

Summary
Embryonic stem cells (ESCs) enable rapid proliferation that also causes DNA damage. To maintain genomic stabilization during rapid proliferation, ESCs must have an efficient system to repress genotoxic stress. Here, we show that withdrawal of leukemia inhibitory factor (LIF), which maintains the self-renewal capability of mouse ESCs (mESCs), significantly inhibits the cell proliferation and DNA damage of mESCs and upregulates the expression of miR-590. miR-590 promotes single-strand break (SSB) and double-strand break (DSB) damage repair, thus slowing proliferation of mESCs without influencing stemness. miR-590 directly targets Activin receptor type 2a (Acvr2a) to mediate Activin signaling. We identified the homologous recombination-mediated repair (HRR) gene, Rad51b, as a downstream molecule of the miR-590/Acvr2a pathway regulating the SSB and DSB damage repair and cell cycle. Our study shows that a miR-590/Acvr2a/Rad51b signaling axis ensures the stabilization of mESCs by balancing DNA damage repair and rapid proliferation during self-renewal.
Highlights
-
•
miR-590 promotes DNA damage repair and slows proliferation by targeting Acvr2a
-
•
miR-590/Acvr2a/Rad51b axis balances SSB and DSB damage repair in mESCs
In this article, Kang and colleagues show that miR-590 promotes DNA single-strand break and double-strand break damage repair, slowing proliferation of mESCs by directly targeting Acvr2a. They further identified that Rad51b was regulated by miR-590/Acvr2a pathway and involved in the formation of the miR-590/Acvr2a/Rad51b signaling axis to balance the DNA damage repair and rapid proliferation during mESC self-renewal.
Introduction
Embryonic stem (ES) cells derived from the inner cell mass of the blastocyst have been used to understand early embryonic development (Keller, 2005). The notable characteristic of ESCs is self-renewal that is critically involved in the stimulation of rapid proliferation. In fact, rapid proliferation might protect ESCs from external signals inducing differentiation (Ruiz et al., 2011). However, rapid proliferation would be harmful because it causes successive mitotic division with a long S phase in which DNA is replicating most of the time (Fluckiger et al., 2006; Savatier et al., 2002), and the successive rounds of DNA replication causes many replication errors that may lead to DNA damage (Strumberg et al., 2000; Tichy and Stambrook, 2008). In addition, there is no G1 checkpoint in ESCs (White and Dalton, 2005), which might exacerbate DNA damage during rounds of replication without enough time for repair (Hong and Stambrook, 2004). Consistent with these characteristics, there is a high level of double-strand break (DSB) damage, which is the most toxic type of DNA damage (Valerie and Povirk, 2003), and DSB damage is indicated by the γ-H2AX marker (H2AX becomes phosphorylated on serine 139) in both human ES (hES) cells and mouse ES (mES) cells (Banáth et al., 2009; Chuykin et al., 2008; Momcilovic et al., 2010). Similar to irradiated fibroblast cells, normal mESCs also contain a high frequency of single-strand break (SSB) (Chuykin et al., 2008). However, ESCs still have an integrated genome and stable pluripotency during rapid proliferation (Tichy and Stambrook, 2008; Wang et al., 2008). Additionally, the mutation frequency and mitotic recombination frequency are lower in mESCs than in adult somatic or isogenic mouse embryonic fibroblasts cells (Tichy and Stambrook, 2008). Thus, ESCs must have unique regulatory mechanisms that counteract DNA damage both quickly and efficiently.
Transforming growth factor β (TGF-β) signaling is closely related to DNA damage repair regulation (Mitra et al., 2013). Studies have shown that TGF-β signaling can suppress BRCA1-dependent repair of DSBs (Dubrovska et al., 2005). Activin A, a member of the TGF-β superfamily of cytokines, interacts with Activin type I (ALK 2, ALK 4, or ALK 7) and type II (Acvr2a and Acvr2b) receptors (Bondestam et al., 1999; Donaldson et al., 1992; Robson et al., 2008), and it participates in DNA damage repair. In premalignant cells, DSB damage results in Activin A-dependent induction of Cox-2, which is associated with a high level of γ-H2AX (Carlson et al., 2013; Fordyce et al., 2012). In ductal carcinoma, DNA damage response (shorter telomeres and γ-H2AX foci) is also associated with high level of Activin A (Fordyce et al., 2012). Additionally, in hESCs, Activin A can maintain pluripotency even without feeder layers (Beattie et al., 2005). In mESCs, Activin A signaling can promote cell proliferation (Ogawa et al., 2007). However, it remains unknown whether the maintenance of a stable status in ESCs is also related to the DNA damage repair function of Activin A signaling.
DSB damage is the most toxic type of DNA damage (Valerie and Povirk, 2003). Homologous recombination-mediated repair (HRR) is thought to be used in ESCs to repair DSBs (Hasty et al., 1992; Shrivastav et al., 2008; Smih et al., 1995; Tichy et al., 2010). Rad51 family members, including Rad51, Rad51b, Rad5c, Rad51d, and so on, are evolutionarily conserved proteins that play important roles during HHR (Baumann et al., 1996; Chun et al., 2013; Kawabata et al., 2005; Thacker, 2005). Rad51 paralogs take part in SSB and DSB damage repair (Jensen et al., 2010, 2013). RAD51B is also a protein kinase regulating the function of cell cycle-related genes (Havre et al., 2000) and is a known molecule to promote HRR by participating in the Holliday junction process (Kawabata et al., 2005; Takata et al., 2000). Previous studies have shown that overexpression of Rad51b in Chinese hamster ovary cells causes a G1 delay and UV irradiation hypersensitivity (Havre et al., 1998). TGF-β signaling has also been reported to inhibit DNA damage repair by downregulating the expression of Rad51 in Mv1Lu epithelial cells (Kanamoto et al., 2002). However, it is unknown whether Rad51 paralogs can regulate both the DNA damage repair and cell cycle or even maintain their balance in mESCs. Additionally, the upstream regulators of Rad51 paralogs in mESCs are unclear.
MicroRNAs (miRNAs) are posttranscriptional modulators of gene expression and are connected to the transcriptional regulatory circuitry of mESCs (Marson et al., 2008). Several miRNAs target DNA repair-related factors and influence DNA damage repair. Studies have shown that UV damage promotes miRNA expression in a partially ataxia telangiectasia mutated kinase (ATM)/ataxia telangiectasia and Rad3-related kinase-independent manner (Pothof et al., 2009). Overexpression of miR-24 attenuates H2AX, leading to high sensitivity to irradiation and reduced repair capacity (Lal et al., 2009). miRNAs can also regulate TGF-β signaling. miR-302/367 can modulate BMP signaling, which supports self-renewal by targeting BMP inhibitors in hESCs (Lipchina et al., 2011; Qi et al., 2004). However, there is limited understanding of miRNA modulation of these signaling pathways to regulate DNA damage repair to maintain self-renewal during ESC proliferation, which differs from differentiated cells (Tichy and Stambrook, 2008). In the present study, we found that miR-590 inhibits Activin signaling by directly targeting Acvr2a to inhibit the expression of Rad51b, balancing the DNA damage repair and rapid proliferation of ESCs.
Results
miR-590 Promotes SSB and DSB Damage Repair during Rapid Proliferation of mESCs
Consistent with that, leukemia inhibitory factor (LIF) is critically needed for the self-renewal of mESCs. We found that the rapid proliferation was greatly inhibited in both the cells cultured without LIF, which detected by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay (Figure 1A) and those during the embryonic body (EB) formation, which detected by flow cytometric (FACS) analysis of 5′-bromo-deoxyuridine (BrdU) incorporation (Figure S1A available online). We further found that both of the mature miR-590, namely miR-590-3p and miR-590-5p, were upregulated in cells during the culture without LIF (Figure 1A) and EB formation (Figure S1B), indicating a possible linkage between miR-590 expression and the mESC proliferation. In order to detect that whether miR-590 is associated with the regulation of self-renewal, we transfected pre-miR-590, which can be processed to be miR-590-3p and miR-590-5p (Figure S1C), into mESCs and found that the size of mESC colonies became smaller than control cells examined at 48 hr posttransfection (Figure 1B). We further digested mESC clones by trypsin into single cells and counted the amount of the total cells under the microscope by using blood count board and found that total cells transfected with pre-miR-590 were less than control cells (Figure S1D). Transfection of miR-590-3p or miR-590-5p mimics respectively into mESCs also resulted in a similar phenomenon of pre-miR-590 (Figure S1E). In contrast, clone size of mESCs was larger than control cells after transfected with miR-590-3p or miR-590-5p inhibitor, which can compete for the miR-590 with target mRNAs (Figure 1B). The amount of total cells was also more than control cells (Figure S1D). MTS assay and FACS of BrdU incorporation also showed that miR-590 inhibited proliferation of mESCs posttransfected for 48 hr with pre-miR-590, while cells transfected with miR-590-3p or miR-590-5p inhibitor showed faster proliferation than control cells (Figures 1C and 1D). Further, we found that the size of clones of another kind of mESC line 46C after transfected with pre-miR-590 was also smaller than control mESCs (Figure S1F).
Figure 1.
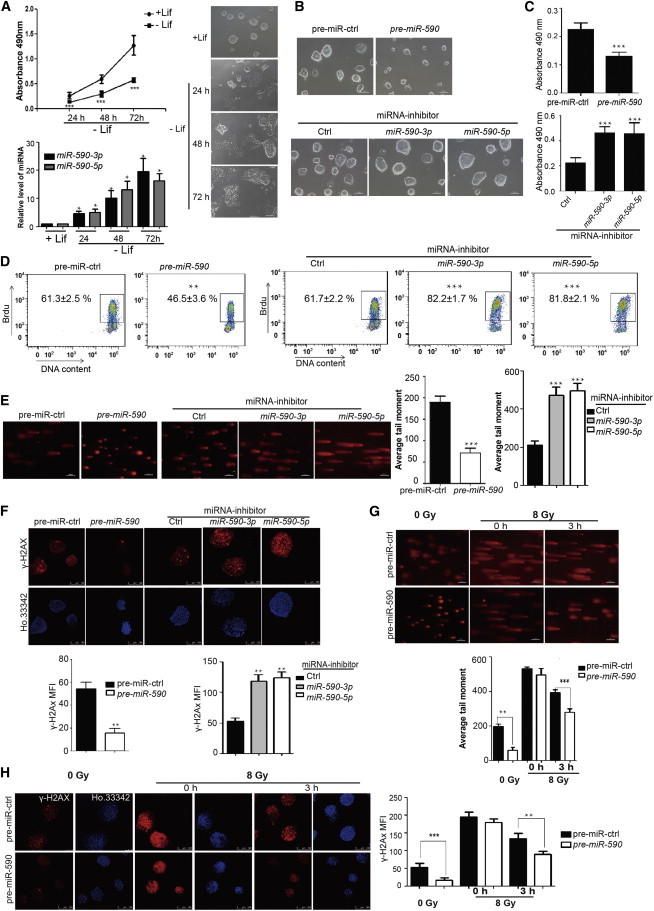
miR-590 Inhibits Rapid Proliferation and Promotes SSB and DSB Damage Repair of mESCs
(A) Analysis of the proliferation of mESCs cultured with or without LIF by MTS cell proliferation assay. (Bottom) The qRT-PCR verification of miR-590-3p and miR-590-5p levels. (Right) The morphology of mESCs cultured with (+LIF) on 24 hr or without LIF (−LIF) on 24, 48, 72 hr. Data shown are means ± SD of six independent experiments (n = 6). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. The scale bar represents 100 μm.
(B) Morphology of colonies of mESCs after transfection with pre-miR-590 or miR-590-3p/5p inhibitor. pre-miR-ctrl indicates pre-miRNA control, which has no homology to any known mammalian gene. Inhibitor control is a random sequence molecule that has no homology to any known mammalian gene. The scale bar represents 100 μm.
(C) Analysis of the proliferation of mESCs transfected with pre-miR-590 or miR-590-3p/5p inhibitor by MTS cell proliferation assay. Data shown are means ± SD of five independent experiments (n = 5). ∗∗∗p < 0.001.
(D) Analysis of the proliferation of mESCs transfected with pre-miR-590 or miR-590-3p/5p inhibitor by FACS analysis of BrdU incorporation. The figure shows the percentage (%) of population the of BrdU-positive cells. Data shown are means ± SD of three independent experiments (n = 3). ∗∗p < 0.01 and ∗∗∗p < 0.001.
(E) Comet assay for SSB damage in mESCs. The scale bar represents 100 μm. Data shown are means ± SD of three independent experiments (n = 3). (Right) The quantification of the average DNA tail moment. ∗∗∗p < 0.001.
(F) Immunofluorescence analysis of γ-H2AX (red) to indicate the state of DSB damage in mESCs. Ho.33342 (Hoechst 33342) represents nuclear staining (blue). The scale bar represents 100 μm. (Bottom) The quantification of the fluorescence of γ-H2AX immunostaining. Data shown are means ± SD of three independent experiments (n = 3). ∗∗p < 0.01. MFI, mean fluorescence intensity.
(G) Comet assay for SSB damage of mESCs with irradiation from X-ray. 0 Gy indicates mESCs not subjected to X-ray irradiation. The scale bar represents 100 μm. The bottom shows the quantification of the average DNA tail moment. Data shown are means ± SD of three independent experiments (n = 3). ∗∗p < 0.01 and ∗∗∗p < 0.001.
(H) Immunofluorescence analysis of γ-H2AX (red) with Ho.33342 (Hoechst 33342) nuclear staining (blue). The scale bar represents 100 μm. (Right) The quantification of the fluorescence of γ-H2AX immunostaining. Data shown are means ± SD of three independent experiments (n = 3). ∗∗p < 0.01 and ∗∗∗p < 0.001.
FACS analysis of cell cycle showed that there was a significant reduction in the proportion of cells in S phase and an increase in the proportion of cells in G1 and G2/M phases in mESCs after transfected with pre-miR-590 for 48 hr (Figure S1G). In contrast, the proportion of cells in S phase was increased and in G1 and G2/M phases was reduced in mESCs transfected with miR-590-3p or miR-590-5p inhibitor (Figure S1G). miR-590 could not induce the apoptosis (Figure S1H) and showed no significant influence on the expression of stemness genes (Figure S1I).
Additionally, we found that miR-590 can regulate the SSB and DSB damage in mESCs. SSB and DSB damage was relieved in mESCs after being transfected with pre-miR-590 detected by the comet assay and the immunostaining of γ-H2AX foci, respectively (Figures 1E and 1F). In contrast, mESCs transfected with the miR-590-3p or miR-590-5p inhibitor showed increased SSB damage (Figure 1E) and DSB (Figure 1F). To investigate whether miR-590 can promote DNA damage repair directly or through regulating proliferation to free up more time to repair DNA, we used X-ray to irradiate mESCs to simulate DSB and SSB damage. We found that after treating with 8 Gy radiation, both mESCs transfected with pre-miR-590 and pre-miR-control showed violent DSB and SSB damage. Three hours later, both groups of mESCs had repaired much DNA damage (Figures 1G and 1H). However, we found that the repair capacity of mESCs transfected with pre-miR-590 was faster than that of control mESCs. The DNA damage was significantly decreased in mESCs transfected with pre-miR-590 compared with control mESCs (Figures 1G and 1H). Trypan blue staining showed that there was no significant dying cell in mESCs after transfected with pre-miR-590 or 0/3 hr postirradiated by 8 Gy X-ray (Figures S1J and S1K). Thus, we concluded that miR-590 may regulate DNA damage repair directly in mESCs. Additionally, the miR-590 regulation of proliferation led us to investigate whether miR-590 connects the cell proliferation and DNA damage repair in mESCs.
miR-590 Regulates the Expression of Rad51b and Cell Cycle-Related Genes
To test whether cell cycle-related, SSB damage repair-related, and DSB damage repair-related genes can be regulated by miR-590, we performed quantitative RT-PCR (qRT-PCR) and western blot analyses examined 48 hr posttransfection, and we found that Rad51b was upregulated in mESCs transfected with pre-miR-590 but that Rad51c, Rad9, and Brca1 showed no significant change (Figure 2A). Additionally, the cell cycle-related genes, Cyclin E and Cyclin B, were downregulated while P21 and Rb1 were upregulated (Figure 2A). Western blot analysis was used to confirm the change in protein levels of the cell cycle-related genes and Rad51b (Figure 2B). In contrast, the miR-590-3p or miR-590-5p inhibitor downregulated the expression of Rad51b, p21, and Rb1 and upregulated the expression of Cyclin B and Cyclin E (Figure 2C). Protein levels of the cell cycle-related genes and Rad51b were detected to confirm the function of miR-590-3p or miR-590-5p inhibitors (Figure 2D). These results showed that miR-590 may mediate downstream genes to regulate DSB damage repair, SSB damage repair, as well as the cell proliferation.
Figure 2.
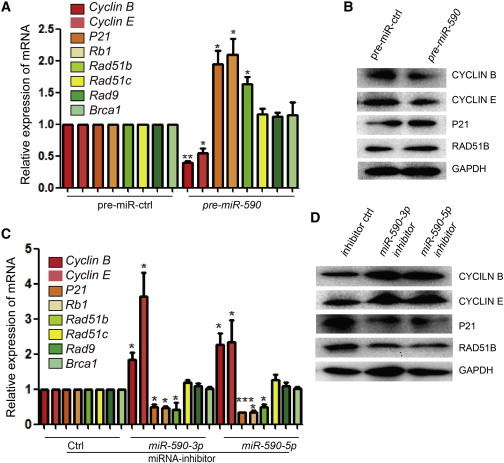
Effects of miR-590 on Cell Cycle and DNA Damage Repair-Related Genes
(A) qRT-PCR verification of transcript levels of the cell cycle regulation and homologous recombination damage repair genes in mESCs transfected with pre-miR-590 or control. Data shown are means ± SD of three independent experiments (n = 3). ∗p < 0.05 and ∗∗p < 0.01.
(B) Western blot analysis for the cell cycle regulation and homologous recombination damage repair genes in mESCs transfected with pre-miR-590 or control.
(C) qRT-PCR verification of mRNA levels of the cell cycle regulation and homologous recombination damage repair genes in mESCs transfected with miR-590-3p/5p inhibitor or control. Data shown are means ± SD of three independent experiments (n = 3). ∗p < 0.05 and ∗∗∗p < 0.001.
(D) Western blot analysis of the cell cycle regulation and homologous recombination damage repair genes in mESCs transfected with miR-590-3p/5p inhibitor or control.
miR-590 Directly Targets Acvr2a to Regulate Activin Signaling in mESCs
We used Targetscan and Miranda to predict the direct target of miRNAs and found that both miR-590-3p and miR-590-5p can target Acvr2a (Figure 3A). Then we found that Acvr2a was downregulated in mESCs cultured without LIF compared with mESCs cultured with LIF (Figure 3A), which was opposite to the upregulation of miR-590 −3p/5p in the mESCs cultured in the same system (Figure 1A). These results thus suggested a negative correlation between Acvr2a and miR-590-3p/5p during the differentiation of mESCs. Furthermore, we performed luciferase reporter assay to analyze whether miR-590-3p or miR-590-5p can target the 3′UTR of Acvr2a mRNA. We generated WT and mutant 3′UTR reporters of Acvr2a and found that both miR-590-3p and miR-590-5p inhibited the luciferase activity of the WT 3′UTR reporter but not the mutant reporters (Figure 3B). Furthermore, we detected miR-590 regulation on endogenous Acvr2a in mESCs. We transfected pre-miR-590 into mESCs and found that the expression of Acvr2a on both the mRNA and protein level was decreased (Figure 3C). To investigate whether miR-590 can influence Activin signaling, we detected the level of p-SMAD2 by western blot analysis and found that mESCs transfected with pre-miR-590 had a lower level of p-SMAD2 than the control group (Figure 3D). Transfection of the miR-590-3p or miR-590-5p inhibitor increased the level of p-SMAD2 (Figure 3E). These results demonstrated that miR-590 inhibits the Activin signaling pathway by directly targeting Acvr2a.
Figure 3.
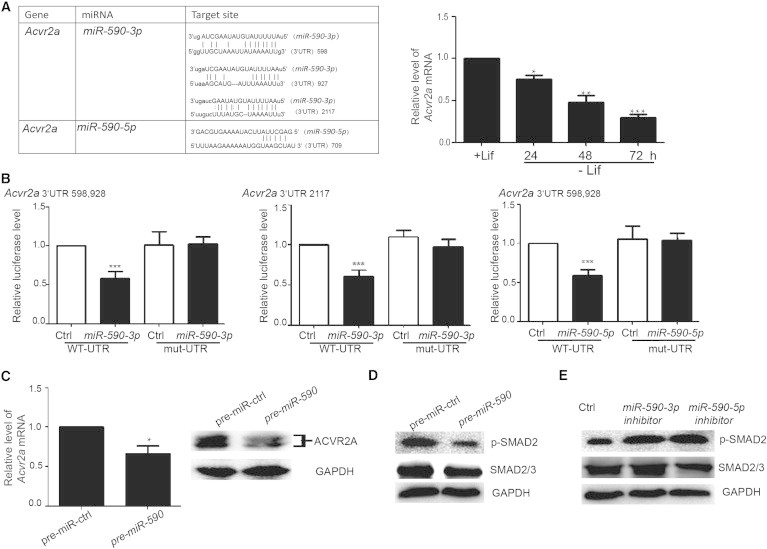
miR-590 Directly Targets Acvr2a to Regulate Activin Signaling in mESCs
(A) Summary of miR-590-3p and miR-590-5p target sites in the 3′UTRs of Acvr2a. In the double strands of the sequence, the upper strand is miR-590-3p or miR-590-5p, and the lower strand is the binding sites of 3′UTR mRNA. The right panel shows the level of Acvr2a expression detected by qRT-PCR. Data shown are means ± SD of three independent experiments (n = 3). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
(B) miR-590-3p and miR-590-5p specifically repress their targets in the luciferase assay. WT-UTR indicates WT 3′ UTR. Mut-UTR indicates the 3′UTR containing the mutant binding site of miR-590. Data shown are means ± SD of three independent experiments (n = 3). ∗∗∗p < 0.001.
(C) Detection of Acvr2a expression on mRNA and protein levels in mESCs. Data shown are means ± SD of three independent experiments (n = 3). ∗p < 0.05.
(D and E) The level of p-SMAD2 was detected by western blot analysis. Glyceraldehyde-3-phosphate dehydrogenase is as an internal control.
The miR-590/Acvr2a Pathway Balances SSB and DSB Damage Repair with Rapid Proliferation of mESCs
We next tested whether the miR-590/Acvr2a pathway can regulate mESC proliferation and DNA damage repair. We constructed the Acvr2a overexpression vector (Figure S2A). Then we found that clone size of mESCs overexpressed Acvr2a was larger than control cells (Figure 4A). The amount of total cells was also more than control cells (Figure S2B). Both the FACS analysis of BrdU incorporation and MTS assay showed the increase of proliferation of mESCs overexpressed by Acvr2a (Figures 4B and 4C). FACS analysis of cell cycle showed that there was a significant increase in the proportion of cells in S phase and a reduction in the proportion of cells in G1 and G2/M phases in mESCs overexpressed by Acvr2a examined at 48 hr posttransfection (Figure S2C).
Figure 4.
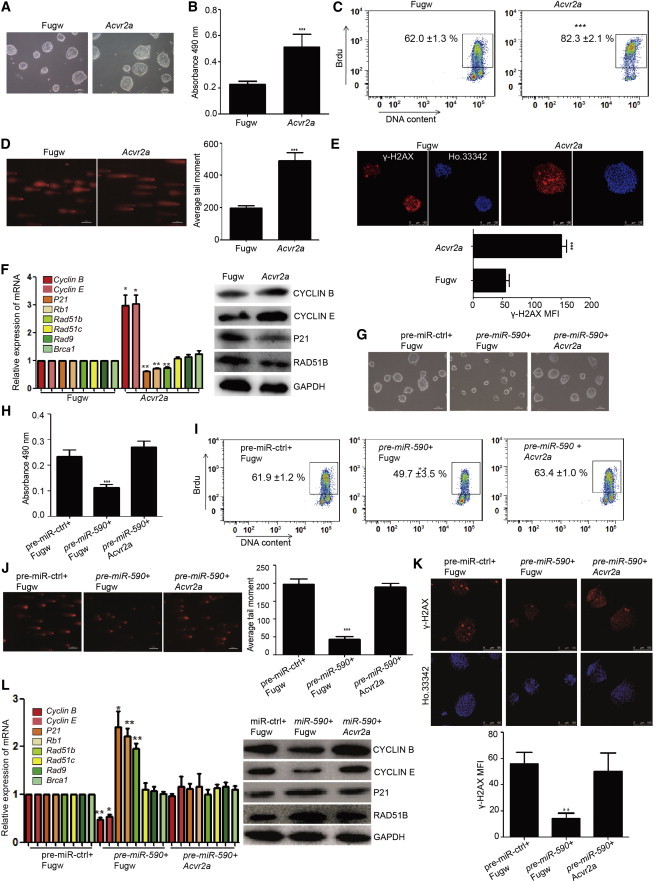
miR-590 Regulates the SSB Damage Repair, DSB Damage Repair, and Proliferation of mESCs by Acvr2a
(A) Morphology of mESCs clones overexpressing Acvr2a. The scale bar represents 100 μm.
(B) Analysis of proliferation of mESCs overexpressed with Acvr2a by MTS cell proliferation assay. Data shown are means ± SD of five independent experiments (n = 5). ∗∗∗p < 0.001.
(C) Analysis of proliferation of mESCs overexpressed with Acvr2a by FACS analysis of BrdU incorporation. The figure means the percentage of the population of BrdU-positive cells. Data shown are means ± SD of three independent experiments (n = 3). ∗∗∗p < 0.001.
(D) Comet assay for SSB damage in mESCs. The scale bar represents 100 μm. Right panel is the quantification of average DNA tail moment. Data shown are means ± SD of three independent experiments (n = 3). ∗∗∗p < 0.001.
(E) Immunofluorescence analysis of γ-H2AX (red) to indicate the state of DSB damage in mESCs. Ho.33342 (Hoechst 33342) represents nuclear staining (blue). The scale bar represents 100 μm. (Bottom) The quantification of the fluorescence of γ-H2AX immunostaining. Data shown are means ± SD of three independent experiments (n = 3). ∗∗∗p < 0.001.
(F) qRT-PCR verification of transcript levels of the cell cycle regulation and homologous recombination damage repair genes in mESCs. The right side shows the protein levels according to western blot analysis. Data shown are means ± SD of three independent experiments (n = 3). ∗p < 0.05 and ∗∗p < 0.01.
(G) Morphology of mESCs clones. The scale bar represents 100 μm.
(H) Cell proliferation assay in the rescue experiment by MTS cell proliferation assay. Data shown are means ± SD of three independent experiments (n = 3). ∗∗∗p < 0.001.
(I) Cell proliferation assay in the rescue experiment by FACS of BrdU incorporation. The figure means the percentage of the population of BrdU-positive cells. Data shown are means ± SD of three independent experiments (n = 3). ∗∗p < 0.01.
(J) Comet assay shows the SSB damage state. The scale bar represents 100 μm. The right panel is the quantification of average DNA tail moment. Data shown are means ± SD of three independent experiments (n = 3). ∗∗∗p < 0.001.
(K) Overexpression of ACVR2a restores DSB damage in mESCs transfected with pre-miR-590. Immunofluorescence analysis of γ-H2AX (red) indicates the state of DSB damage in mESCs with Ho. 33342 (Hoechst 33342) staining for the nucleus (blue). The scale bar represents 100 μm. (Bottom) The quantification of the fluorescence of γ-H2AX immunostaining. Data shown are means ± SD of three independent experiments (n = 3). ∗∗p < 0.01.
(L) qRT-PCR and western blot analysis of the cell cycle- and homologous recombination damage repair-related genes in mESCs. Data shown are means ± SD of three independent experiments (n = 3). ∗p < 0.05 and ∗∗p < 0.01.
We also found that overexpression of Acvr2a caused more SSB (Figure 4D) and DSB damage (Figure 4E) in mESCs compared with the control mESCs, and the overexpression of Acvr2a increased the expression of Cyclin B and Cyclin E as well as decreased the expression of Rad51b, P21, and Rb1 (Figure 4F). As expected, in mESCs with Acvr2a knockdown, clone size is smaller than control cells (Figure S2D). The amount of total cells was also less than control group (Figure S2D). Proliferation was inhibited by downregulation of the protein level of Acvr2a (Figure S2E). There was a significant reduction in the proportion of cells in S phase and an increase in the proportion of cells in G1 and G2/M phases in mESCs with downregulation of Acvr2a (Figure S2F). The SSB and DSB damage were also decreased in mESCs with knockdown of Acvr2a (Figures S2G and S2H).
We next tested whether miR-590 can regulate the DSB, SSB damage repair, and cell proliferation of mESCs by directly targeting Acvr2a. We performed a rescue experiment. The expression level of Acvr2a was detected by qPCR in rescue experiment. We first detected the Acvr2a expression level of the rescue experiment (Figure S3A) and found that the influence on clone size (Figure 4G), cell proliferation ((Figures 4H, 4I, and S3B), and cell cycle (Figure S3C) of mESCs transfected with pre-miR-590 could be restored by overexpressing Acvr2a. We also showed that overexpression of Acvr2a in mESCs transfected with pre-miR-590 could restore the state of excessive SSB and DSB DNA damage (Figures 4J and 4K) to a similar level of control mESCs. The cell cycle-related genes and Rad51b were also restored by overexpressing Acvr2a in mESCs transfected with pre-miR-590 (Figure 4L). These results showed that miR-590 regulated the DNA damage repair and cell proliferation of mESCs through directly targeting Acvr2a.
Rad51b Promotes SSB and DSB Damage Repair in mESCs to Slow the Cell Proliferation
We found that Rad51b can be regulated by the miR-590/Acvr2a pathway. To determine whether Rad51b can inhibit the mES proliferation and repair SSB and DSB damage, we downregulated the expression of Rad51b in mESCs. The colonies of mESCs with knockdown of Rad51b were larger than control cells (Figure 5A). The total number of cells was more than control cells (Figure S4A). The proliferation was increased by downregulating Rad51b in mESCs detected by MTS assay and FACS analysis of BrdU incorporation (Figures 5B and 5C). We also found that knockdown of Rad51b resulted in a reduced fraction of cells in G1 and G2/M phases as well as an increased fraction of cells in S phase (Figure S4B). The SSB and DSB damage was significantly increased after knockdown of Rad51b (Figures 5D and 5E). In mESCs with Rad51b knockdown, the expression levels of Cyclin B and Cyclin E were upregulated, and the expression levels of P21 and Rb1 were downregulated (Figure 5F). These results showed that Rad51b can regulate both the DNA damage repair and cell proliferation in mESCs to balance the rapid proliferation and DNA damage repair.
Figure 5.
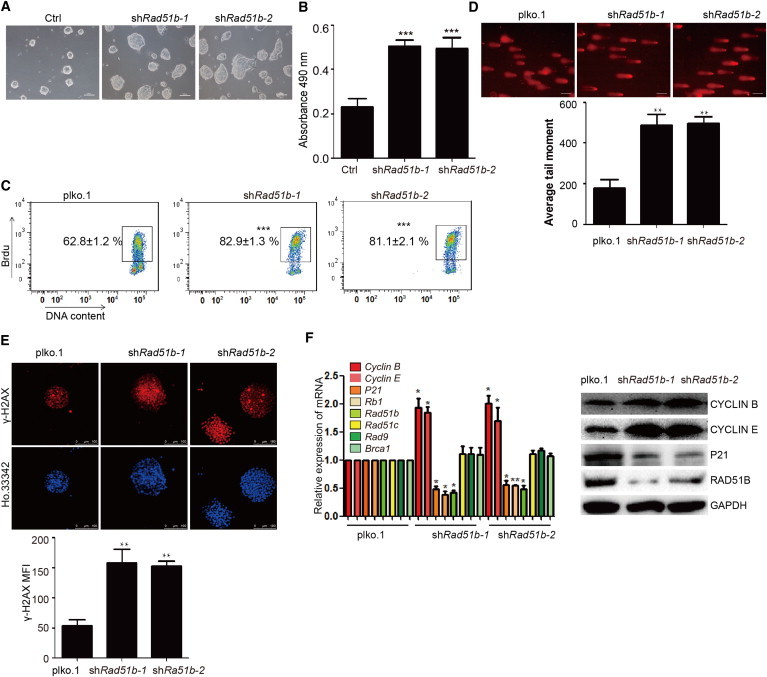
Rad51b Promotes SSB and DSB Damage Repair in mESCs and Slows the Cell Proliferation
(A) Morphology of mESC clones with knockdown of Rad51b compared with the control group. The scale bar represents 100 μm.
(B) Cell proliferation assay by MTS cell proliferation assay. Data shown are means ± SD of three independent experiments (n = 3). ∗∗∗p < 0.001.
(C) Cell proliferation assay by FACS of BrdU incorporation. The figure means the percentage of the population of BrdU-positive cells in total cells. Figure shows the percentage of the population of BrdU-positive cells. Data shown are means ± SD of three independent experiments (n = 3). ∗∗∗p < 0.001.
(D) Knockdown of Rad51b increases the SSB damage in mESCs as detected by the comet assay. The scale bar represents 100 μm. Bottom panel shows the quantification of average DNA tail moment. Data shown are means ± SD of three independent experiments (n = 3). ∗∗p < 0.01.
(E) Immunofluorescence analysis of γ-H2AX (red) indicates the state of DSB damage. γ-H2AX was increased by knockdown of Rad51b in mESCs. Ho.33342 (Hoechst 33342) represents nuclear staining (blue). The scale bar represents 100 μm. (Bottom) The quantification of the fluorescence of γ-H2AX immunostaining. Data shown are means ± SD of three independent experiments (n = 3). ∗∗p < 0.01.
(F) qRT-PCR and western blot indicating the levels of the cell cycle- and homologous recombination damage repair-related genes in mESCs. Data shown are means ± SD of three independent experiments (n = 3). ∗p < 0.05 and ∗∗p < 0.01.
The miR-590/Acvr2a/Rad51b Pathway Balances the Rapid Proliferation and DNA Damage Repair of mESCs
To confirm whether Rad51b is the functional component of the miR-590/Acvr2a pathway, we performed rescue experiments. We downregulated the expression of Rad51b in mESCs transfected with pre-miR-590 and found that the size of clones (Figure 6A) and amount of total cells (Figure S5A) were restored compared with mESCs transfected only with pre-miR-590. MTS assay and BrdU incorporation assay showed the rapid proliferation of mESCs was restored by downregulating Rad51b compared with mESCs transfected only with pre-miR-590 (Figures 6B and 6C). FACS analysis of cell cycle showed that the fraction of cell cycle in mESCs with exogenous pre-miR-590 transfection was restored by knockdown of Rad51b (Figure S5B). The state of SSB and DSB damage in mESCs transfected with pre-miR-590 was also restored by knockdown of Rad51b (Figures 6D and 6E). The levels of cell cycle-related genes and Rad51b in the rescue group were similar to those in the control group (Figure 6F). These results showed that Rad51b is the downstream functional factor of miR-590 and regulates both DNA damage and cell proliferation.
Figure 6.
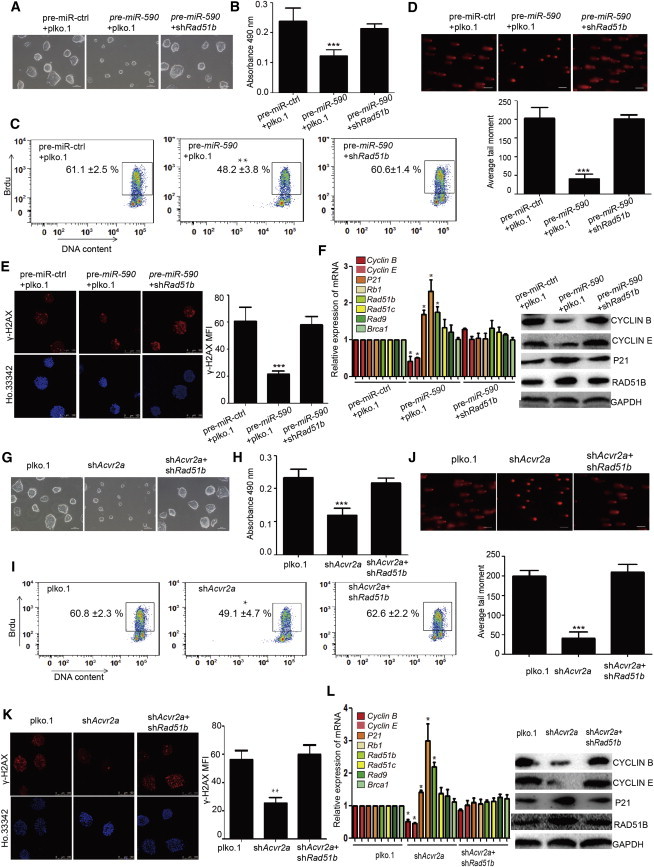
Knockdown of Rad51b Restores the State of Proliferation and DNA Damage in mESCs Overexpressing Pre-miR-590 or Knockdown of Acvr2a
(A) Knockdown of Rad51b can restore the size of mESC clones influenced by miR-590. The scale bar represents 100 μm.
(B) Cell proliferation assay by MTS cell proliferation assay. Data shown are means ± SD of five independent experiments (n = 5). ∗∗∗p < 0.001.
(C) Cell proliferation assay by FACS of BrdU incorporation. The figure means the percentage of the population of BrdU-positive cells. Figure shows the percentage of the population of BrdU-positive cells. Data shown are means ± SD of three independent experiments (n = 3). ∗∗p < 0.01.
(D) The comet assay shows that knockdown of Rad51b relieves the inhibition of SSB damage caused by miR-590. The scale bar represents 100 μm. (Bottom) The quantification of average DNA tail moment. Data shown are means ± SD of five independent experiments (n = 5). ∗∗∗p < 0.001.
(E) Immunofluorescence analysis of γ-H2AX (red) to show DSB damage of mESCs. Nuclei were stained with Ho.33342 (Hoechst 33342) (blue). The scale bar represents 100 μm. The right panel shows the quantification of the fluorescence of γ-H2AX immunostaining. Data shown are means ± SD of three independent experiments (n = 3). ∗∗∗p < 0.001.
(F) qRT-PCR indicated that the levels of the cell cycle- and homologous recombination damage repair-related genes were rescued by downregulating Rad51b in mESCs transfected with pre-miR-590. The right side shows the protein levels detected by western blot analysis. ∗p < 0.05 and ∗∗p < 0.01.
(G) Knockdown of Rad51b restores the size mESC clones containing knockdown of Acvr2a. The scale bar represents 100 μm.
(H) MTS cell proliferation assay. Data shown are means ± SD of five independent experiments (n = 5). ∗∗∗p < 0.001.
(I) FACS analysis of BrdU incorporation. The figure means the percentage of the population of BrdU-positive cells. Figure shows the percentage of the population of BrdU-positive cells. Data shown are means ± SD of three independent experiments (n = 3). ∗p < 0.05.
(J) The comet assay. The scale bar represents 100 μm. (Bottom) The quantification of average DNA tail moment. Data shown are means ± SD of three independent experiments (n = 3). ∗∗∗p < 0.001.
(K) Immunofluorescence analysis of γ-H2AX (red). Nuclei were stained with Ho.33342 (Hoechst 33342) (blue). The scale bar represents 100 μm. The right panel shows the quantification of the fluorescence of γ-H2AX immunostaining. Data shown are means ± SD of three independent experiments (n = 3). ∗∗p < 0.01.
(L) qRT-PCR of the cell cycle- and homologous recombination damage repair-related genes in mESCs. The right side shows the protein levels as detected by western blot analysis. Data shown are means ± SD of three independent experiments (n = 3). ∗p < 0.05.
Furthermore, we performed a rescue experiment to test the regulation of Rad51b by Acvr2a. We found that knockdown of Rad51b restored the size of clones and the amount of total cells of mESCs with Acvr2a knockdown (Figures 6G and S5C). Additionally, the capacity of proliferation (Figures 6H and 6I) and fraction of cell cycle (Figure S5D) in Acvr2a knockdown mESCs was also restored after downregulation of Rad51b.
We also found that downregulation of Rad51b restored the DSB and SSB damage in Acvr2a knockdown mESCs, which was similar to the control mESCs (Figures 6J and 6K). The expression levels of cell cycle-related genes and Rad51B were restored to similar level of the control group (Figure 6L). These results confirmed that Rad51b is a downstream functional factor of Acvr2a and that the miR-590/Acvr2a/Rad51b pathway balances the rapid proliferation with DSB and SSB damage repair in mESCs to maintain the self-renewal and genome integrity.
Discussion
Rapid proliferation can maintain the self-renewal of ESCs, but it can also result in DNA damage that leads to catastrophic mutations affecting the differentiation of organisms and causing mutations to be passed to progeny (Savatier et al., 2002; Tichy and Stambrook, 2008). In response to DNA damage stress, cells generally slow or arrest cell cycle progression to repair or prevent the transmission of damage to daughter cells (Ishikawa et al., 2006). Cell cycle and DNA damage repair can be regulated during mES differentiation. During differentiation, mESCs accumulate in G1 phase and exhibit a cell cycle lengthened from 8–10 hr to more than 16 hr (White and Dalton, 2005). Additionally, DNA damage, especially DSB damage, is decreased during differentiation (Turinetto et al., 2012). Thus, there should be a system balancing the paradox of rapid proliferation and DNA damage repair to guarantee the normal self-renewal, pluripotency, and genomic stabilization in ESCs (Tichy, 2011). Here, we found that miR-590 balances DNA damage repair and proliferation by affecting the expression of Rad51b in mESCs. Importantly, Activin signaling regulated by Acvr2a mediates the balancing regulation of miR-590 on Rad51b. Furthermore, the miR-590/Acvr2a/Rad51b axis regulates the balance of rapid proliferation and DNA damage repair in mESCs.
The DNA damage repair system involves a complex network in which miRNAs play important roles (Chen and Rajewsky, 2007; Song et al., 2011; Wan et al., 2011). For example, miR-24 and miR-138 can downregulate H2AX, leading to high sensitivity to radiation and weak repair capacity (Lal et al., 2009). miR-182 suppresses DSB damage repair by targeting Brca1 (Moskwa et al., 2011). A previous study has also shown that miR-590 participates in the regulation of cell proliferation and death. miR-590-3p regulates neuronal death in patients with Alzheimer disease and frontotemporal lobar degeneration (Villa et al., 2011). In bladder cancer, miR-590-3p inhibits cell proliferation and migration (Mo et al., 2013). In acute myeloid leukemia, miR-590-5p can be regulated by interleukin-3 to affect the growth of cells (Favreau and Sathyanarayana, 2012). These studies suggest that miR-590 widely participates in cell growth of different cell types. However, the function of miR-590 in mESCs remains unknown. We found that overexpression of miR-590 significantly inhibited the proliferation by upregulating the expression of P21 and Rb1 as well as downregulating the expression of Cyclin E and Cyclin B, but the overexpression of miR-590 did not affect the pluripotency of mESCs. This result was similar to that of a previous study where no significant induction of differentiation of mESCs occurred with overexpression of P21 and Rb (Li et al., 2012). LIF signaling is important for mESCs to maintain rapid proliferation (Furue et al., 2005). In our study, we also found that during the process of culture withdrawing LIF, the proliferation rate of mESCs was slower, and miR-590-3p/5p were upregulated, suggesting a possible important role of miR-590 during mESC proliferation regulation. Studies have shown that ESCs have robust DNA damage repair capacity, which includes nucleotide excision repair (Saretzki et al., 2004), mismatch repair (Saretzki et al., 2004), and DSB repair (Donoho et al., 1998; Smih et al., 1995). DSB damage is considered to be the most toxic type of DNA damage (Valerie and Povirk, 2003), which may be caused by the replication fork collapsing during rapid and continuous proliferation in ESCs (Saretzki et al., 2004). We found that overexpression of miR-590 significantly decreased DNA damage of mESCs by detecting the state of SSB and DSB damage. Our results suggested that miR-590 might be the link between rapid proliferation and DNA damage repair in mESCs. Furthermore, we found that miR-590 can accelerate the process of DNA damage repair after irradiation by X-ray but that miR-590 does not weaken the radiation sensitivity. These results showed that miR-590 not only influences the proliferation and cell cycle to assist the process of damage repair but also participates in DNA damage repair, especially for SSB and DSB repair.
Activin signaling has been reported to be crucial in maintaining ESC pluripotency and proliferation (Beattie et al., 2005; Ogawa et al., 2007). Activin performs its function through Acvr2a, which is required for binding Activin to cause downstream p-SMAD2 activation, thus initiating Activin signaling. Activin signaling has also been reported to participate in the DNA damage response. A study has also shown that Activin signaling potentiates epithelial support cell proliferation by Acvr2a and Acvr2b. Blocking Activin-Acvr2a/b signaling inhibits epithelial support cell proliferation, whereas an Activin receptor agonist increases proliferation (McCullar et al., 2010). Specific knockout of Activin βA in fetal Leydig cells represses sertoli cell proliferation (Archambeault and Yao, 2010). In our study, we found that the level of Acvr2a expression can mediate the strength of Activin signaling and regulate the efficiency of DSB damage repair, SSB damage repair, and cell proliferation in mESCs. Overexpression of Acvr2a reduced the repair of DNA damage and promoted the proliferation at a faster rate. Knockdown of Acvr2a resulted in the opposite phenomenon. We further found that the level of Acvr2a was higher in mESCs cultured in medium with LIF, which contrasted the expression of miR-590. More importantly, we determined that Acvr2a is the target gene of miR-590 in mESCs. miR-590 regulates the amount of Acvr2a on cell membranes to regulate Activin signaling in mESCs, thereby balancing mESC proliferation and DNA damage repair.
There are two major repair methods for DSB damage repair, namely HRR and nonhomologous end joining (NHEJ). HRR results in error-free repair, but NHEJ sometimes leads to errors. During HRR, DSB damage repair uses a template containing hundreds of base pairs of sequence homology, and usually the sister chromatids are available to serve as templates (Haber, 2000; Morrison et al., 2000; Takata et al., 1998). The S phase of ESCs has a long duration, so most of the ESC genomes would have sister chromatids, which assures the process of HHR (Tichy and Stambrook, 2008). Rad51b has been reported to play an important role in the HRR process to repair DSB, and Rad51b is also a protein kinase that can regulate cell cycle-related genes (Havre et al., 2000; Shrivastav et al., 2008; Stordal and Davey, 2009). In our study, we found that Rad51b is also important for mESCs. Knockdown of Rad51b results in inefficient DNA damage repair and a faster cell cycle, which creates a vicious circle of producing more DSB and SSB damage. We further found that Rad51B can be regulated by the miR-590-Acvr2a pathway. Rad51b can be upregulated by miR-590 or knockdown of Acvr2a in mESCs. Moreover, knockdown of Rad51b attenuated the effects caused by knockdown of Acvr2a or overexpression of miR-590. Our results suggested that miR-590/Acvr2a/Rad51b signaling axis balances the coexistence of DNA damage repair and rapid proliferation, assuring the stabilization of mESCs. Our study provides insights into the regulation of proliferation and DNA damage repair during mESC self-renewal, and it suggests that an ESC-related signaling pathway not only maintains the stabilization of mESC self-renewal or pluripotency but also may be the regulator or responder of DNA damage repair.
Experimental Procedures
mESC Culture
mESCs (E14) were cultured on feeder-free, gelatin-coated plates with Dulbecco’s modified Eagle’s medium (Hyclone) supplemented with LIF, 15% fetal bovine serum (GIBCO), 1% nonessential amino acids (Invitrogen, Life Technologies), 1% Gln (Invitrogen), and 0.18% β-mercaptoethanol (Sigma). mESCs were cultured at 37°C in a humidified 5% CO2 atmosphere. 46C is another kind of mESC line that cultures in the same condition as E14.
Comet Assay
A comet assay was performed according to the comet assay kit (Cwbio).
All other methods can be found in the Supplemental Information.
Acknowledgments
This work was supported by grants obtained from the Ministry of Science and Technology (grant numbers 2011CB965100, 2013CB967600, 2011CBA01100, 2012CB966603, and 2013CB967401), the National Natural Science Foundation of China (grant numbers 91219305, 31210103905, 31371510, 31101061, 31171432, 81170499, 31201107, 31301208, and 81201599), the Science and Technology Commission of Shanghai Municipality (grant number 12ZR1450900), IRT1168 from Ministry of Education, sponsored by Shanghai Rising-Star Program (14QA1403900), the Specialized Research Fund for the Doctoral Program of Higher Education (20110072110039), support of the “Chen Guang” project from the Shanghai Municipal Education Commission and Shanghai Education Development Foundation (12CG19), and the Fundamental Research Funds for the Central Universities (2000219099).
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information
References
- Archambeault D.R., Yao H.H. Activin A, a product of fetal Leydig cells, is a unique paracrine regulator of Sertoli cell proliferation and fetal testis cord expansion. Proc. Natl. Acad. Sci. USA. 2010;107:10526–10531. doi: 10.1073/pnas.1000318107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banáth J.P., Bañuelos C.A., Klokov D., MacPhail S.M., Lansdorp P.M., Olive P.L. Explanation for excessive DNA single-strand breaks and endogenous repair foci in pluripotent mouse embryonic stem cells. Exp. Cell Res. 2009;315:1505–1520. doi: 10.1016/j.yexcr.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Baumann P., Benson F.E., West S.C. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell. 1996;87:757–766. doi: 10.1016/s0092-8674(00)81394-x. [DOI] [PubMed] [Google Scholar]
- Beattie G.M., Lopez A.D., Bucay N., Hinton A., Firpo M.T., King C.C., Hayek A. Activin A maintains pluripotency of human embryonic stem cells in the absence of feeder layers. Stem Cells. 2005;23:489–495. doi: 10.1634/stemcells.2004-0279. [DOI] [PubMed] [Google Scholar]
- Bondestam J., Horelli-Kuitunen N., Hildén K., Ritvos O., Aaltonen J. Assignment of ACVR2 and ACVR2B the human activin receptor type II and IIB genes to chromosome bands 2q22.2—>q23.3 and 3p22 and the human follistatin gene (FST) to chromosome 5q11.2 by FISH. Cytogenet. Cell Genet. 1999;87:219–220. doi: 10.1159/000015429. [DOI] [PubMed] [Google Scholar]
- Carlson B.C., Hofer M.D., Ballek N., Yang X.J., Meeks J.J., Gonzalez C.M. Protein markers of malignant potential in penile and vulvar lichen sclerosus. J. Urol. 2013;190:399–406. doi: 10.1016/j.juro.2013.01.102. [DOI] [PubMed] [Google Scholar]
- Chen K., Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 2007;8:93–103. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- Chun J., Buechelmaier E.S., Powell S.N. Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway. Mol. Cell. Biol. 2013;33:387–395. doi: 10.1128/MCB.00465-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuykin I.A., Lianguzova M.S., Pospelova T.V., Pospelov V.A. Activation of DNA damage response signaling in mouse embryonic stem cells. Cell Cycle. 2008;7:2922–2928. doi: 10.4161/cc.7.18.6699. [DOI] [PubMed] [Google Scholar]
- Donaldson C.J., Mathews L.S., Vale W.W. Molecular cloning and binding properties of the human type II activin receptor. Biochem. Biophys. Res. Commun. 1992;184:310–316. doi: 10.1016/0006-291x(92)91194-u. [DOI] [PubMed] [Google Scholar]
- Donoho G., Jasin M., Berg P. Analysis of gene targeting and intrachromosomal homologous recombination stimulated by genomic double-strand breaks in mouse embryonic stem cells. Mol. Cell. Biol. 1998;18:4070–4078. doi: 10.1128/mcb.18.7.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrovska A., Kanamoto T., Lomnytska M., Heldin C.H., Volodko N., Souchelnytskyi S. TGFbeta1/Smad3 counteracts BRCA1-dependent repair of DNA damage. Oncogene. 2005;24:2289–2297. doi: 10.1038/sj.onc.1208443. [DOI] [PubMed] [Google Scholar]
- Favreau A.J., Sathyanarayana P. miR-590-5p, miR-219-5p, miR-15b and miR-628-5p are commonly regulated by IL-3, GM-CSF and G-CSF in acute myeloid leukemia. Leuk. Res. 2012;36:334–341. doi: 10.1016/j.leukres.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluckiger A.C., Marcy G., Marchand M., Négre D., Cosset F.L., Mitalipov S., Wolf D., Savatier P., Dehay C. Cell cycle features of primate embryonic stem cells. Stem Cells. 2006;24:547–556. doi: 10.1634/stemcells.2005-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordyce C.A., Patten K.T., Fessenden T.B., DeFilippis R., Hwang E.S., Zhao J., Tlsty T.D. Cell-extrinsic consequences of epithelial stress: activation of protumorigenic tissue phenotypes. Breast Cancer Res. 2012;14:R155. doi: 10.1186/bcr3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furue M., Okamoto T., Hayashi Y., Okochi H., Fujimoto M., Myoishi Y., Abe T., Ohnuma K., Sato G.H., Asashima M., Sato J.D. Leukemia inhibitory factor as an anti-apoptotic mitogen for pluripotent mouse embryonic stem cells in a serum-free medium without feeder cells. In Vitro Cell. Dev. Biol. Anim. 2005;41:19–28. doi: 10.1290/0502010.1. [DOI] [PubMed] [Google Scholar]
- Haber J.E. Partners and pathwaysrepairing a double-strand break. Trends Genet. 2000;16:259–264. doi: 10.1016/s0168-9525(00)02022-9. [DOI] [PubMed] [Google Scholar]
- Hasty P., Rivera-Pérez J., Bradley A. The role and fate of DNA ends for homologous recombination in embryonic stem cells. Mol. Cell. Biol. 1992;12:2464–2474. doi: 10.1128/mcb.12.6.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havre P.A., Rice M.C., Noe M., Kmiec E.B. The human REC2/RAD51B gene acts as a DNA damage sensor by inducing G1 delay and hypersensitivity to ultraviolet irradiation. Cancer Res. 1998;58:4733–4739. [PubMed] [Google Scholar]
- Havre P.A., Rice M., Ramos R., Kmiec E.B. HsRec2/Rad51L1, a protein influencing cell cycle progression, has protein kinase activity. Exp. Cell Res. 2000;254:33–44. doi: 10.1006/excr.1999.4725. [DOI] [PubMed] [Google Scholar]
- Hong Y., Stambrook P.J. Restoration of an absent G1 arrest and protection from apoptosis in embryonic stem cells after ionizing radiation. Proc. Natl. Acad. Sci. USA. 2004;101:14443–14448. doi: 10.1073/pnas.0401346101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa K., Ishii H., Saito T. DNA damage-dependent cell cycle checkpoints and genomic stability. DNA Cell Biol. 2006;25:406–411. doi: 10.1089/dna.2006.25.406. [DOI] [PubMed] [Google Scholar]
- Jensen R.B., Carreira A., Kowalczykowski S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010;467:678–683. doi: 10.1038/nature09399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen R.B., Ozes A., Kim T., Estep A., Kowalczykowski S.C. BRCA2 is epistatic to the RAD51 paralogs in response to DNA damage. DNA Repair (Amst.) 2013;12:306–311. doi: 10.1016/j.dnarep.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamoto T., Hellman U., Heldin C.H., Souchelnytskyi S. Functional proteomics of transforming growth factor-beta1-stimulated Mv1Lu epithelial cells: Rad51 as a target of TGFbeta1-dependent regulation of DNA repair. EMBO J. 2002;21:1219–1230. doi: 10.1093/emboj/21.5.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata M., Kawabata T., Nishibori M. Role of recA/RAD51 family proteins in mammals. Acta Med. Okayama. 2005;59:1–9. doi: 10.18926/AMO/31987. [DOI] [PubMed] [Google Scholar]
- Keller G. Embryonic stem cell differentiation: emergence of a new era in biology and medicine. Genes Dev. 2005;19:1129–1155. doi: 10.1101/gad.1303605. [DOI] [PubMed] [Google Scholar]
- Lal A., Pan Y., Navarro F., Dykxhoorn D.M., Moreau L., Meire E., Bentwich Z., Lieberman J., Chowdhury D. miR-24-mediated downregulation of H2AX suppresses DNA repair in terminally differentiated blood cells. Nat. Struct. Mol. Biol. 2009;16:492–498. doi: 10.1038/nsmb.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li V.C., Ballabeni A., Kirschner M.W. Gap 1 phase length and mouse embryonic stem cell self-renewal. Proc. Natl. Acad. Sci. USA. 2012;109:12550–12555. doi: 10.1073/pnas.1206740109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipchina I., Elkabetz Y., Hafner M., Sheridan R., Mihailovic A., Tuschl T., Sander C., Studer L., Betel D. Genome-wide identification of microRNA targets in human ES cells reveals a role for miR-302 in modulating BMP response. Genes Dev. 2011;25:2173–2186. doi: 10.1101/gad.17221311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson A., Levine S.S., Cole M.F., Frampton G.M., Brambrink T., Johnstone S., Guenther M.G., Johnston W.K., Wernig M., Newman J. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–533. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullar J.S., Ty S., Campbell S., Oesterle E.C. Activin potentiates proliferation in mature avian auditory sensory epithelium. J. Neurosci. 2010;30:478–490. doi: 10.1523/JNEUROSCI.5154-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra D., Fernandez P., Bian L., Song N., Li F., Han G., Wang X.J. Smad4 loss in mouse keratinocytes leads to increased susceptibility to UV carcinogenesis with reduced Ercc1-mediated DNA repair. J. Invest. Dermatol. 2013;133:2609–2616. doi: 10.1038/jid.2013.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo M., Peng F., Wang L., Peng L., Lan G., Yu S. Roles of mitochondrial transcription factor A and microRNA-590-3p in the development of bladder cancer. Oncol. Lett. 2013;6:617–623. doi: 10.3892/ol.2013.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momcilovic O., Knobloch L., Fornsaglio J., Varum S., Easley C., Schatten G. DNA damage responses in human induced pluripotent stem cells and embryonic stem cells. PLoS ONE. 2010;5:e13410. doi: 10.1371/journal.pone.0013410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison C., Sonoda E., Takao N., Shinohara A., Yamamoto K., Takeda S. The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J. 2000;19:463–471. doi: 10.1093/emboj/19.3.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskwa P., Buffa F.M., Pan Y., Panchakshari R., Gottipati P., Muschel R.J., Beech J., Kulshrestha R., Abdelmohsen K., Weinstock D.M. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol. Cell. 2011;41:210–220. doi: 10.1016/j.molcel.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa K., Saito A., Matsui H., Suzuki H., Ohtsuka S., Shimosato D., Morishita Y., Watabe T., Niwa H., Miyazono K. Activin-Nodal signaling is involved in propagation of mouse embryonic stem cells. J. Cell Sci. 2007;120:55–65. doi: 10.1242/jcs.03296. [DOI] [PubMed] [Google Scholar]
- Pothof J., Verkaik N.S., van IJcken W., Wiemer E.A., Ta V.T., van der Horst G.T., Jaspers N.G., van Gent D.C., Hoeijmakers J.H., Persengiev S.P. MicroRNA-mediated gene silencing modulates the UV-induced DNA-damage response. EMBO J. 2009;28:2090–2099. doi: 10.1038/emboj.2009.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X., Li T.G., Hao J., Hu J., Wang J., Simmons H., Miura S., Mishina Y., Zhao G.Q. BMP4 supports self-renewal of embryonic stem cells by inhibiting mitogen-activated protein kinase pathways. Proc. Natl. Acad. Sci. USA. 2004;101:6027–6032. doi: 10.1073/pnas.0401367101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson N.C., Phillips D.J., McAlpine T., Shin A., Svobodova S., Toy T., Pillay V., Kirkpatrick N., Zanker D., Wilson K. Activin-A: a novel dendritic cell-derived cytokine that potently attenuates CD40 ligand-specific cytokine and chemokine production. Blood. 2008;111:2733–2743. doi: 10.1182/blood-2007-03-080994. [DOI] [PubMed] [Google Scholar]
- Ruiz S., Panopoulos A.D., Herrerías A., Bissig K.D., Lutz M., Berggren W.T., Verma I.M., Izpisua Belmonte J.C. A high proliferation rate is required for cell reprogramming and maintenance of human embryonic stem cell identity. Curr. Biol. 2011;21:45–52. doi: 10.1016/j.cub.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saretzki G., Armstrong L., Leake A., Lako M., von Zglinicki T. Stress defense in murine embryonic stem cells is superior to that of various differentiated murine cells. Stem Cells. 2004;22:962–971. doi: 10.1634/stemcells.22-6-962. [DOI] [PubMed] [Google Scholar]
- Savatier P., Lapillonne H., Jirmanova L., Vitelli L., Samarut J. Analysis of the cell cycle in mouse embryonic stem cells. Methods Mol. Biol. 2002;185:27–33. doi: 10.1385/1-59259-241-4:27. [DOI] [PubMed] [Google Scholar]
- Shrivastav M., De Haro L.P., Nickoloff J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–147. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- Smih F., Rouet P., Romanienko P.J., Jasin M. Double-strand breaks at the target locus stimulate gene targeting in embryonic stem cells. Nucleic Acids Res. 1995;23:5012–5019. doi: 10.1093/nar/23.24.5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L., Lin C., Wu Z., Gong H., Zeng Y., Wu J., Li M., Li J. miR-18a impairs DNA damage response through downregulation of ataxia telangiectasia mutated (ATM) kinase. PLoS ONE. 2011;6:e25454. doi: 10.1371/journal.pone.0025454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stordal B., Davey R. ERCC1 expression and RAD51B activity correlate with cell cycle response to platinum drug treatment not DNA repair. Cancer Chemother. Pharmacol. 2009;63:661–672. doi: 10.1007/s00280-008-0783-x. [DOI] [PubMed] [Google Scholar]
- Strumberg D., Pilon A.A., Smith M., Hickey R., Malkas L., Pommier Y. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol. Cell. Biol. 2000;20:3977–3987. doi: 10.1128/mcb.20.11.3977-3987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M., Sasaki M.S., Sonoda E., Morrison C., Hashimoto M., Utsumi H., Yamaguchi-Iwai Y., Shinohara A., Takeda S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998;17:5497–5508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M., Sasaki M.S., Sonoda E., Fukushima T., Morrison C., Albala J.S., Swagemakers S.M., Kanaar R., Thompson L.H., Takeda S. The Rad51 paralog Rad51B promotes homologous recombinational repair. Mol. Cell. Biol. 2000;20:6476–6482. doi: 10.1128/mcb.20.17.6476-6482.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker J. The RAD51 gene family, genetic instability and cancer. Cancer Lett. 2005;219:125–135. doi: 10.1016/j.canlet.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Tichy E.D. Mechanisms maintaining genomic integrity in embryonic stem cells and induced pluripotent stem cells. Exp. Biol. Med. (Maywood) 2011;236:987–996. doi: 10.1258/ebm.2011.011107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichy E.D., Stambrook P.J. DNA repair in murine embryonic stem cells and differentiated cells. Exp. Cell Res. 2008;314:1929–1936. doi: 10.1016/j.yexcr.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichy E.D., Pillai R., Deng L., Liang L., Tischfield J., Schwemberger S.J., Babcock G.F., Stambrook P.J. Mouse embryonic stem cells, but not somatic cells, predominantly use homologous recombination to repair double-strand DNA breaks. Stem Cells Dev. 2010;19:1699–1711. doi: 10.1089/scd.2010.0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turinetto V., Orlando L., Sanchez-Ripoll Y., Kumpfmueller B., Storm M.P., Porcedda P., Minieri V., Saviozzi S., Accomasso L., Cibrario Rocchietti E. High basal γH2AX levels sustain self-renewal of mouse embryonic and induced pluripotent stem cells. Stem Cells. 2012;30:1414–1423. doi: 10.1002/stem.1133. [DOI] [PubMed] [Google Scholar]
- Valerie K., Povirk L.F. Regulation and mechanisms of mammalian double-strand break repair. Oncogene. 2003;22:5792–5812. doi: 10.1038/sj.onc.1206679. [DOI] [PubMed] [Google Scholar]
- Villa C., Fenoglio C., De Riz M., Clerici F., Marcone A., Benussi L., Ghidoni R., Gallone S., Cortini F., Serpente M. Role of hnRNP-A1 and miR-590-3p in neuronal death: genetics and expression analysis in patients with Alzheimer disease and frontotemporal lobar degeneration. Rejuvenation Res. 2011;14:275–281. doi: 10.1089/rej.2010.1123. [DOI] [PubMed] [Google Scholar]
- Wan G., Mathur R., Hu X., Zhang X., Lu X. miRNA response to DNA damage. Trends Biochem. Sci. 2011;36:478–484. doi: 10.1016/j.tibs.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Baskerville S., Shenoy A., Babiarz J.E., Baehner L., Blelloch R. Embryonic stem cell-specific microRNAs regulate the G1-S transition and promote rapid proliferation. Nat. Genet. 2008;40:1478–1483. doi: 10.1038/ng.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J., Dalton S. Cell cycle control of embryonic stem cells. Stem Cell Rev. 2005;1:131–138. doi: 10.1385/SCR:1:2:131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.