

Summary
miR-133a and miR-1 are known as muscle-specific microRNAs that are involved in cardiac development and pathophysiology. We have shown that both miR-1 and miR-133a are early and progressively upregulated during in vitro cardiac differentiation of adult cardiac progenitor cells (CPCs), but only miR-133a expression was enhanced under in vitro oxidative stress. miR-1 was demonstrated to favor differentiation of CPCs, whereas miR-133a overexpression protected CPCs against cell death, targeting, among others, the proapoptotic genes Bim and Bmf. miR-133a-CPCs clearly improved cardiac function in a rat myocardial infarction model by reducing fibrosis and hypertrophy and increasing vascularization and cardiomyocyte proliferation. The beneficial effects of miR-133a-CPCs seem to correlate with the upregulated expression of several relevant paracrine factors and the plausible cooperative secretion of miR-133a via exosomal transport. Finally, an in vitro heart muscle model confirmed the antiapoptotic effects of miR-133a-CPCs, favoring the structuration and contractile functionality of the artificial tissue.
Highlights
-
•
miR-1 and miR-133a have a role in adult cardiac progenitor cells (CPCs)
-
•
miR-133a-CPCs protect cardiac function
-
•
miR-133a-CPCs increase vascularization and protect against hypertrophy
-
•
miR-133a is enriched in CPCs-derived exosomes
Bernad and colleagues address the role of miR-1 and miR-133a in adult CPCs. Both are upregulated during cardiac differentiation, but only miR-133 protects from oxidative stress. In a myocardial infarction rat model, they demonstrate that miR-133a-CPCs transplantation increases vasculogenesis and protects from fibrosis and hypertrophy. The study also suggests a combined enhanced growth factor secretion and exosomal contribution of miR-133a.
Introduction
Cardiovascular diseases are the major cause of illness and death in developed countries, and transplantation of progenitor cells has emerged as a bona fide therapeutic option for this group of diseases. Currently, cell therapy approaches with diverse types of mature or stem cell populations have produced modest improvements (Sanganalmath and Bolli, 2013). Among them, resident cardiac stem/progenitor cells (CSCs/CPCs) are a promising option (Chugh et al., 2012, Malliaras et al., 2014). Many experimental adult CPC populations have been isolated, based on a specific surface marker profile or their differentiation potential. The most studied are c-KIT+, SCA-1+, cardiosphere-derived cells, and cardiac mesoangioblasts, all of which have demonstrated regenerative potential in vivo (Beltrami et al., 2003, Galvez et al., 2008, Malliaras et al., 2012, Oh et al., 2003, Tateishi et al., 2007). However, transplanted cells die in large numbers in the infarcted myocardium shortly after transplantation, presenting a hurdle to improving heart function through stem cell therapy (Robey et al., 2008). The infarct microenvironment is characterized by hypoxia, oxidative stress, and proapoptotic and inflammatory factors (Hori and Nishida, 2009); thus, a deeper understanding of how transplanted progenitor cells engraft and survive in this hostile context is required to enhance their clinical usefulness.
microRNAs (miRNAs) are small noncoding RNAs of 22 nucleotides that control gene expression chiefly by translational repression (Lee et al., 2004). miRNA expression is spatially and temporally regulated during development, and they can participate in the regulation of pluripotent stem cell lineage commitment (Ivey et al., 2008). miR-1 and miR-133a belong to the MyomiR group of muscle-specific miRNAs. They regulate heart and skeletal muscle biology and play an important role in heart development (Liu et al., 2008, Matkovich et al., 2010). Both miRNAs are transcribed from the same locus and are processed as a bicistronic transcript. This locus is duplicated in the genome, although mature miRNA forms are identical from both loci (Townley-Tilson et al., 2010). Their expression is controlled by serum response factor (SRF), MEF2, and MyoD transcription factors, which bind to sequences located upstream from the miRNAs and/or to their intergenic sequences (Liu et al., 2008, Zhao et al., 2005). Overexpression of miR-1 mediates cardiomyocyte withdrawal from the cell cycle through the targeting of Hand2. Conversely, mice deficient for miR-1-2 have an elevated number of postnatal cardiomyocytes proliferating and present an enlargement of ventricular walls (Zhao et al., 2007). Double knockout mice for both mature forms of miR-133a exhibit excessive cardiomyocyte proliferation and increased apoptosis associated with an elevated expression of their targets SRF and cyclin D2, leading to ventricular-septal defects (Zhao et al., 2007). However, overexpression of miR-133a in postnatal cardiomyocytes (Matkovich et al., 2010) or under the control of a muscle-specific promoter (Deng et al., 2011) has no significant effect.
miR-1 and miR-133a have been implicated in pathological processes such as hypertrophy (Liu et al., 2008, Matkovich et al., 2010, Zhao et al., 2005, 2007), and their levels may define a predisease stage. A recent meta-analysis (comprising 15 studies and 2,136 patients) demonstrated the validity of miR-133a and miR-499 levels in plasma or serum as a diagnostic biomarker of myocardial infarction (MI) (Cheng et al., 2014). However, analysis of surgical samples of the right atrial myocardium of patients showed that miR-133 expression decreased significantly as the severity of heart failure increased; no correlation was found for miR-1 (Danowski et al., 2013). Furthermore, miR-133a is downregulated in response to transaortic constriction-induced hypertrophy and after MI in mice, rats, pigs, and humans (Carè et al., 2007, Hullinger et al., 2012, He et al., 2011) and also in diabetic cardiomyopathy (Chen et al., 2014, Yildirim et al., 2013). Downregulation of miR-133a has been also associated with several vascular pathologies such as atherosclerosis, intracranial aneurysms, and arterial calcification (Gao et al., 2014, Jiang et al., 2013, Liao et al., 2013).
miR-1 and miR-133a have been demonstrated to influence embryonic stem (ES) cell fate; in fact, overexpression of each miR in mouse and human ES cells promotes mesoderm specification while suppressing other lineages (Ivey et al., 2008). Ivey and Srivastava (2010) showed that forced miR-1 expression alone was sufficient to drive expression of cardiogenic markers, while miR-133a overexpression blocked this process. Controversially, other studies have described that miR-1 overexpression reduces the expression of cardiac markers in mouse ES cells (Takaya et al., 2009). Therefore, thorough knowledge about how miR-1 and miR-133a influence cardiac differentiation of pluripotent and multipotent cells is needed. In addition, apart from their central role in heart function, they are also implicated in a regulatory network of miRNAs controlling osteoblastic and chondrogenic differentiation of MSCs (Li et al., 2008, Wang et al., 2012). Collectively, these examples illustrate the putative central role of these muscle-specific miRNAs in cellular homeostasis.
In this work, we describe a role for miR-133a in the survival regulation of a Sca-1+ CPC population, which shares features with other populations of mesenchymal stem cells (MSCs) in terms of surface marker profile and differentiation potential. Interestingly, miR-133a promotes survival of CPCs under oxidative stress by decreasing caspase 3 activity and targeting the proapoptotic genes Bim and Bmf. Furthermore, transplantation of miR-133a-CPCs significantly improves heart function in murine models of acute MI by reducing cardiac hypertrophy and cardiomyocyte apoptosis and increasing bFgf, Vegf, Igf1, and Hgf expression in vitro. Finally, we describe that the majority of secreted miR-133a is localized in the exosomal fraction of miR-133-CPCs.
Results
Role of miR-1 and miR-133a in Adult CPCs
CPCs from adult mouse hearts accounted for 7%–20% of the myocyte-depleted small cells from the myocardium (Figures S1A and S1B available online) and endowed cardiac, endothelial, and smooth-muscle differentiation capacity (Figures S1C and S2). In addition, mouse CPCs also demonstrated MSC-like differential potential (Figures S1D and S1E) as previously shown for human counterparts (Moscoso et al., 2013). miR-1 and miR-133a expression was under the detection level in undifferentiated CPCs, but exposure to cardiac differentiation conditions (Figure 1A) or coculture with neonatal rat cardiomyocytes (NRCMs; Figure 1B) progressively increased the expression of miR-1 and miR-133a.
Figure 1.
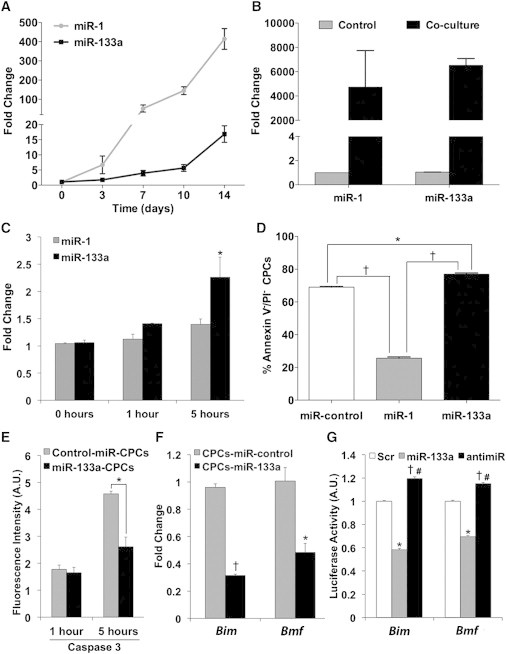
Role of miR-1 and miR-133a in CPC Differentiation and Response to Oxidative Stress
(A) Quantitative RT-PCR analysis of miR-1 and miR-133a expression in CPCs exposed to cardiac differentiation medium.
(B) miR-1 and miR-133a expression in CPCs after 7 day co-culture with NRCMs.
(C) miR-1 and miR-133a expression in CPCs after treatment with H2O2 (n = 3, ∗p < 0.05 versus miR-133a at 0 hr).
(D) Fluorescence-activated cell sorting (FACS) analysis of live CPCs (annexin V−/PI− cells) after 5 hr of H2O2 treatment (n = 3; †p < 0.01, ∗p < 0.05).
(E) Caspase 3 activity measured by FACS (mean fluorescence intensity) 1 and 5 hr after H2O2 treatment (n = 3; ∗p < 0.005).
(F) Gene expression analysis of Bim and Bmf after H2O2 treatment (5 hr; n = 3; †p < 0.00005, ∗p < 0.005).
(G) 3′UTR reporter assay for Bim and Bmf. Renilla luciferase activity was assayed 24 hr after transfection, and the values were normalized to the activity of firefly luciferase encoded in the same vector (n = 3; Bim: ∗p < 0.00005, †p < 0.001 versus Scr, #p < 0.00005 versus miR-133a; Bmf: ∗p < 0.0001, †p < 0.001 versus Scr, #p < 0.00005 versus miR-133a).
(C–G) Data are mean ± SEM, n = 3 experiments. See also Figures S3 and S4.
To analyze the influence of both myomiRs in CPCs differentiation capacity, we transduced them with retroviral vectors encoding miR-1, miR-133a, or a control miRNA (Figure S3A). After dexamethasone (Dex) induction, we analyzed Nkx2.5 and Troponin T expression as early and late cardiac markers, respectively. Interestingly, whereas miR-1-CPCs showed a significant increase in Nkx2.5 expression, both in basal conditions and after cardiac induction with Dex, miR-133a-CPCs did not display any significant modification (Figure S3B). Evaluation of levels of Troponin T, after induction (Dex), rendered a similar result (Figure S3C). This strongly suggests that forced expression of miR-1 could be favoring cardiac commitment and differentiation of CPCs, whereas overexpression of miR-133a does not seem to modulate significantly these processes. Neither of the transduced miRNAs affected the proliferation, as measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay or 3H-thymidine incorporation (Figures S3D and S3E) or apoptosis rates (data not shown) of CPCs, in basal conditions.
In an in vitro model of induced oxidative stress injury, only miR-133a was significantly upregulated upon treatment of CPCs with H2O2 (200 μM; 1–5 hr) (Figure 1C). Importantly, miR-1-CPCs displayed increased apoptosis in comparison with both control-miR-CPCs and miR-133a-CPCs (Figures 1D, S4A, and S4B). A significant increase in caspase 3 activity in control cells was prevented in miR-133a-CPCs (Figure 1E); a comparable result was obtained by measurement of cytosolic cytochrome c (Figure S4C).
Bioinformatic Prediction of miR-133a Targets
Bioinformatic analysis of putative target genes for miR-133a by Ingenuity Pathway Analysis software predicted cell death as the main biological function affected at organ and cellular level and hypertrophy, fibrosis, and apoptosis as the main toxic processes altered. We found common targets as Bmf (Bcl-2 modifying factor) and Bim (Bcl2l11), which are potent proapoptotic factors (reviewed by Piñon et al., 2008), a serine threonine kinase Stk4 (formerly Mst1) activated by oxidative stress (Cottini et al., 2014, Del Re et al., 2014), and finally Foxo1 that has been described as a critical promotor of cardiomyocyte survival upon oxidative stress conditions (Puthanveetil et al., 2013). Particularly interesting is the recent implication of Bmf in oxidative stress-induced apoptosis of hepatic progenitor populations after activation of transforming growth factor β (TGF-β) signaling (Martínez-Palacián et al., 2013), which was predicted in our analysis as putative upstream regulator of Bmf, Bim, and Foxo1 (see Supplemental Experimental Procedures; Figures S5 and S6A).
Quantitative RT-PCR (RT-qPCR) analysis of miR-133a-CPCs versus miR-control-CPCs, challenged with H2O2, showed that although the expression of all these predicted targets was effectively downregulated, Bim and Bmf were specially affected (Figures 1F and S6B). To confirm whether miR-133a could modulate directly the expression of Bim and Bmf, we cloned separately a 1kb fragment of their 3′UTRs containing predicted binding sites for miR-133a, downstream of the Renilla luciferase reporter and transfected 293T cells with the constructs. The cotransfection with a synthetic precursor of miR-133a significantly reduced luciferase activity in both cases with respect to a scramble miRNA, while cotransfection with a miR-133a inhibitor abolished this reduction (Figure 1G).
miR-133a-CPCs Protect Cardiac Function
To evaluate the potential protective effect of miR-133a in vivo, GFP-tagged CPCs (105) expressing control miRNA or miR-133a were injected into the infarct border zone in a MI model in immunosuppressed rats (Figure 2A). Cardiac function was monitored by echocardiography 1 day before MI and 2 and 4 weeks after MI and CPC/vehicle injection (Figure 2B). Compared with vehicle injection, animals injected with CPCs showed significantly greater fractional area change (FAC) at 2 weeks after MI, but no significant differences were observed between rats injected with control-miR-CPCs and miR-133a-CPCs. However, at 4 weeks after MI, animals injected with miR-133a-CPCs showed significantly greater FAC compared with animals injected with either vehicle or control-miR-CPCs (Figure 2C; Table S3). Similarly, fractional shortening (FS) at 2 weeks after MI was increased by injection with CPCs, independently of miRNA expression, but at 4 weeks after MI, this increase was sustained only in rats injected with miR-133a-CPCs (Figure 2D; Table S3). No GFP+ cells could be found on heart sections 4 weeks after transplant (Figure 2E); similar results were obtained using a syngeneic mouse MI model, in which GFP+ CPCs could be detected at low numbers within the first postinjection days, although a clear advantage for miR-133a-CPCs was not evidenced (unpublished data).
Figure 2.
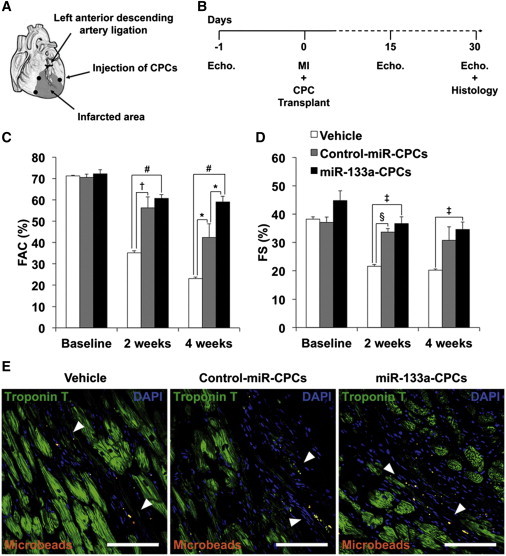
CPC Transplantation in a Rat Model of MI
(A) Diagram of in vivo transplantation of CPCs. After ligation of the left descending coronary artery, vehicle, control-miR-CPCs or miR-133a-CPCs were injected together with red fluorescent microbeads as a marker for injected material, into three locations at the infarct border.
(B) Experimental timeline of echocardiography analysis.
(C and D) Echocardiography measurements of FAC (C) and FS (D) (hearts: vehicle n = 5; control-miR–CPCs n = 6; miR-133a–CPCs n = 11. ∗p < 0.05, †p < 0.01, ‡p < 0.005, §p < 0.0005, #p < 0.000005).
(E) Detection of fluorescent microbeads (white arrowheads) in the infarct border zone (white bars are 100 μm). Data are mean ± SEM. See also Table S3.
Morphometric analysis of explanted hearts 4 weeks after MI showed severe left ventricle chamber dilatation and infarct wall thinning in vehicle-treated animals (Figure 3A; Table S4). Transplantation of control-miR-CPCs significantly decreased the scar area with respect to vehicle-treated animals, although no significant differences were found in infarcted wall thickness or viable myocardium inside the risk area (Figures 3B–3E). However, transplantation with miR-133a-CPCs further decreased the scar area and infarcted wall thinning compared with vehicle and control-miR-CPCs (Figures 3B and 3C). While the risk area decreased with respect to vehicle-treated animals, with no significant differences between the two CPC-injected groups (Figure 3D), the amount of viable myocardium inside the risk area (Figure 3E) was significantly higher in hearts injected with miR-133a-CPCs.
Figure 3.
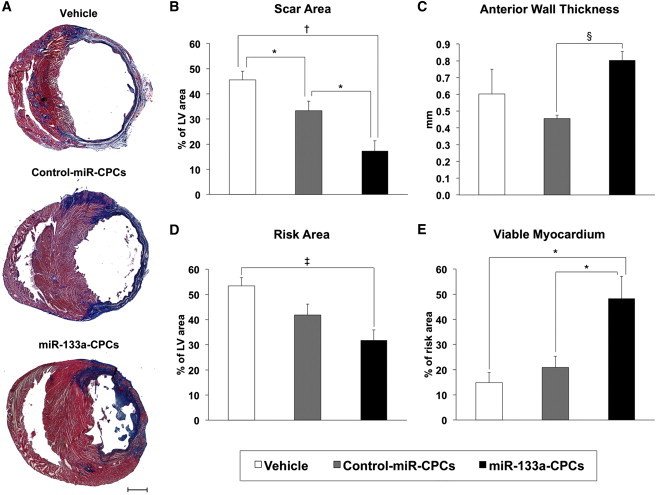
Morphometric Analysis
(A) Representative Masson’s trichrome-stained myocardial sections of infarcted rat hearts 4 weeks after injection with vehicle, control-miR-CPCs, or miR-133a-CPCs. Scar tissue and viable myocardium are identified as blue and red, respectively (the scale bar represents 2 mm).
(B–E) Left ventricle morphometric parameters. (B) Scar area. (C) Anterior wall thickness. (D) Risk area and (E) viable myocardium inside the risk area (hearts: vehicle n = 5; control-miR-CPCs n = 7; miR-133a-CPCs n = 7. ∗p < 0.05, †p < 0.005, ‡p < 0.001, §p < 0.00005).
Data are mean ± SEM. See also Table S4.
CPCs Increase Vascularization and Protect against Hypertrophy
Hearts from animals treated with control-miR-CPCs or miR-133a-CPCs contained a higher density of α-smooth muscle actin (α-SMA)+ blood vessels in the infarct border zone compared with vehicle-treated animals (Figure 4B), but there were not significant differences in the remote zone (Figure 4A). Nonetheless, the density of α-SMA+ blood vessels in the border zone did not differ between hearts injected with control-miR-CPCs or miR-133a-CPCs (Figure 4D). We also investigated the capacity of CPCs to promote new cardiomyocyte formation (Figure 4C). Even though CPC injection did not significantly increase the total number of EdU+ nuclei, both control-miR-CPCs and miR-133a-CPCs significantly increased the percentage of DNA-replicating cardiomyocytes (EdU+/troponin T+ cells) 4 weeks after MI, although this increase was smaller for animals injected with miR-133a-CPCs (Figure 4E).
Figure 4.
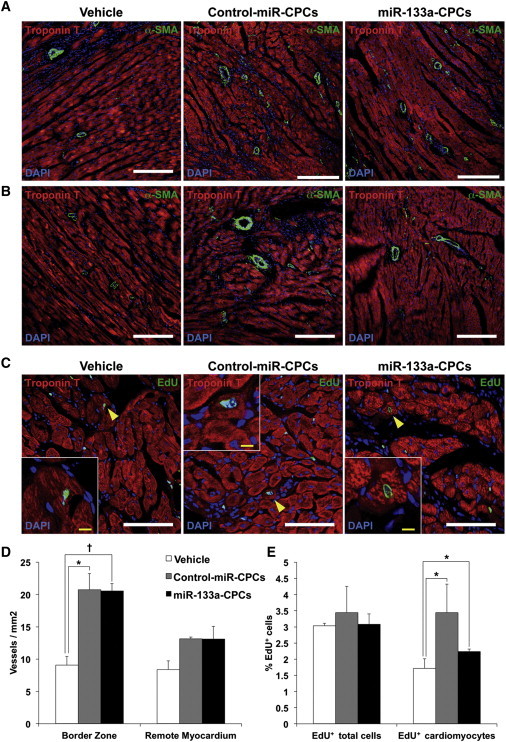
Effect of CPC Transplantation on Vascular Density and Cardiomyocyte Proliferation
(A and B) Representative immunofluorescence on rat heart sections taken from the infarction zone (A) and the healthy myocardium (B) 4 weeks after MI and injection with vehicle, control-miR-CPCs, or miR-133a-CPCs. Sections were stained for α-SMA and troponin T to reveal the density of α-SMA+ blood vessels (white bars are 200 μm).
(C) Representative sections from border zone, stained for EdU and troponin T (yellow arrowheads indicate EdU+/troponin T+ cardiomyocytes; white bars indicate 100 μm, and yellow bars show 10 μm).
(D) Quantification of α-SMA+ blood vessels/mm2 (hearts: vehicle n = 3; control-miR–CPCs n = 3; miR-133a-CPCs, n = 4, ∗p < 0.05, †p < 0.005).
(E) Quantification of EdU+ total cells and EdU+/troponin T+ cardiomyocytes (hearts: vehicle n = 3; control-miR-CPCs n = 3; miR-133a-CPCs, n = 3, ∗p < 0.05).
Data are mean ± SEM.
We also measured the extent of hypertrophy in the border zone of infarcted rat hearts (Figure 5A). Cardiomyocyte cross-sectional area was significantly lower in animals injected with control-miR-CPCs compared with vehicle-injected animals (Figure 5B), and this reduction was higher with miR-133a-CPCs. Pathological hypertrophy is linked to the activation of a fetal cardiac gene program (Liu et al., 2008). An assessment of key fetal and adult cardiac genes expression revealed that animals treated with miR-133a-CPCs showed a significant reduction in the expression ratios of β-Mhc/α-Mhc and, more pronouncedly, in the ANP/BNP ratio (Figure 5C) when compared with animals treated with control-miR-CPCs, suggesting that miR-133a-CPCs protected the heart from pathological hypertrophy.
Figure 5.
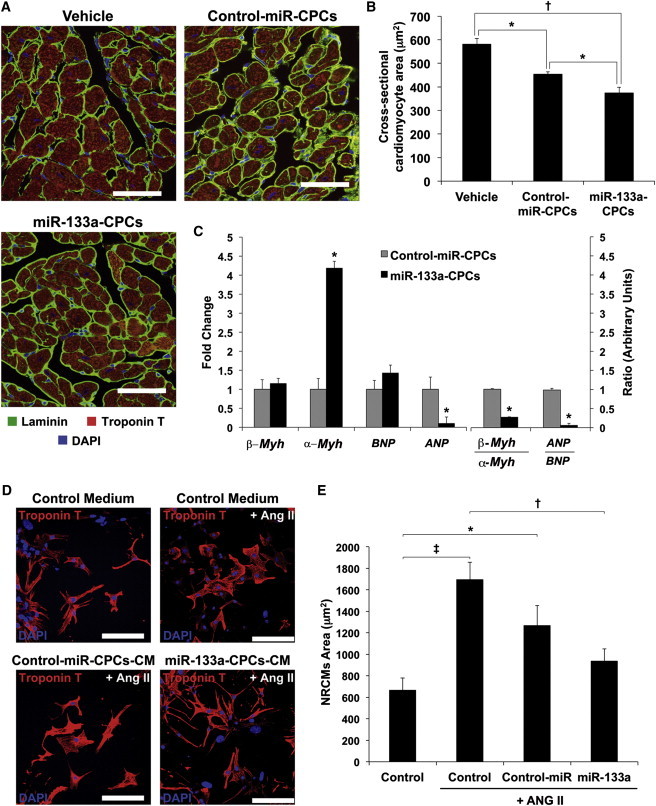
Effect of miR-133a-CPCs on Cardiac Hypertrophy Parameters
(A) Representative immunofluorescence on rat heart sections taken from the infarct border zone 4 weeks after injection with vehicle, control-miR-CPCs, or miR-133a-CPCs. Sections were stained for laminin and troponin T in order to determine cardiomyocyte cross-sectional area (scale bars represent 50 μm).
(B) Quantification of cardiomyocyte cross-sectional (hearts: vehicle n = 3; control-miR-CPCs n = 8; miR-133a-CPCs, n = 8, ∗p < 0.05, †p < 0.001).
(C) RT-qPCR analysis of fetal and adult cardiac genes at 4 weeks after MI (hearts: control-miR-CPCs, n = 8, miR-133a-CPCs, n = 6; ∗p < 0.05).
(D and E) Effect of CPC-CM on ANGII-induced hypertrophy in NRCMs in vitro. (D) Representative troponin T staining in the presence of control medium, control-miR-CPC-CM or miR-133a-CPC-CM (bars, 100 μm). (E) Quantification of cardiomyocyte area (n = 3 experiments; ∗p < 0.05, †p < 0.01, ‡p < 0.005).
Data are mean ± SEM.
We tested conditioned medium (CM) from CPCs in an in vitro model of angiotensin II (ANG II)-induced hypertrophy in NRCMs (Figures 5D and 5E). ANG II-stimulated NRCMs cultured in control medium are larger than nonstimulated cells (Figure 5D). In contrast, culture of NRCMs in CM from miR-133a-CPCs exhibited a significant decrease in ANGII-induced hypertrophy, and cardiomyocyte area was comparable to nonstimulated cells (Figure 5E). All together, these results suggested that miR-133-CPC transplantation would improve cardiac function mainly by protecting resident cardiomyocytes from stress conditions rather than inducing regeneration of the endogenous damaged population, which is consistent with other reports using cell therapy approaches (Wang et al., 2009).
To test this hypothesis, we cocultured control-miR-CPCs and miR-133a-CPCs with NRCMs in serum-free medium to mimic stress conditions. Analysis of double-stained TUNEL+/troponin T+ cardiomyocytes revealed that coculture with miR-133a-CPCs significantly and specifically reduced (>40%) the number of apoptotic NRCMs after 7 days (Figures 6A and 6B). In order to determine whether this effect required direct cell-to-cell interaction, we cultured NRCMs overnight with CM from control-miR-CPCs or miR-133a-CPCs, followed by exposure to H2O2 (200 μM) for 2 hr. CM from miR-133a-CPCs significantly reduced activation of caspases 3 and 9 (Figure 6C). Growth factor expression analysis showed that Vegf and bFgf levels, after H2O2 treatment for 1 and 5 hr, were significantly increased in miR-133-CPCs (Figure 6D), suggesting that higher release of these cytokines could mediate their protective effects via paracrine actions.
Figure 6.
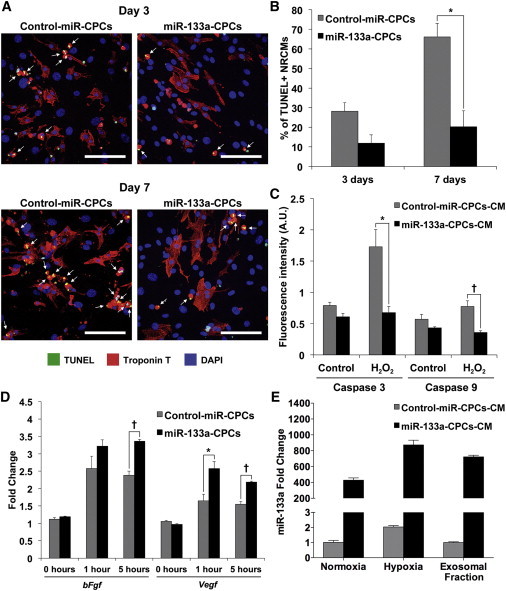
Effect of miR-133a-CPCs on Cardiomyocyte Apoptosis
(A) Representative immunofluorescence images of NRCMs cocultured with control-miR-CPCs or miR-133a-CPCs and serum starved for 3 or 7 days. Apoptosis was detected by TUNEL assay (yellow spots indicated by white arrows). Troponin T, red; DAPI-stained nuclei, blue; scale bars represent 100 μm).
(B) Quantification of apoptotic cardiomyocytes (TUNEL+/troponin T+ cells) after 3 and 7 days of serum starvation (n = 3 experiments; ∗p < 0.05).
(C) Effect of CPC-CM on H2O2-induced cardiomyocyte apoptosis (caspase 3/caspase 9 activity; n = 3 experiments, two replicates per experiment; ∗p < 0.01, †p < 0.005).
(D) RT-qPCR analysis of bFgf and Vegf during H2O2-induced apoptosis in CPCs (1–5 hr, n = 3 experiments, ∗p < 0.05, †p < 0.005).
(E) RT-qPCR analysis of miR-133a in CPC-CM after 48 hr in normoxia (20% O2) and hypoxia (1% O2) and in the purified exosomal fraction (normoxia), n = 3 experiments.
Data are mean ± SEM.
Recent studies have solidly established exosomes as paracrine effectors transferring functional molecules between cells in many physiological and pathological processes (Bang et al., 2014). In human CPCs (hCPCs), a critical role for exosomes has been associated to their capacity for improving cardiac function after injury (Barile et al., 2014, Ibrahim et al., 2014). Based on these evidences, we analyzed the levels of miR-133a present in the CM (48 hr) from miR-133a-CPCs and control-miR-CPCs cultures, in basal conditions (normoxia) and after hypoxia (Figure 6E), which simulates the situation after MI. CM from miR-133a-CPCs contained very high amounts of miR-133a compared with those detected in CM from control-miR-CPCs. The differences were comparable to those found in the cellular fraction (Figure S3A). Furthermore, hypoxia (1% O2) induced the increase of miR-133a levels secreted to the medium by 2-fold in both CPC populations. The analysis of purified exosomal fraction revealed that exosomes derived from control-miR-CPCs and miR-133a-CPCs were highly enriched in miR-133a, in relation to the whole CM.
Simulation of miR-133a-CPCs Protection in Engineered Heart Muscle
To investigate the responsiveness of heart muscle to miR-133a-CPCs in a controllable fashion, we used an in vitro heart muscle model, termed engineered heart muscle (EHM) (Tiburcy et al., 2011, Zimmermann et al., 2000, 2002). We generated cocultures of CPCs-EHMs by adding CPCs to isolated primary rat cardiomyocytes (84% ± 3% actinin-α1-positive, n = 4) in a 1:3 ratio (Figure 7A). In comparison with miR-control-EHM (containing control-miR-CPCs), the contractile force of miR-133a-EHM (containing miR-133a-CPCs) was significantly enhanced after addition of calcium to evoke contraction (Figure 7B). Given the demonstrated extensive cardiomyocyte death occurring during the first 3 days of EHM cultures (Tiburcy et al., 2011), we hypothesized that miR-133a-CPCs could protect cardiomyocytes against apoptosis in this period, leading to an increased number of viable cardiomyocytes at day 12 of culture. Indeed, we confirmed that caspase 3 activation, at day 3 of EHM culture, was reduced in miR-133a-EHM (Figures 7C and 7D). Consequently, we detected a higher percentage of cardiomyocytes in miR-133a EHM at day 12 when compared with miR-control cultures (Figures 7E and 7F). To clarify the nature of the support provided by miR-133a-CPCs to tissue formation, we analyzed the expression of growth factors. Fgf, Hgf, Igf-1, and Vegf levels were all increased in miR-133a-EHMs (Figures 7G–7J). Specially in early culture stages, we did not find a relevant modulation of additional prosurvival factors for cardiomyocytes as Sdf-1, a critical factor for the in vivo regeneration capacity of cardiospheres-derived cells (CDCs) (Malliaras et al., 2014) or Sirt-1, which protects the heart against oxidative stress when upregulated at moderate levels (Alcendor et al., 2007), but we found a clear upregulation of S100a4 throughout the whole culture (Figure S7). S100a4 has been shown to inhibit apoptosis and increase the survival capacity of cardiomyocytes after injury (Schneider et al., 2007). Accordingly, we found a highly significant difference (in early stages) in Bmf expression and a small not-significant difference in levels of Bcl-2, Bcl-xl, and Bim, suggesting that while Bim would be only relevant in CPCs, Bmf might mediate the protective role of miR-133 against stress-induced apoptosis, both in CPCs and cardiomyocytes.
Figure 7.
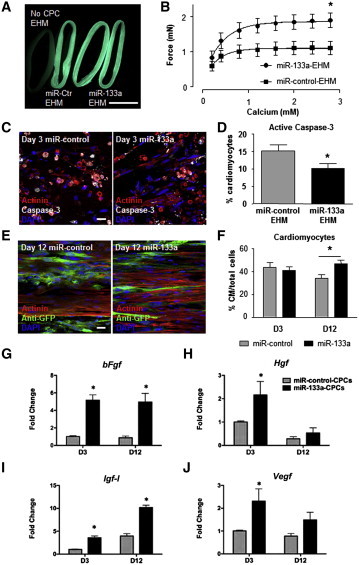
miR-133a-CPCs Enhance the Functionality of EHM
(A) GFP fluorescence on day 12 of culture of EHM with GFP-labeled miR-control-CPCs and miR-133a-CPCs compared with EHM without CPCs (left). The scale bar represents 5 mm.
(B) Calcium-induced contractile force of miR-control- or miR-133a-CPC EHM, n = 6 experiments, ∗p < 0.05.
(C) Representative staining of day 3 EHM cocultures with miR-control-CPCs (left) and miR-133a-CPCs (right) (actinin-α1, red; activated caspase 3, white; DAPI-stained nuclei, blue; scale bar represents 20 μm).
(D) Quantification of caspase 3-positive cardiomyocytes on day 3 of miR-control and miR-133a-CPCs-EHM cocultures; n > 1,000 cells from three experiments.
(E) Representative staining of cardiomyocytes and GFP+-CPCs on day 12 of cocultured EHM (actinin-α1, red; anti-GFP, green; DAPI-stained nuclei, blue; scale bar represents 20 μm).
(F) Quantification of cardiomyocyte percentage on days 3 and 12 of miR-control-CPCs and miR-133a-CPC-EHM cocultures, n > 1,000 cells/group from two experiments (day 3) and three experiments (day 12, ∗p < 0.05).
(G–J) Expression of bFgf, Hgf, Igf-1 and Vegf in miR-control-CPCs (gray bars) and miR-133a-CPCs (black bars) in EHM cocultures at day 3 and day 12 (n = 3 experiments per group, ∗p < 0.05).
Data are mean ± SEM. See also Figure S7.
Discussion
Cardiac differentiation of Sca-1+ CPCs is associated with a progressive increase in the expression of miR-1 and miR-133a. Although overexpression of these miRNAs had little influence on proliferation or apoptosis of CPCs in basal conditions, miR-1 increased the level of apoptosis promoted by H2O2, while miR-133 increased the number of viable cells under the same conditions. This is consistent with prior findings demonstrating that expression of miR-1 is induced after exposure to H2O2, in both a dose- and time-dependent manner and that several cell types overexpressing miR-1 are more vulnerable to H2O2-induced oxidative stress (Chen et al., 2012). The antiapoptotic action of miR-133a has not been reported previously in stem cells or CSCs/CPCs. In cardiomyocytes, although the role of miR-133a during in vitro apoptosis has been questioned, it has been validated in in vivo studies (Liu et al., 2008, Matkovich et al., 2010). Some reports have recently suggested that caspase 9 might be a direct target of miR-133a since in vivo infusion of this miRNA after MI reduces caspase 9 activity and decreases the number of apoptotic cardiomyocytes (He et al., 2011).
Bioinformatic analysis of putative miR-133 target genes rendered candidates mainly related to cell death and heart damage events such as hypertrophy, fibrosis, and apoptosis. Among them, Bim, Bmf, Stk4, and Foxo1 were involved in many of these processes. RT-qPCR analysis of miR-133a-CPCs versus miR-control-CPCs, treated with H2O2, demonstrated the potent proapoptotic factors Bim and Bmf were the most significantly downregulated. Among all BH3-only proteins known to date, Bim is the best described concerning its biological function, whereas little is known about its closest relative Bmf, which seems to play a more restricted role by supporting Bim in some cell death processes (Piñon et al., 2008). Recently, it was demonstrated that in mouse and human hematopoietic stem and progenitor cells, low expression of Bim or Bmf provokes a similar effect to overexpression of Bcl-2 and that their downregulation inhibits apoptosis, favoring HSC long-term engraftment (Labi et al., 2013). Furthermore, Bim is an important target for a miRNA cocktail (miR-21, miR-24, and miR-221) that significantly improves survival of untreated Sca1+CPCs (Hu et al., 2011). In agreement with these data, our results suggest that miR-133 protects CPCs from oxidative stress-induced apoptosis, at least in part, through targeting of Bmf and Bim. In addition, we found an increase of expression of bFgf and Vegf in miR-133-CPCs after treatment with H2O2 and a clear downregulation of caspase 3 and Bmf what would support the antioxidative role in miR-133 in CPCs.
GFP+ CPCs or GFP+ CPC-derived cells were not detected in rats 4 weeks after injection in infarcted hearts. This result is consistent with other previous studies where no or very few cells were found 1 month after cell administration. Overall, this implies that direct differentiation of implanted cells is not responsible for the improved cardiac function; accordingly, paracrine stimulation of endogenous repair has been proposed in cell therapy studies (Malliaras et al., 2012, Tang et al., 2010). This correlates with our findings that CPC transplantation increases vascular density and proliferation rate of resident cardiomyocytes. Our main hypothesis that miR-133a enhances CPCs-paracrine effects is supported by the upregulation of bFgf and Vegf in miR-133-CPCs upon oxidative stress, which might be an indirect consequence of the increased survival of CPCs. In the EHM model, we found that both growth factors were identically upregulated in the CPCs, as well as we found increased levels of Igf-1 and Hgf, being this last factor recently described as protective for hepatic stem cell/progenitor populations subjected to oxidative stress-induced apoptosis (Martínez-Palacián et al., 2013). In the EHM, CPCs were retained during culture, and the addition of miR-133a-CPCs enhanced the contractile capacity. This is at least partially achieved by reduced cardiomyocyte cell death during the initial 3 days of EHM culture and a probable direct effect of increased secretion of Vegf, bFgf, and Igf1 on contractility promotion (Cilvik et al., 2013, Troncoso et al., 2014, Zentilin et al., 2010). The first 3 days of EHM culture represent a vulnerable phase accompanied by substantial cell death, mainly by apoptosis of cardiomyocytes (Tiburcy et al., 2011). Antiapoptotic factors are particularly helpful during this early EHM culture phase (Naito et al., 2006, Vantler et al., 2010). Compared with miR-control-CPCs, miR-133a-CPCs reduced cardiomyocyte apoptosis and enabled a higher percentage of cardiomyocytes in longer cultures. Interestingly, inclusion of unmodified CPCs also provided protection, resulting in lower rates of apoptosis (∼15%) when compared with standard EHM cultures without CPCs (∼35%) (Tiburcy et al., 2011). These lower rates may be explained by the increased expression of cardioprotective growth factors (e.g., Igf-1), which are at least partly responsible for the beneficial effects of CSCs/CPCs and MSCs transplanted into ischemic heart (Malliaras et al., 2012, Wang et al., 2009). Other prosurvival factor incremented in miR-133a-EHM was S100a4, which has been shown to promote protection against apoptosis and improve cell survival rate in injured myocardium (Schneider et al., 2007). Our own data with the EHM model appear to concur with all of these favorable effects.
Our findings showing an increase of growth factors expression in miR133-CPCs upon stress conditioning and the amelioration of cardiac functions after miR133-CPCs transplantation post-AMI are in agreement with a recent publication that demonstrates that previous supplementation (“priming”) of SCA1+/CD31+ cells with recombinant IGF-1+HGF increases their engraftment capacity and survival rate, promoting angiogenesis and favoring regeneration in response to the hostile microenvironment of an infarcted heart, after their transplantation (Wang et al., 2014). Our results demonstrate that miR-133a, without affecting proliferation/differentiation potential, promotes CPCs survival and significantly increases their capacity to protect the heart against hypertrophy and apoptosis after MI. Recently, it has been proposed that exosomes, which resulted to be highly enriched in specific miRNAs, play a critical role in hCPC-driven improvement of cardiac function after injury (Barile et al., 2014). Exosomes secreted by CDCs also contain a distinctive repertoire of miRNAs (Ibrahim et al., 2014) and inhibit apoptosis and promote proliferation of cardiomyocytes, while enhancing angiogenesis. All of these recent evidences support the phenotype that we observed with miR-133a-CPCs. We confirmed that secreted miR-133a was mainly incorporated into the exosomal fraction of miR-133a-CPCs-CM, suggesting that direct transfer of the miRNA might mediate part of miR-133a-CPCs paracrine actions on the damaged tissue. However, the high levels of miR-133a already present in mature cardiomyocytes makes unlikely that CPC-exosomal contribution of miR-133a may provide additional significant effects on damaged cardiomyocytes.
Finally, it has been recently demonstrated that the combination of miR-133a with cardiac core transcriptional factors (Gata4, Mef2c, and Tbx5) or GMT plus Mesp1 and Myocd significantly improves direct cardiac reprogramming from human and mouse fibroblasts (Muraoka et al., 2014). Our results on the role of miR-133a in CPCs could be fully compatible with its plausible contribution to transient progenitor survival during the reprogramming process.
In summary, we believe that miR-133a protective actions integrate the rescue of endogenous CPCs, early activated progenitors as angioblasts (Malliaras et al., 2014) or endothelial cells, and the secretome/exosome-mediated repair of damaged resident cardiomyocytes via a complex combination of secreted miRNAs and growth and survival factors. Our work provides a detailed dissection of the mechanisms activated by miR-133-CPC transplantation and establishes the basis for a future improvement of therapeutic use of CPCs for regenerating injured myocardium after infarct.
Experimental Procedures
In Vitro Experiments with CPCs
Adult mouse hearts (C57BL/6 mice, 8–12 weeks old) were treated with collagenase type II (Worthington), and SCA-1+ Lin− cells were isolated using a Lineage Depletion kit and Sca-1 microbeads (Miltenyi Biotech). For preparation of CM, cardiomyocytes were cultured for 48 hr in fetal bovine serum-free M199 (apoptosis and hypertrophy experiments). For apoptosis experiments, cells were treated for 1–5 hr with 200 μmol/l H2O2, and samples were collected for gene expression analysis (RT-PCR) or apoptosis quantification (caspase 3/caspase 9; MBL International).
miR-1 and miR-133a Overexpression
pLenti4 lentiviral vectors were subcloned with the Gateway system (Invitrogen) from pcDNA vectors obtained with a BLOCK-iT Pol II miR RNAi Expression Vector Kit with EmGFP (Invitrogen) and designed for the expression of miR-1, miR-133a, and a nontargeting control miRNA. Retroviral vectors (MiR-Vec) used for the expression of miR-133a, and a control-miRNA, were purchased from Geneservice (Source BioScience). The pRRL.CMV.IRES.GFP lentiviral vector was used to trace GFP+ cells. Supernatants containing lentiviral or retroviral particles were obtained at high titer at the CNIC Viral Vector Facility.
EHM Cultures
EHM culture is a biomimetic in vitro heart model with cardiac muscle properties. EHM cocultures were prepared with CPCs and enriched NRCMs (1:3 ratio, total 2.5 × 106 cells per EHM) and maintained as previously described (Tiburcy et al., 2011, Zimmermann et al., 2000, 2002).
Statistical Analysis
Data are generally expressed as mean ± SEM. Data were analyzed with Student’s t test for paired experiments and with two-way ANOVA for multiple group comparisons. Analyses were conducted with GraphPad Prism 5.0, and values of p < 0.05 were considered statistically significant.
Author Contributions
A.I. and I.M. performed most of the experimental research; I.V., V.B., and A.D.-J. collaborated in several stages of the initial characterization on CPC populations. S.C. has collaborated in the final experiments requested by reviewers and manuscript adaptation; E.L., M.T., and W.-H.Z. designed and developed the EHM studies. I.J.N.-G., I.C., A.R.-S., and P.S., were involved in the analyses of CPC activity in the murine models of acute cardiac infarct. A.B. coordinated the study and was responsible for manuscript writing and fund raising.
Acknowledgments
This work was supported by grants to A.B. from the Ministry of Economy and Competition (SAF2012-3432, PLE2009-0147, and PSE-010000-2009-3), the Comunidad Autónoma de Madrid (S2011/BMD-2420), ISCIII (RD12/0019/0018). W.-H.Z. is supported by the DZHK (German Center for Cardiovascular Research), the German Federal Ministry for Science and Education (BMBF FKZ 13GW0007A, joint program with the California Institute of Renerative Medicine), the German Research Foundation (DFG ZI 708/7-1, 8-1, 10-1, SFB 1002 TP C04 and TP S), and the NIH (U01 HL099997). A.B. and W.-H.Z. have been both supported by the European Commission (FP7-HEALTH-2009/CARE-MI). A.I. and I.M. were postdoctoral fellows funded by the Ministry of Economy and Competition. S.C. is a contracted scientist funded by ISCIII. The CNIC and CNB are supported by the Ministry of Economy and Competition. We thank Iria Sánchez, Tamara Córdoba, Erica López, and Carmen Albo for technical help, Candelas Carreiro for logistics support, Marta Ramón for secretarial assistance, and Kenneth McCreath for help with manuscript editing.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information
References
- Alcendor R.R., Gao S., Zhai P., Zablocki D., Holle E., Yu X., Tian B., Wagner T., Vatner S.F., Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- Bang C., Batkai S., Dangwal S., Gupta S.K., Foinquinos A., Holzmann A., Just A., Remke J., Zimmer K., Zeug A. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Invest. 2014;124:2136–2146. doi: 10.1172/JCI70577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barile L., Lionetti V., Cervio E., Matteucci M., Gherghiceanu M., Popescu L.M., Torre T., Siclari F., Moccetti T., Vassalli G. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc. Res. 2014;103:530–541. doi: 10.1093/cvr/cvu167. [DOI] [PubMed] [Google Scholar]
- Beltrami A.P., Barlucchi L., Torella D., Baker M., Limana F., Chimenti S., Kasahara H., Rota M., Musso E., Urbanek K. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- Carè A., Catalucci D., Felicetti F., Bonci D., Addario A., Gallo P., Bang M.L., Segnalini P., Gu Y., Dalton N.D. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- Chen T., Ding G., Jin Z., Wagner M.B., Yuan Z. Insulin ameliorates miR-1-induced injury in H9c2 cells under oxidative stress via Akt activation. Mol. Cell. Biochem. 2012;369:167–174. doi: 10.1007/s11010-012-1379-7. [DOI] [PubMed] [Google Scholar]
- Chen S., Puthanveetil P., Feng B., Matkovich S.J., Dorn G.W., 2nd, Chakrabarti S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J. Cell. Mol. Med. 2014;18:415–421. doi: 10.1111/jcmm.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C., Wang Q., You W., Chen M., Xia J. MiRNAs as biomarkers of myocardial infarction: a meta-analysis. PLoS ONE. 2014;9:e88566. doi: 10.1371/journal.pone.0088566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugh A.R., Beache G.M., Loughran J.H., Mewton N., Elmore J.B., Kajstura J., Pappas P., Tatooles A., Stoddard M.F., Lima J.A. Administration of cardiac stem cells in patients with ischemic cardiomyopathy: the SCIPIO trial: surgical aspects and interim analysis of myocardial function and viability by magnetic resonance. Circulation. 2012;126(11, Suppl 1):S54–S64. doi: 10.1161/CIRCULATIONAHA.112.092627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cilvik S.N., Wang J.I., Lavine K.J., Uchida K., Castro A., Gierasch C.M., Weinheimer C.J., House S.L., Kovacs A., Nichols C.G., Ornitz D.M. Fibroblast growth factor receptor 1 signaling in adult cardiomyocytes increases contractility and results in a hypertrophic cardiomyopathy. PLoS ONE. 2013;8:e82979. doi: 10.1371/journal.pone.0082979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottini F., Hideshima T., Xu C., Sattler M., Dori M., Agnelli L., ten Hacken E., Bertilaccio M.T., Antonini E., Neri A. Rescue of Hippo coactivator YAP1 triggers DNA damage-induced apoptosis in hematological cancers. Nat. Med. 2014;20:599–606. doi: 10.1038/nm.3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danowski N., Manthey I., Jakob H.G., Siffert W., Peters J., Frey U.H. Decreased expression of miR-133a but not of miR-1 is associated with signs of heart failure in patients undergoing coronary bypass surgery. Cardiology. 2013;125:125–130. doi: 10.1159/000348563. [DOI] [PubMed] [Google Scholar]
- Del Re D.P., Matsuda T., Zhai P., Maejima Y., Jain M.R., Liu T., Li H., Hsu C.P., Sadoshima J. Mst1 promotes cardiac myocyte apoptosis through phosphorylation and inhibition of Bcl-xL. Mol. Cell. 2014;54:639–650. doi: 10.1016/j.molcel.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z., Chen J.F., Wang D.Z. Transgenic overexpression of miR-133a in skeletal muscle. BMC Musculoskelet. Disord. 2011;12:115. doi: 10.1186/1471-2474-12-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez B.G., Sampaolesi M., Barbuti A., Crespi A., Covarello D., Brunelli S., Dellavalle A., Crippa S., Balconi G., Cuccovillo I. Cardiac mesoangioblasts are committed, self-renewable progenitors, associated with small vessels of juvenile mouse ventricle. Cell Death Differ. 2008;15:1417–1428. doi: 10.1038/cdd.2008.75. [DOI] [PubMed] [Google Scholar]
- Gao S., Wassler M., Zhang L., Li Y., Wang J., Zhang Y., Shelat H., Williams J., Geng Y.J. MicroRNA-133a regulates insulin-like growth factor-1 receptor expression and vascular smooth muscle cell proliferation in murine atherosclerosis. Atherosclerosis. 2014;232:171–179. doi: 10.1016/j.atherosclerosis.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B., Xiao J., Ren A.J., Zhang Y.F., Zhang H., Chen M., Xie B., Gao X.G., Wang Y.W. Role of miR-1 and miR-133a in myocardial ischemic postconditioning. J. Biomed. Sci. 2011;18:22. doi: 10.1186/1423-0127-18-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori M., Nishida K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc. Res. 2009;81:457–464. doi: 10.1093/cvr/cvn335. [DOI] [PubMed] [Google Scholar]
- Hu S., Huang M., Nguyen P.K., Gong Y., Li Z., Jia F., Lan F., Liu J., Nag D., Robbins R.C., Wu J.C. Novel microRNA prosurvival cocktail for improving engraftment and function of cardiac progenitor cell transplantation. Circulation. 2011;124(11, Suppl.):S27–S34. doi: 10.1161/CIRCULATIONAHA.111.017954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hullinger T.G., Montgomery R.L., Seto A.G., Dickinson B.A., Semus H.M., Lynch J.M., Dalby C.M., Robinson K., Stack C., Latimer P.A. Inhibition of miR-15 protects against cardiac ischemic injury. Circ. Res. 2012;110:71–81. doi: 10.1161/CIRCRESAHA.111.244442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim A.G., Cheng K., Marbán E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Rep. 2014;2:606–619. doi: 10.1016/j.stemcr.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivey K.N., Srivastava D. MicroRNAs as regulators of differentiation and cell fate decisions. Cell Stem Cell. 2010;7:36–41. doi: 10.1016/j.stem.2010.06.012. [DOI] [PubMed] [Google Scholar]
- Ivey K.N., Muth A., Arnold J., King F.W., Yeh R.F., Fish J.E., Hsiao E.C., Schwartz R.J., Conklin B.R., Bernstein H.S., Srivastava D. MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell. 2008;2:219–229. doi: 10.1016/j.stem.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Zhang M., He H., Chen J., Zeng H., Li J., Duan R. MicroRNA/mRNA profiling and regulatory network of intracranial aneurysm. BMC Med. Genomics. 2013;6:36. doi: 10.1186/1755-8794-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labi V., Bertele D., Woess C., Tischner D., Bock F.J., Schwemmers S., Pahl H.L., Geley S., Kunze M., Niemeyer C.M. Haematopoietic stem cell survival and transplantation efficacy is limited by the BH3-only proteins Bim and Bmf. EMBO Mol. Med. 2013;5:122–136. doi: 10.1002/emmm.201201235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R., Feinbaum R., Ambros V. A short history of a short RNA. Cell. 2004;116:S89–92. doi: 10.1016/s0092-8674(04)00035-2. 1 p following S96. [DOI] [PubMed] [Google Scholar]
- Li Z., Hassan M.Q., Volinia S., van Wijnen A.J., Stein J.L., Croce C.M., Lian J.B., Stein G.S. A microRNA signature for a BMP2-induced osteoblast lineage commitment program. Proc. Natl. Acad. Sci. USA. 2008;105:13906–13911. doi: 10.1073/pnas.0804438105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X.B., Zhang Z.Y., Yuan K., Liu Y., Feng X., Cui R.R., Hu Y.R., Yuan Z.S., Gu L., Li S.J. MiR-133a modulates osteogenic differentiation of vascular smooth muscle cells. Endocrinology. 2013;154:3344–3352. doi: 10.1210/en.2012-2236. [DOI] [PubMed] [Google Scholar]
- Liu N., Bezprozvannaya S., Williams A.H., Qi X., Richardson J.A., Bassel-Duby R., Olson E.N. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22:3242–3254. doi: 10.1101/gad.1738708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malliaras K., Li T.S., Luthringer D., Terrovitis J., Cheng K., Chakravarty T., Galang G., Zhang Y., Schoenhoff F., Van Eyk J. Safety and efficacy of allogeneic cell therapy in infarcted rats transplanted with mismatched cardiosphere-derived cells. Circulation. 2012;125:100–112. doi: 10.1161/CIRCULATIONAHA.111.042598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malliaras K., Makkar R.R., Smith R.R., Cheng K., Wu E., Bonow R.O., Marbán L., Mendizabal A., Cingolani E., Johnston P.V. Intracoronary cardiosphere-derived cells after myocardial infarction: evidence of therapeutic regeneration in the final 1-year results of the CADUCEUS trial (CArdiosphere-Derived aUtologous stem CElls to reverse ventricUlar dySfunction) J. Am. Coll. Cardiol. 2014;63:110–122. doi: 10.1016/j.jacc.2013.08.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Palacián A., del Castillo G., Suárez-Causado A., García-Álvaro M., de Morena-Frutos D., Fernández M., Roncero C., Fabregat I., Herrera B., Sánchez A. Mouse hepatic oval cells require Met-dependent PI3K to impair TGF-β-induced oxidative stress and apoptosis. PLoS ONE. 2013;8:e53108. doi: 10.1371/journal.pone.0053108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matkovich S.J., Wang W., Tu Y., Eschenbacher W.H., Dorn L.E., Condorelli G., Diwan A., Nerbonne J.M., Dorn G.W., 2nd MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ. Res. 2010;106:166–175. doi: 10.1161/CIRCRESAHA.109.202176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscoso I., Tejados N., Barreiro O., Sepúlveda P., Izarra A., Calvo E., Dorronsoro A., Salcedo J.M., Sádaba R., Díez-Juan A. Podocalyxin-like protein 1 is a relevant marker for human c-kit(pos) cardiac stem cells. J. Tissue Eng. Regen. Med. 2013 doi: 10.1002/term.1795. Published online July 30, 2013. [DOI] [PubMed] [Google Scholar]
- Muraoka N., Yamakawa H., Miyamoto K., Sadahiro T., Umei T., Isomi M., Nakashima H., Akiyama M., Wada R., Inagawa K. MiR-133 promotes cardiac reprogramming by directly repressing Snai1 and silencing fibroblast signatures. EMBO J. 2014;33:1565–1581. doi: 10.15252/embj.201387605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito H., Melnychenko I., Didié M., Schneiderbanger K., Schubert P., Rosenkranz S., Eschenhagen T., Zimmermann W.H. Optimizing engineered heart tissue for therapeutic applications as surrogate heart muscle. Circulation. 2006;114(1 Suppl.):I72–I78. doi: 10.1161/CIRCULATIONAHA.105.001560. [DOI] [PubMed] [Google Scholar]
- Oh H., Bradfute S.B., Gallardo T.D., Nakamura T., Gaussin V., Mishina Y., Pocius J., Michael L.H., Behringer R.R., Garry D.J. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc. Natl. Acad. Sci. USA. 2003;100:12313–12318. doi: 10.1073/pnas.2132126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñon J.D., Labi V., Egle A., Villunger A. Bim and Bmf in tissue homeostasis and malignant disease. Oncogene. 2008;27(Suppl 1):S41–S52. doi: 10.1038/onc.2009.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthanveetil P., Wan A., Rodrigues B. FoxO1 is crucial for sustaining cardiomyocyte metabolism and cell survival. Cardiovasc. Res. 2013;97:393–403. doi: 10.1093/cvr/cvs426. [DOI] [PubMed] [Google Scholar]
- Robey T.E., Saiget M.K., Reinecke H., Murry C.E. Systems approaches to preventing transplanted cell death in cardiac repair. J. Mol. Cell. Cardiol. 2008;45:567–581. doi: 10.1016/j.yjmcc.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanganalmath S.K., Bolli R. Cell therapy for heart failure: a comprehensive overview of experimental and clinical studies, current challenges, and future directions. Circ. Res. 2013;113:810–834. doi: 10.1161/CIRCRESAHA.113.300219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M., Kostin S., Strøm C.C., Aplin M., Lyngbaek S., Theilade J., Grigorian M., Andersen C.B., Lukanidin E., Lerche Hansen J., Sheikh S.P. S100A4 is upregulated in injured myocardium and promotes growth and survival of cardiac myocytes. Cardiovasc. Res. 2007;75:40–50. doi: 10.1016/j.cardiores.2007.03.027. [DOI] [PubMed] [Google Scholar]
- Takaya T., Ono K., Kawamura T., Takanabe R., Kaichi S., Morimoto T., Wada H., Kita T., Shimatsu A., Hasegawa K. MicroRNA-1 and MicroRNA-133 in spontaneous myocardial differentiation of mouse embryonic stem cells. Circ. J. 2009;73:1492–1497. doi: 10.1253/circj.cj-08-1032. [DOI] [PubMed] [Google Scholar]
- Tang X.L., Rokosh G., Sanganalmath S.K., Yuan F., Sato H., Mu J., Dai S., Li C., Chen N., Peng Y. Intracoronary administration of cardiac progenitor cells alleviates left ventricular dysfunction in rats with a 30-day-old infarction. Circulation. 2010;121:293–305. doi: 10.1161/CIRCULATIONAHA.109.871905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi K., Ashihara E., Takehara N., Nomura T., Honsho S., Nakagami T., Morikawa S., Takahashi T., Ueyama T., Matsubara H., Oh H. Clonally amplified cardiac stem cells are regulated by Sca-1 signaling for efficient cardiovascular regeneration. J. Cell Sci. 2007;120:1791–1800. doi: 10.1242/jcs.006122. [DOI] [PubMed] [Google Scholar]
- Tiburcy M., Didié M., Boy O., Christalla P., Döker S., Naito H., Karikkineth B.C., El-Armouche A., Grimm M., Nose M. Terminal differentiation, advanced organotypic maturation, and modeling of hypertrophic growth in engineered heart tissue. Circ. Res. 2011;109:1105–1114. doi: 10.1161/CIRCRESAHA.111.251843. [DOI] [PubMed] [Google Scholar]
- Townley-Tilson W.H., Callis T.E., Wang D. MicroRNAs 1, 133, and 206: critical factors of skeletal and cardiac muscle development, function, and disease. Int. J. Biochem. Cell Biol. 2010;42:1252–1255. doi: 10.1016/j.biocel.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troncoso R., Ibarra C., Vicencio J.M., Jaimovich E., Lavandero S. New insights into IGF-1 signaling in the heart. Trends Endocrinol. Metab. 2014;25:128–137. doi: 10.1016/j.tem.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Vantler M., Karikkineth B.C., Naito H., Tiburcy M., Didié M., Nose M., Rosenkranz S., Zimmermann W.H. PDGF-BB protects cardiomyocytes from apoptosis and improves contractile function of engineered heart tissue. J. Mol. Cell. Cardiol. 2010;48:1316–1323. doi: 10.1016/j.yjmcc.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Wang X., Zhao T., Huang W., Wang T., Qian J., Xu M., Kranias E.G., Wang Y., Fan G.C. Hsp20-engineered mesenchymal stem cells are resistant to oxidative stress via enhanced activation of Akt and increased secretion of growth factors. Stem Cells. 2009;27:3021–3031. doi: 10.1002/stem.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Li L., Moore B.T., Peng X.H., Fang X., Lappe J.M., Recker R.R., Xiao P. MiR-133a in human circulating monocytes: a potential biomarker associated with postmenopausal osteoporosis. PLoS ONE. 2012;7:e34641. doi: 10.1371/journal.pone.0034641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Li Q., Hu Q., Suntharalingam P., From A.H., Zhang J. Intra-myocardial injection of both growth factors and heart derived Sca-1+/CD31- cells attenuates post-MI LV remodeling more than does cell transplantation alone: neither intervention enhances functionally significant cardiomyocyte regeneration. PLoS ONE. 2014;9:e95247. doi: 10.1371/journal.pone.0095247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim S.S., Akman D., Catalucci D., Turan B. Relationship between downregulation of miRNAs and increase of oxidative stress in the development of diabetic cardiac dysfunction: junctin as a target protein of miR-1. Cell Biochem. Biophys. 2013;67:1397–1408. doi: 10.1007/s12013-013-9672-y. [DOI] [PubMed] [Google Scholar]
- Zentilin L., Puligadda U., Lionetti V., Zacchigna S., Collesi C., Pattarini L., Ruozi G., Camporesi S., Sinagra G., Pepe M. Cardiomyocyte VEGFR-1 activation by VEGF-B induces compensatory hypertrophy and preserves cardiac function after myocardial infarction. FASEB J. 2010;24:1467–1478. doi: 10.1096/fj.09-143180. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Samal E., Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–220. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Ransom J.F., Li A., Vedantham V., von Drehle M., Muth A.N., Tsuchihashi T., McManus M.T., Schwartz R.J., Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- Zimmermann W.H., Fink C., Kralisch D., Remmers U., Weil J., Eschenhagen T. Three-dimensional engineered heart tissue from neonatal rat cardiac myocytes. Biotechnol. Bioeng. 2000;68:106–114. [PubMed] [Google Scholar]
- Zimmermann W.H., Schneiderbanger K., Schubert P., Didié M., Münzel F., Heubach J.F., Kostin S., Neuhuber W.L., Eschenhagen T. Tissue engineering of a differentiated cardiac muscle construct. Circ. Res. 2002;90:223–230. doi: 10.1161/hh0202.103644. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.