

Summary
Hematopoietic stem cells (HSCs) are characterized by the capacity for self-renewal and the ability to reconstitute the entire hematopoietic compartment. Thrombopoietin maintains adult HSCs in a quiescent state through the induction of cell cycle inhibitors p57Kip2 and p19INK4d. Using the p19INK4d−/− mouse model, we investigated the role of p19INK4d in basal and stress-induced hematopoiesis. We demonstrate that p19INK4d is involved in the regulation of HSC quiescence by inhibition of the G0/G1 cell cycle transition. Under genotoxic stress conditions, the absence of p19INK4d in HSCs leads to accelerated cell cycle exit, accumulation of DNA double-strand breaks, and apoptosis when cells progress to the S/G2-M stages of the cell cycle. Moreover, p19INK4d controls the HSC microenvironment through negative regulation of megakaryopoiesis. Deletion of p19INK4d results in megakaryocyte hyperproliferation and increased transforming growth factor β1 secretion. This leads to fibrosis in the bone marrow and spleen, followed by loss of HSCs during aging.
Highlights
-
•
p19INK4d regulates HSC quiescence through inhibition of the G0/G1 transition
-
•
p19INK4d protects HSC from DNA damage and apoptosis during genotoxic stress
-
•
Absence of p19INK4d leads to MK amplification, splenomegaly, and fibrosis development
-
•
p19INK4d controls HSC pool through microenvironment
Hematopoietic stem cells (HSCs) are characterized by the self-renewal capacity and the ability to reconstitute the entire hematopoietic compartment. Raslova and colleagues demonstrate that p19INK4d regulates HSC quiescence through inhibition of the G0/G1 transition and protects HSC from apoptosis during genotoxic stress. They show that p19INK4d controls HSC pool through microenvironment, and its absence leads to MK amplification, splenomegaly, and fibrosis development.
Introduction
Hematopoietic stem cells (HSCs) possess the capacity for self-renewal and multilineage differentiation that underlies the maintenance and reconstitution of the entire hematopoietic compartment. In the bone marrow (BM), the majority of HSCs remain quiescent in the G0 phase of the cell cycle. Upon exposure to stress, the number of mature cells in the blood circulation is reduced, causing quiescent HSCs to enter the cell cycle and replenish the hematopoietic system.
Accumulating evidence has demonstrated that quiescence is an active process regulated by intrinsic factors, including numerous transcription factors, as well as environmental cues, including the Notch, Wnt, and Sonic hedgehog signaling pathways. Cytokines also play a major role in regulating the HSC cell cycle. For example, thrombopoietin (TPO), the primary regulator of megakaryocyte (MK) differentiation, is required for the maintenance of adult HSC quiescence, via induction of the cell cycle inhibitors, p57Kip2 and p19INK4d (Qian et al., 2007; Yoshihara et al., 2007). TGF-β1 can also enforce HSC quiescence by inducing p57Kip2 expression (Scandura et al., 2004; Yamazaki and Nakauchi, 2009).
Cyclin-dependent kinase inhibitors (CDKIs) directly control the cell cycle by inhibiting cell cycle entry. They are divided into two groups: the INK4 family and the Cip/Kip family.
Cip/Kip proteins are expressed at higher levels in HSCs than in progenitor cells (Passegué et al., 2005). The role of p21Cip1 in HSCs is restricted to cell cycle regulation under stress conditions (van Os et al., 2007). p27Kip1 deficiency does not affect HSC numbers or HSC self-renewal, but alters the proliferation of progenitor cells (Cheng et al., 2000a). p57Kip2 is an important regulator of hematopoiesis in the aorta gonads mesonephros region, where HSCs emerge (Mascarenhas et al., 2009). Inducible loss of p57Kip2 in hematopoietic cells has demonstrated the critical role of this CDKI in the maintenance of HSC quiescence (Matsumoto et al., 2011).
More recent studies have implicated INK4 members in the control of HSC functions. p16INK4a expression is repressed by EZH1 in young animals (Hidalgo et al., 2012). Its expression increases with age, contributing to the decreased self-renewal, homing, and repopulating activities of HSCs in response to stress (Janzen et al., 2006). However, the role of p16INK4a in regulating steady-state HSC aging in vivo appears to be less important (Attema et al., 2009).
p18INK4c is also involved in the senescence of HSCs. In its absence, the number of cycling HSCs increases, although the overall self-renewal capacity of the HSC compartment remains unchanged (Yuan et al., 2006). In a sense, p18INK4c deletion mimics HSC aging, and it may, paradoxically, have an opposite role to p16INK4a and p21Cip1.
Prior evidence for the importance of p19INK4d in HSC cell cycle regulation was reported using the Thpo−/− mouse model. These mice exhibit a significant decrease in HSC numbers that correlates with decreased expression of p19INK4d and p57Kip2 (Qian et al., 2007; Yoshihara et al., 2007). p19INK4d plays a role in the development of the cerebral cortex (Zindy et al., 1999), controls mouse spermatogenesis (Zindy et al., 2001), and is involved in macrophage differentiation (Adachi et al., 1997). We previously demonstrated that by linking endomitotic arrest and terminal maturation p19INK4d is implicated in megakaryopoiesis (Gilles et al., 2008). In addition to its role in cell cycle and differentiation, in neuroblastoma cells, p19INK4d is also important for DNA repair and resistance to apoptosis in response to diverse forms of genotoxic stress (Ceruti et al., 2009). Interestingly, sensory hair cells lacking p19INK4d aberrantly re-enter the cell cycle and subsequently undergo apoptosis. This supports the notion that p19INK4d is essential for maintenance of their postmitotic state (Chen et al., 2003) and that p19INK4d therefore acts as an antiapoptotic regulator.
Although a number of studies suggest that p19INK4d is implicated in HSC biology, its precise role remains unclear. Using a p19INK4d−/− mouse model, we demonstrate that p19INK4d is involved in the regulation of HSC quiescence. Under conditions of genotoxic stress, the absence of p19INK4d leads to HSC apoptosis during the S/G2-M phases of the cell cycle. We also report that p19INK4d participates in control of the HSC microenvironment during aging, as its absence results in BM and spleen fibrosis, and loss of HSCs.
Results
Loss of p19INK4d Affects HSC Numbers
The expression profile of different CDKIs has previously been reported in C57BL/6 mice (Passegué et al., 2005). Since the p19INK4d−/− mice in this study are in a mixed C57BL/6 × Sv129j genetic background, we first compared the expression level of all CDKIs in mice with a mixed genetic background with that of pure C57BL/6 mice. In long-term (LT) HSCs, purified as the CD48−CD150+ (SLAM) cell subpopulation of primitive Lin−SCA1+C-KIT+ (LSK) BM cells, p19INK4d transcript levels were similar in both genetic backgrounds. Slight differences in transcript levels were observed in both the immature and more mature progenitor populations (Figure 1A). The levels of other CDKIs expressed during hematopoiesis were similar between C57BL/6 and C57BL/6 × Sv129j mice (Figures S1A–S1D available online). We observed relatively high levels of p27Kip1 and p19INK4d transcripts in SLAM cells compared with p18INK4c, p21Cip1 and p57Kip2, suggesting that p27Kip1 and p19INK4d have important roles in quiescent HSCs.
Figure 1.
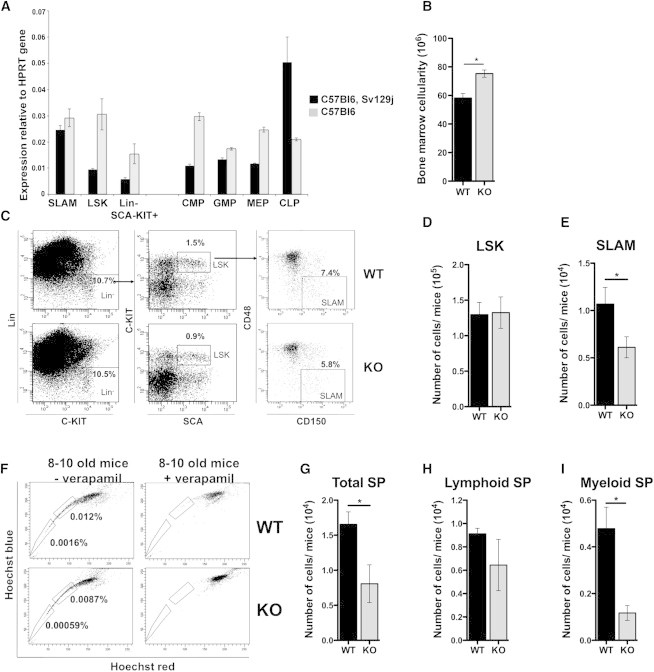
p19INK4d−/− Mice Exhibit a Defect in the HSC Compartment
(A) mRNA expression level of p19INK4d in different populations of C57BL/6 and C57BL/6-Sv129j mice: SLAM (Lin−SCA1+C-KIT+CD48−CD150+), LSK, myeloid progenitors (Lin−SCA1−C-KIT+), CLP (Lin−SCA1lowC-KITlowCD127+THY-1−), CMP (Lin−SCA1−C-KIT+FCγR−CD34+), GMP (Lin−SCA1−C-KITt+FCγR+CD34+), and MEP (Lin−SCA1−C-KIT+FCγR−CD34−). Each population represents a pool derived from 10 mice. Data are normalized to HPRT transcript levels and represent the mean ± SEM of triplicates.
(B) BM cellularity (n = 10).
(C–E) Immunophenotype (C) and number of LSK (D) and SLAM (E) cells (n = 8).
(F–I) Immunophenotype (F) and number of total SP (G), lymphoid SP (H), and myeloid SP (I) populations (n = 4).
Mice that were 8 to 10 weeks old were used. KO, p19INK4d−/−. Data represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, unpaired t test. See also Figure S1.
To investigate the function of p19INK4d, we first analyzed the total number of BM cells in 8- to 10-week-old mice. A 1.23-fold increase in total BM cells was observed in p19INK4d−/− mice (Figure 1B). No significant difference was observed in the absolute number of LSK cells (Figures 1C and 1D). In contrast, a significant decrease (1.75-fold) in the absolute number of SLAM cells (Figures 1C and 1E) was observed for p19INK4d−/− mice. This result was confirmed by characterizing the LSK side population (SP). The upper SP is biased toward lymphoid differentiation and the lower SP toward myeloid differentiation (Challen et al., 2010) (Figure 1F). We confirmed a 2.1-fold decrease in HSC numbers (total SP population) (Figure 1G). No difference was observed in the lymphoid-biased HSC population (Figure 1H), although myeloid-biased HSCs were decreased 4.1-fold in p19INK4d−/− mice (Figure 1I).
p19INK4d Is Involved in the Regulation of HSC Quiescence by Inhibition of the G0/G1 Transition
To study the role of p19INK4d in the cell cycle, we stained LSK and SLAM cells with anti-Ki-67 and Hoechst (Figure 2A). A significant acceleration in the G0/G1 transition was observed in p19INK4d−/− mice, without any significant change in the S/G2-M phases (Figures 2B and 2C). Using LT bromodeoxyuridine (BrdU) incorporation (Figure 2D), a 1.3- and 1.9-fold decrease in the percentage of BrdU-positive cells was observed in LSK and SLAM cells at day (D) 8. The effect on BrdU incorporation was even more pronounced at D11 and D21, particularly for SLAM cells (Figures 2E and 2F). These experiments revealed an increased G0/G1 transition (Figure 2C), with a decreased progression to S/G2-M phases for p19INK4d−/− HSCs. Since no increase in the number of LSK cells was detected, we investigated whether the absence of p19INK4d was associated with apoptosis in cycling LSK and SLAM cells (Figure 2G). We observed a significant increase in the percentage of annexin-V+/BrdU+ cells in the LSK population at D11 (1.4-fold) and D21 (1.6-fold) in p19INK4d−/− mice (Figure 2H), which was even more pronounced in the SLAM population (3.5- and 1.7-fold at D11 and D21, respectively; Figure 2I). These results suggest that p19INK4d protects immature progenitors and, to an even greater extent, HSCs from apoptosis in the S/G2-M phases of the cell cycle.
Figure 2.
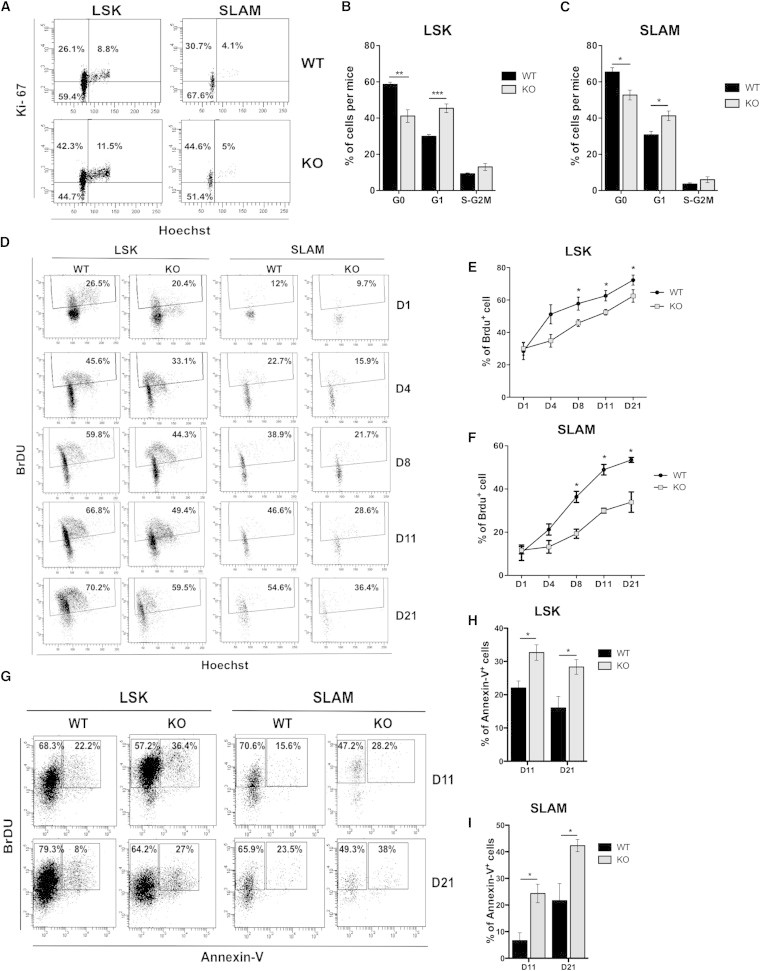
Absence of p19INK4d Leads to Accelerated Exit of HSCs from Quiescence and Increased Apoptosis in S/G2-M Cell Cycle Phases
(A–C) Cell cycle analysis by Ki-67/Hoechst costaining. Immunophenotype (A) and frequencies of cell cycle phases in LSK (B) and SLAM (C) populations (n = 4) are shown.
(D–F) In vivo cell proliferation assays post-BrdU administration in mice by BrdU/Hoechst costaining. Immunophenotype (D) and frequencies of cell cycle phases in LSK (E) and SLAM (F) populations are shown. One of three independent experiments with similar results is presented. For each experiment, three mice were analyzed per point.
(G–I) Apoptosis assessment at D11 or D21 post-BrdU administration. Immunophenotype (G) and frequencies of annexin-V+ cells in LSK (H) and SLAM (I) populations are shown. One of two independent experiments with similar results is presented. For each experiment, four mice were analyzed per data point.
Mice that were 8 to 10 weeks old were used. KO, p19INK4d−/−. Data represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, unpaired t test.
p19INK4d Plays a Crucial Role in Stress-Induced Hematopoiesis
To investigate the consequence of this increased HSC cell cycle progression that is followed by enhanced apoptosis in p19INK4d−/− mice during hematopoietic stress, we examined the effect of stress-inducing compounds, 5-fluorouracil (5-FU), hydroxyurea (HU), and menadione.
p19INK4d−/− mice exhibited a markedly increased mortality after weekly injections of 5-FU (Figure 3A). To confirm that the increased mortality is related to a more rapid exhaustion of p19INK4d−/− HSCs as a consequence of the enhanced G0/G1 transition that is followed by apoptosis, we analyzed the cell cycle of WT and p19INK4d−/− HSC/Ps 4 days after 5-FU injection (Figure 3B). As expected, we observed a substantial decrease in quiescent p19INK4d−/− LSK (2.6-fold) and SLAM (11.6-fold) cell numbers, with a concomitant increase in G1 phase cells, whereas no differences in the number of cells in S/G2-M phases were observed (Figures 3C and 3D).
Figure 3.
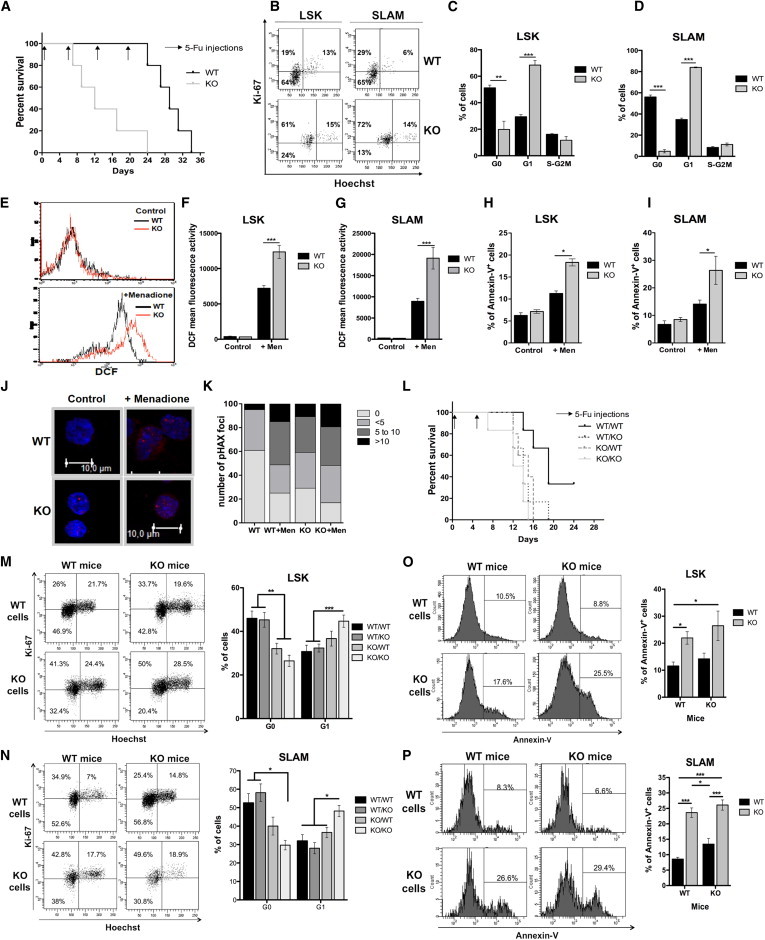
Hematopoietic Stress in p19INK4d−/− Mice Leads to a Defect in Survival due to Increased HSC Exhaustion and Apoptosis
(A) Survival rate of mice injected weekly with 5-FU. One of two independent experiments with similar results is presented. For each experiment, five mice were injected.
(B–D) Cell cycle analysis by Ki-67/Hoechst costaining four days after 5-FU injection. Immunophenotype (B) and cell cycle distribution in LSK (C) and SLAM (D) populations are shown (n = 4).
(E–G) Immunophenotype of DCF staining (E) and DCF mean fluorescence in LSK (F) and SLAM (G) populations before and after oxidative stress induction by menadione (n = 4).
(H and I) Annexin-V staining before and after oxidative stress induction by menadione in LSK (H) and SLAM (I) populations (n = 4).
(J and K) P-H2AX staining in LSK cells before and after oxidative stress induction with menadione. Representative pictures (J) are shown. Nuclei are stained with DAPI (blue) and DNA-DSBs with an anti-P-H2AX antibody (red). (K) The number of P-H2AX foci per 100 LSK cells.
(L) Survival rate of sublethally irradiated mice reconstituted with 2 × 105 Lin− cells and injected with 5-FU 1 month after reconstitution. One of two independent experiments with similar results is presented. For each experiment, six mice were injected.
(M and N) The distribution of cells in the cell cycle was determined by Ki-67/Hoechst costaining 2 days after 5-FU injection. Immunophenotype and frequencies of cell cycle phases in LSK (M) and SLAM (N) populations in 2-month-old transplanted lethally irradiated mice are shown (n = 6).
(O and P) Annexin-V staining two days after 5-FU treatment in LSK (O) and SLAM (P) populations (n = 4).
Mice that were 8 to 10 weeks old were used. KO, p19INK4d−/−. Data represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, unpaired t test. See also Figure S2.
To investigate the role of p19INK4d under other stress conditions, we treated Lin− cells with menadione and measured the endogenous levels of reactive oxygen species (ROS). ROS levels were identical in WT and p19INK4d−/− LSK and SLAM cells under basal conditions (Figures 3E–3G). Following menadione treatment, ROS levels were greatly increased in LSK and SLAM cells of both WT and p19INK4d−/− mice; however, levels were significantly higher in the p19INK4d−/− genetic background (1.7- and 2.2-fold increase in LSK and SLAM populations, respectively) (Figures 3E–3G). In accordance with the fact that high ROS levels increase DNA damage and subsequently induce apoptosis, we observed a 1.75- and 1.8-fold increase in annexin-V+ cells in p19INK4d−/− LSK and SLAM populations, respectively (Figures 3H and 3I). Analysis of the level of phospho-H2AX (P-H2AX), a biomarker of DNA double-strand breaks (DNA-DSBs), revealed that in comparison to WT cells, the number of large P-H2AX foci in p19INK4d−/− LSK cells was already higher prior to stress induction and further increased following the induction of oxidative stress (Figures 3J and 3K).
To determine the role of p19INK4d during another type of stress, we treated WT and p19INK4d−/− mice with HU for 3 consecutive days and subsequently analyzed cell cycle and apoptosis in the LSK and SLAM populations (Figures S2A–S2D). As expected, a slight increase in the percentage of S phase (Figures S2A and S2B) and apoptotic (Figure S2C) cells was observed after HU treatment of both WT and p19INK4d−/− mice. However, no difference in the percentage of annexin-V+ LSK and SLAM cells was observed between WT and p19INK4d−/− mice after HU treatment (Figure S2D). Together, these results suggest that p19INK4d is predominantly involved in the response to genotoxic and oxidative stresses, but not to replicative stress induced by HU treatment.
Finally, to determine whether the effect of 5-FU on HSCs was cell autonomous or related to the microenvironment, we transplanted either p19INK4d−/− or WT hematopoietic Lin− cells into lethally irradiated recipients of both genetic backgrounds in order to obtain chimeric mice. One month after hematopoietic reconstitution, 5-FU was injected twice and mortality was monitored as described previously. As expected, all p19INK4d−/−/p19INK4d−/− and p19INK4d−/−/WT mice died after treatment with 5-FU (150 mg/kg), while survival of WT/WT mice was higher, in the range of 20%–35%. Surprisingly, all chimeric WT/p19INK4d−/− mice also died, but with delayed kinetics compared with the p19INK4d−/−/p19INK4d−/− mice, suggestive of a defect in the hematopoietic microenvironment of p19INK4d−/− mice (Figure 3L).
To differentiate the cell-autonomous and environmental effects of p19INK4d on the cell cycle (Figures 3M and 3N) and apoptosis (Figures 3O and 3P) following induction of stress, a lower dose of 5-FU (50 mg/kg) was injected into the chimeric mice 2 months after reconstitution. Within 48 hr, a more rapid exhaustion of quiescent p19INK4d−/− LSK (Figure 3M) and SLAM (Figure 3N) cells was observed compared with WT cells, regardless of the genetic background of the recipient mice. This cell cycle re-entry was accompanied by an increased percentage of p19INK4d−/− cells in G1 and S/G2-M phases (Figures 3M, 3N, S2E, and S2F). Increased apoptosis was also detected in both LSK (Figure 3O) and SLAM (Figure 3P) p19INK4d−/− cells compared with WT cells, regardless of the genetic environment. These results clearly demonstrate the intrinsic role of p19INK4d in maintaining HSC quiescence and in protecting cells from apoptosis following genotoxic stress.
In conclusion, under conditions of stress, p19INK4d loss leads to enhanced cell cycle progression, accumulation of ROS, and increased DNA damage and apoptotic cell death in a cell autonomous manner.
p19INK4d−/− BM Cells Exhibit High Reconstitutive Activity, but the p19INK4d−/− Stroma Displays a Defective HSC Supportive Activity
To better assess the defect in the microenvironment, we tested the reconstitutive capacities of p19INK4d−/− BM Lin− cells in competitive transplantation assays (Figure 4A). At 15-weeks postengraftment, the increase in the fraction of p19INK4d−/− SLAM, LSK, and myeloid cells was 9.9-, 2.26-, and 2.75-fold, respectively (Figure 4B). This result strongly supports the hypothesis that HSCs, which are less numerous in p19INK4d−/− mice, exhibit a higher repopulating capacity than WT HSCs when injected into WT recipients. This paradoxical result further implies that the BM microenvironment is defective in p19INK4d−/− mice. To test this hypothesis, adherent stromal BM cells were isolated from p19INK4d−/− and WT mice, and their capacity to maintain LT culture initiating cells (LTC-ICs) was assessed (Figure 4C). WT and p19INK4d−/− cells exhibited a 2.3- and 2.6-fold reduction, respectively, in the number of LTC-ICs on the p19INK4d−/− stroma compared with the WT stroma (Figure 4D). No marked difference in LTC-IC content was observed between WT and p19INK4d−/− mice, regardless of the stroma used. This demonstrates that p19INK4d is also required for maintenance of the hematopoietic function of the BM microenvironment. To confirm this result in vivo, reconstitution of lethally irradiated WT or p19INK4d−/− mice with WT or p19INK4d−/− donor total BM cells was performed. In this assay, WT and p19INK4d−/− BM cells have similar irradiation protective properties in WT recipients. However, a defect in survival was observed in p19INK4d−/− recipient mice transplanted with WT BM, and this occurred to an even greater extent with p19INK4d−/− BM cells (Figure 4E). These results clearly demonstrate an important role for p19INK4d in the regulation of both the BM environment and HSCs.
Figure 4.
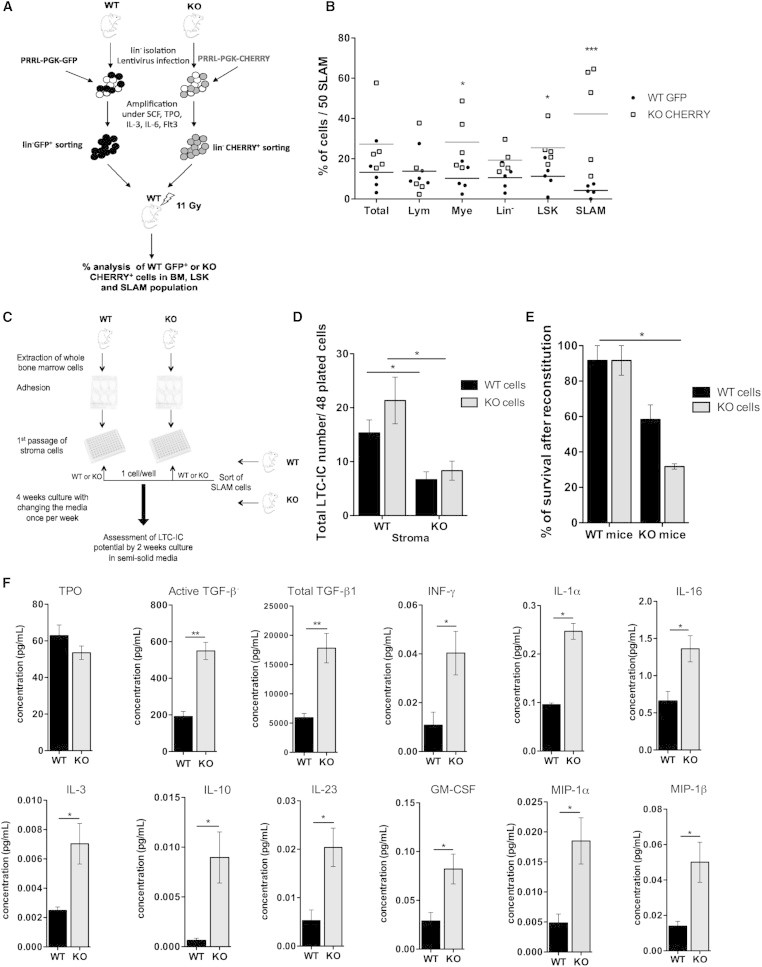
The Hematopoietic Supportive Function of the Microenvironment Is Altered by p19INK4d Loss
(A and B) Competitive engraftment between Lin−GFP+WT cells and Lin−CHERRY+KO cells in recipient lethally irradiated WT mice. The experimental protocol (A) and percentage of WT GFP+ and KO CHERRY+ BM-derived cells (B) are shown in the following hematopoietic populations: myeloid (GR1+CD11b+), T lymphoid (CD3+), Lin−, LSK, and SLAM per 50 transplanted SLAM cells at 15 weeks postengraftment. Each dot represents data from a single mouse (n = 5).
(C and D) LTC-IC growth of WT and KO SLAM cells on WT and KO stroma. Experimental protocol (C) and number of LTC-IC/48 plated cells (D) (n = 3) are shown.
(E) Potential of WT and KO HSC engraftment in WT and KO mice evaluated as the percentage of survival after reconstitution. One of two independent experiments with similar results is presented (n = 6).
(F) Level of different cytokines/chemokines in BM supernatants, (n = 5) for TPO and TGF-β1, (n = 3) for interferon γ (IFN-γ), interleukin 1α (IL-1α), IL-16, IL-3, IL-10, IL-23, granulomonocyte stem call factor, and macrophage inflammatory protein 1α (MIP-1α) and MIP-1β.
Mice that were 8 to 10 weeks old mice were used. KO, p19INK4d−/−. Data represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, unpaired t test.
Hematopoietic tissue maintenance under homeostatic conditions is controlled by a large number of soluble cytokines, growth factors, cell-cell interaction molecules, and extracellular matrix components. We determined the levels of two pivotal growth factors in fluid extracted from the BM of WT and p19INK4d−/− mice that exert positive (TPO) or negative (transforming growth factor β1 [TGF-β1]) regulation HSC numbers. No differences in TPO levels were observed, while TGF-β1 levels were significantly increased in p19INK4d−/− mice. The levels of several other inflammatory cytokines were also significantly increased in p19INK4d−/− mice (Figure 4F).
A Defect in the MK Lineage Is Involved in the Defective Microenvironment and Leads to Progressive Fibrosis and Osteosclerosis in p19INK4d−/− Mice
MKs play an important role in the BM niche via secretion of cytokines such as TGF-β1 and by cell-cell interactions. This led us to perform a detailed study of the MK lineage in p19INK4d−/− mice during aging, as this is a biological process that is known to modify HSC properties. While no differences were observed in MK-P numbers of 8- to 10-week-old mice compared with WT mice, the frequency of MK-Ps at 20–25 weeks was increased 2.3-fold in the BM and 6.1-fold in the spleen of p19INK4d−/− mice (Figures 5A and 5B). In contrast, no differences were observed for all other BM myeloid progenitors during aging (Figures S3A and S3B). In peripheral blood, a significant increase in red blood cells and lymphocytes detected in WT mice was not observed in p19INK4d−/− mice (Figures S3C–S3F). In contrast, the slight increase in platelet counts observed in young p19INK4d−/− animals was more pronounced (1.35-fold) in p19INK4d−/− mice that were older than 91 weeks (Figure S3F).
Figure 5.
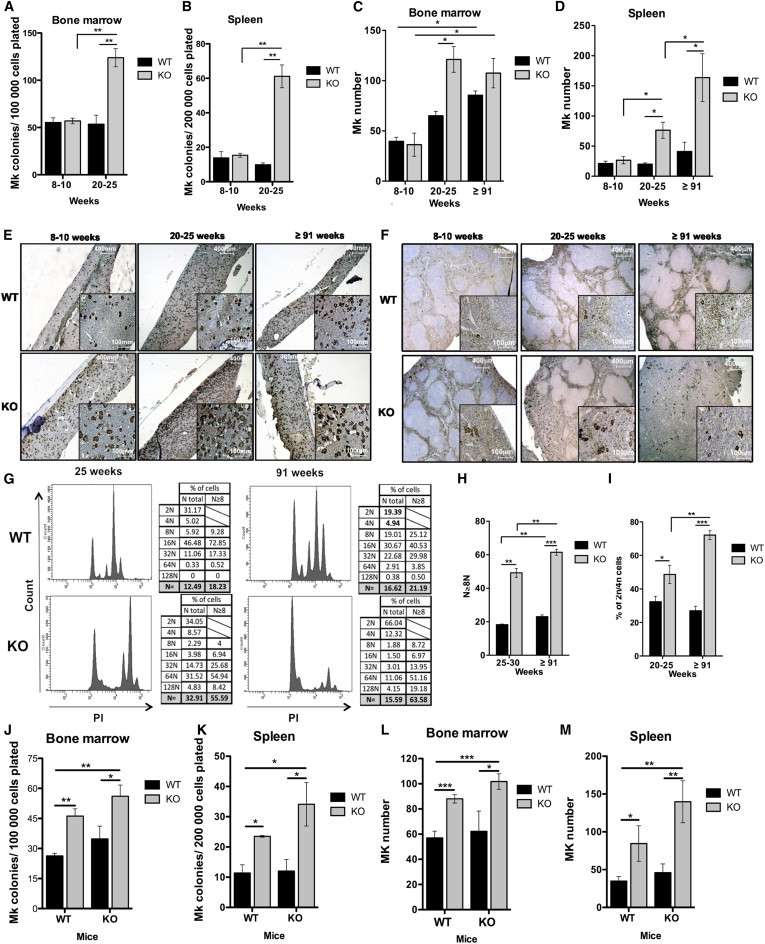
Absence of p19INK4d Leads to a Progressive Age-Dependent Increase in the Number of MKs in BM and Spleen of Mice
(A and B) Number of MK-Ps derived from total BM cells (A) and total spleen cells (B) of 8- to 10-week-old (n = 3) and 20- to 25-week-old (n = 4) mice cultured in fibrin clots. Experiments were performed in triplicate for each biological replicate.
(C–F) Counts of MKs identified by vWF staining in BM (C) and spleen (D) of 8- to 10-week-old (n = 3), 20- to 25-week-old (n = 4) and in >91-week-old (n = 6) mice. The number of MKs was determined on equal areas of three independent image frames for each mouse. Representative pictures are shown for BM (E) and spleen (F).
(G) Cell cycle was measured by PI staining (middle panel) within the MK (CD41+) population (left panel). Total and ≥8N MK ploidy level (right panel) was calculated for 20- to 25-week-old and >91-week-old mice.
(H) Mean ploidy level calculated for ≥8N MK.
(I) Percentage of 2N/4N MK cells in BM of 20- to 25-week-old and >91-week-old mice. Data represent mean ± SEM (n = 4) for 20- to 25-week-old and (n = 3) for >91-week-old mice.
(J and K) Number of MK-P derived from total BM cells (J) and total spleen cells (K) in 8-month-old transplanted WT and KO mice cultured in fibrin clots. Experiments were performed in triplicate for each biological replicate.
(L and M) Counts of MKs identified by vWF staining in BM (L) and spleen (M) in 8-month-old transplanted WT and KO mice (n = 5). The MK number was determined on equal areas of three independent image frames for each mouse.
KO, p19INK4d−/−. Data represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, unpaired t test. See also Figure S3.
To determine whether the increased MK-P number has consequences on the total MK cell population, we performed a histological examination of BM and spleen from mice of different ages. Accumulation of clustered, polylobulated MKs (Figure S3G) in the BM and spreading of the red pulp (Figure S3H) in the spleen of p19INK4d−/− mice were detected during aging. von Willebrand factor (vWF) staining of BM (Figures 5C and 5E) and spleen (Figures 5D and 5F) revealed that the total MK number increased progressively with age in BM of WT mice. However, this increase occurred more rapidly in the BM of p19INK4d−/− mice, reaching a plateau at 20–25 weeks (3.4-fold increase) (Figure 5C). A 6.2-fold increase in the number of MKs was observed in the spleen of aged 19INK4d−/− mice, whereas MK numbers remained constant in the spleen of aged WT mice (Figure 5D). Since p19INK4d regulates MK ploidization arrest (Gilles et al., 2008), we next investigated MK ploidy (Figure 5G). Regardless of age, the modal ploidy level remained 64N in p19INK4d−/− mice compared with 16N for WT MKs (Figure 5H). However, a progressive increase in the proliferative 2N/4N MK population was noted in BM during aging (Figure 5I). Overall these results demonstrate that p19INK4d is involved not only in the arrest of ploidization, but also in regulation of the cell cycle in MK-Ps, thereby explaining the increase in MKs with age.
To determine whether the increase in MKs observed with aging in p19INK4d−/− mice was due exclusively to an intrinsic role of p19INK4d, MK-Ps, and MKs were quantified in 8-month-old chimeric mice. A significant increase in the number of MK-Ps in both BM (Figure 5J) and spleen (Figure 5K) was detected when WT or p19INK4d−/− recipient mice were reconstituted with p19INK4d−/− cells. The same profile was observed for MK numbers in both BM (Figure 5L) and spleen (Figure 5M). These results indicate that MK amplification is due to an intrinsic role of p19INK4d.
The loss of p19INK4d therefore mimics hematopoietic stress, which is normally associated with hematopoietic development in the spleen and induction of splenomegaly. Accordingly, the spleen size did not change for WT mice during aging, whereas a 1.4-fold increase in spleen size for p19INK4d−/− mice was already observed at 8–10 weeks of age. This effect became more pronounced as a function of age (Figure 6A) and was associated with a parallel increase in spleen cellularity (Figure 6B). To determine the role of the microenvironment in the splenomegaly, spleen weights were determined for 8-month-old chimeric mice. A significant increase in spleen weight was detected when WT and p19INK4d−/− recipient mice were reconstituted with p19INK4d−/− cells (Figure 6C), suggesting an intrinsic role of p19INK4d in extramedullary hematopoiesis.
Figure 6.
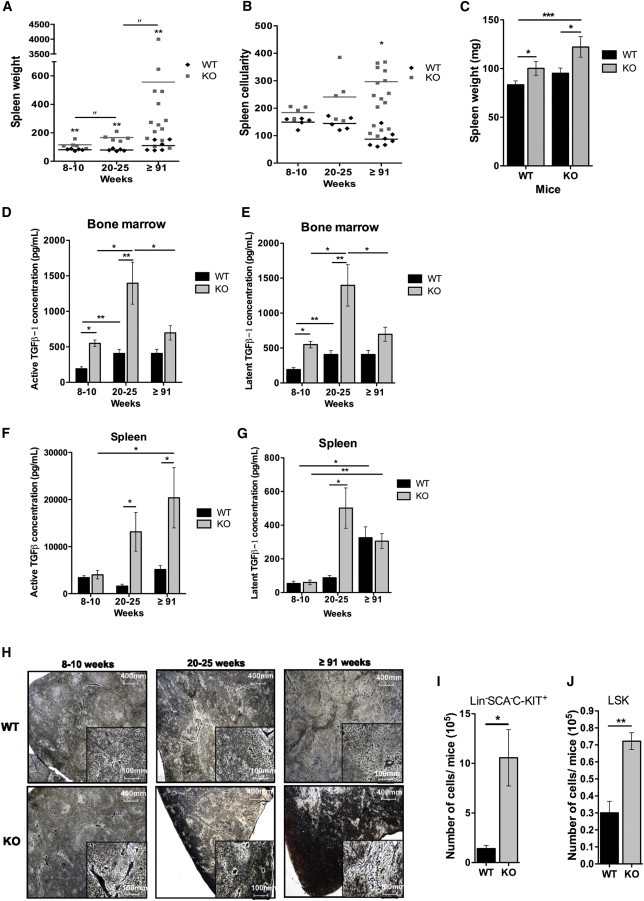
Megakaryopoiesis and Environment Defects in the Absence of p19INK4d Lead to the Development of Fibrosis
(A and B) Spleen weight (A) and cellularity (B) of 8- to 10-week-old (n = 5), 20- to 25-week-old (n = 5) and >91-week-old (n = 6 for WT and n = 14 for KO) mice. Each dot represents data from a single mouse. The ∗ shows the differences between WT and KO mice, and the ″ shows differences between 8- to 10-week-old and 20- to 25-week-old mice or between 8- to 10-week-old and >91-week-old KO mice.
(C) Spleen weights of 8-month-old transplanted mice (n = 5). Data represent mean ± SEM.
(D and E) Dosage by ELISA of total TGF-β1 in fluids extracted from BM (D) and spleen (E) of 8- to 10-week-old (n = 5), 20- to 25-week-old (n = 3) and >91-week-old (n = 3 for WT and n = 8 for KO) mice.
(F and G) Dosage of latent TGF-β1 in fluids extracted from BM (F) and spleen (G) of 8- to 10-week-old (n = 5), 20- to 25-week-old (n = 3) and >91-week-old (n = 3 for WT and n = 8 for KO) mice.
(H) Data represent mean ± SEM. Representative images of 8- to 10-week-old, 20- to 25-week-old, and >91-week-old mice spleens stained for reticulin, highlighting the fibrotic status of the tissue.
(I and J) Number of Lin−SCA1−C-KIT+ (I) and LSK (J) cells in peripheral blood of >91-week-old mice. Data represent mean ± SEM (n = 4).
KO: p19INK4d−/−. Data represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, unpaired t test. See also Figure S4.
An enhanced MK mass is typically associated with increased TGF-β1 secretion. The levels of active and latent TGF-β1 in the extracellular fluids from BM and spleen directly paralleled MK numbers in BM and spleen during aging (Figures 6D–6G). Nonetheless, the level of active TGFβ1 increased in the BM of p19INK4d−/− mice until 20–25 weeks of age, after which it decreased again (Figure 6D). In the spleen, a strong increase was also already observed at 20–25 weeks (Figure 6F). The presence of active TGF-β1 in tissues is usually associated with fibrosis. Increased reticulin fiber deposition was observed during aging in p19INK4d−/− mice. In mice older than 91 weeks, fibrosis was detected in the spleens of all p19INK4d−/− mice (Figure 6H) and in the BM of 20% of mice (Figure S4A). Osteosclerosis could also be observed in these mice (Figure S4B, upper panels). Consistent with the changes observed in myelofibrosis, we also detected an increase in circulating myeloid (Lin−Sca1−Kit+) progenitors (Figure 6I) and LSK cells (Figure 6J) in older mice.
To investigate whether fibrosis was a consequence of abnormal hematopoiesis, we transplanted a pool of total BM and spleen cells from p19INK4d−/− mice with myelofibrosis and a spleen weight of 4 g into lethally irradiated WT recipients. At 6 weeks posttransplantation, splenomegaly was detected in primary recipients and in a less pronounced manner also in secondary recipients (Figure S4C). No osteosclerosis or fibrosis of the BM was observed (Figure S4B). However, fibrosis was detected in the spleen of both primary and secondary recipients, and this correlated with the increased MK numbers (Figure S4D).
These results indicate that the disease is transplantable and that it originates in the hematopoietic system.
p19INK4d Regulates HSC Number during Aging
p19INK4d loss increased MK mass and resulted in elevated TGF-β1 secretion and fibrosis during aging. The TGF-β1 pathway has previously been reported to regulate the HSC pool (Scandura et al., 2004; Yamazaki and Nakauchi, 2009), while DNA damage accumulates in HSCs during aging (Rossi et al., 2007). As we demonstrated above, p19INK4d protects HSCs from apoptosis during genotoxic stress. Loss of p19INK4d could then alter the HSC pool during aging in both an intrinsic and extrinsic manner. Therefore, the number of LSK and SLAM cells was studied in aged animals (Figure 7A). No difference was observed in LSK cells during aging (Figure 7B). In agreement with previously published work (Beerman et al., 2010), we observed a 3-fold increase in SLAM cell numbers in WT mice more than 48 weeks old compared with 8- to 10-week-old mice, while only a slight increase was observed in the SLAM population of p19INK4d−/− mice (Figure 7C). No difference in the total clonogenic potential between WT and p19INK4d−/− SLAM cells from mice that were more than 48 weeks old was detected by in vitro assay on OP9/OP9Δ1 cell lines (Figure 7D).
Figure 7.
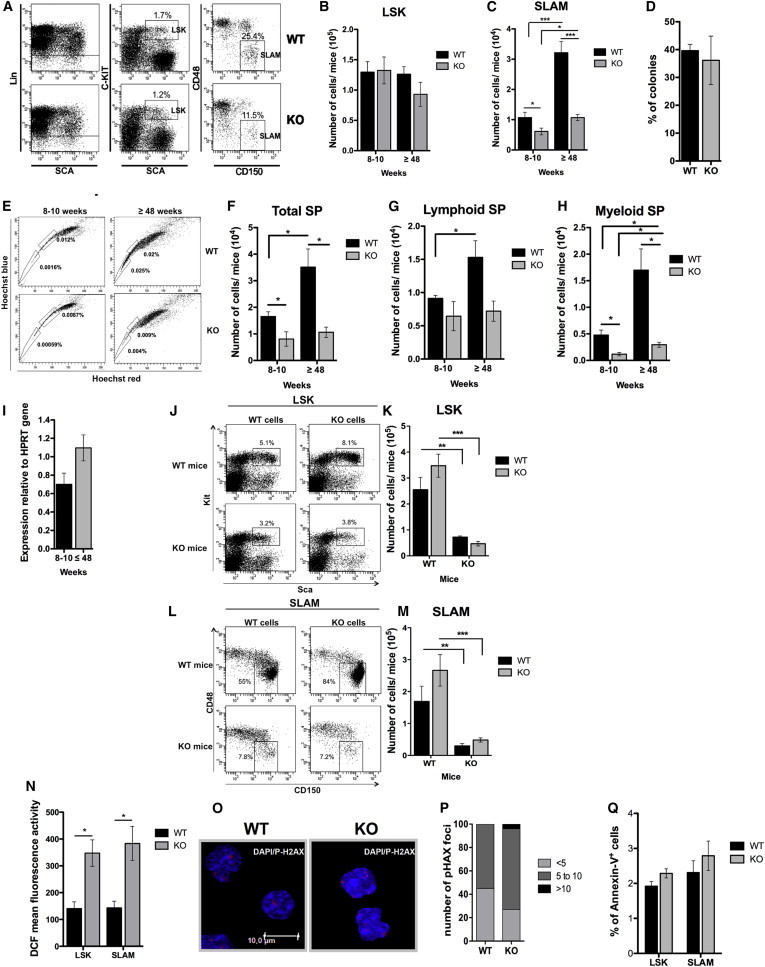
Defect in HSC Amplification during Aging Is Primarily due to Environmental Effects Induced by p19INK4d Loss
(A–C) Immunophenotype (A) and number of LSK (B) and SLAM (C) cells in 8- to 10-week-old (n = 8) and >48-week-old mice (n = 4).
(D) Assessment of biological functions in HSCs of >48-week-old mice (n = 3). For each condition, 144 SLAM cells were plated at one cell/well on OP9/OP9Δ1 stromal cells.
(E–H) Immunophenotype (E) and number of total SP (F), lymphoid SP (G), and myeloid SP (H) cells in 8- to 10-week-old (n = 4) and >48-week-old (n = 3) WT and KO mice.
(I) p19INK4d transcript level in WT SLAM cells during aging. Data are normalized to HPRT levels and represent mean ± SEM of triplicate experiments.
(J–M) Immunophenotype (J and L) and number of LSK (K) and SLAM (M) cells in 8-month-old transplanted mice, KO mice transplanted with WT cells (n = 3), WT mice transplanted with WT cells (n = 4), WT mice transplanted with KO cells (n = 5), and KO mice transplanted with KO cells (n = 5).
(N) DCF mean fluorescence in LSK and SLAM cells in >48-week-old mice (n = 4).
(O and P) P-H2AX staining in LSK cells in 48-week-old mice. Representative pictures (O) are shown. Nuclei are stained with DAPI (blue), and DNA-DSBs are stained by an anti-P-H2AX antibody (red). (P) The number of P-H2AX foci per 100 LSK cells.
(Q) Apoptosis assessment in LSK and SLAM populations in >48-week-old mice (n = 4).
KO: p19INK4d−/−. Data represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, unpaired t test.
The proportion of HSCs with myeloid potential increases with age, and this occurs at the expense of lymphoid potential (Beerman et al., 2010). Investigation of the myeloid versus lymphoid potential during aging (Figure 7E) confirmed an increase in HSC number in WT mice (2.2-fold), but not in p19INK4d−/− mice (Figure 7F). In 8- to 10-week-old WT and p19INK4d−/− mice, the lymphoid SP subpopulation was increased compared with its myeloid counterpart (Figures 7G and 7H). In WT mice, the proportion of myeloid SP increased 3.6-fold in old mice compared with young mice, and it exceeded the lymphoid SP fraction by 1.15-fold (Figures 7G and 7H). Moreover, p19INK4d expression increased during aging in WT mice (Figure 7I). In contrast, the myeloid SP population in p19INK4d−/− mice was only slightly amplified during aging, and no inversion in the ratio of lymphoid and myeloid SP was detected (Figures 7G and 7H).
To determine whether the age-related decrease of HSCs in p19INK4d−/− mice is due to intrinsic and/or extrinsic effects of p19INK4d, the numbers of LSK and SLAM from 8 month-old chimeric mice were analyzed (Figures 7J and 7L). As expected, the p19INK4d−/− recipient mice that were reconstituted with p19INK4d−/− cells (KO/KO) exhibited a 5.5- and 3.5-fold decrease in LSK (Figure 7K) and SLAM cell numbers (Figure 7M), respectively, compared with WT mice transplanted with WT cells (WT/WT). Surprisingly, when p19INK4d−/− recipient mice were transplanted with WT cells (WT/KO), we observed respective decreases of 3.5- and 5.7-fold in LSK and SLAM cell numbers compared with WT/WT. Furthermore, when WT mice were transplanted with p19INK4d−/− cells (KO/WT), a nonsignificant increase in LSK and SLAM numbers was observed compared with WT/WT. This suggests that there is increased turnover of p19INK4d−/− cells in a WT environment.
These results clearly show that the absence of age-associated HSC amplification in a p19INK4d−/− background is due exclusively to environmental effects that are caused by the loss of p19INK4d.
Finally, we analyzed DNA-damage during aging. We observed an increase in ROS accumulation in p19INK4d−/− LSK and SLAM cells (Figure 7N) that was associated with a slight increase in P-H2AX accumulation in p19INK4d−/− LSK cells (Figures 7O and 7P). However, no increase in apoptosis was detected in p19INK4d−/− LSK and SLAM cells (Figure 7Q). These results therefore indicate that p19INK4d plays a crucial extrinsic role in the accumulation of HSCs with myeloid potential.
Discussion
We report here that the p19INK4d CDKI plays at least two important roles in regulation of the HSC pool. One of these is cell autonomous and manifests itself under conditions of stress. The second role relates to the microenvironment and underscores the role of p19INK4d in mediating age-related effects on the HSC pool.
Cell Autonomous Role of p19INK4d in HSCs under Basal and Stress Conditions
The first preliminary evidence for a role of p19INK4d in the regulation of HSC quiescence came from the observation that Thpo−/− mice exhibited a decreased number of dormant HSCs that was associated with decreased p19INK4d and p57CDKN1c expression (Qian et al., 2007). Our studies revealed that p19INK4d−/− mice displayed a slight decrease in the absolute number of HSCs, which decreased further with age. This was particularly the case for HSCs with myeloid potential. As previously described in other cellular models, our in vivo study of HSC proliferation demonstrates increased apoptosis in the absence of p19INK4d. An association between loss of p19INK4d and re-entry into the cell cycle followed by apoptosis has been reported for neurons (Zindy et al., 1999) and terminally differentiated hair cells (Chen et al., 2003). However, in our study, apoptosis in the HSC compartment was measured after in vivo administration of BrdU, which may inherently induce stress. To further investigate the role of p19INK4d in this context, we induced three types of stress. First, we used 5-FU, which induces both replicative and genotoxic stresses in quiescent HSCs. Second, we used menadione, which is a genotoxic agent that induces DNA damage through elevated ROS levels. Third, we used HU, which induces replicative rather than genotoxic stress. Our results clearly demonstrate that 5-FU injection leads to enhanced mortality of p19INK4d−/− mice that is associated with a marked increase in apoptosis following the exit of HSCs from quiescence. The same outcome was observed when p19INK4d−/− cells were transplanted into WT recipients, thereby confirming a cell-autonomous role of p19INK4d. In light of the massive apoptosis in the S/G2-M phases that occurs following menadione treatment, and the absence of apoptosis after HU treatment, these results indicate that p19INK4d is required to protect HSCs from apoptosis under conditions of genotoxic stress, but not the replicative stress induced by HU. Additional experiments are necessary, however, to clearly define the roles of p19INK4d after induction of these two types of stress. Previous studies have demonstrated that p19INK4d contributes to DNA repair, apoptotic, and checkpoint mechanisms in order to maintain integrity of the genome under conditions of genotoxic stress, in a manner that is independent of CDK4/6 inhibition (Ogara et al., 2013; Scassa et al., 2007). Our data suggest that HSC cell cycle entry is enhanced by p19INK4d loss as a result of CDK4/6-dependent functions but that CDK4/6-independent p19INK4d functions contribute to reduced HSC numbers following increased apoptosis in the S/G2-M phases of the cell cycle. This mechanism may operate preferentially during genotoxic stress because p19INK4d−/− HSCs have normal reconstitutive capacities in BM transplant experiments.
Extrinsic Role of p19INK4d in the BM Niche: Development of Myelofibrosis in the Absence of p19INK4d
The effect of p19INK4d on the HSC compartment is, however, not purely cell autonomous. A significant decrease in the number of HSCs was observed in p19INK4d−/− mice even in the absence of stress induction, but p19INK4d−/− BM Lin− cells display a competitive advantage compared with controls after transplantation into WT recipients. This strongly suggests that the HSC microenvironment is also defective in p19INK4d−/− mice.
There is evidence that MKs play an important role in HSC regulation. We previously reported that p19INK4d regulates MK ploidy (Gilles et al., 2008). By further investigating the MK lineage, we also detected an increase in MK-P and mature MK numbers in p19INK4d−/− mice with an increased proportion of proliferative 2N/4N MKs during aging. Moreover, we demonstrated that the number of MKs increased with age in the BM of WT mice, but did not change in the spleen. In contrast, MK numbers increased in the spleens of p19INK4d−/− mice after reaching a plateau in the BM, leading to splenomegaly and fibrosis in both the BM and spleen. This phenotype was accompanied by increased levels of total and bioactive TGF-β1 in the extracellular fluids of the BM and spleen. MK and platelet α granules are the principal storage sites for TGF-β1, and myelofibrosis occurring in myeloid disorders is often associated with a marked MK hyperplasia and increased TGF-β1 release into the BM microenvironment. Interestingly, there was a striking similarity between the phenotype of aged p19INK4d−/− mice and human primary myelofibrosis (PMF), which is characterized by splenomegaly and myelofibrosis with osteomyelosclerosis. Similar to PMF, an increase in circulating HSPs was observed in p19INK4d−/− animals. Human PMF has been associated with acquired mutations in signaling molecules such as JAK2, MPL, and CALR, all of which lead to JAK/STAT pathway activation (Klampfl et al., 2013; Nangalia et al., 2013). How these mutations contribute to the myelofibrosis development has not been completely elucidated. There is, however, strong evidence that the myelofibrosis is a response to the in situ release of myeloid-derived factors such as TGF-β1, platelet-derived growth factor, vascular endothelial growth factor, and other proinflammatory cytokines. It is likely that MKs are the key cells in the development of myelofibrosis because increased proliferation of dysplastic MKs is a hallmark of PMF, with the onset of fibrosis occurring around dysplastic MK clusters. This is further supported by different mouse models of myelofibrosis, such as JAK2V617F (Lacout et al., 2006), MPLW515L/A (Pikman et al., 2006), TPOhigh (Villeval et al., 1997), or GATA1low (Vannucchi et al., 2002) systems. GATA1 is one of the major cofactors of the transcription factor RUNX1 in MKs (Tijssen et al., 2011). We have previously shown that p19INK4d is directly regulated by RUNX1 in MKs (Gilles et al., 2008), suggesting that p19INK4d expression could be altered in MK progenitors of GATA1low mice.
We provide evidence that, as in human PMF, the fibrosis observed in p19INK4d−/− mice is related to abnormal hematopoiesis. Indeed, splenomegaly and increased MK numbers were detected in recipient WT mice transplanted with p19INK4d−/− cells, but not in p19INK4d−/− recipient mice transplanted with WT cells. This suggests that the development of fibrosis is dependent on p19INK4−/− hematopoietic cells. However, our in vitro experiments indicate that p19INK4d−/− stroma also has a decreased capacity to sustain hematopoiesis. This is reminiscent of the in vitro stromal defects observed in myeloid malignancies, including PMF. Whether this defect is related to stromal epigenetic modifications due to abnormal myeloid proliferation or to a primary intrinsic defect in the BM environment, however, remains controversial. Regardless, the phenotype of p19INK4d−/− mice underscores the role of MKs not only in the development of myelofibrosis, but also in the regulation of the hematopoietic niche.
During their life span, HSCs undergo changes in the proportions of different subpopulations. HSC aging is characterized by an imbalance in the myelo-lymphopoiesis ratio, leading to amplification of myeloid-biased HSCs, progressive loss of lymphoid-biased HSCs (Challen et al., 2010), and accumulation of DNA damage (Rossi et al., 2007). The higher self-renewal capacity of myeloid HSCs that is associated with the accumulation of DNA damage with age likely contributes to the increased frequency of myeloid malignancies observed in elderly individuals. We demonstrate here that p19INK4d loss enhances accumulation of DNA-DSBs during aging, although the major effect of p19INK4d on the amplification of myeloid-biased HSCs during aging is clearly related to the microenvironment. Further studies to determine the precise mechanism by which this occurs are required. One potential explanation could be an age-associated increase in TGF-β1 levels produced by MKs. High levels of TGF-β1 may contribute to decreased numbers of HSCs (Scandura et al., 2004) observed in p19INK4d−/− animals during aging. On the other hand and in contrast to our observations in the absence of p19INK4d, it has been shown that TGF-β1 may expand myeloid-biased HSCs and inhibit the lymphoid-biased HSC population that occurs with aging (Challen et al., 2010). However, we demonstrate that increasing p19INK4d expression in HSCs during aging is clearly associated with an increase in myeloid-biased HSCs, indicating that TGF-β1 may regulate myeloid-biased HSCs through p19INK4d.
In conclusion, we report a mechanism by which fibrosis can be induced by loss of a protein, p19INK4d, that plays a crucial role in regulating the MK cell cycle. Whether p19INK4d could be an indirect target of signaling molecules that have been genetically altered in human PMF remains to be investigated. The results presented here also underscore the protective role of p19INK4d during genotoxic stress and demonstrate that p19INK4d plays an important role in hematopoiesis, including the changes that occur during aging. This study hence opens avenues of research for the delineation of this gene’s precise function in HSCs and their microenvironmental niche.
Experimental Procedures
Mice
Heterozygous p19INK4d(+/−) (C57BL/6 × Sv129j) mice (Zindy et al., 2000) were obtained from Martine Roussel. WT and p19INK4d−/− mice were obtained by intercrossing heterozygous mice. All animal experiments were approved by the local Ethical Committee of University Paris-Sud no. 26 (authorized project no. 2012-060).
Flow Cytometry
LSK, progenitors, or mature cells were analyzed on FACS LSR II or FACS CANTO II (BD) and sorted on MoFlo or Influx flow cytometers (BD). SP staining and MK ploidy analyses were performed as previously described (Goodell et al., 1996; Gilles et al., 2008). HSC cell cycle analysis was performed with either Ki-67/Hoechst 33342 or BrdU/Hoechst 33342 costaining.
Reconstitution Assays
To study the role of the environment in hematopoietic reconstitution, p19INK4d−/− and WT mice were lethally irradiated (11 Gy). After 24 hr, the mice were reconstituted with 2 × 106 WT or p19INK4d−/− total BM cells. The reconstitution potential was evaluated as survival rate and cell cycle and apoptosis assays were performed. To evaluate the reconstitution advantage of p19INK4d−/− or WT cells, competitive transplantation was performed with 5 × 104 WT-GFP: 5 × 104 p19INK4d−/− CHERRY cells. Blood was sampled once per month to follow hematopoietic reconstitution. Mice were euthanized 3.5 months after transplantation. BM cells were collected, and different populations were analyzed by flow cytometry to determine the proportion of GFP+ and CHERRY+ cells.
In primary and secondary transplantation assays, 5 × 106 cells from whole spleen of myelofibrotic mice were injected intraorbitally into lethally irradiated recipient WT mice. For secondary transplantation assays, spleen cells were collected 2 months after transplantation. Half of the spleen and one femur were retained for each mouse in order to perform immunohistochemistry.
Treatment for Myelosuppression by Administration of 5-FU
Mice were injected intraperitoneally with 5-FU (Fluorouracil, Dakota Pharm) at 150 mg/kg body weight for survival rates of mice and at 250 or 50 mg/kg body weight for cell cycle analysis in nontransplanted or transplanted mice, respectively.
Stress Induction with HU Treatment
Mice were treated with HU (100 mg/kg/day) by intraperitoneal injection for three consecutive days, and cell cycle analysis and apoptosis assays were subsequently performed.
Oxidative Stress Induction
Lin− cells were incubated with or without menadione (10 mM) for 1 hr, and LSKs were then stained to measure ROS levels. ROS production was monitored by flow cytometry using 2′,7′ dichlorodihydrofluorescin diacetate (DCFH2-DA).
Statistical Analysis
Results were evaluated by performing unpaired t tests using GraphPad Prism version 5.0 software (GraphPad Software). Results are presented as means ± SEM. ∗p < 0.05 was determined significant, and ∗∗p < 0.01 or ∗∗∗p < 0.001 were considered highly significant; “n =” stands for individual mice in all experiments.
Acknowledgments
This work was supported by French grants from the Agence Nationale de la Recherche (H.R.), the Ligue Nationale contre le Cancer (équipe labellisée 2009 [W.V.] and 2013 [H.R.]). M.H. was supported by the ARC. We thank P. Rameau and Y. Lecluse for flow cell analysis and sorting and O. Bawa for immunohistochemistry (IRCIV, Institut Gustave Roussy). We are grateful to Professor R. Skoda (University Hospital of Basel) and Professor J.D. Crispino (Northwestern University) for helpful discussions, and Dr. B.J. Thompson (Northwestern University) and Dr. K. Rice (Hospital St. Louis) for English editing.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information
References
- Adachi M., Roussel M.F., Havenith K., Sherr C.J. Features of macrophage differentiation induced by p19INK4d, a specific inhibitor of cyclin D-dependent kinases. Blood. 1997;90:126–137. [PubMed] [Google Scholar]
- Attema J.L., Pronk C.J., Norddahl G.L., Nygren J.M., Bryder D. Hematopoietic stem cell ageing is uncoupled from p16 INK4A-mediated senescence. Oncogene. 2009;28:2238–2243. doi: 10.1038/onc.2009.94. [DOI] [PubMed] [Google Scholar]
- Beerman I., Bhattacharya D., Zandi S., Sigvardsson M., Weissman I.L., Bryder D., Rossi D.J. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl. Acad. Sci. USA. 2010;107:5465–5470. doi: 10.1073/pnas.1000834107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceruti J.M., Scassa M.E., Marazita M.C., Carcagno A.C., Sirkin P.F., Cánepa E.T. Transcriptional upregulation of p19INK4d upon diverse genotoxic stress is critical for optimal DNA damage response. Int. J. Biochem. Cell Biol. 2009;41:1344–1353. doi: 10.1016/j.biocel.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Challen G.A., Boles N.C., Chambers S.M., Goodell M.A. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell. 2010;6:265–278. doi: 10.1016/j.stem.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P., Zindy F., Abdala C., Liu F., Li X., Roussel M.F., Segil N. Progressive hearing loss in mice lacking the cyclin-dependent kinase inhibitor Ink4d. Nat. Cell Biol. 2003;5:422–426. doi: 10.1038/ncb976. [DOI] [PubMed] [Google Scholar]
- Cheng T., Rodrigues N., Dombkowski D., Stier S., Scadden D.T. Stem cell repopulation efficiency but not pool size is governed by p27(kip1) Nat. Med. 2000;6:1235–1240. doi: 10.1038/81335. [DOI] [PubMed] [Google Scholar]
- Gilles L., Guièze R., Bluteau D., Cordette-Lagarde V., Lacout C., Favier R., Larbret F., Debili N., Vainchenker W., Raslova H. P19INK4D links endomitotic arrest and megakaryocyte maturation and is regulated by AML-1. Blood. 2008;111:4081–4091. doi: 10.1182/blood-2007-09-113266. [DOI] [PubMed] [Google Scholar]
- Goodell M.A., Brose K., Paradis G., Conner A.S., Mulligan R.C. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 1996;183:1797–1806. doi: 10.1084/jem.183.4.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo I., Herrera-Merchan A., Ligos J.M., Carramolino L., Nuñez J., Martinez F., Dominguez O., Torres M., Gonzalez S. Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence-like cell cycle arrest. Cell Stem Cell. 2012;11:649–662. doi: 10.1016/j.stem.2012.08.001. [DOI] [PubMed] [Google Scholar]
- Janzen V., Forkert R., Fleming H.E., Saito Y., Waring M.T., Dombkowski D.M., Cheng T., DePinho R.A., Sharpless N.E., Scadden D.T. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- Klampfl T., Gisslinger H., Harutyunyan A.S., Nivarthi H., Rumi E., Milosevic J.D., Them N.C., Berg T., Gisslinger B., Pietra D. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013;369:2379–2390. doi: 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- Lacout C., Pisani D.F., Tulliez M., Gachelin F.M., Vainchenker W., Villeval J.L. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108:1652–1660. doi: 10.1182/blood-2006-02-002030. [DOI] [PubMed] [Google Scholar]
- Mascarenhas M.I., Parker A., Dzierzak E., Ottersbach K. Identification of novel regulators of hematopoietic stem cell development through refinement of stem cell localization and expression profiling. Blood. 2009;114:4645–4653. doi: 10.1182/blood-2009-06-230037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A., Takeishi S., Kanie T., Susaki E., Onoyama I., Tateishi Y., Nakayama K., Nakayama K.I. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell. 2011;9:262–271. doi: 10.1016/j.stem.2011.06.014. [DOI] [PubMed] [Google Scholar]
- Nangalia J., Massie C.E., Baxter E.J., Nice F.L., Gundem G., Wedge D.C., Avezov E., Li J., Kollmann K., Kent D.G. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013;369:2391–2405. doi: 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogara M.F., Sirkin P.F., Carcagno A.L., Marazita M.C., Sonzogni S.V., Ceruti J.M., Cánepa E.T. Chromatin relaxation-mediated induction of p19INK4d increases the ability of cells to repair damaged DNA. PLoS ONE. 2013;8:e61143. doi: 10.1371/journal.pone.0061143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passegué E., Wagers A.J., Giuriato S., Anderson W.C., Weissman I.L. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med. 2005;202:1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikman Y., Lee B.H., Mercher T., McDowell E., Ebert B.L., Gozo M., Cuker A., Wernig G., Moore S., Galinsky I. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H., Buza-Vidas N., Hyland C.D., Jensen C.T., Antonchuk J., Månsson R., Thoren L.A., Ekblom M., Alexander W.S., Jacobsen S.E. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell. 2007;1:671–684. doi: 10.1016/j.stem.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Rossi D.J., Bryder D., Seita J., Nussenzweig A., Hoeijmakers J., Weissman I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447:725–729. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- Scandura J.M., Boccuni P., Massagué J., Nimer S.D. Transforming growth factor beta-induced cell cycle arrest of human hematopoietic cells requires p57KIP2 up-regulation. Proc. Natl. Acad. Sci. USA. 2004;101:15231–15236. doi: 10.1073/pnas.0406771101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scassa M.E., Marazita M.C., Ceruti J.M., Carcagno A.L., Sirkin P.F., González-Cid M., Pignataro O.P., Cánepa E.T. Cell cycle inhibitor, p19INK4d, promotes cell survival and decreases chromosomal aberrations after genotoxic insult due to enhanced DNA repair. DNA Repair (Amst.) 2007;6:626–638. doi: 10.1016/j.dnarep.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Tijssen M.R., Cvejic A., Joshi A., Hannah R.L., Ferreira R., Forrai A., Bellissimo D.C., Oram S.H., Smethurst P.A., Wilson N.K. Genome-wide analysis of simultaneous GATA1/2, RUNX1, FLI1, and SCL binding in megakaryocytes identifies hematopoietic regulators. Dev. Cell. 2011;20:597–609. doi: 10.1016/j.devcel.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Os R., Kamminga L.M., Ausema A., Bystrykh L.V., Draijer D.P., van Pelt K., Dontje B., de Haan G. A Limited role for p21Cip1/Waf1 in maintaining normal hematopoietic stem cell functioning. Stem Cells. 2007;25:836–843. doi: 10.1634/stemcells.2006-0631. [DOI] [PubMed] [Google Scholar]
- Vannucchi A.M., Bianchi L., Cellai C., Paoletti F., Rana R.A., Lorenzini R., Migliaccio G., Migliaccio A.R. Development of myelofibrosis in mice genetically impaired for GATA-1 expression (GATA-1(low) mice) Blood. 2002;100:1123–1132. doi: 10.1182/blood-2002-06-1913. [DOI] [PubMed] [Google Scholar]
- Villeval J.L., Cohen-Solal K., Tulliez M., Giraudier S., Guichard J., Burstein S.A., Cramer E.M., Vainchenker W., Wendling F. High thrombopoietin production by hematopoietic cells induces a fatal myeloproliferative syndrome in mice. Blood. 1997;90:4369–4383. [PubMed] [Google Scholar]
- Yamazaki S., Nakauchi H. Insights into signaling and function of hematopoietic stem cells at the single-cell level. Curr. Opin. Hematol. 2009;16:255–258. doi: 10.1097/MOH.0b013e32832c6705. [DOI] [PubMed] [Google Scholar]
- Yoshihara H., Arai F., Hosokawa K., Hagiwara T., Takubo K., Nakamura Y., Gomei Y., Iwasaki H., Matsuoka S., Miyamoto K. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell. 2007;1:685–697. doi: 10.1016/j.stem.2007.10.020. [DOI] [PubMed] [Google Scholar]
- Yuan Y., Yu H., Boyer M.J., Song X., Cao S., Shen H., Cheng T. Hematopoietic stem cells are not the direct target of spontaneous leukemic transformation in p18(INK4C)-null reconstituted mice. Cancer Res. 2006;66:343–351. doi: 10.1158/0008-5472.CAN-05-2945. [DOI] [PubMed] [Google Scholar]
- Zindy F., Cunningham J.J., Sherr C.J., Jogal S., Smeyne R.J., Roussel M.F. Postnatal neuronal proliferation in mice lacking Ink4d and Kip1 inhibitors of cyclin-dependent kinases. Proc. Natl. Acad. Sci. USA. 1999;96:13462–13467. doi: 10.1073/pnas.96.23.13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F., van Deursen J., Grosveld G., Sherr C.J., Roussel M.F. INK4d-deficient mice are fertile despite testicular atrophy. Mol. Cell. Biol. 2000;20:372–378. doi: 10.1128/mcb.20.1.372-378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F., den Besten W., Chen B., Rehg J.E., Latres E., Barbacid M., Pollard J.W., Sherr C.J., Cohen P.E., Roussel M.F. Control of spermatogenesis in mice by the cyclin D-dependent kinase inhibitors p18(Ink4c) and p19(Ink4d) Mol. Cell. Biol. 2001;21:3244–3255. doi: 10.1128/MCB.21.9.3244-3255.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.