

Abstract
Background
Embryonic neurogenesis and differentiation in the hypothalamic feeding circuitry is under the control of a variety of diffused morphogens and intrinsic transcription factors, leading to the unique structural and functional characteristics of each nucleus.
Scope of review
The transcriptional regulation of the development of feeding neuroendocrine systems during the period of embryonic neurogenesis and differentiation will be reviewed here, with a special emphasis on genetic and environmental manipulations that yield an adverse metabolic phenotype.
Major conclusions
Emerging data suggest that developmental mechanisms can be perturbed not only by genetic manipulation, but also by manipulations to maternal nutrition during the gestational period, leading to long-lasting behavioral, neurobiological, and metabolic consequences. Leptin is neurotrophic in the embryonic brain, and given that it varies in proportion to maternal energy balance, may mediate these effects through an interaction with the mechanisms of hypothalamic development.
Keywords: Leptin, Developmental origins of health and disease, Hypothalamic neurogenesis, Basic helix-loop helix
Homeostatic control over feeding and energy balance is regulated in large part by the hypothalamus, specifically a network of nuclei comprising the arcuate nucleus (ARC), ventromedial hypothalamus (VMH), paraventricular nucleus (PVN) and the lateral hypothalamus (LH). Work over the past decade has made it clear that the perinatal life is a critical period for the organization and development of the feeding circuitry. Genetic disruptions to leptin signaling, as well as altered maternal nutrition can permanently disrupt this organizational process leading to an obesity-prone phenotype [1]. Prenatal life is also a critical period during which a wide range of genetic and environmental manipulations can alter the structure and function of the feeding circuitry, leading to adverse metabolic outcomes. This review is intended to summarize our current understanding of embryonic hypothalamic neurogenesis and differentiation, and apply this to the context of developmental programming. Understanding the various genetic and transcriptional mechanisms that govern normal development may point to novel directions in the study of developmental programming and the role of hormonal and environmental influences in development.
1. Neurogenesis and differentiation in the developing hypothalamus
Neural tube development can be divided into stages, the first of which is regionalization. During the period prior to embryonic day 10 (E10), the diencephalic region is separated from surrounding regions by the influence of organizing signals such as Wingless/integrins (Wnts), Sonic hedgehog (Shh), Bone morphogenetic proteins (Bmps), and fibroblast growth factors (Fgfs), culminating ultimately in the induction of the NK2 homeobox transcription factor Nkx2.1 — a key marker of hypothalamic tissue (for review, see Ref. [2]). The subsequent neurogenic period encompasses E12-16 [3,4]. During the neurogenic period, neural stem cells in the ventricular zone give rise to neural precursors which, under the influence of factors discussed in this review, go on to assume their mature phenotype and position. Neurons, astrocytes, and oligodendrocytes all derive from the same progenitor pool, so the proneural transcription factors that characterize the neurogenic period simultaneously drive neurogenesis and suppress gliogenesis (for review, see Ref. [5]). This review will begin by covering medially situated nuclei, though it should be noted that neurogenesis in the hypothalamus proceeds in an ‘outside-in’ fashion, with laterally situated nuclei being born earlier [6] (Figure 1).
Figure 1.
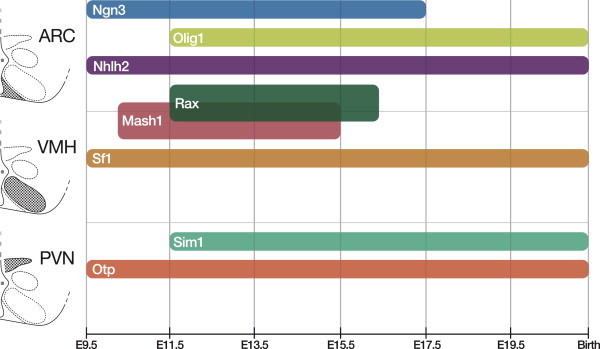
Expression timeline of transcription factors directly implicated in the generation of the hypothalamic feeding nuclei.
2. Arcuate nucleus
The ARC feeding circuitry is characterized by two non-coextensive neural populations expressing either Pro-opiomelanocortin (POMC) or Neuropeptide Y (NPY) and Agouti-related peptide (AgRP). Neural progenitors destined to partake in feeding circuitry may become either orexigenic NPY/AgRP neurons or anorexigenic POMC neurons, and the opposing nature of these populations brings the issue of cell fate decisions into sharp relief. POMC mRNA is first detected in the ventral hypothalamus at E10.5, and NPY expression between E13.5-14.5 [7]. In the period following E13.5, there is a drastic decrease in the number of POMC neurons [7]. This decrease cannot be accounted for by apoptosis, rather it seems that these cells begin to assume a NPY/AgRP phenotype [7]. It is not known what governs this change in cell fate, but it is clear that these two populations are intimately linked during development (Table 1).
Table 1.
Effect of global and conditional transcription factor knockout models on ARC neurons.
| Gene | Type | Model | Effect on POMC neurons | Effect on NPY/AgRP neurons | Ref |
|---|---|---|---|---|---|
| Rax | Homeobox transcription factor | Six3::Cre;Raxflox/null | x | [10] | |
| Bmpr1a | Receptor | Olig1cre/+BMPR1afx/− | − | + | [14] |
| Ngn3 | bHLH transcription factor | Ngn−/− | − | + | [18,17] |
| Ngn3 | bHLH transcription factor | Nkx2.1iCre/+;Ngn3flox/flox | − | = | [19] |
| Nhlh2 | bHLH transcription factor | Nhlh2−/− | = | − | [22,24,25] |
| Nhlh2 | bHLH transcription factor | POMC EGFP/GnRH Cre/NSCL-2ΔloxP/ΔloxP |
− | [27] | |
| Mash1 | bHLH transcription factor | Mash1−/− | − | − | [28] |
| Mash1 | bHLH transcription factor | Mash1+/− | = | + | [28] |
| Rbpjκ | Notch cofactor | Rbpjκfl/fl Nkx2.1-cre+/cre | + | + | [29] |
| Notch1 | Receptor | RosaNotchICD/+ Nkx2.1cre+/cre |
x | x | [29] |
x — Absent/ablated; + Increased; − Decreased; = No change.
The ARC develops in the context of a number of homeobox and basic helix-loop-helix (bHLH) transcription factors with temporally distinct patterns of expression. Retinal and anterior neural fold homeobox (Rax), a homeobox transcription factor traditionally defined by its involvement in early retinal development, is critical for the formation of the ventral neuroendocrine hypothalamus [8]. Rax is expressed in the ventricular zone medial to POMC expressing cells at E12.5, the expected location and time of a transcription factor involved in the generation of these cells [9]. Using Cre-mediated lineage tracing, Lu et al., found evidence that Nkx2.1-expressing neurons in the ARC derived from Rax-expressing lineages [10]. Mice lacking Rax in SIX homeobox 3 (Six3)-expressing cells do not show ventral hypothalamic Nkx2.1 expression, nor do they ever express POMC [10]. This appears to be due to a reassignment of cell fate, as the VMH comes to ectopically express GABAergic neural marker Gad67 in lieu of the expected terminal markers. When Rax is specifically knocked out in Shh-expressing cells, the formation of the VMH is uniquely affected — these animals retain approximately normal patterns of POMC expression [10].
The Oligodendrocyte transcription factor family (Olig1 & Olig2) are bHLH transcription factors expressed medially in the developing CNS, with the highest densities found in the periventricular regions [11,12]. Contrary to what is suggested by their names, Olig1 and Olig2 have been implicated not only in oligodendrogenesis but also in neurogenesis [11,13]. Lineage tracing experiments indicate that a number of POMC and NPY cells derive from Olig1 progenitors, and that the majority of Olig1 expressing cells also express Bone morphogenetic protein receptor 1A (Bmpr1A) [14]. When Bmpr1A is knocked out in Olig1 progenitors, affected mice fail to gain weight when transitioning to solid food [15]. These mice have significantly fewer POMC neurons, and yet significantly greater numbers of AgRP neurons [14]. Neither of these outcomes depend on a difference in neurogenesis, leaving changes in cell fate as a possible mechanism.
The Neurogenins (Ngn1-3) are another family of bHLH transcription factors important in ARC development. Ngn3 expression begins as early as E9.5 and extinguishes by E17.5 [16,17]. Cells co-expressing Ngn3 and POMC are not seen during development, but fate mapping studies show that many of the earliest born (ca. E10.5) POMC neurons arose from Ngn3-expressing progenitors — later born POMC neurons arise from non-Ngn3 expressing progenitors [17]. A large fraction of NPY neurons also appear to have descended from Ngn3-expressing progenitors [17]. Ngn3−/− mice show a substantial reduction in the number of POMC neurons born between E10.5-13.5, though this number rebounds somewhat by E17.5 [17]. On the other hand, NPY neuron counts are dramatically increased in Ngn3−/− mice, a difference that appears as soon as NPY begins to be expressed [18,17]. Mutant mice do not show any changes in overall cell count, implying a change in cell fate as opposed to altered rates of neurogenesis or apoptosis [17].
This work has recently been elaborated upon with the generation of a conditional mutant strain of mice in which Ngn3 is knocked out only in cells that express the ventral diencephalic marker Nkx2.1 [19]. In contrast to Ngn3−/− mice, the conditional mutants survive until adulthood, however they develop early-onset obesity and hyperphagia [19]. Similar to Ngn3−/−, conditional knockouts show a substantial reduction in the number of POMC neurons beginning at E10.5 that continues into adulthood. Curiously, the number of Cocaine- and amphetamine-regulated transcript (CART) cells remains at levels equivalent to wild-type mice, as do the number of estrogen receptor-α and dynorphin-expressing cells. Because CART and POMC expression is almost entirely coextensive in the wild-type ARC, these results suggest that the effects of Ngn3 ablation specifically affect POMC transcription without affecting the number of cells that could have expressed POMC. Interestingly, the increases in NPY cell count seen in the full knockout mice are not seen in the conditional mutants [19]. Given that NPY neurons were shown to be embryonically derived from POMC-expressing progenitors, with a changeover and corresponding reduction in the number of POMC expressing cells occurring sometime after E13.5 [7], the study of Ngn3 raises several interesting questions about the embryonic origins of these cell populations. It seems that the ratio of these two opposing groups is plastic, and factors such as Ngn3 may play a role in fine-tuning that ratio.
Members of the Nescient helix-loop-helix (Nhlh) transcription factor family are critical in the development of both feeding and reproductive circuits in the hypothalamus. Nhlh2 (also known as NSCL-2) is expressed in the developing nervous system as early as E9.5 [20,21]. Mice lacking Nhlh2 are hypogonadal, virtually infertile, and develop adult-onset obesity secondary to reduced spontaneous locomotion [22,23]. When this model was originally characterized, Nissl staining revealed an approximately 25% reduction in overall cell number in the ARC of both male and female Nhlh2−/− mice [22]. These animals were later revealed to be deficient in GnRH and NPY-expressing cells [24]. Initially, anti-ACTH immunohistochemistry appeared to reveal a deficit in POMC neurons [22], though subsequent investigation revealed no difference in POMC mRNA [25]. This discrepancy can be accounted for by the role of Nhlh2 in the regulation of Prohormone convertase (Pc1/3) expression [25–27], the absence of which leads to a post-transcriptional deficit in biologically active POMC cleavage products like Adrenocorticotropic hormone (ACTH) and α-melanocyte stimulating hormone (α-MSH). This issue was recently made more complex by the discovery that POMC neurons are indeed reduced in mice when Nhlh2 is knocked out specifically in GnRH-expressing neurons [27]. This difference becomes greater in magnitude as a function of age [27], lending importance to the study of POMC regulation over the lifespan.
Mash1 (also known as Achaete-scute complex 1 (Ascl1)) is homologous to the Achaete-scute genes found in drosophila. Mash1 is expressed in the retrochiasmic hypothalamic basal plate, as well as in the ventralmost regions of the nearby alar plate. Mash1−/− mice show impaired neurogenesis in the ventral diencephalon, impaired expression of other bHLH transcription factors such as NeuroD, Ngn3, and Nhlh-2, as well as substantial reductions in the number of both NPY and POMC expressing neurons by ED17.5 [28]. Importantly, neither NPY nor POMC expression is completely absent in Mash1−/− embryos, adding more evidence to the existence of multiple pools of progenitors. The situation becomes more complex in heterozygous mutants, as Mash1+/− mice show an 18% increase in NPY neurons and approximately normal numbers of POMC neurons [28]. bHLH transcription factors are occasionally interchangeable and this seems to be the case with Mash1, for when Ngn2 is expressed under the control of the Mash1 promoter, POMC expression is recovered by E12.5 [28]. The timecourse of this result suggests that the control of POMC expression may become more promiscuous as development progresses, allowing for a higher level of redundancy.
The Notch signaling pathway has recently come into focus in hypothalamic development. Mice lacking Rbpjκ, a key downstream effector of Notch signaling, in Nkx2.1-expressing cells show an expansion of the ARC and an increased number of POMC neurons [29]. Mash1 expression is increased in these animals, and this could account for the aberrant POMC expression, as Mash1 is an important factor in the differentiation of POMC neurons [28]. The number of NPY-expressing neurons is also increased, and again the results are suggestive of a role of Mash1 [29]. Mice with a constitutively active Notch1 intracellular domain show a reduction in ARC area and a complete absence of POMC and NPY neurons [29], mirroring the effects of Mash1 knockout [28].
The population of POMC neurons in the ARC is a heterogenous group. POMC neurons may assume either a glutamatergic or GABAergic phenotype [30], some are primarily responsive to leptin [31], others to insulin [32], and there are semi-overlapping pools of POMC neurons that respond to estrogens as well [33,34]. A given neuron's phenotype and role in metabolism cannot be inferred solely from its expression of POMC (or any other neuropeptide) — a complete understanding of that neuron's development requires knowledge of its complement of receptors and ability to participate in signaling cascades, as well as its synaptic connections. Unless a special effort is made, it is impossible to tell whether a give neuron has the expected phenotype (beyond merely expressing a certain neuropeptide), or whether it possesses latent defects. Future research into the development of ARC feeding circuits would benefit from exploring not only the number of POMC and NPY/AgRP neurons, but also their complement of receptors and neurotransmitters, for these aspects of cell fate are presumably also established during the neurogenic period.
3. Ventromedial hypothalamus
Situated lateral to the ARC, the VMH is the other major constituent of the mediobasal hypothalamus, and a key node in the circuits that govern reproduction, feeding, and glucose homeostasis. Steroidogenic factor 1 (Sf1) is a nuclear transcription factor originally identified for its role in adrenal and gonadal development. Early studies of embryo-wide Sf1 expression revealed that it is also expressed within the developing prosencephalon in a region that eventually becomes the VMH [37]. Sf1 expression begins to appear in the brain as early as E9.5 [38], and remains confined entirely within the VMH through the extent of development. A VMH-specific enhancer is found in intron 6 of the Sf1 gene affording a mechanism for the strict adherence of Sf1 to the boundaries of its nucleus [39]. This spatial restriction provides an excellent opportunity for studying VMH function, as it offers a genetic target that (within the CNS) is exclusive to cells of the adult VMH. Mice possessing homozygous disruptions to the Sf1 gene show massive hypocellularity in the VMH, with sparing of the other hypothalamic nuclei [40,41]. These mice also show aberrant patterns of ERα and NPY expression in the presumptive VMH, with these neurons taking on a much more scattered, medially biased arrangement than normal [42,41]. Birthdate labeling experiments using the thymidine analog 5-bromo-2′-deoxyuridine (BrDU) between E10.5-15.5 show that VMH hypocellularity in Sf1−/− mice is not due to differences in neurogenesis [43,44], leaving a deficit in organization or coalescence of the nucleus as a possible mechanism. Evidence of this is seen in the expression of Nkx2.1, which is normally down-regulated as hypothalamic nuclei establish their terminal identities. Nkx2.1 expression lingers in the presumptive VMH of Sf1−/− mice [43], suggesting that the nucleus never reaches a mature state. On the other hand, Brain-derived neurotrophic factor (Bdnf) expression in the VMH is reduced in Sf1−/− mice, as are efferent projections to anterior hypothalamic and limbic structures [43].
Given that Sf1 is critical for the development of most of the cell types of the VMH, it is reasonable to expect that its transcription factor activity is somewhat selective for genes that are characteristic of the VMH. Sf1 binding sites are found upstream of many genes including: Fezf1, Nkx2.2, A2bp1 and others [45]. Morpholino-based knockdown of Fezf1 and A2bp1 in zebrafish leads to substantial malformation of the ventral hypothalamus, attesting to the possibility that these genes are important downstream mediators of Sf1's effects [45]. Further study of these genes in mouse models will likely be required to solidify their importance in VMH development. Interestingly, some of these transcripts continue to be expressed outside of the VMH in Sf1−/− mice, for example Nkx2.2 and A2bp1 continue to be expressed in the zona limitans intrathalamica (ZLI) and dorsal thalamus. It seems, therefore, that the role Sf1 plays as a transcription factor in the developing VMH is substituted for by other factors elsewhere in the developing brain.
Being the two major constituents of the mediobasal hypothalamus, there is a fair bit of overlap between factors contributing to the development of the ARC and VMH. For example, Rax-expressing progenitors occupy the ventricular zone medial to Sf1 expressing cells at E12.5, and lineage tracing indicates that Sf1 neurons derive from Rax-expressing progenitors [10]. Animals lacking Rax in Shh-expressing progenitors have an unusual pattern of Sf1 expression in the VMH as compared to wild-type mice, with approximately normal Sf1 expression in the ventral and dorsal regions of the VMH, but a complete lack of expression in a strip of tissue intermediate to these domains [10]. vGlut2 and Nkx2.1 are expressed in the VMH at P0, but these transcripts are also absent in a band of tissue midway through the dorsoventral extent of the VMH. On the other hand, Gad67 — normally a marker of DMH neurons — was ectopically expressed in the very same band of tissue, suggesting a possible reassignment of cell identity [10].
Mice lacking Mash1 show a number of developmental aberrations in the ventral hypothalamus. Sf1 neurons are largely absent in these mice at E10.5, though the generation of these neurons does recover somewhat by ED12.5 [28]. Ultimately the VMH is strikingly hypocellular in these animals by E17.5, an effect that shows a dose-dependent relationship with the Mash1 zygosity [28]. When Ngn2 is knocked into the Mash1 locus, mice show a recovery in neurogenesis in the VMH but Sf1 expression does not fully recover [28], indicating a level of specificity in the regulation of Sf1 expression. Of course, Mash1 is also active in the nearby ARC, and indeed at E10.5 all POMC neurons also express Sf1 [28]. The ARC and VMH clearly resolve themselves into functionally and anatomically distinct nuclei by adulthood, but how this occurs remains uncertain.
4. Paraventricular hypothalamus
The PVN is one of the main recipients of projections arising from POMC and AgRP/NPY neurons in the ARC. These neurons innervate the various neurosecretory neurons and interneurons of the PVN to bring about behavioral and homeostatic changes associated with the hormonal status sensed by the ARC. Orthopedia (Otp) is a homeobox transcription factor named because its homeodomain sequence in mice possesses features in common with the Otx and Antennapedia sequences in Drosophila. Otp−/− mice are severely growth retarded, and invariably die by day 3 of postnatal life. These mice have severe deficits in PVN and Supraoptic nucleus (SON) formation, as evidenced by a generalized lack of cells in these regions, as well as a large reduction in the PVN and SON marker Calbindin D28K as compared to heterozygous mutants [46,47]. Homozygous mutants showed a complete absence of thyrotropin-releasing hormone (TRH), corticotrophin-releasing hormone (CRH), somatostatin, oxytocin (OT), and vasopressin (AVP) cells [46,47]. This phenotype may be partly accounted for by reductions in neurogenesis between E10-12.5 [46]. Alterations in neural migration are also evident in this model, as cells labeled with the null Otp allele seem to migrate ventrolaterally to a position not seen under normal conditions [46].
The Single-minded homolog 1 gene (Sim1) is a bHLH PAS domain transcription factor. The PAS domain, so named for its presence in Period, Aryl hydrocarbon receptor nuclear translocator, and Sim proteins allows for dimerization between other proteins in this family [48]. Sim1 is expressed throughout the PVN and SON of adult mice; its pattern of expression does not appear to segregate magnocellular and parvocellular compartments in the PVN [48]. Sim1−/− mice show hypocellularity in the PVN and SON, though they maintain normal numbers of cells in the other nearby nuclei. Magnocellular neuropeptides AVP and OT are absent in these animals, as are parvocellular products CRH and TRH [48]. Sim1 is co-expressed with the related bHLH-PAS transcription factor Aryl hydrocarbon receptor nuclear translocator 2 (Arnt2) in the PVN and SON, forming heterodimerized transcription factors that are essential for development of these regions [49,50]. Sim1 and Arnt2 transcription begin during cell migration out of the ventricular zone, after the final cell division is completed [49,48]. Interestingly, while the PVN and SON do not form if either of Sim1 or Arnt2 is missing, certain other domains that express one or the other do remain, suggesting that neuroendocrine development of these regions involves different combinations of bHLH heterodimers [49]. The homeodomain transcription factor Otp is likely upstream of Sim1, as Otp expression remains normal in Sim1−/− mice, but Sim1 expression is disrupted in Otp−/− mice [46]. The complete absence of Sim1 is lethal, but beyond that there is a dose–response relationship between the gene and an obese phenotype. Sim1 haploinsufficiency leads to obesity in both humans and mice [51,52]. Haploinsufficient mice show evidence of compensatory physiological changes including hyperleptinemia, hyperinsulinemia, elevated energy expenditure [53], and increased POMC expression in the ARC [54]. These mice show hypocellularity in the PVN and a marked decrease in both parvocellular and magnocellular-derived hormones [52].
5. Lateral hypothalamus
The two varieties of LH neurons of particular interest in the study of feeding are those that express orexin and those that express Melanin-concentrating hormone (Mch). Orexin neurons are born almost entirely on E12 [55], though Orexin mRNA does not appear until E18 [56]. Mch neurons are born earlier by comparison, with a neurogenic peak at E11 [57]. Mch mRNA is first expressed in rats at E13, protein expression is detected soon after, with a progressive increase in expression as embryonic development progresses [58]. Mch neurons develop under the influence of Sonic hedgehog (Shh), Nkx2.1, as well as Nkx2.2 [57]. Deletion of Shh leads to a deficit in both Mch and Orexin neurons [59,57]. Unlike the previous nuclei mentioned, there has yet to emerge a transcription factor or morphogenic signal that clearly regulates the formation of the LH. The apparent paucity of embryological research into this nucleus may be due to the fact that it is a rather large and heterogenous region and the neurons that relate to feeding make up a relatively small percent of the LH. Factors that govern the formation and migration of these neurons may be particularly subtle.
6. Early-life influences on developmental processes
Hypothalamic development is readily affected by genetic manipulations, with outcomes ranging from mild hypocellularity and altered differentiation to the complete absence of one or more cell populations. Manipulations that affect the prenatal environment also affect hypothalamic development — a phenomenon known as developmental programming. While genetic and programming models often differ in the severity of their outcomes, they may have a similar mechanistic basis. In order to explore this, a brief review of programming models known to affect hypothalamic morphology is in order.
7. Maternal food restriction
Exposing pregnant animals to food restriction during gestation leads to offspring that demonstrate relative hyperphagia. This is accompanied by a reconfiguration of hypothalamic peptide expression to support a hyperphagic phenotype, including reduced POMC expression in the ARC [60], and increased NPY expression in the ARC upon postnatal exposure to a high-fat diet [61]. Interestingly, these animals also demonstrated a generalized hypocellularity in the ARC as revealed by hematoxylin/eosin staining [60]. These animals also showed reductions in POMC and NPY immunopositive neurons — a result that mirrors the observed alterations in mRNA expression [60]. Desai et al. found evidence that prenatal food restriction (50% of ad lib from E10 to term) leads to decreased neurogenesis and retarded migration in the hypothalamus over the late embryonic period in rats [62]. In another study, offspring of food restricted dams showed evidence of increased proliferation in the ARC, VMH, PVN and median eminence [63]. The earliest proliferative timepoint investigated here was E19, long after the critical period for generating feeding-relevant neurons. The result, therefore, may reflect an increase in the rate of the gliogenesis that normally takes place later in development.
8. Maternal obesity
Rats prenatally exposed to a high fat diet show increased body weight and hyperleptinemia in adulthood [64]. P1 offspring of obese dams had decreased NPY, POMC, and ObRB expression in the ARC, as well as decreased expression of the intracellular leptin signalers STAT3 and SOC3 [65]. Differences in neuropeptide expression are also seen in other feeding nuclei. P0 pups of high-fat diet consuming dams had marked increases in the number of Orexin and MCH mRNA expressing cells in the LH, as well as increases in Galanin, Enkephalin, and Dynorphin expression in the PVN [66]. Interestingly, these changes were accompanied by evidence of increased neural proliferation in the PVN and LH, as measured by the incorporation of BrDU given at timepoints ranging from E11-E15 [66]. Interestingly, BrDU labeled neurons showed a high rate of co-localization with Orexin and MCH mRNA in the LH, and with Galanin, Enkephalin and Dynorphin in the PVN, indicating that the surplus neurons assumed an orexigenic neuropeptidergic identity in these feeding areas [66].
9. Maternal insulin resistance
Maternal diabetes is often a confound in studies of maternal obesity, but it can be studied on its own and there is great benefit in doing so. Indeed, both human and animal data suggest that maternal diabetes can predispose offspring to obesity and metabolic disorders independent of maternal obesity [35]. Maternal diabetes in mice realized through injection of streptozotocin (STZ) on E5.5 leads to obesity, hyperphagia, and hyperglycemia in offspring [36]. This is accompanied by attenuated central leptin sensitivity, and paradoxically elevated numbers of POMC expressing neurons [36]. In fact, similar effects on POMC neuron population are seen in offspring of dams heterozygous for a functioning insulin receptor [35]. NPY/AgRP neurons remain at normal levels in both models [35,36]. Neurogenesis in this model has not yet been studied in depth, so the origin of these surplus POMC neurons remains to be discovered.
10. Maternal protein deprivation
Maternal protein deprivation is typically accomplished against the background of an isocaloric diet and can be interpreted as a model of low-quality nutrition, rather than frank under-nutrition. The phenotype of offspring subjected to maternal protein restriction is not unlike that seen in other developmental models, with adult-onset obesity and glucose intolerance being key features [67,68]. Protein restriction leads to reduced brain volume in general, though reductions in regional volume and density are not universal: many regions appear to remain unchanged. In offspring of dams fed an isocaloric, 8% protein diet, morphological alterations of specific hypothalamic nuclei were variable. The PVN, for example, showed a decrease in volume but an increase in density. The VMH, in contrast, showed increased volume and increased density [69]. The volume and cell density of the ARC remained at levels equivalent to control animals in this study, however protein restricted offspring did have significantly fewer NPY immunopositive cells [69]. A detailed survey of embryonic neurogenesis and differentiation remains to be conducted using this model, though these morphological changes do raise interesting questions.
11. Leptin as a neurotrophic factor
The emerging evidence that prenatal nutrition can alter hypothalamic development in specific ways raises the question of how maternal nutrition is communicated to the fetus. Leptin is an ideal candidate to fill such a role, given that it is a hormonal signal of energy balance with known postnatal neurotrophic effects [70]. The rodent placenta produces little leptin, yet is permeable to leptin derived from the maternal side of circulation [71]. Circulating leptin in pregnant rats gradually climbs over the course of pregnancy, reaching a peak at the end of gestation [72,71]. This dynamic is correlated with increased placental expression of short isoforms of the leptin receptor, thereby offering a mechanism for the increase in transplacental exchange [71]. In rats, term fetuses of obese dams consuming a high-fat diet had elevated serum insulin and leptin as compared to control fetuses of non-obese dams [73], suggesting that fetal leptin does reflect maternal energy balance.
The embryonic brain is sensitive to leptin, and can respond to it in meaningful ways. ObRb is expressed throughout the rodent brain as early as E10.5, though hypothalamic expression does not begin until ED14 [74,75]. Leptin's physiological and behavioral effects develop over time. The JAK2/STAT3 signaling pathway regulated by leptin develops gradually in rodents, reaching adult levels of function by P13 [76]. The prenatal signaling pathways activated by leptin are somewhat less studied, but several investigations have begun to do so. Primary cultures derived from ED18 mouse hypothalamus do not show STAT3 phosphorylation upon leptin exposure, but do show decreased NPY and POMC expression [77]. Interestingly, the JAK2/STAT3 pathway is not generally disabled in these cells, as ciliary neurotrophic factor (CNTF) exposure can still induce pSTAT3 [77].
Emerging data from in vivo and in vitro experiments strongly suggest that leptin acts as a neurotrophic factor. Progenitor cells of the ventricular zones express ObRb mRNA as early as E12.5 [74]. Mice possessing the ob/ob mutation that deprives them of secreted leptin have significantly reduced brain weight, volume, and cortical volume as compared to wild-type animals [78]. Functional leptin signaling plays a role in cortical development, as ob/ob mice show reduced neurogenesis beginning on ED14 and cortical hypocellularity by E16 [79]. This is likely attributable to a neurotrophic effect of leptin, as neurosphere cells derived from the embryonic cortex showed increased proliferation when exposed to leptin in the culture medium [79]. Neurons throughout the hypothalamus of these animals show reduced cross-sectional areas, though the LH is not affected in this way, and is thus unique among the nuclei examined [78]. The ob/ob mutation is also associated with mild hypocellularity in both the ARC and VMH in the C57BL/6J background strain [80]. The absence of an effect in the LH may be due to its lateral position and the fact that the cells of the LH are, on average, born earlier than those of the medial nuclei. Since ObRb expression in the developing hypothalamus does not begin until ED14, it could be that neurons born before that time-point are simply unaffected by the presence or absence of leptin, or that leptin affects cell fate but not neurogenesis itself.
Leptin is capable of inducing neurosphere proliferation in tissue derived from the embryonic diencephalon, and this process is dependent on intact Notch, STAT3, and ERK signaling [81]. The neurotrophic potency of leptin can be modified by early-life under-nutrition. Neurosphere cultures harvested from newborn rat pups exposed to gestational food restriction show reduced proliferation under standard conditions, as well as an impaired neurotrophic response to leptin added to the culture medium [62]. These findings neatly complement evidence of reduced embryonic neurogenesis in undernourished fetuses [62]. The JAK2/STAT3 pathway is generally thought to be hyporesponsive to leptin during the early phases of neurodevelopment [77], yet inhibition of STAT3 signaling severely curtails leptin's ability to induce nestin expression, indicating a role for JAK/STAT3 signaling in leptin's neurotrophic effects [81]. It may be the case that because the neural progenitor population is relatively small compared to the population of the hypothalamus in general, localized pSTAT3 responses in the ventricular zone were obscured in broader investigations. On the other hand, the neurotrophic effects of leptin may be owed to some form of cross-talk with other JAK2/STAT3-based neurotrophins, which function normally throughout development [77].
Taken together, these results show that embryonic neurogenesis (or the conditions of adult hypo- and hypercellularity that are used to infer the rate of embryonic neurogenesis) is positively correlated with gestational nutrition. That is to say, conditions of nutritional excess lead to increased rates of proliferation and adult hypercellularity, while conditions of privation lead to the opposite. By the virtue of link between maternal nutrition and fetal leptin exposure, as well as the evidence that leptin acts as a neurotrophic factor, there seems to be sufficient circumstantial evidence to implicate leptin in the genesis of these effects. To be sure, leptin is not the only possible factor in this equation. The glucocorticoids, for example have known effects on developmental programming [82], but since they impair the passage of leptin across the placental barrier [71], leptin may be the more fundamental factor. What remains to be determined, then, is how leptin might act on the developmental processes known to govern embryonic neurogenesis, migration, and differentiation in the various hypothalamic nuclei known to regulate food intake.
12. Leptin interacts with proneural transcription factors and morphogens
Positioned as it is at the intersection of prenatal nutrition and neurogenesis, the theory that leptin signaling unites the two has a good deal of esthetic appeal. However, a thoroughly characterized model of how gestational hypo- or hyperleptinemia could act on the developmental machinery to bring about the changes it is implicated in has not yet emerged. The following section will review the available evidence supporting such a link. Because the relationship between leptin and the mediators of neurodevelopment has not been thoroughly explored in the embryonic brain, it will be necessary, in this instance, to venture beyond this context and explore the putative relationship in adulthood, or in other systems. Extrapolations such as these are not intended to provide direct evidence for the posited link between leptin and neurodevelopment, but rather to demonstrate that such a link as at least theoretically possible, and therefore a worthy avenue for future research.
The Bmp family of morphogens have a plausible link to the developmental effects of leptin. A growing body of evidence suggests the existence of a novel form of interaction between the two signalers. Bmp4, like leptin, is capable of activating STAT3. This occurs indirectly through a novel signal transduction pathway involving the serine–threonine kinase FKBP12/rapamycin-associated protein (FRAP), the ultimate result of which appears to allow for a synergistic interaction between Bmp4 and signalers that exploit the JAK/STAT3 pathway (Cntf, etc.) [83]. The extent to which leptin interacts with this pathway is presently unknown, but given that this form of signal integration is capable of regulating cellular fate, it would be an interesting area for future research. Recent evidence further supports the possibility of an interaction between Bmp signaling and leptin. Injection of leptin into P7 mice leads to an increase in pSMAD expression in the ARC, indicating a leptin-induced activation of the canonical Bmp signal transduction pathway [14]. While this has not yet been tried in the embryonic brain, it seems reasonable to suspect that the same interaction can occur there as well. This is especially interesting in light of the recently discovered importance of Bmp signaling in regulation the pre- and post-natal development of the ARC feeding circuitry [14].
There is limited evidence that leptin can affect the expression of Shh. One study showed that leptin can induce Shh and Ptc expression in primary rat hepatic stellate cells through interactions between the ObRb receptor and the PI3K/Akt signaling pathway [84]. This pathway is active in the adult [85] and neonatal brain, but does not have a role in the postnatal neurotrophic effects of leptin [86]. Given Shh's role in promoting the formation of the ventral diencephalon, and the fact that several of the bHLH transcription factors that govern hypothalamic neurogenesis are downstream of hedgehog signaling [87], it is tempting to speculate that leptin's pro-neurogenic effects on the embryonic mediobasal hypothalamus involve Shh in some fashion. This becomes a complicated matter to test in vivo, as models of maternal obesity presumably raise serum cholesterol, and post-translational modification of the Shh peptide with cholesterol is an important regulator of its function [88], thereby confounding the effects of cholesterol and leptin [66].
A great deal of the refinement and differentiation that takes place throughout the embryonic hypothalamus is regulated by homeobox transcription factors, and emerging evidence points to a link between these and leptin signaling. Nkx2.1 is critical for the development of the ventral hypothalamus [89], and continues to be expressed into adulthood [90,91]. Nkx2.1 is upregulated by fasting in adult animals [91], and down-regulated by both central and peripheral leptin injection [90]. In the adult brain, Nkx2.1 serves to regulate the expression of feeding peptides by upregulating AgRP and downregulating POMC. In the embryonic brain, this regulatory property may in fact be important in terminal differentiation of these cell populations. Bsx, another hypothalamically expressed homeobox transcription factor [92] was recently shown to be regulated by energy status in adulthood, increasing under the influence of exogenous ghrelin or fasting, and decreasing in response to leptin or refeeding [93]. Bsx is co-expressed with AgRP and NPY in the ARC, and appears to be critical in regulating their constitutive and fasting-induced gene expression [94].
Some bHLH transcription factors are also known to be regulated by leptin in the adult. Of those investigated, Sim1 presents a relatively strong case. Peripheral leptin is capable of upregulating PVN Sim1 expression in the adult [54]. Fetal leptin may do the same thing, and if that is the case then this relationship could mean that gestational undernourishment and the hyperphagic phenotype it can produce in offspring mimics, in some capacity, Sim1 haploinsufficiency. The bHLH transcription factor Nhlh2 is expressed in the ARC and PVN into adulthood, and is strongly implicated in energy balance regulation, having been shown in several [95,96] (but not all [97]) studies to be regulated acutely by leptin in adult animals. Nhlh2 is often colocalized with POMC in ARC neurons [25], however it does not directly affect POMC mRNA expression. Rather, Nhlh2 regulates the expression of the PC 1/3 enzymes that cleave the POMC prepropetide into its biologically active forms [26,25]. Importantly, Nhlh2 is a dimerization partner for STAT3, its availability in developing neurons may regulate the transcriptional response to leptin in adulthood and in early life. This is likely of particular importance for STAT3-dependent neurodevelopmental processes such as the postnatal outgrowth of axons from POMC expressing ARC neurons [86].
13. Conclusion
The successful implementation of adult homeostatic function depends on the successful development of the hypothalamus, and in an era of epidemic obesity it is crucial to thorough understand that development. The genetic control of embryonic neurogenesis and differentiation is increasingly well understood, and with this understanding comes a raft of new opportunities for exploring the mechanisms of developmental programming. This review explores the possibility that leptin is a novel neurotrophic factor acting as a bridge between these domains. Further research along these lines offers exciting implications for a deeper understanding of the development of metabolic systems.
Acknowledgements
This work was supported by an Ontario Graduate Scholarship to H.M., as well as a Carleton University Research Award and National Sciences and Engineering Research Council of Canada Grant to A.A.. We thank Dr. Barbara Woodside for her reading of earlier versions of this manuscript.
Contributor Information
Harry MacKay, Email: harry.m@gmail.com.
Alfonso Abizaid, Email: alfonso_abizaid@carleton.ca.
Conflict of interest
None declared.
References
- 1.Coupe B., Bouret S.G. Development of the hypothalamic melanocortin system. Frontiers in Endocrinology. 2013;4:38. doi: 10.3389/fendo.2013.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinez-Ferre A., Martinez S. Molecular regionalization of the diencephalon. Frontiers in Neuroscience. 2012;6:73. doi: 10.3389/fnins.2012.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ifft J. An autoradiographic study of the time of final division of neurons in rat hypothalamic nuclei. Journal of Comparative Neurology. 1972;144:193–204. doi: 10.1002/cne.901440204. [DOI] [PubMed] [Google Scholar]
- 4.Ishii Y., Bouret S.G. Embryonic birthdate of hypothalamic leptin-activated neurons in mice. Endocrinology. 2012;153:3657–3667. doi: 10.1210/en.2012-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rowitch D.H., Kriegstein A.R. Developmental genetics of vertebrate glial-cell specification. Nature. 2010;468:214–222. doi: 10.1038/nature09611. [DOI] [PubMed] [Google Scholar]
- 6.Altman J., Bayer S. Development of the diencephalon in the rat. Correlation of the embryonic development of the hypothalamus with the time of origin of its neurons. Journal of Comparative Neurology. 1978;182:973–993. doi: 10.1002/cne.901820512. [DOI] [PubMed] [Google Scholar]
- 7.Padilla S., Carmody J., Zeltser L. Pomc-expressing progenitors give rise to antagonistic populations in hypothalamic feeding circuits. Nature Medicine. 2010;16:403–405. doi: 10.1038/nm.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L., Mathers P., Jamrich M. Function of Rx, but not Pax6, is essential for the formation of retinal progenitor cells in mice. Genesis. 2000;28:135–142. [PubMed] [Google Scholar]
- 9.Muranishi Y., Terada K., Furukawa T. An essential role for Rax in retina and neuroendocrine system development. Development, Growth & Differentiation. 2012;54:341–348. doi: 10.1111/j.1440-169X.2012.01337.x. [DOI] [PubMed] [Google Scholar]
- 10.Lu F., Kar D., Gruenig N., Zhang Z.W., Cousins N., Rodgers H.M. Rax is a selector gene for mediobasal hypothalamic cell types. The Journal of Neuroscience. 2013;33:259–272. doi: 10.1523/JNEUROSCI.0913-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takebayashi H., Yoshida S., Sugimori M., Kosako H., Kominami R., Nakafuku M. Dynamic expression of basic helix-loop-helix Olig family members: implication of Olig2 in neuron and oligodendrocyte differentiation and identification of a new member, Olig3. Mechanisms of Development. 2000;99:143–148. doi: 10.1016/s0925-4773(00)00466-4. [DOI] [PubMed] [Google Scholar]
- 12.Zhou Q., Wang S., Anderson D.J. Identification of a novel family of oligodendrocyte lineage-specific basic helix-loop-helix transcription factors. Neuron. 2000;25:331–343. doi: 10.1016/s0896-6273(00)80898-3. [DOI] [PubMed] [Google Scholar]
- 13.Lu Q., Sun T., Zhu Z., Ma N., Garcia M. Common developmental requirement for Olig function indicates and motor neuron/oligodendrocyte connection. Cell. 2002;109:75–86. doi: 10.1016/s0092-8674(02)00678-5. [DOI] [PubMed] [Google Scholar]
- 14.Peng C.-Y., Mukhopadhyay A., Jarrett J.C., Yoshikawa K., Kessler J.A. BMP receptor 1A regulates development of 18 hypothalamic circuits critical for feeding behavior. The Journal of Neuroscience. 2012;32:17211–17224. doi: 10.1523/JNEUROSCI.2484-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samanta J., Burke G.M., McGuire T., Pisarek A.J., Mukhopadhyay A., Mishina Y. BMPR1a signaling determines numbers of oligodendrocytes and calbindin-expressing interneurons in the cortex. The Journal of Neuroscience. 2007;27:7397–7407. doi: 10.1523/JNEUROSCI.1434-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sommer L., Ma Q., Anderson D.J. Neurogenins, a novel family of atonal-related bHLH transcription factors, are putative mammalian neuronal determination genes that reveal progenitor cell heterogeneity in the developing CNS and PNS. Molecular and Cellular Neuroscience. 1996;241:221–241. doi: 10.1006/mcne.1996.0060. [DOI] [PubMed] [Google Scholar]
- 17.Pelling M., Anthwal N., McNay D., Gradwohl G., Leiter A.B., Guillemot F. Differential requirements for neurogenin 3 in the development of POMC and NPY neurons in the hypothalamus. Developmental Biology. 2011;349:406–416. doi: 10.1016/j.ydbio.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Arai Y., Gradwohl G., Kameda Y. Expression of neuropeptide Y and agouti-related peptide in the hypothalamic arcuate nucleus of newborn neurogenin3 null mutant mice. Cell and Tissue Research. 2010;340:137–145. doi: 10.1007/s00441-009-0925-4. [DOI] [PubMed] [Google Scholar]
- 19.Anthwal N., Pelling M., Claxton S., Mellitzer G., Collin C., Kessaris N. Conditional deletion of Neurogenin3 using Nkx2.1Cre results in a mouse model for the central control of feeding, activity and obesity. Disease Models & Mechanisms. 2013;6:1133–1145. doi: 10.1242/dmm.011916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Begley C.G., Lipkowitz S., Göbel V., Mahon K.A., Bertness V., Green A.R. Molecular characterization of NSCL, a gene encoding a helix-loop-helix protein expressed in the developing nervous system. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:38–42. doi: 10.1073/pnas.89.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Göbel V., Lipkowitz S., Kozak C., Kirsch I. NSCL-2: a basic domain helix-loop-helix gene expressed in early neurogenesis. Cell Growth & Differentiation. 1992;3:143–148. [PubMed] [Google Scholar]
- 22.Good D., Porter F., Mahon K., Parlow A., Westphal H., Kirsch I. Hypogonadism and obesity in mice with a targeted deletion of the Nhlh2 gene. Nature Genetics. 1997;15:397–401. doi: 10.1038/ng0497-397. [DOI] [PubMed] [Google Scholar]
- 23.Coyle C.A., Jing E., Hosmer T., Powers J.B., Wade G., Good D.J. Reduced voluntary activity precedes adult-onset obesity in Nhlh2 knockout mice. Physiology & Behavior. 2002;77:387–402. doi: 10.1016/s0031-9384(02)00885-5. [DOI] [PubMed] [Google Scholar]
- 24.Cogliati T., Delgado-Romero P., Norwitz E.R., Guduric-Fuchs J., Kaiser U.B., Wray S. Pubertal impairment in Nhlh2 null mice is associated with hypothalamic and pituitary deficiencies. Molecular Endocrinology. 2007;21:3013–3027. doi: 10.1210/me.2005-0337. [DOI] [PubMed] [Google Scholar]
- 25.Jing E., Nillni E.A., Sanchez V.C., Stuart R.C., Good D.J. Deletion of the Nhlh2 transcription factor decreases the levels of the anorexigenic peptides alpha melanocyte-stimulating hormone and thyrotropin-releasing hormone and implicates prohormone convertases I and II in obesity. Endocrinology. 2004;145:1503–1513. doi: 10.1210/en.2003-0834. [DOI] [PubMed] [Google Scholar]
- 26.Fox D.L., Good D.J. Nescient helix-loop-helix 2 interacts with signal transducer and activator of transcription 3 to regulate transcription of prohormone convertase 1/3. Molecular Endocrinology. 2008;22:1438–1448. doi: 10.1210/me.2008-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmid T., Günther S., Mendler L., Braun T. Loss of NSCL-2 in gonadotropin releasing hormone neurons leads to reduction of pro-opiomelanocortin neurons in specific hypothalamic nuclei and causes visceral obesity. The Journal of Neuroscience. 2013;33:10459–10470. doi: 10.1523/JNEUROSCI.5287-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McNay D.E.G., Pelling M., Claxton S., Guillemot F., Ang S.-L. Mash1 is required for generic and subtype differentiation of hypothalamic neuroendocrine cells. Molecular Endocrinology. 2006;20:1623–1632. doi: 10.1210/me.2005-0518. [DOI] [PubMed] [Google Scholar]
- 29.Aujla P.K., Naratadam G.T., Xu L., Raetzman L.T. Notch/Rbpjκ signaling regulates progenitor maintenance and differentiation of hypothalamic arcuate neurons. Development (Cambridge, England) 2013;140:3511–3521. doi: 10.1242/dev.098681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wittmann G., Hrabovszky E., Lechan R.M. Distinct glutamatergic and GABAergic subsets of hypothalamic pro-opiomelanocortin neurons revealed by in situ hybridization in male rats and mice. The Journal of Comparative Neurology. 2013;521:3287–3302. doi: 10.1002/cne.23350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Münzberg H., Huo L., Nillni E.A., Hollenberg A.N., Bjørbaek C. Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin. Endocrinology. 2003;144:2121–2131. doi: 10.1210/en.2002-221037. [DOI] [PubMed] [Google Scholar]
- 32.Williams K.W., Margatho L.O., Lee C.E., Choi M., Lee S., Scott M.M. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2010;30:2472–2479. doi: 10.1523/JNEUROSCI.3118-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Y., Nedungadi T.P., Zhu L., Sobhani N., Irani B.G., Davis K.E. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metabolism. 2011;14:453–465. doi: 10.1016/j.cmet.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Souza F.S.J., Nasif S., López-Leal R., Levi D.H., Low M.J., Rubinsten M. The estrogen receptor α colocalizes with proopiomelanocortin in hypothalamic neurons and binds to a conserved motif present in the neuron-specific enhancer nPE2. European Journal of Pharmacology. 2011;660:181–187. doi: 10.1016/j.ejphar.2010.10.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carmody J.S., Wan P., Accili D., Zeltser L.M., Leibel R.L. Respective contributions of maternal insulin resistance and diet to metabolic and hypothalamic phenotypes of progeny. Obesity. 2011;19:492–499. doi: 10.1038/oby.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steculorum S.M., Bouret S.G. Maternal diabetes compromises the organization of hypothalamic feeding circuits and impairs leptin sensitivity in offspring. Endocrinology. 2011;152:4171–4179. doi: 10.1210/en.2011-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ikeda Y., Shen W.H., Ingraham H.A., Parker K.L. Developmental expression of mouse steroidogenic factor-1, an essential regulator of the steroid hydroxylases. Molecular Endocrinology. 1994;8:654–662. doi: 10.1210/mend.8.5.8058073. [DOI] [PubMed] [Google Scholar]
- 38.Ikeda Y., Takeda Y., Shikayama T., Mukai T., Hisano S., Morohashi K.I. Comparative localization of Dax-1 and Ad4BP/SF-1 during development of the hypothalamic-pituitary-gonadal axis suggests their closely related and distinct functions. Developmental Dynamics. 2001;220:363–376. doi: 10.1002/dvdy.1116. [DOI] [PubMed] [Google Scholar]
- 39.Shima Y., Zubair M., Ishihara S., Shinohara Y., Oka S., Kimura S. Ventromedial hypothalamic nucleus-specific enhancer of Ad4BP/SF-1 gene. Molecular Endocrinology. 2005;19:2812–2823. doi: 10.1210/me.2004-0431. [DOI] [PubMed] [Google Scholar]
- 40.Ikeda Y., Luo X., Abbud R., Nilson J.H., Parker K.L. The nuclear receptor steroidogenic factor 1 is essential for the formation of the ventromedial hypothalamic nucleus. Molecular Endocrinology. 1995;9:478–486. doi: 10.1210/mend.9.4.7659091. [DOI] [PubMed] [Google Scholar]
- 41.Majdic G., Young M., Gomez-Sanchez E., Anderson P., Szczepaniak L.S., Dobbins R.L. Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology. 2002;143:607–614. doi: 10.1210/endo.143.2.8652. [DOI] [PubMed] [Google Scholar]
- 42.Dellovade T.L., Young M., Ross E.P., Henderson R., Caron K., Parker K. Disruption of the gene encoding SF-1 alters the distribution of hypothalamic neuronal phenotypes. The Journal of Comparative Neurology. 2000;423:579–589. doi: 10.1002/1096-9861(20000807)423:4<579::aid-cne4>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 43.Tran P.V., Lee M.B., Marín O., Xu B., Jones K.R., Reichardt L.F. Requirement of the orphan nuclear receptor SF-1 in terminal differentiation of ventromedial hypothalamic neurons. Molecular and Cellular Neuroscience. 2003;22:441–453. doi: 10.1016/S1044-7431(03)00027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davis A.M., Seney M.L., Stallings N.R., Zhao L., Parker K.L., Tobet S.A. Loss of steroidogenic factor 1 alters cellular topography in the mouse ventromedial nucleus of the hypothalamus. Journal of Neurobiology. 2004;60:424–436. doi: 10.1002/neu.20030. [DOI] [PubMed] [Google Scholar]
- 45.Kurrasch D.M., Cheung C.C., Lee F.Y., Tran P.V., Hata K., Ingraham H.A. The neonatal ventromedial hypothalamus transcriptome reveals novel markers with spatially distinct patterning. The Journal of Neuroscience. 2007;27:13624–13634. doi: 10.1523/JNEUROSCI.2858-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Acampora D. Progressive impairment of developing neuroendocrine cell lineages in the hypothalamus of mice lacking the Orthopedia gene. Genes & Development. 1999:2787–2800. doi: 10.1101/gad.13.21.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang W., Lufkin T. The murine Otp homeobox gene plays an essential role in the specification of neuronal cell lineages in the developing hypothalamus. Developmental Biology. 2000;227:432–449. doi: 10.1006/dbio.2000.9902. [DOI] [PubMed] [Google Scholar]
- 48.Michaud J.L., Rosenquist T., May N.R., Fan C.-M. Development of neuroendocrine lineages requires the bHLH-PAS transcription factor SIM1. Genes & Development. 1998;12:3264–3275. doi: 10.1101/gad.12.20.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michaud J.L., DeRossi C., May N.R., Holdener B.C., Fan C.M. ARNT2 acts as the dimerization partner of SIM1 for the development of the hypothalamus. Mechanisms of Development. 2000;90:253–261. doi: 10.1016/s0925-4773(99)00328-7. [DOI] [PubMed] [Google Scholar]
- 50.Keith B., Adelman D.M., Simon M.C. Targeted mutation of the murine arylhydrocarbon receptor nuclear translocator 2 (Arnt2) gene reveals partial redundancy with Arnt. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6692–6697. doi: 10.1073/pnas.121494298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holder J.L., Butte N.F., Zinn A.R. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Human Molecular Genetics. 2000;9:101–108. doi: 10.1093/hmg/9.1.101. [DOI] [PubMed] [Google Scholar]
- 52.Kublaoui B.M., Gemelli T., Tolson K.P., Wang Y., Zinn A.R. Oxytocin deficiency mediates hyperphagic obesity of Sim1 haploinsufficient mice. Molecular Endocrinology. 2008;22:1723–1734. doi: 10.1210/me.2008-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Michaud J.L., Boucher F., Melnyk A., Gauthier F., Goshu E., Lévy E. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Human Molecular Genetics. 2001;10:1465–1473. doi: 10.1093/hmg/10.14.1465. [DOI] [PubMed] [Google Scholar]
- 54.Kublaoui B.M., Holder J.L., Gemelli T., Zinn A.R. Sim1 haploinsufficiency impairs melanocortin-mediated anorexia and activation of paraventricular nucleus neurons. Molecular Endocrinology (Baltimore, Md.) 2006;20:2483–2492. doi: 10.1210/me.2005-0483. [DOI] [PubMed] [Google Scholar]
- 55.Amiot C., Brischoux F., Colard C., La Roche A., Fellmann D., Risold P.Y. Hypocretin/orexin-containing neurons are produced in one sharp peak in the developing ventral diencephalon. The European Journal of Neuroscience. 2005;22:531–534. doi: 10.1111/j.1460-9568.2005.04224.x. [DOI] [PubMed] [Google Scholar]
- 56.Steininger T.L., Kilduff T.S., Behan M., Benca R.M., Landry C.F. Comparison of hypocretin/orexin and melanin-concentrating hormone neurons and axonal projections in the embryonic and postnatal rat brain. Journal of Chemical Neuroanatomy. 2004;27:165–181. doi: 10.1016/j.jchemneu.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 57.Croizier S., Amiot C., Chen X., Presse F., Nahon J.-L., Wu J.Y. Development of posterior hypothalamic neurons enlightens a switch in the prosencephalic basic plan. PLoS One. 2011;6:e28574. doi: 10.1371/journal.pone.0028574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brischoux F., Fellmann D., Risold P.Y. Ontogenetic development of the diencephalic MCH neurons: a hypothalamic ‘MCH area’ hypothesis. The European Journal of Neuroscience. 2001;13:1733–1744. doi: 10.1046/j.0953-816x.2001.01552.x. [DOI] [PubMed] [Google Scholar]
- 59.Szabó N.-E., Zhao T., Cankaya M., Theil T., Zhou X., Alvarez-Bolado G. Role of neuroepithelial Sonic hedgehog in hypothalamic patterning. The Journal of Neuroscience. 2009;29:6989–7002. doi: 10.1523/JNEUROSCI.1089-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Garcia A., Palou M., Priego T. Moderate caloric restriction during gestation results in lower arcuate nucleus NPY- and αMSH-neurons and impairs hypothalamic response to fed/fasting conditions in weaned rats. Diabetes, Obesity and Metabolism. 2010:403–413. doi: 10.1111/j.1463-1326.2009.01174.x. [DOI] [PubMed] [Google Scholar]
- 61.Ikenasio-Thorpe B.A., Breier B.H., Vickers M.H., Fraser M. Prenatal influences on susceptibility to diet-induced obesity are mediated by altered neuroendocrine gene expression. The Journal of Endocrinology. 2007;193:31–37. doi: 10.1677/joe.1.07017. [DOI] [PubMed] [Google Scholar]
- 62.Desai M., Li T., Ross M.G. Hypothalamic neurosphere progenitor cells in low birth-weight rat newborns: neurotrophic effects of leptin and insulin. Brain Research. 2011;1378:29–42. doi: 10.1016/j.brainres.2010.12.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Coupé B., Dutriez-Casteloot I., Breton C., Lefèvre F., Mairesse J., Dickes-Coopman A. Perinatal undernutrition modifies cell proliferation and brain-derived neurotrophic factor levels during critical time-windows for hypothalamic and hippocampal development in the male rat. Journal of Neuroendocrinology. 2009;21:40–48. doi: 10.1111/j.1365-2826.2008.01806.x. [DOI] [PubMed] [Google Scholar]
- 64.Chen H., Simar D., Morris M.J. Hypothalamic neuroendocrine circuitry is programmed by maternal obesity: interaction with postnatal nutritional environment. PLoS One. 2009;4:e6259. doi: 10.1371/journal.pone.0006259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morris M.J., Chen H. Established maternal obesity in the rat reprograms hypothalamic appetite regulators and leptin signaling at birth. International Journal of Obesity (2005) 2009;33:115–122. doi: 10.1038/ijo.2008.213. [DOI] [PubMed] [Google Scholar]
- 66.Chang G.-Q., Gaysinskaya V., Karatayev O., Leibowitz S.F. Maternal high-fat diet and fetal programming: increased proliferation of hypothalamic peptide-producing neurons that increase risk for overeating and obesity. The Journal of Neuroscience. 2008;28:12107–12119. doi: 10.1523/JNEUROSCI.2642-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu X.-m., Kong J., Song W.-w., Lu Y. Glucose metabolic and gluconeogenic pathways disturbance in the intrauterine growth restricted adult male rats. Chinese Medical Sciences Journal. 2009;24:208–212. doi: 10.1016/s1001-9294(10)60003-x. [DOI] [PubMed] [Google Scholar]
- 68.Orozco-Solís R., Matos R.J.B., Guzmán-Quevedo O., Lopes de Souza S., Bihouée A., Houlgatte R. Nutritional programming in the rat is linked to long-lasting changes in nutrient sensing and energy homeostasis in the hypothalamus. PLoS One. 2010;5:e13537. doi: 10.1371/journal.pone.0013537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Plagemann A., Harder T., Rake A., Melchior K., Rohde W., Dörner G. Hypothalamic nuclei are malformed in weanling offspring of low protein malnourished rat dams. The Journal of Nutrition. 2000;130:2582–2589. doi: 10.1093/jn/130.10.2582. [DOI] [PubMed] [Google Scholar]
- 70.Bouret S.G., Draper S.J., Simerly R.B. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science. 2004;304:108–110. doi: 10.1126/science.1095004. [DOI] [PubMed] [Google Scholar]
- 71.Smith J.T. Leptin distribution and metabolism in the pregnant rat: transplacental leptin passage increases in late gestation but is reduced by excess glucocorticoids. Endocrinology. 2003;144:3024–3030. doi: 10.1210/en.2003-0145. [DOI] [PubMed] [Google Scholar]
- 72.Szczepankiewicz D., Wojciechowicz T., Kaczmarek P., Nowak K.W. Leptin and its receptors in the course of pregnancy in the rat. International Journal of Molecular Medicine. 2006;17:95–99. [PubMed] [Google Scholar]
- 73.Gupta A., Srinivasan M., Thamadilok S., Patel M.S. Hypothalamic alterations in fetuses of high fat diet-fed obese female rats. The Journal of Endocrinology. 2009;200:293–300. doi: 10.1677/JOE-08-0429. [DOI] [PubMed] [Google Scholar]
- 74.Udagawa J., Hatta T., Naora H., Otani H. Expression of the long form of leptin receptor (Ob-Rb) mRNA in the brain of mouse embryos and newborn mice. Brain Research. 2000;868:251–258. doi: 10.1016/s0006-8993(00)02334-9. [DOI] [PubMed] [Google Scholar]
- 75.Beloosesky R., Gayle D., Amidi F., Ahanya S., Desai M., Ross M. Ontogenic expression of putative feeding peptides in the rat fetal brain and placenta. Nutritional Neuroscience. 2006;9:33–40. doi: 10.1080/10284150600630676. [DOI] [PubMed] [Google Scholar]
- 76.Frontini A., Bertolotti P., Tonello C., Valerio A., Nisoli E., Cinti S. Leptin-dependent STAT3 phosphorylation in postnatal mouse hypothalamus. Brain Research. 2008;1215:105–115. doi: 10.1016/j.brainres.2008.03.078. [DOI] [PubMed] [Google Scholar]
- 77.Carlo A., Pyrski M., Loudes C., Faivre-Baumann A., Epelbaum J., Williams L.M. Leptin sensitivity in the developing rat hypothalamus. Endocrinology. 2007;148:6073–6082. doi: 10.1210/en.2007-0822. [DOI] [PubMed] [Google Scholar]
- 78.Bereiter D., Jeanrenaud B. Altered neuroanatomical organization in the central nervous system of the genetically obese (ob/ob) mouse. Brain Research. 1979;165:249–260. doi: 10.1016/0006-8993(79)90557-2. [DOI] [PubMed] [Google Scholar]
- 79.Udagawa J., Hashimoto R., Suzuki H., Hatta T., Sotomaru Y., Hioki K. The role of leptin in the development of the cerebral cortex in mouse embryos. Endocrinology. 2006;147:647–658. doi: 10.1210/en.2005-0791. [DOI] [PubMed] [Google Scholar]
- 80.Garris D.R. Morphometric analysis of obesity (ob/ob)- and diabetes (db/db)-associated hypothalamic neuronal degeneration in C57BL/KsJ mice. Brain Research. 1989;501:162–170. doi: 10.1016/0006-8993(89)91037-8. [DOI] [PubMed] [Google Scholar]
- 81.Desai M., Li T., Ross M.G. Fetal hypothalamic neuroprogenitor cell culture: preferential differentiation paths induced by leptin and insulin. Endocrinology. 2011;152:3192–3201. doi: 10.1210/en.2010-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cottrell E.C., Holmes M.C., Livingstone D.E., Kenyon C.J., Seckl J.R. Reconciling the nutritional and glucocorticoid hypotheses of fetal programming. FASEB Journal. 2012;26:1866–1874. doi: 10.1096/fj.12-203489. [DOI] [PubMed] [Google Scholar]
- 83.Rajan P., Panchision D.M., Newell L.F., McKay R.D.G. BMPs signal alternately through a SMAD or FRAP-STAT pathway to regulate fate choice in CNS stem cells. The Journal of Cell Biology. 2003;161:911–921. doi: 10.1083/jcb.200211021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Choi S.S., Syn W.-K., Karaca G.F., Omenetti A., Moylan C.A., Witek R.P. Leptin promotes the myofibroblastic phenotype in hepatic stellate cells by activating the hedgehog pathway. The Journal of Biological Chemistry. 2010;285:36551–36560. doi: 10.1074/jbc.M110.168542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hill J., Williams K., Ye C., Luo J., Balthasar N., Coppari R. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. The Journal of Clinical Investigation. 2008;118 doi: 10.1172/JCI32964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bouret S.G., Bates S.H., Chen S., Myers M.G., Simerly R.B. Distinct roles for specific leptin receptor signals in the development of hypothalamic feeding circuits. The Journal of Neuroscience. 2012;32:1244–1252. doi: 10.1523/JNEUROSCI.2277-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rohr K.B., Barth K.A., Varga Z.M., Wilson S.W. The nodal pathway acts upstream of hedgehog signaling to specify ventral telencephalic identity. Neuron. 2001;29:341–351. doi: 10.1016/s0896-6273(01)00210-0. [DOI] [PubMed] [Google Scholar]
- 88.Incardona J.P., Roelink H. The role of cholesterol in Shh signaling and teratogen-induced holoprosencephaly. Cellular and Molecular Life Sciences. 2000;57:1709–1719. doi: 10.1007/PL00000653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pera E.M., Kessel M. Demarcation of ventral territories by the homeobox gene NKX2.1 during early chick development. Development Genes and Evolution. 1998;208:168–171. doi: 10.1007/s004270050170. [DOI] [PubMed] [Google Scholar]
- 90.Kim J.G., Park B.S., Yun C.H., Kim H.J., Kang S.S., D'Elia A.V. Thyroid transcription factor-1 regulates feeding behavior via melanocortin pathway in the hypothalamus. Diabetes. 2011;60:710–719. doi: 10.2337/db10-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim J.G., Nam-Goong I.S., Yun C.H., Jeong J.K., Kim E.S., Park J.J. TTF-1, a homeodomain-containing transcription factor, regulates feeding behavior in the rat hypothalamus. Biochemical and Biophysical Research Communications. 2006;349:969–975. doi: 10.1016/j.bbrc.2006.08.147. [DOI] [PubMed] [Google Scholar]
- 92.Cremona M., Colombo E., Andreazzoli M., Cossu G., Broccoli V. Bsx, an evolutionary conserved Brain Specific homeoboX gene expressed in the septum, epiphysis, mammillary bodies and arcuate nucleus. Gene Expression Patterns. 2004;4:47–51. doi: 10.1016/s1567-133x(03)00151-0. [DOI] [PubMed] [Google Scholar]
- 93.Nogueiras R., Lopez M., Lage R., Perez-Tilve D., Pfluger P., Mendieta-Zeron H. Bsx, a novel hypothalamic factor linking feeding with locomotor activity, is regulated by energy availability. Endocrinology. 2008;149:3009–3015. doi: 10.1210/en.2007-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sakkou M., Wiedmer P., Anlag K., Hamm A., Seuntjens E., Ettwiller L. A role for brain-specific homeobox factor Bsx in the control of hyperphagia and locomotory behavior. Cell Metabolism. 2007;5:450–463. doi: 10.1016/j.cmet.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 95.Vella K.R., Burnside A.S., Brennan K.M., Good D.J. Expression of the hypothalamic transcription factor Nhlh2 is dependent on energy availability. Journal of Neuroendocrinology. 2007;19:499–510. doi: 10.1111/j.1365-2826.2007.01556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Al Rayyan N., Zhang J., Burnside A.S., Good D.J. Leptin signaling regulates hypothalamic expression of nescient helix-loop-helix 2 (Nhlh2) through signal transducer and activator 3 (Stat3) Molecular and Cellular Endocrinology. 2014;384:134–142. doi: 10.1016/j.mce.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nilaweera K.N., Ellis C., Barrett P., Mercer J.G., Morgan P.J. Hypothalamic bHLH transcription factors are novel candidates in the regulation of energy balance. The European Journal of Neuroscience. 2002;15:644–650. doi: 10.1046/j.1460-9568.2002.01894.x. [DOI] [PubMed] [Google Scholar]