

Abstract
Cerebrotendinous xanthomatosis (CTX) OMIM#213700 is a rare autosomal-recessive lipid storage disease caused by mutations in the CYP27A1 gene; this gene codes for the mitochondrial enzyme sterol 27-hydroxylase, which is involved in bile acid synthesis. The CYP27A1 gene is located on chromosome 2q33-qter and contains nine exons. A CYP27A1 mutation leads to decreased synthesis of bile acid, excess production of cholestanol, and consequent accumulation of cholestanol in tissues. Currently there is no consensus on the prevalence of CTX, one estimate being <5/100,000 worldwide. The prevalence of CTX due to the CYP27A1 mutation R362C alone is approximately 1/50,000 in Caucasians. Patients with CTX have an average age of 35 years at the time of diagnosis and a diagnostic delay of 16 years. Clinical signs and symptoms include adult-onset progressive neurological dysfunction (i.e., ataxia, dystonia, dementia, epilepsy, psychiatric disorders,peripheral neuropathy, and myopathy) and premature non-neurologic manifestations (i.e., tendon xanthomas, childhood-onset cataracts, infantile-onset diarrhea, premature atherosclerosis, osteoporosis, and respiratory insufficiency). Juvenile cataracts, progressive neurologic dysfunction, and mild pulmonary insufficiency are unique symptoms that distinguish CTX from other lipid storage disorders including familial dysbetalipoproteinemia, homozygous familial hypercholesterolemia, and sitosterolemia, all of which might also present with xanthomas and cardiovascular diseases. Brain magnetic resonance imaging (MRI) shows bilateral lesions in the dentate nucleus of the cerebellum and mild white matter lesions. The classical symptoms and signs, namely elevated levels of cholestanol and bile alcohols in serum and urine, brain MRI, and the mutation in the CYP27A1 gene confirm the diagnosis of CTX. Early diagnosis and long-term treatment with chenodeoxycholic acid (750 mg/d) improve neurological symptoms and contribute to a better prognosis.
Keywords: Cerebrotendinous xanthomatosis, Xanthoma, CYP27A1, Sterol 27-hydroxylase, Chenodeoxycholic acid, Cholic acid
Introduction
Definition, history, epidemiology
Cerebrotendinous xanthomatosis (CTX; OMIM#213700) is a rare autosomal-recessive inborn disorder of bile acid metabolism due to mutations in the CYP27A1 gene(OMIM *606530) located on chromosome 2q33-qter, leading to increased deposition of cholesterol and cholestanol in multiple tissues [1]. Since Bogaert’s first report in 1937 of a case of CTX, more than several hundred cases have been reported worldwide [2,3]. CTX may have a higher prevalence than usually recognized. There are no consensus data on the prevalence of CTX, the estimated rate being <5/100,000 worldwide [4]. The prevalence of this disease varies with country and ethnic group; the prevalence of CTX due to the CYP27A1 mutation R362C alone is 1/800,000 individuals in Spain and is approximately 1/50,000 in Caucasians [4,5].
Etiology and biochemical pathogenesis
In 1974, Setoguchi et al. first found that the decrease in bile acid synthesis in patients with CTX resulted from impaired oxidation of the cholesterol side-chain, which suggested that CTX is linked to a disorder of bile acid synthesis [6]. Patients with CTX lack mitochondrial sterol 27-hydroxylase (EC 1.14.13.15). This enzyme is located on the inner membranes of the mitochondria, is expressed in almost all cells of the body, and is an important enzyme in both the alternative and classic bile acid synthesis pathways [7,8]. Cholesterol 7α-hydroxylase (CYP7A1) is the rate-limiting enzyme in the classic pathway. In the alternative pathway, sterol 27-hydroxylase (CYP27A1) oxidizes cholesterol to 27-hydroxycholesterol, which is subsequently hydroxylated by oxysterol 7α-hydroxylase, leading in humans mostly to the formation of chenodeoxycholic acid (CDCA) [8-10]. A deficiency of sterol 27-hydroxylase leads to reduced production of CDCA and subsequently to upregulation of cholesterol 7α-hydroxylase. Upregulation of the rate-limiting enzyme in the classic bile acid pathway results in elevated levels of 7α-hydroxy-4-cholesten-3-one, an efficient precursor to cholestanol [3]. Most of the cholestanol accumulated in patients with CTX is derived from 7α-hydroxylated metabolites of cholesterol, 7α-hydroxy-4-cholesten-3-one being the most important [3,11]. Development and progression of CTX are secondary to the further efficient conversion of 7α-hydroxy-4-cholesten-3-one into cholestanol and bile alcohols by two different pathways [3,12] (Figure 1).
Figure 1.
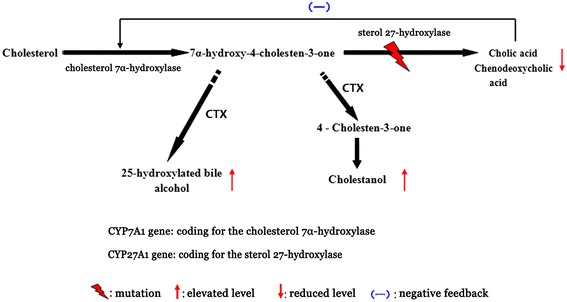
Metabolic pathway involved in cerebrotendinous xanthomatosis (CTX; modified from Chales and Bjorkhem [ 10 , 74 ] ). Due to a mutation in the CYP27A1 gene, cholesterol cannot be converted into bile acids, but is instead converted into cholestanol and bile alcohol. Providing chenodeoxycholic acid exogenously has a negative feedback effect that reduces synthesis of bile acid, thus preventing accumulation of cholestanol.
General symptoms
Patients with CTX present diverse manifestations with multi-organ involvement and a broad range of neurological and non-neurological symptoms [13]. For example, intractable infantile-onset diarrhea and psychomotor retardation are common coexisting clinical features of CTX [14,15]. The mean age at onset of symptoms in patients with CTX is 19 years, but the average age at the time of diagnosis is 35 years (range 23–44), thus representing a diagnostic delay of 16 years (range 2–34) [5].
Central nervous system involvement
Central nervous system symptoms and signs commonly present and sometimes constitute the initial manifestations in patients with CTX [16-22]. Epilepsy and Parkinsonism are the initial neurological features of CTX [19,20]. In a retrospective study involving 25 patients in Spain, Pilo-de-la-Fuente et al. divided the neurological manifestations into two main clinical subgroups, the classic form (cerebellar and supratentorial symptoms) and the spinal form (chronic myelopathy) [5]. The range of neurological features of CTX reported in the literature is broad; these features include intellectual disability, dementia, psychiatric symptoms (i.e., behavioral changes, depression, agitation, hallucination, and suicide attempts), pyramidal signs, progressive ataxia, dystonia, and palatal myoclonus [16,17,19-21,23-26].
Ocular system involvement
Childhood-onset cataract is a typical sign of CTX [13-15]. This has been emphasized as an early symptom preceding neurological signs and tendon xanthoma, and is considered useful for early diagnosis. Cataracts and optic disk paleness are also the common ocular features in adults with CTX [21]. Other ocular abnormalities in CTX include retinal vessel sclerosis and cholesterol-like deposits [21,27].
Cardiovascular system involvement
Premature atherosclerosis and cardiovascular disease have been reported among the multiple clinical manifestations of CTX [28]. Patients with CTX suffered from severe premature atherosclerosis in spite of normal serum cholesterol concentrations [29]. Blood lipid analysis in patients with CTX revealed dramatically high levels of 27-hydroxycholesterol and low levels of high-density lipoprotein cholesterol,which place patients with CTX at a high risk of suffering from cardiovascular disease [30].
Skeletal system involvement
Osteoporosis and repeated bone fractures are also common clinical manifestations of patients with CTX. Low bone mass in the patient with severe gait disturbances increases the risk of accidental falls and bone fractures [31-33]. Federico et al. discovered that serum calcium, phosphate, and vitamin D metabolites in CTX patients were normal, but the total body bone mineral density was low and intestinal radiocalcium absorption was decreased [33]. However, the underlying pathogenesis is still unknown. There is no correlation between the severity of osteoporosis and biochemical parameters (i.e. cholestanol level, phenotypic data, and disability associated with neurological dysfunction) in patients with CTX [32].
Pulmonary system involvement
Kawabata et al. discovered accumulations of foamy and giant cells engorged with cholestanol in bronchoalveolar lavage fluids and lung biopsy of patients with CTX, which demonstrated that the lungs are involved in CTX [34]. Patients with CTX with pulmonary involvement may have no clinical pulmonary symptoms (e.g., shortness of breath, chest distress) and no disturbance in pulmonary function tests [34]. Based on the results of positron emission tomography (PET), we found a high-density lesion (12 × 14 mm) and a cyst with a gas-fluid level (16 × 20 mm) in the lung of a patient with CTX (Figure 2). Pulmonary lymphangioleiomyomatosis was detected in a patient with CTX, but whether there is an association between these two rare diseases is unknown [35].
Figure 2.
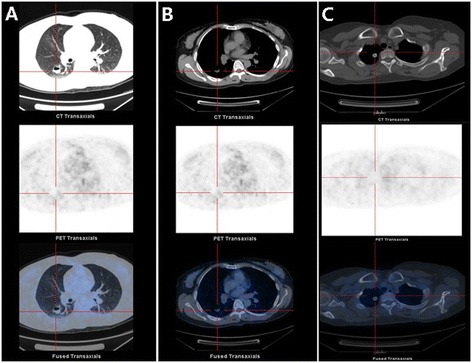
Positron emission tomography (PET) imaging results in CTX: pulmonary system involvement. PET reveals a cyst with a gas-fluid level (16 × 20 mm, A and B) and a high-density lesion (12 × 14 mm, C) in the lung.
Enterohepatic system involvement
In a large series of 32 patients with CTX studied by the Verrips et al., 50% had chronic and intractable diarrhea, which began in childhood [36]. Ninety-two percent of the patients with CTX in another large retrospective study in Spain had chronic diarrhea [5]. However, the gastro-intestinal examinations in many of the patients with diarrhea were normal [37]. Diarrhea in patients with CTX disappears a shortly after the start of CDCA therapy; however, the underlying mechanism is unknown [15,36,37]. This may be partly because bile alcohols produced in CTX are replaced by CDCA, which is an excellent micelle-forming bile acid, thereby improving fat and fat-soluble vitamin absorption and the attendant diarrhea. Some patients with CTX also develop neonatal cholestatic jaundice, cholecystic polypus, and gallstones [21,38].
Peripheral nervous system and muscle involvement
Ginanneschi et al. revealed that 74.2% of patients with CTX (n =35) showed peripheral nerve abnormalities [39]. Demyelination and remyelination, features of axonal degeneration, can be found in patients with CTX [40]. Mild myopathic changes and ultrastructural abnormalities in mitochondria are observed in muscle lesions [41]. Of note, the presence of tendon xanthomas is not necessary for the diagnosis of CTX because not all patients have visible tendon xanthomas.
Laboratory findings
The biochemical abnormalities in CTX include a plasma cholestanol concentration five- to ten-fold greater than normal (330 ± 30 μg/dL), a urine bile alcohol concentration of 14,000 ± 3,500 nmol/L, and a plasma bile alcohol concentration more than 500- to 1,000-fold greater than normal (8.48 ± 3.67 nmol/L). The biochemical abnormalities that distinguish CTX from other diseases with xanthomas include: high plasma cholestanol concentration, normal-to-low plasma cholesterol concentration, decreased CDCA level, and increased levels of cholestanol and apolipoprotein B in cerebrospinal fluid [42]. An elevated plasma 5-α-cholestanol concentration detected by gas chromatography–mass spectrometry (GC-MS) is a biomarker for CTX [43].
Instrumental examinations
Transcranial magnetic stimulation (TMS) is a useful tool for detecting corticospinal tract damage and for evaluating improvements in pyramidal function after CDCA therapy [44]. In CTX, abnormalities are seen in neurophysiological examinations such as visual evoked potential (VEP), somatosensory evoked potential (SSEP), brainstem auditory evoked potential (BAEP), and nerve conduction velocity (NCV), tests that have been widely described [5,39,45-47].
Imaging
Imaging studies have a significant role to play in prompt diagnosis.
First, the brain MRI reveals cerebellar atrophy, white matter signal alterations, and symmetric hyperintensities in the dentate nuclei [42,48-53] (Figure 3). Gray matter (GM) and white matter (WM) volume are diffusely decreased in patients with CTX [54,55]. In additional to conventional MRI, diffusion tensor imaging (DTI) and voxel-based morphometry (VBM) can provide complementary information about the involvement of GM and WM in CTX [54]. DTI is a useful tool for detecting white matter tract changes, because it is sensitive to water diffusion. VBM as an unbiased brain quantitative method that can be used to delineate volume losses in GM and WM [56]. Several previous magnetic resonance spectroscopy (MRS) studies revealed increased lactate and lipid peaks in FLAIR-hypointense lesions and decreases in N-acetylaspartate (NAA) peaks diffusely [49,51,57,58]. SPECT imaging reveals regional cerebral blood flow (rCBF) changes in multiple brain lobes before and after therapy, which might be a useful tool for monitoring the response to therapy in patients with CTX [54,59,60]. SPECT imaging can assess mitochondrial status and presynaptic dopaminergic function (using the special photographic developers 99mTc-sestamibi and 123I-FP-CIT, respectively) associated with Parkinsonian symptoms in CTX [61,62]. Using 18F-6-fluoro-L-dopa for PET analysis, Kuwabara et al. found reduced uptake of dopamine into the putamen in a patient with both CTX and hemiparkinsonism, suggesting a dysfunction of presynaptic dopaminergic neurons, which is not classical in CTX [63]. The PET images exhibited remarkable differences in basal brain metabolic rate between patients with CTX and normal volunteers (Figure 4).
Figure 3.
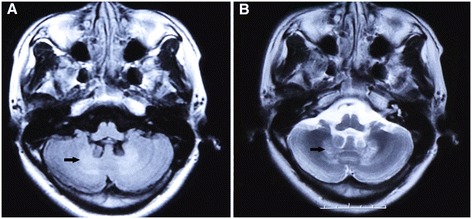
MRI results: brain. Brain MRI shows T1-weighted (A, arrow) and T2-weighted (B, arrow) hyperintensities in the dentate nuclei.
Figure 4.
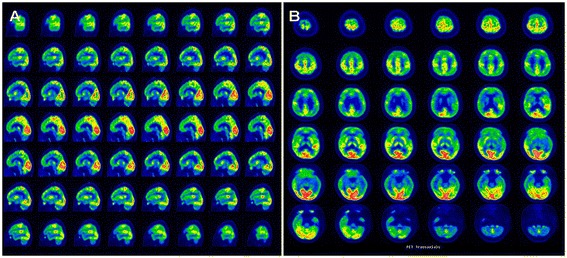
PET imaging results: brain. Remarkable abnormalities are seen in the basal brain metabolic rate in a patient with CTX. PET reveals hypometabolism in cerebral lobes (especially in the frontal and temporal lobes) in sagittal section (A), and in axial section (B).
Second, MRI of both ankles shows fusiform thickening and heterogeneous signals [24] (Figure 5). Moreover, PET analysis using 18F-2-deoxy-2-fluoro-glucose shows abnormally high radioactivity in the Achilles tendons and adjacent regions (Standardized Uptake Value 8.7-13.6) (Figure 6).
Figure 5.
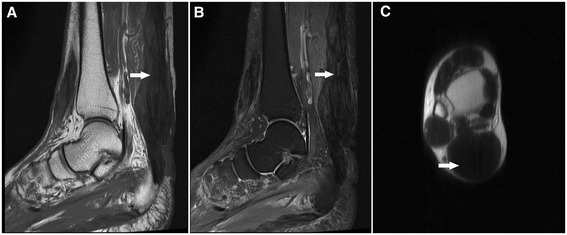
5 MRI results: tendon. MRI of both ankles. Sagittal long-T1-weighted image (A, arrow) and short-T2-weighted image (B, arrow), and axial long-T1-weighted image (C, arrow) of fusiform thickenings in the Achilles tendons in a patient with CTX.
Figure 6.

PET imaging results: tendon. Unusually high radioactivity is found in both Achilles tendons of a patient with CTX. PET shows abnormal soft-tissue thickening in a CT window in coronal section (A) and in axial section (D), and unusually high radioactivity in Achilles tendons and adjacent regions in PET (B and E) and fusion windows (C and F).
Third, radiological images of the lung have been reported in several cases with CTX. The lesions in the lungs manifest as diffuse, nodular, infiltrated, and fibrotic shadows, which suggests interstitial pulmonary dysfunction or xanthomatous lesions [21,34,35].
Pathology
Macroscopic section of the brain shows brain atrophy with multiple yellowish deposits in the plexus choroideus and in brain white matter [64]. Under microscopic examination, pathological findings in the central nervous system in CTX include multiple dispersed lipid crystal clefts and granulomatous lesions in the cerebellar hemispheres, demyelination and perivascular accumulation of foamy macrophages in the globus pallidus, and extracellular deposition of homogeneous myelin-like material in periventricular areas [52,64-66]. Demyelination, gliosis, and involvement of the long tracts of the spinal cord have also been described [52,67]. Nerve biopsy reveals primary axonal degeneration, demyelination, and remyelination [40,68-70]. The pathological findings from needle aspiration and autopsy of the lungs of patients with CTX reveal granulomatous materials, foamy cells, and intracellular accumulations of foreign bodies [34,35,71]. Histopathology of the tendon masses shows an accumulation of xanthoma cells and multiple, dispersed lipid crystal clefts [70] (Figure 7). Under electron microscopic examination, ultrastructural abnormalities are found in the lesions, including subsarcolemmal accumulation in mitochondria and swollen sarcoplasmic reticulum [46].
Figure 7.
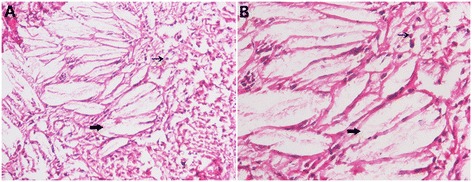
Histology: tendon. HE staining of the tendon masses reveals accumulation of xanthoma cells (fine arrows) and dispersed lipid crystal clefts (coarse arrows). A, 100×; B, 200 × .
Genetic analysis
The gene coding for sterol 27-hydroxylase was first located on the q33-qter interval of human chromosome 2 and mouse chromosome 1 by the group of Russell [1]. DNA sequence analysis of CYP27A1 predicted a human sterol 27-hydroxylase consisting of a 33 amino-acid mitochondrial signal sequence followed by 498 amino acids [1]. Various mutations in all nine exons and in introns 2,4,6,7, and 8 of the CYP27A1 gene have been described worldwide [8]. Fifty percent of mutations in CYP27A1 have been detected in the region of exons 6–8, 16% in exon 2, and 14% in exon 4 [8,72]. Various mutation types, including missense (approximately 45%), nonsense (approximately 20%), splice site (18%), deletion (14%), and insertion (2%) have been detected in all nine exons of CYP27A1 [2]. No genotype-phenotype correlations have been identified in CTX [2,5,73]. It is notable that CYP27A1 was identified as a candidate gene for sporadic amyotrophic lateral sclerosis in a large genome-wide screening study [74,75].
Differential diagnosis
Sitosterolemia
Sitosterolemia (OMIM#210250), is a very rare inherited sterol storage disease caused by mutations in the adenosine triphosphate-binding cassette (ABC) transporter genes ABCG5 and ABCG8, which are located on chromosome 2p21 and expressed at the membrane of enterohepatic cells [76-78]. Around 100 sitosterolemia cases have been reported in the literature [79]. Increased absorption and decreased excretion of plant sterols are the metabolic characteristics of sitosterolemia [22,80]. Clinical manifestations are characterized by extensive tuberous and tendonous xanthomas, premature atherosclerosis, hemolytic anemia, arthritis, and thrombocytopenic purpura [79,81,82]. Laboratory findings included high plasma phytosterol concentration and normal to mildly elevated plasma cholesterol concentrations [22,79]. Combined treatment with ezetimibe and a low plant-sterol diet can be effective in reducing plant sterol levels in the plasma, promoting xanthoma regression, improving the cardiovascular and hematological signs [79,83].
Dyslipidemias
Familial hypercholesterolemia (FH; OMIM#143890) is an autosomal-dominant disease caused by a deficiency in low-density lipoprotein (LDL) receptor (ADH1) activity or in LDL-related genes (ApoB and PCSK9), which leads to obviously elevated LDL cholesterol (LDL-C) and triglyceride concentrations [10,84]. The prevalence of FH is estimated to be 1/300-500 (heterozygous type) worldwide [84,85]. The underlying pathogenesis of FH is the decreased clearance of apolipoprotein B-containing lipoproteins and their subsequent accumulation in multiple tissues [22]. The diagnostic criteria for FH in adults (>15 years of age) include hyper-LDL cholesterolemia (untreated LDL-C level ≥180 mg/dL), tendonous xanthomas (xanthoma on the backs of the hands, elbows, knees, etc.) or xanthoma tuberosum, and a family history of FH or premature coronary artery disease [84]. Familial dysbetalipoproteinemia may present with palmar crease xanthomas, but familial hypercholesterolemia presents with intertriginous xanthomas in children, and sitosterolemia and CTX present with tendonous xanthomas in adults [86]. Lifestyle modification and high-potency statins should be the first-line treatment and can delay the onset of coronary artery disease [84,87].
Neurologic symptoms and diarrhea, which are important features of CTX, are non-existent in patients with sitosterolemia or FH [22]. Note also that the morphology of xanthomatosis is variable. Depending on the clinical morphology, localization, development, and progression of this lipid storage disorder, xanthomas can present as either eruptive, tuberous, tendonous, or planar [10,86]. CTX can be distinguished from other lipid storage disorders by its specific clinical features such as childhood-onset cataracts, progressive neurologic symptoms, mild pulmonary insufficiency, increased plasma cholestanol levels, and the results of the SSEP assessment [10,22].
Other inborn errors of bile acid metabolism
Other disorders of bile acid synthesis and metabolism include cholesterol 7α-hydroxylase deficiency (mutation in the CYP7A1 gene), 3β-hydroxy-C27-steroid oxidoreductase deficiency,and 2-methylacyl-CoA racemase deficiency [88]. Patients with cholesterol 7α-hydroxylase deficiency have elevated serum cholesterol concentrations and are unresponsive to hydroxymethylglutaryl (HMG)-CoA reductase inhibitor therapy. Neonates with 3-β-hydroxy-C27-steroid oxidoreductase deficiency show hepatomegaly, mild steatorrhea, elevated serum ALT and AST, hyperbilirubinemia, and normal serum γ-glutamyl transpeptidase [89]. Defects in 2-methylacyl-CoA racemase have profound effects on both the bile acid and the fatty acid pathways. Patients may present with a sensorimotor neuropathy in adults and with a fat-soluble vitamin deficiency, hematochezia, and cholestatic liver disease in infants [90]. Bile acid therapy with cholic acid (10 to 15 mg · kg−1 · day−1) has also been proved effective in treating the above three metabolic disorders.
Diagnosis
Early detection and diagnosis of CTX is crucial because early and long-term treatment of CTX with CDCA improves neurological symptoms and even reverses the progression of the disease [5,23,38,53,77]. However, an obvious delay between symptom onset and diagnosis is prevalent [5,38].
The diagnosis of CTX is mainly based on clinical findings, biochemical testing, neuroimaging, and molecular genetic analysis. A diagnosis of CTX should be considered for patients with xanthomas and neurological symptoms starting in childhood. It should be noted that the symptoms might also start in adulthood [91]. The biochemical abnormalities of patients with CTX in the laboratory examination include elevated plasma cholestanol level and increased levels of bile alcohols in urine associated with a diminished biliary concentration of chenodeoxycholic acid [22]. An elevated plasma level of cholestanol is a feature of CTX [10,22,43]. It was reported that the serum levels of 7-α-hydroxy-4-cholesten-3-one and cholesta-4,6-dien-3-one in patients with CTX were 100 times higher than normal [92]. A new and sensitive multi-analyte blood test with liquid chromatography–electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) methodology can be used to quantify plasma ketostetrol bile acid precursors such as 7-α-hydroxy-4-cholesten-3-one and 5-α-cholestanol [43]. With the development of genetic analysis, MRI and other neuroimaging technologies are not crucial for the diagnosis of CTX, but could provide complementary clinical information [93].
On the basis of a pool analysis of a screened international CTX series, Mignarri et al. developed an effective suspicion index for early diagnosis composed of weighted scores related to indicators such as family history and systemic and neurological characteristics [38]. The indicators were classified as very strong (score = 100), strong (score = 50), or moderate (score = 25). Childhood-onset cataracts, diarrhea, and neonatal cholestatic jaundice in combination with neurological features and dentate nucleus abnormalities in MRI were demonstrated to be strong indicators. Tendon xanthomas were considered very strong indicators. Plasma cholestanol examination with a total score ≥100 is requisite, as is CYP27A1 gene analysis with a total sore ≥200 or the existence of one very strong or four strong indicators. Using this efficient diagnostic tool, the investigators achieved a diagnostic age in their study of only 10.6 ± 9.8 years, which compares favorably to the previous average age at diagnosis of 35 years (p <0.01) [38].
Management and prognosis
The management of CTX includes replacement therapy, surgery, and other symptomatic therapy. Our knowledge of the pathogenesis of CTX suggests that replacement therapy involving bile acid supplementation can restore bile acid synthesis by reducing plasma cholestanol levels and eliminating bile alcohols. Moreover, the benefits could be enhanced by administration of HMG-CoA reductase inhibitors [22,94]. The potential mechanism of bile acid therapy may be exogenous inhibition of bile acid production by activating the bile acid negative feedback mechanism. This would inhibit production of the intermediate 7-α-hydroxy-4-cholesten-3-one, thereby normalizing cholestanol concentration and preventing the accumulation of cholestanol in tissues.
Replacement therapy involves administration of bile acids such as CDCA, ursodeoxycholic acid (UDCA), cholic acid, or taurocholic acid [95-97]. Compared to administration of UDCA or taurocholic acid, CDCA treatment (750 mg/d) is the therapy of choice for treating the neurological and non-neurological symptoms of CTX, but cholic acid is also efficient for non-neurological symptoms [39,41,44,73,96]. Bile acid therapy with cholic acid has also proven its effectiveness in treating other lipid metabolic disorders [88]. Administration of CDCA (750 mg/d) or cholic acid can normalize plasma cholestanol and improve non-neurological symptoms in many individual CTX cases, but only CDCA can improve the neurological symptoms in patients with CTX [5,23,95,97]. Long-term replacement therapy with CDCA can increase bone mineral content and improve intestinal absorption of vitamin D by activating the bile acid negative feedback mechanism [32]. Combination therapy with CDCA (300 mg/d) and pravastatin (10 mg/d) can improve lipoprotein metabolism, inhibit cholesterol synthesis, and reduce plasma levels of cholestanol and plant sterols [98]. The efficacy of treatment with HMG-CoA reductase inhibitors alone is controversial, and some adverse effects such as hepatic dysfunction and rhabdomyolysis may be observed [98,99]. Other possible treatments that lack reliable clinical validation include vitamin E supplementation, low-density lipoprotein apheresis, and liver transplantation [5,21,24,100]. Surgical excision of bilateral tendon may worsen the gait imbalance and cannot prevent the deterioration of neurologically affected patients [22]. Due to the diverse manifestations and signs of CTX, symptomatic therapy is essential: antidepressant medication in case of depression [24], antiepileptic therapy in case of convulsive seizures [19], levodopa in case of parkinsonism, and botulinum toxin in case of dystonia [61,101,102].
Beginning treatment with bile acid therapy as early as possible is crucial for preventing neurological damage and deterioration in patients with CTX [5,21,23,32,95,103]. In a large series of 25 patients with CTX, 60% of patients continued to deteriorate and 20% died in spite of the long-term administration of CDCA, but survival was related to age at diagnosis [5]. Ginanneschi et al. revealed that CDCA treatment improved nerve conduction velocity and promoted myelin synthesis in nerve fibers with residual unaffected axons in a series of 35 patients with polyneuropathy, and the therapeutic effect depended largely on the extent of irreversible structural damage [39]. Neurophysiological follow-up research might be recommended when patients with CTX present clinical symptoms of peripheral neuropathy [45].
Discussion and unresolved questions
Monitoring plasma cholestanol levels can be used to assess the biochemical effects of CDCA and cholic acid in patients with CTX before and after treatment. However, serum cholestanol level has no correlation with clinical features [44]. A possible explanation is that increased cholestanol level is not the only factor important for pathogenesis in CTX. Further studies are required to understand any other underlying mechanisms and to provide reasonable explanations. Cerebral WM lesions and cerebellar vacuolation have been described in a patient with CTX with progressive ataxia [49,75]. White matter changes in patients with CTX suggest the coexistence of demyelinating and axonopathic lesions in CTX [45]. Moreover, further studies are needed to discover why some patients with CTX develop WM lesions in the brain.
Many clinical studies of CTX have been published, but basic animal research on the pathogenesis of CTX is still not commonly reported. The CYP27A1 gene knockout mice do not present with xanthomas in brain or tendon. However, mice may be able to compensate for the loss of the alternative bile acid synthesis pathway [8,104]. Transgenic mice overexpressing the CYP27A1 gene do not show increased synthesis of bile acid [8]. The reason for the obvious difference between the animal models and humans with CTX is not known with certainty. The relationship between the deposition of cholestanol and the development of xanthomas deserves further study.
Conclusion
CTX is an inherited lipid metabolic disorder with diverse manifestations. The classical symptoms and signs, namely elevated levels of cholestanol and bile alcohols in serum and urine, cranial magnetic resonance imaging, and mutation in the CYP27A1 gene, confirm the diagnosis. Patients with CTX have an average diagnosed age of 35 years and a diagnostic delay of 16 years. Early diagnosis and long-term treatment with CDCA (750 mg/d) can improve neurological symptoms and contribute to a better prognosis.
Consent
Written informed consent was obtained from the patient for the publication of this report and any accompanying images.
Acknowledgements
We thank Bouhari Altine for the language editing and critical revision of the manuscript.
Abbreviations
- CTX
Cerebrotendinous xanthomatosis
- CYP7A1
Cholesterol 7α-hydroxylase
- CYP27A1
Sterol 27-hydroxylase
- CDCA
Chenodeoxycholic acid
- GC-MS
Gas chromatography–mass spectrometry
- TMS
Transcranial magnetic stimulation
- VEP
Visual evoked potentials
- SSEP
Somatosensory evoked potentials
- BAEP
Brainstem auditory evoked potentials
- NCV
Nerve conduction velocity
- MRI
Magnetic resonance imaging
- DTI
Diffusion tensor imaging
- VBM
Voxel-based morphometry
- GM
Gray matter
- WM
White matter
- NAA
N-acetylaspartate
- PET
Positron emission tomography
- ALS
Amyotrophic lateral sclerosis
- ABC
Adenosine triphosphate-binding cassette
- LDL
Low density lipoprotein
- LDL-C
LDL cholesterol
- FH
Familial hypercholesterolemia
- LC-ESI-MS/MS
Liquid chromatography–electrospray ionization-tandem mass spectrometry
- UCDA
Ursodeoxycholic acid
Footnotes
Shuke Nie and Guiqin Chen contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SN and GC wrote the paper and revised the manuscript. XC: language editing and critical revision of the manuscript. YZ: final approval of the revision. All authors have read and approved the final manuscript.
Contributor Information
Shuke Nie, Email: nieshuke200554@163.com.
Guiqin Chen, Email: chenguiqin12345@163.com.
Xuebing Cao, Email: caoxuebing@126.com.
Yunjian Zhang, Email: zhangyunjian66@126.com.
References
- 1.Cali JJ, Hsieh CL, Francke U, Russell DW. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biol Chem. 1991;266:7779–7783. [PMC free article] [PubMed] [Google Scholar]
- 2.Gallus GN, Dotti MT, Federico A. Clinical and molecular diagnosis of cerebrotendinous xanthomatosis with a review of the mutations in the CYP27A1 gene. Neurol Sci. 2006;27:143–149. doi: 10.1007/s10072-006-0618-7. [DOI] [PubMed] [Google Scholar]
- 3.Bjorkhem I, Hansson M. Cerebrotendinous xanthomatosis: an inborn error in bile acid synthesis with defined mutations but still a challenge. Biochem Biophys Res Commun. 2010;396:46–49. doi: 10.1016/j.bbrc.2010.02.140. [DOI] [PubMed] [Google Scholar]
- 4.Lorincz MT, Rainier S, Thomas D, Fink JK. Cerebrotendinous xanthomatosis - Possible higher prevalence than previously recognized. Arch Neurol. 2005;62:1459–1463. doi: 10.1001/archneur.62.9.1459. [DOI] [PubMed] [Google Scholar]
- 5.Pilo-de-la-Fuente B, Jimenez-Escrig A, Lorenzo JR, Pardo J, Arias M, Ares-Luque A, Duarte J, Muniz-Perez S, Sobrido MJ. Cerebrotendinous xanthomatosis in Spain: clinical, prognostic, and genetic survey. Eur J Neurol. 2011;18:1203–1211. doi: 10.1111/j.1468-1331.2011.03439.x. [DOI] [PubMed] [Google Scholar]
- 6.Setoguchi T, Salen G, Tint GS, Mosbach EH. A biochemical abnormality in cerebrotendinous xanthomatosis. Impairment of bile acid biosynthesis associated with incomplete degradation of the cholesterol side chain. J Clin Invest. 1974;53:1393–1401. doi: 10.1172/JCI107688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 8.Lorbek G, Lewinska M, Rozman D. Cytochrome P450s in the synthesis of cholesterol and bile acids - from mouse models to human diseases. Febs J. 2012;279:1516–1533. doi: 10.1111/j.1742-4658.2011.08432.x. [DOI] [PubMed] [Google Scholar]
- 9.Duane WC, Javitt NB. 27-hydroxycholesterol: production rates in normal human subjects. J Lipid Res. 1999;40:1194–1199. [PubMed] [Google Scholar]
- 10.Chales G, Coiffier G, Guggenbuhl P. Miscellaneous non-inflammatory musculoskeletal conditions. Rare thesaurismosis and xanthomatosis. Best Pract Res Clin Rheumatol. 2011;25:683–701. doi: 10.1016/j.berh.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 11.Skrede S, Bjorkhem I, Buchmann MS, Hopen G, Fausa O. A novel pathway for biosynthesis of cholestanol with 7 alpha-hydroxylated C27-steroids as intermediates, and its importance for the accumulation of cholestanol in cerebrotendinous xanthomatosis. J Clin Invest. 1985;75:448–455. doi: 10.1172/JCI111719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panzenboeck U, Andersson U, Hansson M, Sattler W, Meaney S, Bjorkhem I. On the mechanism of cerebral accumulation of cholestanol in patients with cerebrotendinous xanthomatosis. J Lipid Res. 2007;48:1167–1174. doi: 10.1194/jlr.M700027-JLR200. [DOI] [PubMed] [Google Scholar]
- 13.Keren Z, Falik-Zaccai TC. Cerebrotendinous xanthomatosis (CTX): a treatable lipid storage disease. Pediatr Endocrinol Rev. 2009;7:6–11. [PubMed] [Google Scholar]
- 14.van Heijst AFJ, Verrips A, Wevers RA, Cruysberg JRM, Renier WO, Tolboom JJM. Treatment and follow-up of children with cerebrotendinous xanthomatosis. Eur J Pediatr. 1998;157:313–316. doi: 10.1007/s004310050818. [DOI] [PubMed] [Google Scholar]
- 15.Cruysberg JR, Wevers RA, Tolboom JJ. Juvenile cataract associated with chronic diarrhea in pediatric cerebrotendinous xanthomatosis. Am J Ophthalmol. 1991;112:606–607. doi: 10.1016/S0002-9394(14)76874-6. [DOI] [PubMed] [Google Scholar]
- 16.Guyant-Marechal L, Verrips A, Girard C, Wevers RA, Zijlstra F, Sistermans E, Vera P, Campion D, Hannequin D. Unusual cerebrotendinous xanthomatosis with fronto-temporal dementia phenotype. Am J Med Genet A. 2005;139A:114–117. doi: 10.1002/ajmg.a.30797. [DOI] [PubMed] [Google Scholar]
- 17.Sedel F, Baumann N, Turpin JC, Lyon-Caen O, Saudubray JM, Cohen D. Psychiatric manifestations revealing inborn errors of metabolism in adolescents and adults. J Inherit Metab Dis. 2007;30:631–641. doi: 10.1007/s10545-007-0661-4. [DOI] [PubMed] [Google Scholar]
- 18.Kalidas K, Behrouz R. Inherited metabolic disorders and cerebral infarction. Expert Rev Neurother. 2008;8:1731–1741. doi: 10.1586/14737175.8.11.1731. [DOI] [PubMed] [Google Scholar]
- 19.Pedroso JL, Pinto WB, Souza PV, Santos LT, Abud IC, Avelino MA, Barsottini OG. Early-onset epilepsy as the main neurological manifestation of cerebrotendinous xanthomatosis. Epilepsy Behav. 2012;24:380–381. doi: 10.1016/j.yebeh.2012.04.121. [DOI] [PubMed] [Google Scholar]
- 20.Ohno T, Kobayashi S, Hayashi M, Sakurai M, Kanazawa I. Diphenylpyraline-responsive parkinsonism in cerebrotendinous xanthomatosis: long-term follow up of three patients. J Neurol Sci. 2001;182:95–97. doi: 10.1016/S0022-510X(00)00441-X. [DOI] [PubMed] [Google Scholar]
- 21.Federico A, Dotti MT. Cerebrotendinous xanthomatosis: clinical manifestations, diagnostic criteria, pathogenesis, and therapy. J Child Neurol. 2003;18:633–638. doi: 10.1177/08830738030180091001. [DOI] [PubMed] [Google Scholar]
- 22.Moghadasian MH, Salen G, Frohlich JJ, Scudamore CH. Cerebrotendinous xanthomatosis: a rare disease with diverse manifestations. Arch Neurol. 2002;59:527–529. doi: 10.1001/archneur.59.4.527. [DOI] [PubMed] [Google Scholar]
- 23.Berginer VM, Salen G, Shefer S. Long-term treatment of cerebrotendinous xanthomatosis with chenodeoxycholic acid. N Engl J Med. 1984;311:1649–1652. doi: 10.1056/NEJM198412273112601. [DOI] [PubMed] [Google Scholar]
- 24.Chen Q, Liu W, Jiang B, Yu R, Li X, Li H. Fluoxetine-responsive depression in a Chinese cerebrotendinous xanthomatosis. Gen Hosp Psychiatry. 2012;34(578):e571–e574. doi: 10.1016/j.genhosppsych.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 25.Lee Y, Lin PY, Chiu NM, Chang WN, Wen JK. Cerebrotendinous xanthomatosis with psychiatric disorders: report of three siblings and literature review. Chang Gung Med J. 2002;25:334–340. [PubMed] [Google Scholar]
- 26.Lagarde J, Roze E, Apartis E, Pothalil D, Sedel F, Couvert P, Vidailhet M, Degos B. Myoclonus and dystonia in cerebrotendinous xanthomatosis. Mov Disord. 2012;27:1805–1810. doi: 10.1002/mds.25206. [DOI] [PubMed] [Google Scholar]
- 27.Cruysberg JR, Wevers RA, van Engelen BG, Pinckers A, van Spreeken A, Tolboom JJ. Ocular and systemic manifestations of cerebrotendinous xanthomatosis. Am J Ophthalmol. 1995;120:597–604. doi: 10.1016/S0002-9394(14)72206-8. [DOI] [PubMed] [Google Scholar]
- 28.Dotti MT, Mondillo S, Plewnia K, Agricola E, Federico A. Cerebrotendinous xanthomatosis: evidence of lipomatous hypertrophy of the atrial septum. J Neurol. 1998;245:723–726. doi: 10.1007/s004150050274. [DOI] [PubMed] [Google Scholar]
- 29.Bjorkhem I, Andersson O, Diczfalusy U, Sevastik B, Xiu RJ, Duan C, Lund E. Atherosclerosis and sterol 27-hydroxylase: evidence for a role of this enzyme in elimination of cholesterol from human macrophages. Proc Natl Acad Sci U S A. 1994;91:8592–8596. doi: 10.1073/pnas.91.18.8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weingartner O, Laufs U, Bohm M, Lutjohann D. An alternative pathway of reverse cholesterol transport: the oxysterol 27-hydroxycholesterol. Atherosclerosis. 2010;209:39–41. doi: 10.1016/j.atherosclerosis.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 31.Kuriyama M, Fujiyama J, Kubota R, Nakagawa M, Osame M. Osteoporosis and increased bone fractures in cerebrotendinous xanthomatosis. Metabolism. 1993;42:1497–1498. doi: 10.1016/0026-0495(93)90205-3. [DOI] [PubMed] [Google Scholar]
- 32.Martini G, Mignarri A, Ruvio M, Valenti R, Franci B, Del Puppo M, Federico A, Nuti R, Dotti MT. Long-term bone density evaluation in cerebrotendinous xanthomatosis: evidence of improvement after chenodeoxycholic acid treatment. Calcif Tissue Int. 2013;92:282–286. doi: 10.1007/s00223-012-9677-3. [DOI] [PubMed] [Google Scholar]
- 33.Federico A, Dotti MT, Lore F, Nuti R. Cerebrotendinous xanthomatosis: pathophysiological study on bone metabolism. J Neurol Sci. 1993;115:67–70. doi: 10.1016/0022-510X(93)90068-A. [DOI] [PubMed] [Google Scholar]
- 34.Kawabata M, Kuriyama M, Mori S, Sakashita I, Osame M. Pulmonary manifestations in cerebrotendinous xanthomatosis. Intern Med. 1998;37:922–926. doi: 10.2169/internalmedicine.37.922. [DOI] [PubMed] [Google Scholar]
- 35.Dormans TP, Verrips A, Bulten J, Cox N. Pulmonary lymphangioleiomyomatosis and cerebrotendinous xanthomatosis: is there a link? Chest. 1997;112:273–274. doi: 10.1378/chest.112.1.273. [DOI] [PubMed] [Google Scholar]
- 36.Verrips A, van Engelen BG, Wevers RA, van Geel BM, Cruysberg JR, van den Heuvel LP, Keyser A, Gabreels FJ. Presence of diarrhea and absence of tendon xanthomas in patients with cerebrotendinous xanthomatosis. Arch Neurol. 2000;57:520–524. doi: 10.1001/archneur.57.4.520. [DOI] [PubMed] [Google Scholar]
- 37.van Heijst AF, Wevers RA, Tangerman A, Cruysberg JR, Renier WO, Tolboom JJ. Chronic diarrhea as a dominating symptom in two children with cerebrotendinous xanthomatosis. Acta Paediatr (Oslo, Norway: 1992) 1996;85:932–936. doi: 10.1111/j.1651-2227.1996.tb14189.x. [DOI] [PubMed] [Google Scholar]
- 38.Mignarri A, Gallus GN, Dotti MT, Federico A. A suspicion index for early diagnosis and treatment of cerebrotendinous xanthomatosis. J Inherit Metab Dis. 2014;37:421–429. doi: 10.1007/s10545-013-9674-3. [DOI] [PubMed] [Google Scholar]
- 39.Ginanneschi F, Mignarri A, Mondelli M, Gallus GN, Del Puppo M, Giorgi S, Federico A, Rossi A, Dotti MT. Polyneuropathy in cerebrotendinous xanthomatosis and response to treatment with chenodeoxycholic acid. J Neurol. 2013;260:268–274. doi: 10.1007/s00415-012-6630-3. [DOI] [PubMed] [Google Scholar]
- 40.Ben Hamida M, Chabbi N, Ben Hamida C, Mhiri C, Kallel R. [Peripheral neuropathy in a sporadic case of cerebrotendinous xanthomatosis]. Neuropathie peripherique dans un cas sporadique de xanthomatose cerebrotendineuse. Rev Neurol. 1991;147:385–388. [PubMed] [Google Scholar]
- 41.Federico A, Dotti MT, Volpi N. Muscle mitochondrial changes in cerebrotendinous xanthomatosis. Ann Neurol. 1991;30:734–735. doi: 10.1002/ana.410300517. [DOI] [PubMed] [Google Scholar]
- 42.Federico A, Dotti MT. Cerebrotendinous xanthomatosis. Neurology. 2001;57:1743. doi: 10.1212/WNL.57.9.1743. [DOI] [PubMed] [Google Scholar]
- 43.DeBarber AE, Luo J, Giugliani R, Souza CFM, Chiang JP-W, Merkens LS, Pappu AS, Steiner RD. A useful multi-analyte blood test for cerebrotendinous xanthomatosis. Clin Biochem. 2014;47:860–863. doi: 10.1016/j.clinbiochem.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mignarri A, Rossi S, Ballerini M, Gallus GN, Del Puppo M, Galluzzi P, Federico A, Dotti MT. Clinical relevance and neurophysiological correlates of spasticity in cerebrotendinous xanthomatosis. J Neurol. 2011;258:783–790. doi: 10.1007/s00415-010-5829-4. [DOI] [PubMed] [Google Scholar]
- 45.Pilo B, de Blas G, Sobrido MJ, Navarro C, Grandas F, Barrero FJ, Moya MA, Jimenez-Escrig A. Neurophysiological study in cerebrotendinous xanthomatosis. Muscle Nerve. 2011;43:531–536. doi: 10.1002/mus.21905. [DOI] [PubMed] [Google Scholar]
- 46.Chen S-F, Tsai N-W, Chang C-C, Lu C-H, Huang C-R, Chuang Y-C, Chang W-N. Neuromuscular abnormality and autonomic dysfunction in patients with cerebrotendinous xanthomatosis. BMC Neurol. 2011;11:63. doi: 10.1186/1471-2377-11-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mondelli M, Rossi A, Scarpini C, Dotti MT, Federico A. Evoked potentials in cerebrotendinous xanthomatosis and effect induced by chenodeoxycholic acid. Arch Neurol. 1992;49:469–475. doi: 10.1001/archneur.1992.00530290051011. [DOI] [PubMed] [Google Scholar]
- 48.Vanrietvelde F, Lemmerling M, Mespreuve M, Crevits L, De Reuck J, Kunnen M. MRI of the brain in cerebrotendinous xanthomatosis (van Bogaert-Scherer-Epstein disease) Eur Radiol. 2000;10:576–578. doi: 10.1007/s003300050964. [DOI] [PubMed] [Google Scholar]
- 49.Mignarri A, Dotti MT, Del Puppo M, Gallus GN, Giorgio A, Cerase A, Monti L. Cerebrotendinous xanthomatosis with progressive cerebellar vacuolation: six-year MRI follow-up. Neuroradiology. 2012;54:649–651. doi: 10.1007/s00234-012-1026-8. [DOI] [PubMed] [Google Scholar]
- 50.Mehta BP, Shmerling RH. Teaching neuroimage: cerebrotendinous xanthomatosis. Neurology. 2008;71:e4. doi: 10.1212/01.wnl.0000316806.01406.74. [DOI] [PubMed] [Google Scholar]
- 51.De Stefano N, Dotti MT, Mortilla M, Federico A. Magnetic resonance imaging and spectroscopic changes in brains of patients with cerebrotendinous xanthomatosis. Brain: J Neurol. 2001;124:121–131. doi: 10.1093/brain/124.1.121. [DOI] [PubMed] [Google Scholar]
- 52.Barkhof F, Verrips A, Wesseling P, van Der Knaap MS, van Engelen BG, Gabreels FJ, Keyser A, Wevers RA, Valk J. Cerebrotendinous xanthomatosis: the spectrum of imaging findings and the correlation with neuropathologic findings. Radiology. 2000;217:869–876. doi: 10.1148/radiology.217.3.r00dc03869. [DOI] [PubMed] [Google Scholar]
- 53.Clemen CS, Spottke EA, Lutjohann D, Urbach H, von Bergmann K, Klockgether T, Dodel R. Cerebrotendinous xanthomatosis: a treatable ataxia. Neurology. 2005;64:1476. doi: 10.1212/01.WNL.0000158671.84150.8F. [DOI] [PubMed] [Google Scholar]
- 54.Chang C-C, Lui C-C, Wang J-J, Huang S-H, Lu C-H, Chen C, Chen C-F, Tu M-C, Huang C-W, Chang W-N. Multi-parametric neuroimaging evaluation of cerebrotendinous xanthomatosis and its correlation with neuropsychological presentations. BMC Neurol. 2010;10:59. doi: 10.1186/1471-2377-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guerrera S, Stromillo ML, Mignarri A, Battaglini M, Marino S, Di Perri C, Federico A, Dotti MT, De Stefano N. Clinical relevance of brain volume changes in patients with cerebrotendinous xanthomatosis. J Neurol Neurosurg Psychiatry. 2010;81:1189–1193. doi: 10.1136/jnnp.2009.203364. [DOI] [PubMed] [Google Scholar]
- 56.Le Bihan D. Looking into the functional architecture of the brain with diffusion MRI. Nat Rev Neurosci. 2003;4:469–480. doi: 10.1038/nrn1119. [DOI] [PubMed] [Google Scholar]
- 57.Embirucu EK, Otaduy MCG, Taneja AK, Leite CC, Kok F, Lucato LT. MR spectroscopy detects lipid peaks in cerebrotendinous xanthomatosis. AJNR Am J Neuroradiol. 2010;31:1347–1349. doi: 10.3174/ajnr.A1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seidel S, Kasprian G, Prayer D, Krssak M, Sycha T, Auff E. Visualisation of treatment response in a case of cerebrotendinous xanthomatosis. J Neurol Neurosurg Psychiatry. 2011;82:703–704. doi: 10.1136/jnnp.2009.196444. [DOI] [PubMed] [Google Scholar]
- 59.Caroppo P, D’Agata F, Mignarri A, Stromillo ML, Dotti MT, Mongini T. Brain metabolism changes after therapy with chenodeoxycholic acid in a case of cerebrotendinous xanthomatosis. Neurol Sci: Offic J Ital Neurol Soc Ital Soc Clin Neurophysiol. 2013;34:1693–1696. doi: 10.1007/s10072-012-1262-z. [DOI] [PubMed] [Google Scholar]
- 60.Selva-O’Callaghan A, Bardes I, Jacas C, Jubany L, Lorenzo-Bosquet C, Cuberas-Borros G, Vilardell-Tarres M. SPECT imaging for brain improvement quantification in a patient with cerebrotendinous xanthomatosis. Clin Nucl Med. 2011;36:38–39. doi: 10.1097/RLU.0b013e3181feed83. [DOI] [PubMed] [Google Scholar]
- 61.Schotsmans K, De Cauwer H, Baets J, Ceyssens S, van den Hauwe L, Deconinck T, Helsen G. Cerebrotendinous xanthomatosis presenting with asymmetric parkinsonism: a case with I-123-FP-CIT SPECT imaging. Acta Neurol Belg. 2012;112:287–289. doi: 10.1007/s13760-012-0064-7. [DOI] [PubMed] [Google Scholar]
- 62.Chen S-F, Chang C-C, Huang S-H, Lu C-H, Chuang Y-C, Pan T-L, Chang W-N. 99mTc-sestamibi thigh SPECT/CT imaging for assessment of myopathy in cerebrotendinous xanthomatosis with histopathological and immunohistochemical correlation. Clin Nucl Med. 2014;39:e202–e207. doi: 10.1097/RLU.0b013e3182a20d8e. [DOI] [PubMed] [Google Scholar]
- 63.Kuwabara K, Hitoshi S, Nukina N, Ishii K, Momose T, Kubota S, Seyama Y, Kanazawa I. PET analysis of a case of cerebrotendinous xanthomatosis presenting hemiparkinsonism. J Neurol Sci. 1996;138:145–149. doi: 10.1016/0022-510X(96)00011-1. [DOI] [PubMed] [Google Scholar]
- 64.Pilo de la Fuente B, Ruiz I, Lopez de Munain A, Jimenez-Escrig A. Cerebrotendinous xanthomatosis: neuropathological findings. J Neurol. 2008;255:839–842. doi: 10.1007/s00415-008-0729-6. [DOI] [PubMed] [Google Scholar]
- 65.Soffer D, Benharroch D, Berginer V. The neuropathology of cerebrotendinous xanthomatosis revisited: a case report and review of the literature. Acta Neuropathol. 1995;90:213–220. doi: 10.1007/BF00294324. [DOI] [PubMed] [Google Scholar]
- 66.Kato H, Koyabu S, Aoki S, Tamai T, Sugawa M, Watanabe M, Shiraishi T. An autopsy case of gallbladder cancer developing in a Japanese man with cerebrotendinous xanthomatosis: genetic analysis of the sterol 27-hydroxylase and p53 genes. Pathology. 2003;35:141–144. doi: 10.1080/0031302031000082241. [DOI] [PubMed] [Google Scholar]
- 67.Van Bogaert L. [The framework of the xanthomatoses and their different types. 2. Secondary xanthomatoses] Rev Med Liege. 1962;17:433–443. [PubMed] [Google Scholar]
- 68.Voiculescu V, Alexianu M, Popescu-Tismana G, Pastia M, Petrovici A, Dan A. Polyneuropathy with lipid deposits in Schwann cells and axonal degeneration in cerebrotendinous xanthomatosis. J Neurol Sci. 1987;82:89–99. doi: 10.1016/0022-510X(87)90009-8. [DOI] [PubMed] [Google Scholar]
- 69.Ohnishi A, Yamashita Y, Goto I, Kuroiwa Y, Murakami S, Ikeda M. De- and remyelination and onion bulb in cerebrotendinous xanthomatosis. Acta Neuropathol. 1979;45:43–45. doi: 10.1007/BF00691803. [DOI] [PubMed] [Google Scholar]
- 70.Tian D, Zhang Z-Q. 2 Novel deletions of the sterol 27-hydroxylase gene in a Chinese family with cerebrotendinous xanthomatosis. BMC Neurol. 2011;11:130. doi: 10.1186/1471-2377-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schimschock JR, Alvord EC, Jr, Swanson PD. Cerebrotendinous xanthomatosis. Clinical and pathological studies. Arch Neurol. 1968;18:688–698. doi: 10.1001/archneur.1968.00470360110011. [DOI] [PubMed] [Google Scholar]
- 72.Schneider H, Lingesleben A, Vogel H-P, Garuti R, Calandra S. A novel mutation in the sterol 27-hydroxylase gene of a woman with autosomal recessive cerebrotendinous xanthomatosis. Orphanet J Rare Dis. 2010;5:27. doi: 10.1186/1750-1172-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verrips A, van Engelen BG, ter Laak H, Gabreels-Festen A, Janssen A, Zwarts M, Wevers RA, Gabreels FJ. Cerebrotendinous xanthomatosis. Controversies about nerve and muscle: observations in ten patients. Neuromuscul Disord: NMD. 2000;10:407–414. doi: 10.1016/S0960-8966(00)00112-7. [DOI] [PubMed] [Google Scholar]
- 74.Diekstra FP, Saris CGJ, van Rheenen W, Franke L, Jansen RC, van Es MA, van Vught PWJ, Blauw HM, Groen EJN, Horvath S, Estrada K, Rivadeneira F, Hofman A, Uitterlinden AG, Robberecht W, Andersen PM, Melki J, Meininger V, Hardiman O, Landers JE, Brown RH, Jr, Shatunov A, Shaw CE, Leigh PN, Al-Chalabi A, Ophoff RA, van den Berg LH, Veldink JH. Mapping of gene expression reveals CYP27A1 as a susceptibility gene for sporadic ALS. PLoS One. 2012;7:e35333. doi: 10.1371/journal.pone.0035333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bjorkhem I. Cerebrotendinous xanthomatosis. Curr Opin Lipidol. 2013;24:283–287. doi: 10.1097/MOL.0b013e328362df13. [DOI] [PubMed] [Google Scholar]
- 76.Tsubakio-Yamamoto K, Nishida M, Nakagawa-Toyama Y, Masuda D, Ohama T, Yamashita S. Current therapy for patients with sitosterolemia-effect of ezetimibe on plant sterol metabolism. J Atheroscler Thromb. 2010;17:891–900. doi: 10.5551/jat.4614. [DOI] [PubMed] [Google Scholar]
- 77.Niu DM, Chong KW, Hsu JH, Wu TJ, Yu HC, Huang CH, Lo MY, Kwok CF, Kratz LE, Ho LT. Clinical observations, molecular genetic analysis, and treatment of sitosterolemia in infants and children. J Inherit Metab Dis. 2010;33:437–443. doi: 10.1007/s10545-010-9126-2. [DOI] [PubMed] [Google Scholar]
- 78.Patel SB, Salen G, Hidaka H, Kwiterovich PO, Stalenhoef AFH, Miettinen TA, Grundy SM, Lee MH, Rubenstein JS, Polymeropoulos MH, Brownstein MJ. Mapping a gene involved in regulating dietary cholesterol absorption - The sitosterolemia locus is found at chromosome 2p21. J Clin Investig. 1998;102:1041–1044. doi: 10.1172/JCI3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Escola-Gil JC, Quesada H, Julve J, Martin-Campos JM, Cedo L, Blanco-Vaca F. Sitosterolemia: diagnosis, investigation, and management. Curr Atheroscler Rep. 2014;16:424. doi: 10.1007/s11883-014-0424-2. [DOI] [PubMed] [Google Scholar]
- 80.Bhattacharyya AK, Connor WE. Beta-sitosterolemia and xanthomatosis. A newly described lipid storage disease in two sisters. J Clin Invest. 1974;53:1033–1043. doi: 10.1172/JCI107640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Merkens LS, Myrie SB, Steiner RD, Mymin D: Sitosterolemia. In GeneReviews(R). Edited by Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K. Seattle (WA): 1993. [PubMed]
- 82.Wang Z, Cao L, Su Y, Wang G, Wang R, Yu Z, Bai X, Ruan C. Specific macrothrombocytopenia/hemolytic anemia associated with sitosterolemia. Am J Hematol. 2014;89:320–324. doi: 10.1002/ajh.23619. [DOI] [PubMed] [Google Scholar]
- 83.Salen G, von Bergmann K, Lutjohann D, Kwiterovich P, Kane J, Patel SB, Musliner T, Stein P, Musser B, Multicenter Sitosterolemia Study G Ezetimibe effectively reduces plasma plant sterols in patients with sitosterolemia. Circulation. 2004;109:966–971. doi: 10.1161/01.CIR.0000116766.31036.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Teramoto T, Sasaki J, Ishibashi S, Birou S, Daida H, Dohi S, Egusa G, Hiro T, Hirobe K, Iida M, Kihara S, Kinoshita M, Maruyama C, Ohta T, Okamura T, Yamashita S, Yokode M, Yokote K, Harada-Shiba M, Arai H, Bujo H, Nohara A, Ohta T, Oikawa S, Okada T, Wakatsuki A. Familial hypercholesterolemia. J Atheroscler Thromb. 2014;21:6–10. doi: 10.5551/jat.20040. [DOI] [PubMed] [Google Scholar]
- 85.Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest. 2003;111:1795–1803. doi: 10.1172/JCI200318925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cruz PD, Jr, East C, Bergstresser PR. Dermal, subcutaneous, and tendon xanthomas: diagnostic markers for specific lipoprotein disorders. J Am Acad Dermatol. 1988;19:95–111. doi: 10.1016/S0190-9622(88)70157-7. [DOI] [PubMed] [Google Scholar]
- 87.Robinson JG, Goldberg AC, National Lipid Association Expert Panel on Familial H Treatment of adults with familial hypercholesterolemia and evidence for treatment: recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S18–S29. doi: 10.1016/j.jacl.2011.03.451. [DOI] [PubMed] [Google Scholar]
- 88.Heubi JE, Setchell KD, Bove KE. Inborn errors of bile acid metabolism. Semin Liver Dis. 2007;27:282–294. doi: 10.1055/s-2007-985073. [DOI] [PubMed] [Google Scholar]
- 89.Clayton PT, Leonard JV, Lawson AM, Setchell KD, Andersson S, Egestad B, Sjovall J. Familial giant cell hepatitis associated with synthesis of 3 beta, 7 alpha-dihydroxy-and 3 beta,7 alpha, 12 alpha-trihydroxy-5-cholenoic acids. J Clin Invest. 1987;79:1031–1038. doi: 10.1172/JCI112915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Setchell KD, Heubi JE, Bove KE, O’Connell NC, Brewsaugh T, Steinberg SJ, Moser A, Squires RH., Jr Liver disease caused by failure to racemize trihydroxycholestanoic acid: gene mutation and effect of bile acid therapy. Gastroenterology. 2003;124:217–232. doi: 10.1053/gast.2003.50017. [DOI] [PubMed] [Google Scholar]
- 91.Lionnet C, Carra C, Ayrignac X, Levade T, Gayraud D, Castelnovo G, Besson G, Androdias G, Vukusic S, Confavreux C, Zaenker C, De Seze J, Collongues N, Blanc F, Tranchant C, Wallon D, Hannequin D, Gerdelat-Mas A, Brassat D, Clanet M, Zephir H, Outteryck O, Vermersch P, Labauge P. [Cerebrotendinous xanthomatosis: a multicentric retrospective study of 15 adults, clinical and paraclinical typical and atypical aspects] Rev Neurol (Paris) 2014;170:445–453. doi: 10.1016/j.neurol.2014.01.675. [DOI] [PubMed] [Google Scholar]
- 92.Bjorkhem I, Skrede S, Buchmann MS, East C, Grundy S. Accumulation of 7 alpha-hydroxy-4-cholesten-3-one and cholesta-4,6-dien-3-one in patients with cerebrotendinous xanthomatosis: effect of treatment with chenodeoxycholic acid. Hepatology. 1987;7:266–271. doi: 10.1002/hep.1840070210. [DOI] [PubMed] [Google Scholar]
- 93.Gallus GN, Dotti MT, Mignarri A, Rufa A, Da Pozzo P, Cardaioli E, Federico A. Four novel CYP27A1 mutations in seven Italian patients with CTX. Eur J Neurol. 2010;17:1259–1262. doi: 10.1111/j.1468-1331.2010.03002.x. [DOI] [PubMed] [Google Scholar]
- 94.Verrips A, Wevers RA, Van Engelen BG, Keyser A, Wolthers BG, Barkhof F, Stalenhoef A, De Graaf R, Janssen-Zijlstra F, Van Spreeken A, Gabreels FJ. Effect of simvastatin in addition to chenodeoxycholic acid in patients with cerebrotendinous xanthomatosis. Metabolism. 1999;48:233–238. doi: 10.1016/S0026-0495(99)90040-9. [DOI] [PubMed] [Google Scholar]
- 95.Pierre G, Setchell K, Blyth J, Preece MA, Chakrapani A, McKiernan P. Prospective treatment of cerebrotendinous xanthomatosis with cholic acid therapy. J Inherit Metab Dis. 2008;31(Suppl 2):S241–S245. doi: 10.1007/s10545-008-0815-z. [DOI] [PubMed] [Google Scholar]
- 96.Koopman BJ, Wolthers BG, van der Molen JC, Waterreus RJ. Bile acid therapies applied to patients suffering from cerebrotendinous xanthomatosis. Clin Chim Acta. 1985;152:115–122. doi: 10.1016/0009-8981(85)90182-2. [DOI] [PubMed] [Google Scholar]
- 97.Setchell KDR, Heubi JE. Defects in bile acid biosynthesis–diagnosis and treatment. J Pediatr Gastroenterol Nutr. 2006;43(Suppl 1):S17–S22. doi: 10.1097/01.mpg.0000226386.79483.7b. [DOI] [PubMed] [Google Scholar]
- 98.Kuriyama M, Tokimura Y, Fujiyama J, Utatsu Y, Osame M. Treatment of cerebrotendinous xanthomatosis: effects of chenodeoxycholic acid, pravastatin, and combined use. J Neurol Sci. 1994;125:22–28. doi: 10.1016/0022-510X(94)90237-2. [DOI] [PubMed] [Google Scholar]
- 99.Federico A, Dotti MT. Treatment of cerebrotendinous xanthomatosis. Neurology. 1994;44:2218. doi: 10.1212/WNL.44.11.2218. [DOI] [PubMed] [Google Scholar]
- 100.Mimura Y, Kuriyama M, Tokimura Y, Fujiyama J, Osame M, Takesako K, Tanaka N. Treatment of cerebrotendinous xanthomatosis with low-density lipoprotein (LDL)-apheresis. J Neurol Sci. 1993;114:227–230. doi: 10.1016/0022-510X(93)90303-G. [DOI] [PubMed] [Google Scholar]
- 101.Su C-S, Chang W-N, Huang S-H, Lui C-C, Pan T-L, Lu C-H, Chuang Y-C, Huang C-R, Tsai N-W, Hsieh M-J, Chang C-C. Cerebrotendinous xanthomatosis patients with and without parkinsonism: clinical characteristics and neuroimaging findings. Mov Disord: Offic J Mov Disord Soc. 2010;25:452–458. doi: 10.1002/mds.22979. [DOI] [PubMed] [Google Scholar]
- 102.Lagarde J, Sedel F, Degos B. Blepharospasm as a new feature of cerebrotendinous xanthomatosis. Parkinsonism Relat Disord. 2013;19:764–765. doi: 10.1016/j.parkreldis.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 103.Yahalom G, Tsabari R, Molshatzki N, Ephraty L, Cohen H, Hassin-Baer S. Neurological outcome in cerebrotendinous xanthomatosis treated with chenodeoxycholic acid: early versus late diagnosis. Clin Neuropharmacol. 2013;36:78–83. doi: 10.1097/WNF.0b013e318288076a. [DOI] [PubMed] [Google Scholar]
- 104.Rosen H, Reshef A, Maeda N, Lippoldt A, Shpizen S, Triger L, Eggertsen G, Bjorkhem I, Leitersdorf E. Markedly reduced bile acid synthesis but maintained levels of cholesterol and vitamin D metabolites in mice with disrupted sterol 27-hydroxylase gene. J Biol Chem. 1998;273:14805–14812. doi: 10.1074/jbc.273.24.14805. [DOI] [PubMed] [Google Scholar]