

Abstract
Aromatase inhibitors are the most important targets in treatment of estrogen-dependent cancers. In order to search for potent steroidal aromatase inhibitors (SAIs) with lower side effects and overcome cellular resistance, comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) were performed on a series of SAIs to build 3D QSAR models. The reliable and predictive CoMFA and CoMSIA models were obtained with statistical results (CoMFA: q2 = 0.636, r2ncv = 0.988, r2pred = 0.658; CoMSIA: q2 = 0.843, r2ncv = 0.989, r2pred = 0.601). This 3D QSAR approach provides significant insights that can be used to develop novel and potent SAIs. In addition, Genetic algorithm with linear assignment of hypermolecular alignment of database (GALAHAD) was used to derive 3D pharmacophore models. The selected pharmacophore model contains two acceptor atoms and four hydrophobic centers, which was used as a 3D query for virtual screening against NCI2000 database. Six hit compounds were obtained and their biological activities were further predicted by the CoMFA and CoMSIA models, which are expected to design potent and novel SAIs.
Keywords: steroidal aromatase inhibitors, 3D QSAR, CoMFA, CoMSIA, pharmacophore, virtual screening
1. Introduction
Aromatase is a cytochrome P-450 dependent enzyme that catalyzes the aromatization of androgens to estrogens. Aromatase inhibitors (AIs) reduce the synthesis of estrogens and offer a therapeutic alternative for the treatment of estrogen-dependent cancers, such as breast cancer [1,2,3]. There are two classes of AIs, steroidal and non-steroidal compounds, which cause potent estrogen suppression [4]. The non-steroidal aromatase inhibitors (NSAIs) are mostly azole type compounds, such as the clinically used anastrozole and letrozole, which compete with the substrate for binding to the enzyme active site [5]. Among steroidal aromatase inhibitors (SAIs), formestane was widely used during the early 1990s, but it is not used nowadays because of the need to administer it by intramuscular injection. Therefore, the orally active exemestane is the main steroidal inhibitor [6]. These SAIs mimic the natural substrate androstenedione and are converted by the enzyme to reactive intermediates, which bind irreversibly to the enzyme active site, resulting in inactivation of aromatase [7]. Despite the success of the third-generation NSAIs (anastrazole and letrozole) and SAIs (exemestane), they still have some major side effects, such as increase of bone loss, joint pain, and heart problems [8]. In addition, after some years of usage they can develop cellular resistance. For these reasons, it is important to search for other potent and specific molecules with lower side effects and which can overcome the resistance phenomena.
Quantitative structure-activity relationship (QSAR) methods have been successfully employed to assist the design of new small molecule drug candidates [9,10,11,12,13,14,15,16]. Comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) are two of the most widely used three-dimensional quantitative structure-activity relationship (3D QSAR) methodologies. CoMFA calculates the energies of steric and electrostatic interactions between the compound and the probe atom at various intersections of a regular 3D lattice according to Lennard-Jones and Coulomb potentials. The resulting energies derived from these two potential functions can be contoured to offer a quantitative spatial description of the molecular properties [17]. CoMSIA introduces the Gaussian function for the distance dependence between the molecular atoms and the probe atom in order to avoid some inherent deficiencies arising from the Lennard-Jones and Coulomb potential functional forms. CoMSIA is applied to gain an insight into how steric fields, electrostatic fields, hydrophobic fields, hydrogen bond donor (HBD) and hydrogen bond acceptor (HBA) influence the activity of inhibitors [18].
Pharmacophore modeling can provide valuable insight into ligand-receptor interactions. Pharmacophore searches are the best option to find a range of chemical structures with viable features. A pharmacophore model can be considered as the ensemble of steric and electrostatic features of different compounds, which are necessary to ensure optimal supramolecular interactions with a specific biological target structure and to trigger or to block its biological response. Thus, pharmacophore modeling is the method of choice for the first round of compound selection. This ability of a pharmacophore model is used to find new classes of inhibitors when one class is known. This is known as “scaffold hopping” [19,20,21].
A series of SAIs, shown in Table 1, have been reported in the recent literatures [22,23,24,25,26,27]. To understand the structural requirements for inhibitory activity and design more potent agents, 3D QSAR studies were performed for the fist time for these SAIs using CoMFA and CoMSIA. In addition, 3D pharmacophore models were created and the selected best model was used as a 3D query for virtual screening against NCI2000 database. The biological activities of hit compounds were further predicted by using CoMFA and CoMSIA models.
Table 1.
Chemical structures and bioactivity values of the steroidal aromatase inhibitors in current study.
| Compound | General Structure | Substituents | IC50 (µM) | pIC50 |
|---|---|---|---|---|
| 1 b | 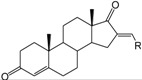 |
 |
5.2 | 5.284 |
| 2 a |  |
22.7 | 4.644 | |
| 3 | 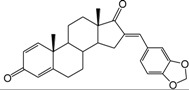 |
6.4 | 5.194 | |
| 4 | 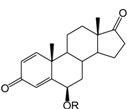 |
–CH3 | 5.2 | 5.284 |
| 5 a | –CH2CH3 | 18.1 | 4.742 | |
| 6 | –CH2C≡CCH3 | 0.1123 | 6.950 | |
| 7 b | –CH2C≡CCH2CH3 | 0.0118 | 7.928 | |
| 8 a | –CH2C≡C(CH2)2CH3 | 0.083 | 7.081 | |
| 9 | –CH2C≡C(CH2)3CH3 | 0.1811 | 6.742 | |
| 10 | –CH2C≡C(CH2)6CH3 | 2.18 | 5.662 | |
| 11 | –CH2C≡CCH2OH | 0.02 | 7.699 | |
| 12 b (Exemestane) | 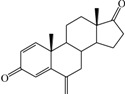 |
0.0501 | 7.300 | |
| 13 b (Formestane) | 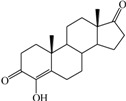 |
0.0486 | 7.313 | |
| 14 a | 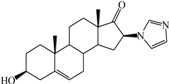 |
3.30 | 5.481 | |
| 15 | 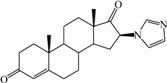 |
0.18 | 6.745 | |
| 16 b | 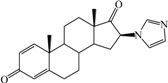 |
0.16 | 6.796 | |
| 17 | 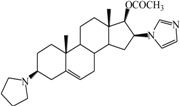 |
4.90 | 5.310 | |
| 18 a | 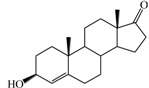 |
0.183 | 6.738 | |
| 19 b | 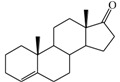 |
0.135 | 6.870 | |
| 20 | 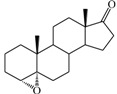 |
0.970 | 6.013 | |
| 21 | 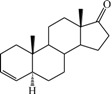 |
0.225 | 6.648 | |
| 22 a | 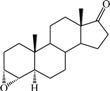 |
0.145 | 6.839 | |
| 23 | 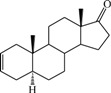 |
1.733 | 5.761 | |
| 24 | 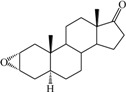 |
1.150 | 5.939 | |
| 25 a | 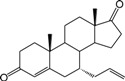 |
0.59 | 6.229 | |
| 26 | 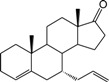 |
0.75 | 6.125 | |
| 27 b | 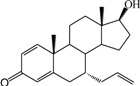 |
0.45 | 6.347 | |
| 28 | 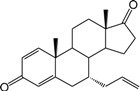 |
0.47 | 6.328 | |
| 29 a | 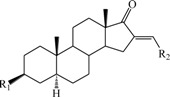 |
1-OH; 2-Ph | 7.27 | 5.138 |
| 30 | 1-OH; 2-MeOPh(p) | 7.13 | 5.147 | |
| 31 | 1-OCOCF3; 2-Ph | 7.12 | 5.148 | |
| 32 | 1-OCOCF3; 2-MeOPh(p) | 7.00 | 5.155 | |
| 33 | 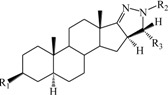 |
1-OCOCF3; 2-COCH3; 3-Ph | 6.57 | 5.182 |
| 34 a | 1-OCOCF3; 2-COCH3; 3-MeOPh(p) | 6.45 | 5.190 | |
| 35 | 1-OCOCF3; 2-COCH2CH3; 3-Ph | 6.72 | 5.173 | |
| 36 | 1-OCOCF3; 2-COCH2CH3; 3-MeOPh(p) | 6.61 | 5.180 | |
| 37 | 1-OH; 2-H; 3-Ph | 6.91 | 5.161 | |
| 38 | 1-OH; 2-H; 3-MeOPh(p) | 6.83 | 5.166 | |
| 39 a | 1-OCOCF3; 2-CH3; 3-Ph | 5.81 | 5.236 | |
| 40 | 1-OCOCF3; 2-CH3; 3-MeOPh(p) | 5.78 | 5.238 | |
| 41 a | 1-OCOCF3; 2-Ph; 3-Ph | 5.67 | 5.246 | |
| 42 | 1-OCOCF3; 2-Ph; 3-MeOPh(p) | 5.45 | 5.264 | |
| 43 | 1-OH; 2-CH3; 3-Ph | 6.34 | 5.198 | |
| 44 | 1-OH; 2-CH3; 3-MeOPh(p) | 6.12 | 5.213 | |
| 45 | 1-OH; 2-Ph; 3-Ph | 6.01 | 5.221 | |
| 46 a | 1-OH; 2-Ph; 3-MeOPh(p) | 5.92 | 5.228 | |
| 47 | 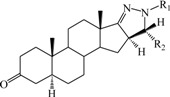 |
1-H; 2-Ph | 5.23 | 5.281 |
| 48 | 1-H; 2-MeOPh(p) | 4.88 | 5.312 | |
| 49 | 1-COCH3; 2-Ph | 4.91 | 5.309 | |
| 50 | 1-COCH3; 2-MeOPh(p) | 4.89 | 5.311 | |
| 51 a | 1-CH3; 2-Ph | 4.78 | 5.321 | |
| 52 | 1-CH3; 2-MeOPh(p) | 4.56 | 5.341 | |
| 53 | 1-Ph; 2-Ph | 4.27 | 5.370 | |
| 54 | 1-Ph; 2-MeOPh(p) | 4.16 | 5.381 | |
| 55 a | 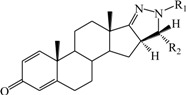 |
1-COCH3; 2-Ph | 2.88 | 5.541 |
| 56 | 1-COCH3; 2-MeOPh(p) | 2.65 | 5.577 | |
| 57 | 1-H; 2-Ph | 3.01 | 5.521 | |
| 58 | 1-H; 2-MeOPh(p) | 2.91 | 5.536 | |
| 59 | 1-CH3; 2-Ph | 2.45 | 5.611 | |
| 60 a | 1-CH3; 2-MeOPh(p) | 2.11 | 5.676 | |
| 61 | 1-Ph; 2-Ph | 1.98 | 5.703 | |
| 62 b | 1-Ph; 2-MeOPh(p) | 1.82 | 5.740 | |
| 63 | 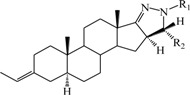 |
1-CH3; 2-Ph | 3.51 | 5.455 |
| 64 | 1-CH3; 2-MeOPh(p) | 3.40 | 5.469 | |
| 65 a | 1-Ph; 2-Ph | 3.34 | 5.476 | |
| 66 | 1-Ph; 2-MeOPh(p) | 3.23 | 5.491 |
a Test-set compounds; and b Compounds used to generate pharmacophore models.
2. Results and Discussion
2.1. CoMFA and CoMSIA Statistical Results
The statistical parameters of standard CoMFA models constructed with steric and electrostatic fields are given in Table 2. The cross-validated coefficient (q2/r2cv), non-cross-validated correlation coefficient (r2ncv), standard error estimate (SEE) and F-statistic values (F) were computed as defined in SYBYL. The cross-validated (leave-one-out) PLS analysis shows a q2 value of 0.636 with ten components and non-cross-validated PLS analysis results in r2ncv of 0.988, SEE of 0.094 and F value of 309.026, which indicates it is a model with high quality. The corresponding field contributions of steric and electrostatic are 0.671 and 0.329, respectively, which means the steric field gives more contribution to the bioactivity than the electrostatic field does.
Table 2.
Summary of CoMFA and CoMSIA results.
| Components | CoMFA | CoMSIA (SA) |
|---|---|---|
| q2 (r2cv) | 0.636 | 0.843 |
| r2ncv | 0.988 | 0.989 |
| SEE | 0.094 | 0.096 |
| F value | 309.026 | 174.304 |
| r2pred | 0.658 | 0.601 |
| No. of compounds | 50 | 50 |
| No. of optimal components | 10 | 17 |
| Contribution | ||
| Steric | 0.671 | 0.677 |
| Electrostatic | 0.329 | – |
| Hydrophobic | – | – |
| H-bond donor | – | – |
| H-bond acceptor | – | 0.323 |
Commonly, in CoMSIA, five different similarity fields (steric, electrostatic, hydrophobic, HBD, and HBA) are calculated. However, for CoMSIA models, the model with global descriptors is not the best model in all probability. Some papers [28,29] have discussed whether the five different descriptor fields in CoMSIA are totally independent of each other. The dependencies of the individual fields usually decrease the signal-to-noise ratio in the data [29] and lower the statistical significance of the results. Therefore, in our study, an optimization of 31 possible combinations of five different descriptor fields was evaluated from the values of the q2, which is shown in Figure 1. The higher the value of q2 shows, the better the model is. With the highest q2 of 0.843, CoMSIA model with the combination of steric and HBA fields (SA) was finally chosen as the best model, which indicates that the steric and HBA fields mainly contribute to the binding affinities. This CoMSIA (SA) model was obtained with seventeen optimal components. The analysis results are summarized in Table 2 (q2 = 0.843, r2ncv = 0.989, F = 174.304, SEE = 0.096). The corresponding field contributions of steric and HBA are 0.677 and 0.323, respectively, which means that the steric field provides more bioactivity contribution than the HBA field does.
Figure 1.

The histogram of 31 possibilities of the CoMSIA field combinations (S = steric, E = electrostatic, H = hydrophobic, D = hydrogen bond donor, A = hydrogen bond acceptor).
2.2. Validation of 3D QSAR Models
In order to validate the obtained 3D QSAR models, r2pred was used to determine the predictive abilities of the CoMFA and CoMSIA models from the 16 compounds (test set), which were not included in the generation of the models. The obtained r2pred of the test set is 0.658, 0.601 for the CoMFA, CoMSIA model, respectively, which indicates that both models have good predictive ability. The observed and predicted pIC50 of the training and test sets by the CoMFA and CoMSIA models are listed in Table 3, and the correlations between the observed and predicted pIC50 of training and test sets are depicted in Figure 2 for CoMFA model, Figure 3 for CoMSIA model, respectively.
Table 3.
Observed and predicted pIC50 of the training and test sets using CoMFA and CoMSIA models.
| Compound | Observed pIC50 | CoMFA | CoMSIA | ||
|---|---|---|---|---|---|
| Pred. | Res. | Pred. | Res. | ||
| 1 | 5.284 | 5.247 | 0.037 | 5.357 | −0.073 |
| 2 a | 4.644 | 5.642 | −0.998 | 5.744 | −1.100 |
| 3 | 5.194 | 5.152 | 0.042 | 5.192 | 0.002 |
| 4 | 5.284 | 5.191 | 0.093 | 5.278 | 0.006 |
| 5 a | 4.742 | 5.314 | −0.572 | 5.822 | −1.080 |
| 6 | 6.950 | 7.149 | −0.199 | 7.064 | −0.114 |
| 7 | 7.928 | 7.778 | 0.150 | 7.867 | 0.061 |
| 8 a | 7.081 | 7.102 | −0.021 | 7.477 | −0.396 |
| 9 | 6.742 | 6.804 | −0.062 | 6.711 | 0.031 |
| 10 | 5.662 | 5.722 | −0.060 | 5.678 | −0.016 |
| 11 | 7.699 | 7.598 | 0.101 | 7.685 | 0.014 |
| 12 | 7.300 | 7.272 | 0.028 | 7.281 | 0.019 |
| 13 | 7.313 | 7.378 | −0.065 | 7.361 | −0.048 |
| 14 a | 5.481 | 6.542 | −1.061 | 6.181 | −0.700 |
| 15 | 6.745 | 6.869 | −0.124 | 6.830 | −0.085 |
| 16 | 6.796 | 6.746 | 0.050 | 6.726 | 0.070 |
| 17 | 5.310 | 5.348 | −0.038 | 5.307 | 0.003 |
| 18 a | 6.738 | 6.668 | 0.070 | 7.123 | −0.385 |
| 19 | 6.870 | 6.811 | 0.059 | 6.888 | −0.018 |
| 20 | 6.013 | 5.965 | 0.048 | 6.062 | −0.049 |
| 21 | 6.648 | 6.436 | 0.212 | 6.267 | 0.381 |
| 22 a | 6.839 | 6.132 | 0.707 | 5.958 | 0.881 |
| 23 | 5.761 | 6.102 | −0.341 | 6.057 | −0.296 |
| 24 | 5.939 | 6.008 | −0.069 | 5.888 | 0.051 |
| 25 a | 6.229 | 6.236 | −0.007 | 6.359 | −0.130 |
| 26 | 6.125 | 6.074 | 0.051 | 6.151 | −0.026 |
| 27 | 6.347 | 6.301 | 0.046 | 6.365 | −0.018 |
| 28 | 6.328 | 6.282 | 0.046 | 6.292 | 0.036 |
| 29 a | 5.138 | 5.202 | −0.064 | 5.084 | 0.054 |
| 30 | 5.147 | 5.186 | −0.039 | 5.122 | 0.025 |
| 31 | 5.148 | 5.165 | −0.017 | 5.111 | 0.037 |
| 32 | 5.155 | 5.154 | 0.001 | 5.148 | 0.007 |
| 33 | 5.182 | 5.169 | 0.013 | 5.184 | −0.002 |
| 34 a | 5.190 | 5.185 | 0.005 | 5.157 | 0.033 |
| 35 | 5.173 | 5.184 | −0.011 | 5.186 | −0.013 |
| 36 | 5.180 | 5.187 | −0.007 | 5.160 | 0.020 |
| 37 | 5.161 | 5.127 | 0.034 | 5.146 | 0.015 |
| 38 | 5.166 | 5.194 | −0.028 | 5.153 | 0.013 |
| 39 a | 5.236 | 5.222 | 0.014 | 5.233 | 0.003 |
| 40 | 5.238 | 5.284 | −0.046 | 5.234 | 0.004 |
| 41 a | 5.246 | 5.237 | 0.009 | 5.273 | −0.027 |
| 42 | 5.264 | 5.291 | −0.027 | 5.282 | −0.018 |
| 43 | 5.198 | 5.156 | 0.042 | 5.200 | −0.002 |
| 44 | 5.213 | 5.222 | −0.009 | 5.209 | 0.004 |
| 45 | 5.221 | 5.177 | 0.044 | 5.236 | −0.015 |
| 46 a | 5.228 | 5.230 | −0.002 | 5.247 | −0.019 |
| 47 | 5.281 | 5.243 | 0.038 | 5.270 | 0.011 |
| 48 | 5.312 | 5.310 | 0.002 | 5.277 | 0.035 |
| 49 | 5.309 | 5.265 | 0.044 | 5.294 | 0.015 |
| 50 | 5.311 | 5.285 | 0.026 | 5.271 | 0.040 |
| 51 a | 5.321 | 5.279 | 0.042 | 5.337 | −0.016 |
| 52 | 5.341 | 5.341 | 0.000 | 5.339 | 0.002 |
| 53 | 5.370 | 5.303 | 0.067 | 5.377 | −0.007 |
| 54 | 5.381 | 5.356 | 0.025 | 5.388 | −0.007 |
| 55 a | 5.541 | 5.626 | −0.085 | 5.603 | −0.062 |
| 56 | 5.577 | 5.640 | −0.063 | 5.574 | 0.003 |
| 57 | 5.521 | 5.594 | −0.073 | 5.565 | −0.044 |
| 58 | 5.536 | 5.656 | −0.120 | 5.569 | −0.033 |
| 59 | 5.611 | 5.633 | −0.022 | 5.638 | −0.027 |
| 60 a | 5.676 | 5.698 | −0.022 | 5.641 | 0.035 |
| 61 | 5.703 | 5.659 | 0.044 | 5.682 | 0.021 |
| 62 | 5.740 | 5.714 | 0.026 | 5.690 | 0.050 |
| 63 | 5.455 | 5.408 | 0.047 | 5.475 | −0.020 |
| 64 | 5.469 | 5.469 | 0.000 | 5.479 | −0.010 |
| 65 a | 5.476 | 5.442 | 0.034 | 5.522 | −0.046 |
| 66 | 5.491 | 5.491 | 0.000 | 5.526 | −0.035 |
a Test-set compounds.
Figure 2.


Plots of observed versus predicted activities of the training set and test set molecules from CoMFA analysis.
Figure 3.


Plots of observed versus predicted activities of the training set and test set molecules from CoMSIA analysis.
2.3. CoMFA Contour Maps
The steric contour map for the CoMFA model with the most active inhibitor compound 7 is shown in Figure 4a, in which the green contours represent regions of high steric bulk tolerance (80% contribution), while the yellow contours represent regions of low steric bulk tolerance (20% contribution). It can be seen that a large green contour near C-6 of the ring B indicates a bulky group in this position is favorable to bioactivity. This is supported by the higher activity of compounds 6–11, which have large substituents in that position, compared with the lower activity of compounds 4–5, which have small substituents in that position. It also can be observed that there are two large yellow contours: one is next to C-3 of the ring A, and another is near C-15 and C-16 of the ring D, which suggests that a bulky group in the two regions will decrease inhibitory activity. It is confirmed by the fact that compound 17 with bulky substitution on C-3 of the ring A has lower bioactivity than compound 14 with small substitution in that position, and all the compounds with bulky groups near C-15 and C-16 of the ring D (compounds 1–3, compounds 29–66) show low bioactivities (pIC50 < 6).
Figure 4.

CoMFA contour maps (standard deviation × coefficient) in combination with compound 7. (a) Steric contour maps: Green contours (80% contribution) refer to sterically favored regions, yellow contours (20% contribution) indicate sterically disfavored regions; and (b) Electrostatic contour maps: Blue contours (80% contribution) refer to regions where positively charged substituents are favored, Red contours (20% contribution) indicate regions where negatively charged substituents are favored.
The CoMFA electrostatic contour map is shown in Figure 4b, in which the red areas are the regions where a negative potential is favorable to activity, while a negative potential is unfavorable in the blue areas. This figure displays two red contours around C-3 of the ring A and one red contour near C-4 of the ring A, which means that the bioactivity can be enhanced if an electronegative atom is present in these areas. It is supported that most of the studied compounds have electronegative atoms on C-3 or C-4 of the ring A. In addition, it also can be confirmed by the fact that compound 22, 24 with oxygen atom between C-3 and C-4 show higher activity than compound 21, 23 with no oxygen atom in that position, respectively.
2.4. CoMSIA Contour Maps
The steric and HBA fields are shown in Figure 5. For each field, the favorable and disfavored contours represent 80% and 20% level contributions, respectively. Figure 5a shows the steric contour map for the CoMSIA model with the most active inhibitor compound 7, in which green regions are sterically favorable and yellow regions are sterically unfavorable. It is clear that the distribution of steric field in CoMSIA is basically consistent with CoMFA results. A little difference is that there is a yellow contour below the large green contour, which indicates a proper bulky substituent on C-6 of the ring B, is favorable to bioactivity while too large substituent in that position is unfavorable. This is supported by the case that the inhibitory activities of compounds 7–10 decline with the augment of the substituents on C-6 of the ring B.
Figure 5.

CoMSIA contour maps (standard deviation × coefficient ) in combination with compound 7. (a) Steric contour maps: Green contours refer to sterically favored regions, yellow contours indicate sterically disfavored regions; and (b) HBA contour maps: Magenta contours show regions where HBA substituents are expected; Red contours refer to areas where HBA substituents are unexpected.
Figure 5b shows the HBA contour maps, in which magenta and red contours represent areas where HBA substituents are favored and disfavored, respectively. There are there magenta contours in this figure: one is next to C-3 of the ring A, another is near C-4 of the ring A, and the third one is close to C-17 of the ring D, which indicates that the activity can be enhanced if the HBA atoms are present in these there positions. This is also consistent with the distribution of electrostatic field in CoMFA. It is supported by the case that all the studied compounds have at least one HBA atom in those positions. It also can be confirmed by the example that compound 13 (Formestane) show quite high bioactivity because of its three HBA atoms in those positions.
2.5. Pharmacophore Generation
Twenty pharmacophore models were generated with default parameters after Genetic Algorithm with Linear Assignment of Hypermolecular Alignment of Database (GALAHAD) run, and their statistical values are listed in Table 4. Each of the obtained models represents a different tradeoff among the conflicting demands of maximizing steric consensus, maximizing pharmacophore consensus, and minimizing energy. All the twenty models had Pareto rank 0, which means no one model is superior to any other one. Model_03 has very high energy, which is recognized that a high-energy value is due to steric clashes [30]. Small value of energy and high values of Specificity, N_hits, Sterics and Mol_Qry are desired for the best model [31]. Therefore, Model_04 was considered to be the best model and its statistical values are shown in Table 4. This model contains two acceptor atoms and four hydrophobic centers, which is shown in Figure 6 and will be converted into a 3D query for the further virtual screening studies. This pharmacophore model is consistent with the results of CoMFA and CoMSIA. Four hydrophobic centers mean the centers of A, B, C and D rings of steroids, and two acceptor atoms next to C-3 of the ring A and C-17 of the ring D indicate that hydrogen bond acceptor atoms in these two positions can enhance the activity.
Table 4.
The statistical values of pharmacophore models after GALAHAD run.
| No. | Specificity | N_hits | Features | Pareto rank | Energy | Sterics | H-bond | Mol_Qry |
|---|---|---|---|---|---|---|---|---|
| Model_01 | 5.09 | 8 | 6 | 0 | 54.31 | 1158.80 | 56.90 | 17.53 |
| Model_02 | 3.67 | 8 | 6 | 0 | 20.85 | 1146.20 | 57.00 | 13.72 |
| Model_03 | 5.09 | 8 | 6 | 0 | 12657.71 | 1151.00 | 56.90 | 28.55 |
| Model_04 a | 5.15 | 8 | 6 | 0 | 8.76 | 1142.60 | 56.00 | 41.94 |
| Model_05 | 4.04 | 8 | 6 | 0 | 7.43 | 1142.30 | 55.50 | 41.25 |
| Model_06 | 4.04 | 8 | 6 | 0 | 5.39 | 1147.30 | 54.10 | 10.74 |
| Model_07 | 4.04 | 8 | 6 | 0 | 7.83 | 1135.50 | 56.60 | 35.13 |
| Model_08 | 4.04 | 8 | 6 | 0 | 8.63 | 1140.00 | 56.60 | 13.95 |
| Model_09 | 4.04 | 8 | 6 | 0 | 11.75 | 1140.40 | 56.60 | 17.87 |
| Model_10 | 4.04 | 8 | 6 | 0 | 7.23 | 1138.90 | 56.10 | 4.67 |
| Model_11 | 4.04 | 8 | 6 | 0 | 16.69 | 1146.70 | 55.50 | 41.25 |
| Model_12 | 5.09 | 8 | 6 | 0 | 24.98 | 1149.10 | 57.00 | 1.67 |
| Model_13 | 4.04 | 8 | 6 | 0 | 12.79 | 1152.80 | 56.60 | 0.00 |
| Model_14 | 4.04 | 8 | 6 | 0 | 29.82 | 1148.80 | 55.50 | 35.13 |
| Model_15 | 4.04 | 8 | 6 | 0 | 38.52 | 1158.60 | 54.50 | 11.47 |
| Model_16 | 5.15 | 8 | 6 | 0 | 7.25 | 1137.40 | 54.40 | 41.94 |
| Model_17 | 5.09 | 8 | 6 | 0 | 49.49 | 1151.90 | 56.90 | 2.81 |
| Model_18 | 3.69 | 8 | 6 | 0 | 7.07 | 1145.50 | 53.00 | 35.13 |
| Model_19 | 3.90 | 8 | 5 | 0 | 28.40 | 1150.00 | 55.90 | 10.03 |
| Model_20 | 4.04 | 8 | 6 | 0 | 9.09 | 1147.40 | 56.60 | 0.00 |
a The selected model (Model_04) is indicated in boldface.
Figure 6.

The selected GALAHAD model includes two acceptor atoms (green) and four hydrophobic centers (cyan). The sphere sizes indicate query tolerances.
2.6. Virtual Screening
The obtained best GALAHAD model (Figure 6) was converted into a UNITY query, which was screened against NCI2000 database. The “flexible database search” option was implemented to perform virtual screening. Primary filters such as Lipinski’s rule of five, Van der Waals bumps, and QFIT (pharmacophoric match between query and the hit compound) were applied to reduce the dataset [32]. The screening of the pharmacophore query yielded six hit compounds that met the specific requirements. The pIC50 values of the six hit compounds were further predicted using the obtained CoMFA and CoMSIA models. Chemical structures and their predicted activity values of the hit compounds are listed in Table 5. Among the hit compounds, compounds NCI 77798 and NCI 79104 show high-predicted pIC50 values by both CoMFA and CoMSIA models (pIC50 > 6.5), which are expected to design potent and novel SAIs.
Table 5.
Chemical structures and their predicted activity values of screened hit compounds.
| Hit Compound | Structure | pIC50 (Predicted by CoMFA) | pIC50 (Predicted by CoMSIA) |
|---|---|---|---|
| NCI 51178 | 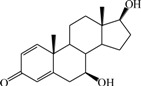 |
6.620 | 5.502 |
| NCI 51181 | 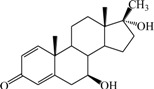 |
6.438 | 5.443 |
| NCI 51183 | 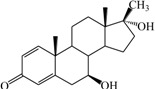 |
6.254 | 5.330 |
| NCI 51184 | 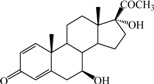 |
6.180 | 5.429 |
| NCI 77798 | 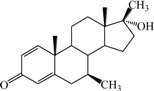 |
6.728 | 6.946 |
| NCI 79104 | 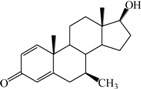 |
6.571 | 7.032 |
3. Experimental Section
3.1. Compounds and Biological Data
Compounds 1–3 [22], compounds 4–13 [23], compounds 14–17 [24], compounds 18–24 [25], compounds 25–28 [26] and compounds 29–66 [27] were used for this analysis, and their structures and bioactivity values are presented in Table 1. The pIC50 (−log IC50) values were used to derive 3D QSAR models. The pIC50 values of the studied compounds cover an interval of more than 3 log units. The whole data set of 66 compounds was divided into two groups in approximate ratio of 4:1; A training set with 50 compounds, a test set with 16 compounds (Table 1). The selection of the training and test sets was done manually such that low, moderate and high activity compounds were present in roughly equal proportions in both sets. The training set was used to build predictive models, while the test set was used to validate the predictive ability of the models.
3.2. Molecular Modeling and Alignment
The 3D QSAR modeling analyses, calculations and visualizations were performed using the SYBYL 7.3 molecular modeling package from Tripos Inc., St. Louis, Mo, USA, installed on Red Hat Linux workstations. Identification of the bioactive conformation is a very important step in a 3D QSAR study [33]. Among the inhibitors, the crystal structure of compound 7 bound with aromatase is available from the protein data bank (PDB code: 4GL7) [23]. Therefore, compound 7 was extracted from the complex and chosen to define the most likely binding conformation, which was modified by adding hydrogen atoms without any change of conformation and was minimized with the following steps: (i) Optimization by Steepest Descent with initial optimization of 200 simplex iterations using Tripos force field and Gasteiger-Marsili charges; (ii) Optimization by conjugate gradient; and (iii) Optimization by BFGS [34]. Three-dimensional structures of the other molecules were constructed from the compound 7. Energy minimizations of each compound were performed according to the above procedure. In the present study, compound 7 was used as a template because of its highest activity and all other compounds were aligned on the basis of the common structure shown in Figure 7. The alignment of training and test compounds is shown in Figure 8.
Figure 7.

Structure of the template molecule (compound 7), common substructure is in bold.
Figure 8.
Alignment of training and test set compounds on compound 7. Molecules are colored in white for common C, blue for N, red for O, green for F atoms, respectively.
3.3. CoMFA and CoMSIA Models
In CoMFA, the steric fields were calculated using a Lennard-Jones potential, while the electrostatic fields were calculated using a Coulombic potential. To calculate the CoMFA fields, a 3D cubic lattice with grid spacing of 2.0 Å in X, Y and Z directions was created automatically by SYBYL. The grid pattern extended 4.0 Å units in all directions beyond the dimensions of each molecule. The steric and electrostatic probe-ligand interaction energies were calculated by using a sp3 carbon probe atom and a +1.0 charge with a distance-dependent dielectric function at each lattice point. The cut-off for energies was set to ± 30 kcal/mol and the electrostatic contributions were ignored at lattice points with maximal steric interactions [17]. In CoMSIA, five different similarity fields (steric, electrostatic, hydrophobic, HBD, and HBA) were calculated. CoMSIA models were also derived with the same lattice box and all five fields were calculated using a probe of charge +1, a radius of 1, hydrophobicity and hydrogen bonding properties of +1, and an attenuation factor of 0.3 for the Gaussian distance-dependent function [18].
3.4. Statistical Analysis
In order to derive 3D QSAR models, CoMFA and CoMSIA descriptors were used as independent variables and the pIC50 values as the dependent variables. PLS method with cross-validation (leave-one-out) was used in SYBYL to determine the optimal numbers of components by using cross-validated coefficient q2 (r2cv). After obtaining the optimal numbers of components, a PLS analysis was performed with no validation and column filtering 2.0 to generate the final model with the training set. The obtained final non-cross-validated correlation coefficient (r2ncv) is a measure of the quality of the model. The predictive capability of the 3D QSAR models was determined from the predictive correlation (r2pred). The predicted activities for the test set were obtained from the model produced by the training set.
3.5. Pharmacophore Hypothesis
The pharmacophore hypothesis was generated using GALAHAD module of SYBYL, which operates in two main stages: The ligands are aligned to each other in internal coordinate space, and then the conformations produced are aligned in Cartesian space. The feature considered in developing the pharmacophore model includes HBD atoms, HBA atoms, hydrophobic and charged centers [35,36,37]. In our study, eight compounds shown in Table 1 were selected to carry out the pharmacophore hypothesis and the genetic algorithm was used to create conformers for all molecules. The compounds selected to generate the pharmacophore hypothesis are highly active and structurally diverse.
4. Conclusions
Aromatase inhibitors have proven to be the most important targets for treatment of estrogen-dependent cancers. In order to search for more potent SAIs with lower side effects and overcome the drug resistance, 3D QSAR studies, pharmacophore modeling and virtual screening were performed. The 3D QSAR techniques, CoMFA and CoMSIA, were applied for the first time to 66 new-synthesized SAIs, and the obtained models show good statistical results, q2 = 0.636, r2ncv = 0.988, r2pred = 0.658 for CoMFA and q2 = 0.843, r2ncv = 0.989, r2pred = 0.601 for CoMSIA, which indicate that both of the models have good quality and predictive ability. CoMFA and CoMSIA contour maps show that a proper bulky group near C-6 is favorable for activity while a bulky group near C-3, C-15 and C-16 is unfavorable, and the hydrogen bond acceptor atoms on C-3, C-4 and C-17 can enhance the bioactivity. In addition, pharmacophore models were derived from eight highly active and structurally diverse compounds using GALAHAD. The obtained best pharmacophore model includes two acceptor atoms and four hydrophobic centers, which was used as a query to search NCI2000 database. Six hit compounds were obtained after the screening of the pharmacophore query, and their pIC50 values were further predicted using the obtained CoMFA and CoMSIA models, which are expected to design potent and novel SAIs. The present 3D QSAR, pharmacophore modeling, and virtual screening approach provide useful information to design and synthesize potent and novel SAIs.
Acknowledgments
The authors gratefully acknowledge the support of this work by the Science and Technology Planning Project of Yunnan Province (No. 2011FZ096).
Statements
As shown in a series of recent publications [38,39,40,41,42,43,44,45] in response to the call in a comprehensive review [46], user-friendly and publicly accessible web-servers represent the future direction for developing practically more useful prediction and modeling methods or demonstrating new and novel findings, we shall make efforts in our future work to provide a web-server for the approach and finding presented in this paper.
Author Contributions
Huiding Xie performed all experiments and data treatments; Writing was done by Huiding Xie, Kaixiong Qiu and Xiaoguang Xie; and management and submission tasks were done by Huiding Xie and Xiaoguang Xie.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Winer E.P., Hudis C., Burstein H.J., Wolff A.C., Pritchard K.I., Ingle J.N., Chlebowski R.T., Gelber R., Edge S.B., Gralow J., et al. American society of clinical oncology technology assessment on the use of aromatase inhibitors as adjuvant therapy for postmenopausal women with hormone receptor-positive breast cancer: Status report 2004. J. Clin. Oncol. 2005;23:619–629. doi: 10.1200/JCO.2005.09.121. [DOI] [PubMed] [Google Scholar]
- 2.Perez E.A. Appraising adjuvant aromatase inhibitor therapy. Oncologist. 2006;11:1058–1069. doi: 10.1634/theoncologist.11-10-1058. [DOI] [PubMed] [Google Scholar]
- 3.Jordan V.C., Brodie A.M.H. Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids. 2007;72:7–25. doi: 10.1016/j.steroids.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brueggemeier R.W., Hackett J.C., Diaz-Cruz E.S. Aromatase inhibitors in the treatment of breast cancer. Endocr. Rev. 2005;26:331–345. doi: 10.1210/er.2004-0015. [DOI] [PubMed] [Google Scholar]
- 5.Recanatini M., Cavalli A., Valenti P. Nonsteroidal aromatase inhibitors: Recent advances. Med. Res. Rev. 2002;22:282–304. doi: 10.1002/med.10010. [DOI] [PubMed] [Google Scholar]
- 6.Seralini G.E., Moslemi S. Aromatase inhibitors: Past, present and future. Mol. Cell Endocrinol. 2001;178:117–131. doi: 10.1016/S0303-7207(01)00433-6. [DOI] [PubMed] [Google Scholar]
- 7.Hong Y., Rashid R., Chen S. Binding features of steroidal and nonsteroidal inhibitors. Steroids. 2011;76:802–806. doi: 10.1016/j.steroids.2011.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dutta U., Pant K. Aromatase inhibitors: Past, present and future in breast cancer therapy. Med. Oncol. 2008;25:113–124. doi: 10.1007/s12032-007-9019-x. [DOI] [PubMed] [Google Scholar]
- 9.Du Q.S., Mezey P.G., Chou K.C. Heuristic molecular lipophilicity potential (HMLP): A 2D QSAR study to LADH of molecular family pyrazole and derivatives. J. Comput. Chem. 2005;26:461–470. doi: 10.1002/jcc.20174. [DOI] [PubMed] [Google Scholar]
- 10.Du Q.S., Huang R.B., Wei Y.T. Multiple field three dimensional quantitative structure-activity relationship (MF-3D QSAR) J. Comput. Chem. 2008;29:211–219. doi: 10.1002/jcc.20776. [DOI] [PubMed] [Google Scholar]
- 11.Du Q.S., Huang R.B. Review: Recent advances in QSAR and their applications in predicting the activities of chemical molecules, peptides and proteins for drug design. Curr. Protein Pept. Sci. 2008;9:248–259. doi: 10.2174/138920308784534005. [DOI] [PubMed] [Google Scholar]
- 12.Du Q.S., Huang R.B., Wei Y.T. Fragment-based quantitative structure-activity relationship (FB QSAR) for fragment-based drug design. J. Comput. Chem. 2009;30:295–304. doi: 10.1002/jcc.21056. [DOI] [PubMed] [Google Scholar]
- 13.Prado-Prado F.J., Gonzalez-Diaz H., de la Vega O.M., Ubeira F.M., Chou K.C. Unified QSAR approach to antimicrobials. Part 3: First multi-tasking QSAR model for Input-Coded prediction, structural back-projection, and complex networks clustering of antiprotozoal compounds. Bioorg. Med. Chem. 2008;16:5871–5880. doi: 10.1016/j.bmc.2008.04.068. [DOI] [PubMed] [Google Scholar]
- 14.Cichero E., Fossa P. Docking-based 3D QSAR analyses of pyrazole derivatives as HIV-1 non-nucleoside reverse transcriptase inhibitors. J. Mol. Model. 2012;18:1573–1582. doi: 10.1007/s00894-011-1190-5. [DOI] [PubMed] [Google Scholar]
- 15.Zhao L.Z., Liu Y.J., Hu S.Y., Zhang H.B. 3D QSAR study of Chk1 kinase inhibitors based on docking. J. Mol. Model. 2012;18:3669–3694. doi: 10.1007/s00894-012-1363-x. [DOI] [PubMed] [Google Scholar]
- 16.Chen J., Yu R., Shen B.Z., Xu Y., Liu Y.F., Zheng H., Yao W.B. Docking-based 3D QSAR modeling of the inhibitors of IMP metallo-β-lactamase. Med. Chem. Res. 2013;22:1730–1739. doi: 10.1007/s00044-012-0172-1. [DOI] [Google Scholar]
- 17.Cramer R.D., Patterson D.E., Bunce J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988;110:5959–5967. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- 18.Klebe G., Abraham U., Mietzner T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994;37:4130–4146. doi: 10.1021/jm00050a010. [DOI] [PubMed] [Google Scholar]
- 19.Sirois S., Wei D.Q., Du Q.S., Chou K.C. Virtual screening for SARS-CoV protease based on KZ7088 pharmacophore points. J. Chem. Inf. Comput. Sci. 2004;44:1111–1122. doi: 10.1021/ci034270n. [DOI] [PubMed] [Google Scholar]
- 20.Liu L., Ma Y., Wang R.L., Xu W.R., Wang S.Q., Chou K.C. Find novel dual-agonist drugs for treating type 2 diabetes by means of cheminformatics. Drug Desi. Dev. Ther. 2013;7:279–287. doi: 10.2147/DDDT.S42113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhatt H.G., Patel P.K. Pharmacophore modeling, virtual screening and 3D QSAR studies of 5-tetrahydroquinolinylidine aminoguanidine derivatives as sodium hydrogen exchanger inhibitors. Bioorg. Med. Chem. Lett. 2012;22:3758–3765. doi: 10.1016/j.bmcl.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 22.Bansal R., Thota S., Karkra N., Minu M., Zimmer C., Hartmann R.W. Synthesis and aromatase inhibitory activity of some new 16E-arylidenosteroids. Bioorg. Chem. 2012;45:36–40. doi: 10.1016/j.bioorg.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 23.Ghosh D., Lo J., Morton D., Valette D., Xi J., Griswold J., Hubbell S., Egbuta C., Jiang W., An J., Davies H.M. Novel aromatase inhibitors by structure-guided design. J. Med. Chem. 2012;55:8464–8476. doi: 10.1021/jm300930n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bansal R., Guleria S., Thota S., Bodhankar S.L., Patwardhan M.R., Zimmer C., Hartmann R.W., Harvey A.L. Design, synthesis and evaluation of novel 16-imidazolyl substituted steroidal derivatives possessing potent diversified pharmacological properties. Steroids. 2012;77:621–629. doi: 10.1016/j.steroids.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 25.Valera C., Tavares da Silva E.J., Amaral C., Correia da Silva G., Baptista T., Alcaro S., Costa G., Carvalho R.A., Teixeira N.A.A., Roleira F.M.F. New structure-activity relationships of A- and D-Ring modified steroidal aromatase inhibitors: Design, synthesis, and biochemical evaluation. J. Med. Chem. 2012;55:3992–4002. doi: 10.1021/jm300262w. [DOI] [PubMed] [Google Scholar]
- 26.Valera C.L., Amaral C., Correia-da-Silva G., Carvalho R.A., Teixeira N.A., Costa S.C., Roleira F.M.F., Tavares-da-Silva E.J. Design, synthesis and biochemical studies of new 7α-allylandrostanes as aromatase inhibitors. Steroids. 2013;78:662–669. doi: 10.1016/j.steroids.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 27.Abdalla M.M., Al-Omar M.A., Bhat M.A., Amr A.E., Al-Mohizea A.M. Steroidal pyrazolines evaluated as aromatase and quinone reductase-2 inhibitors for chemoprevention of cancer. Int. J. Biol. Macromol. 2012;50:1127–1132. doi: 10.1016/j.ijbiomac.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 28.Böhm M., Stürzebecher J., Klebe G. Three-dimensional quantitative structure-activity relationship analyses using comparative molecular field analysis and comparative molecular similarity indices analysis to elucidate selectivity differences of inhibitors binding to trypsin, thrombin, and factor Xa. J. Med. Chem. 1999;42:458–477. doi: 10.1021/jm981062r. [DOI] [PubMed] [Google Scholar]
- 29.Norinder U. Recent progress in CoMFA methodology and related techniques. Perspect. Drug Discov. 1998;3:25–39. doi: 10.1023/A:1017057332537. [DOI] [Google Scholar]
- 30.Dorfman R.J., Smith K.M., Masek B.B., Clark R.D. A knowledge-based approach to generating diverse but energetically representative ensembles of ligand conformers. J. Comput. Aid. Mol. Des. 2008;22:681–691. doi: 10.1007/s10822-007-9156-5. [DOI] [PubMed] [Google Scholar]
- 31.Caballero J. 3D-QSAR (CoMFA and CoMSIA) and pharmacophore (GALAHAD) studies on the differential inhibition of aldose reductase by flavonoid compounds. J. Mol. Graph. Model. 2010;29:363–371. doi: 10.1016/j.jmgm.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 32.Kothandan G., Madhavan T., Gadhe C.G., Cho S.J. A combined 3D QSAR and pharmacophore-based virtual screening for the identification of potent p38 MAP kinase inhibitors: An in silico approach. Med. Chem. Res. 2013;22:1773–1787. doi: 10.1007/s00044-012-0179-7. [DOI] [Google Scholar]
- 33.Thaimattam R., Daga P.R., Banerjee R., Iqbal J. 3D QSAR studies on c-Src kinase inhibitors and docking analyses of a potent dual kinase inhibitor of c-Src and c-Abl kinases. Bioorg. Med. Chem. 2005;13:4704–4712. doi: 10.1016/j.bmc.2005.04.065. [DOI] [PubMed] [Google Scholar]
- 34.Hu R.J., Barbault F., Delamar M., Zhang R.S. Receptor- and ligand-based 3D QSAR study for a series of non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg. Med. Chem. 2009;17:2400–2409. doi: 10.1016/j.bmc.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 35.Richmond N.J., Abrams C.A., Wolohan P.R.N., Abrahamian E., Willett P., Clark R.D. GALAHAD: 1. Pharmacophore identification by hypermolecular alignment of ligands in 3D. J. Comput. Aid. Mol. Des. 2006;20:567–587. doi: 10.1007/s10822-006-9082-y. [DOI] [PubMed] [Google Scholar]
- 36.Shepphird J.K., Clark R.D. A marriage made in torsional space: Using GALAHAD models to drive pharmacophore multiplet searches. J. Comput. Aid. Mol. Des. 2006;20:763–771. doi: 10.1007/s10822-006-9070-2. [DOI] [PubMed] [Google Scholar]
- 37.Andrade C.H., Salum L.B., Pasqualoto K.F.M., Ferreira E.I., Andricopulo A.D. Three-dimensional quantitative structure-activity relationships for a large series of potent antitubercular agents. Lett. Drug Des. Discov. 2008;5:377–387. doi: 10.2174/157018008785777289. [DOI] [Google Scholar]
- 38.Chen W., Feng P.M., Deng E.Z., Lin H., Chou K.C. iTIS-PseTNC: A sequence-based predictor for identifying translation initiation site in human genes using pseudo trinucleotide composition. Anal. Biochem. 2014;462:76–83. doi: 10.1016/j.ab.2014.06.022. [DOI] [PubMed] [Google Scholar]
- 39.Ding H., Deng E.Z., Yuan L.F., Liu L., Lin H., Chen W., Chou K.C. iCTX-Type: A sequence-based predictor for identifying the types of conotoxins in targeting ion channels. Biomed. Res. Int. 2014;2014:286419. doi: 10.1155/2014/286419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fan Y.N., Xiao X., Min J.L., Chou K.C. iNR-Drug: Predicting the interaction of drugs with nuclear receptors in cellular networking. Int. J. Mol. Sci. 2014;15:4915–4937. doi: 10.3390/ijms15034915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo S.H., Deng E.Z., Xu L.Q., Ding H., Lin H., Chen W., Chou K.C. iNuc-PseKNC: A sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics. 2014;30:1522–1529. doi: 10.1093/bioinformatics/btu083. [DOI] [PubMed] [Google Scholar]
- 42.Liu B., Xu J., Lan X., Xu R., Zhou J., Wang X., Chou K.C. iDNA-Prot|dis: Identifying DNA-binding proteins by incorporating amino acid distance-pairs and reduced alphabet profile into the general pseudo amino acid composition. PLoS One. 2014;9:e106691. doi: 10.1371/journal.pone.0106691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiu W.R., Xiao X., Lin W.Z., Chou K.C. iMethyl-PseAAC: Identification of protein methylation sites via a pseudo amino acid composition approach. Biomed. Res. Int. 2014;2014:947416. doi: 10.1155/2014/947416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu Y., Wen X., Shao X.J., Deng N.Y., Chou K.C. iHyd-PseAAC: Predicting hydroxyproline and hydroxylysine in proteins by incorporating dipeptide position-specific propensity into pseudo amino acid composition. Int. J. Mol. Sci. 2014;15:7594–7610. doi: 10.3390/ijms15057594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu Y., Wen X., Wen L.S., Wu L.Y., Deng N.Y., Chou K.C. iNitro-Tyr: Prediction of nitrotyrosine sites in proteins with general pseudo amino acid composition. PLoS One. 2014;9:e105018. doi: 10.1371/journal.pone.0105018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chou K.C. Some remarks on protein attribute prediction and pseudo amino acid composition (50th Anniversary Year Review) J. Theor. Biol. 2011;273:236–247. doi: 10.1016/j.jtbi.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]