

Abstract
LiFePO4 has been intensively investigated as a cathode material in Li-ion batteries, as it can in principle enable the development of high power electrodes. LiFePO4, on the other hand, is inherently “plagued” by poor electronic and ionic conductivity. While the problems with low electron conductivity are partially solved by carbon coating and further by doping or by downsizing the active particles to nanoscale dimensions, poor ionic conductivity is still an issue. To develop colloidally synthesized LiFePO4 nanocrystals (NCs) optimized for high rate applications, we propose here a surface treatment of the NCs. The particles as delivered from the synthesis have a surface passivated with long chain organic surfactants, and therefore can be dispersed only in aprotic solvents such as chloroform or toluene. Glucose that is commonly used as carbon source for carbon-coating procedure is not soluble in these solvents, but it can be dissolved in water. In order to make the NCs hydrophilic, we treated them with lithium hexafluorophosphate (LiPF6), which removes the surfactant ligand shell while preserving the structural and morphological properties of the NCs. Only a roughening of the edges of NCs was observed due to a partial etching of their surface. Electrodes prepared from these platelet NCs (after carbon coating) delivered a capacity of ∼155 mAh/g, ∼135 mAh/g, and ∼125 mAh/g, at 1 C, 5 C, and 10 C, respectively, with significant capacity retention and remarkable rate capability. For example, at 61 C (10.3 A/g), a capacity of ∼70 mAh/g was obtained, and at 122 C (20.7 A/g), the capacity was ∼30 mAh/g. The rate capability and the ease of scalability in the preparation of these surface-treated nanoplatelets make them highly suitable as electrodes in Li-ion batteries.
Keywords: Li ion batteries, nanocrystals, etching, platelets, high rate capability, electrodes
Olivine phosphate LiFePO4 is a notable cathode material in rechargeable Li ion batteries due to its high theoretical capacity (Q = 170 mAh/g according to the reaction: LiFePO4 ↔ FePO4 + Li+ + e– at ∼3.4 V), stability, low cost, environmental compatibility, and safety. Since its discovery,1 this material has been a focal point of battery research. There are two critical issues associated with LiFePO4 as an electrode material, which still hinder its high-power application: its poor electronic2,3 (∼10–10 S/cm to 10–7 S/cm) and poor ionic conductivity4,5 (∼10−16 cm2 s−1 to 10−14 cm2 s−1). Issues with low electron conductivity can be solved partially via carbon coating,6 by doping with various cations (Mg2+, Al3+, Ti4+, W6+, Co2+/3+, Cu1+/2+, Zr4+ and Nb5+),7,8 by reducing the active particle size to nanoscale (<100 nm)9 and by improving interparticle connectivity. However, the ionic conductivity of the active particles remains still an issue.
The kinetics of Li-ion insertion/removal can be optimized by controlling the LiFePO4 crystal morphology, which in principle should improve the ionic conductivity significantly. For example, it was recently reported that carbon-coated ultrathin LiFePO4 nanosheets10 with exposed (010) facets (the main transfer path for Li ions) possess a high rate capability, reaching up to 80 C (1 C ∼170 mA/g) with a specific capacity of ∼70 mAh/g. These sheets have both a large specific surface area and short Li diffusion paths along the [010] crystallographic direction. Similarly, single crystal LiFePO4 nanosheets with thickness between 30 and 60 nm and a capacity of 55 mAh/g at 30 C have been reported.10,11 According to first principle calculations,12 the (010) facets of LiFePO4 are characterized by lower Li insertion/removal redox potential than the other facets, which suggests a possible energetic preference in the extraction of Li from those facets. Therefore, downsizing the thickness of LiFePO4, thus enabling Li ions to access the entire particles volume, in combination with carbon coating, would considerably improve ionic and electronic conductivity in LiFePO4.
In addition to shape engineering, a variety of LiFePO4 surface-modification approaches were exploited to improve the electronic conductivity and Li diffusivity in electrodes. Some examples are the direct coating of LiFePO4 particles (not coated with carbon) with conductive polymers such as polypyrrole13 or with salts such as AlF314,15 and Ti3SiC2.16 Of particular interest is the surface modification of LiFePO4 by anions.17 These methods were found to stabilize the surface Li and Fe sites that have a high activation barrier for charge transfer at the particles surface. In general, issues such as limited rate performance (when carbon is below 10 wt %), irreversible capacity loss at high rates, and high charge transfer resistance across the electrode/electrolyte interface are actually severe bottlenecks for the high-power Li-ion battery technology. These issues call for the need to develop new strategies for NCs surface preparation in electrodes suitable for high-rate applications.
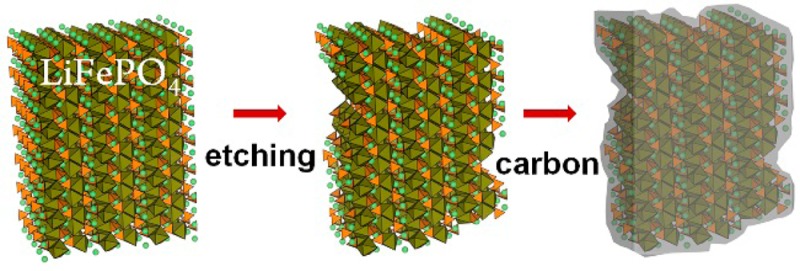
We report here a remarkable high rate capability obtained from platelet-shaped LiFePO4 NCs (∼15 nm thick) by coupling the advantage of the platelet shape (which consists predominantly of exposed (010) facets) with surface etching of the NCs with aqueous LiPF6 solution that strips off their native surfactant ligands and increases the surface area of the NCs. The etched LiFePO4 NCs were subsequently carbon-coated. The electrochemical performances of electrodes made of these NCs (in coin-type 2032 cells with pure Li metal as counter electrode) were examined by electrochemical impedance spectroscopy (EIS), cycling voltammetry (CV), and galvanostatic charge/discharge cycling. The final etched LiFePO4 NCs@C exhibited a considerable high rate capability, most likely due to an intimate contact between the rough etched surface of LiFePO4 and the carbon coating layer.
Results
The LiFePO4 NCs were prepared following a procedure that we recently published elsewhere18 for phospho-olivine type nanoplatelets. Figure 1a reports a scanning electron microscopy (SEM) image of several nanoplatelets, from which it can be seen that they tend to form stacks of face-to-face assembled NCs. The XRD investigation of a dry powder of NCs (Figure 1b) exhibited a diffraction pattern that could be indexed to pure LiFePO4 with orthorhombic olivine-type structure (space group Pnma, PDF card 01-072-7845, a = 10.23 Å, b = 6.00 Å, c = 4.69 Å), indicating the high crystalline nature of the NCs. Figure 1c is a HRTEM image of a single platelet close to the [010] zone axis projection in which (200) and (002) lattice sets are indicated (d-spacing of ∼0.52 nm and ∼0.24 nm, respectively). An enlarged HRTEM view from a [010] orientation, after averaging several regions of the platelet, is reported in Figure 1d, along with the simulated image. The dominant contrast is from the Fe/P columns groups (brown and violet color in the sketch), but some contrast from the lighter Li (green) and O (red) atomic columns is visible. From electron energy loss spectroscopy (EELS) we obtained a Li/Fe ratio of 0.7 (±0.2), a P/Fe ratio of 0.9 (±0.1), and O/Fe ratio of 3.8 (±0.4), close to the expected stoichiometry of LiFePO4. X-ray photoelectron spectroscopy (XPS) data confirm the oxidation state of Fe as +II, as well as the presence of (PO4)3– groups (all analyses are reported in the Supporting Information, SI). An atomistic sketch of a LiFePO4 platelet illustrating the presence of FeO6 octahedra (brown), PO4 tetrahedra (orange), and Li ions (green) arranged along the [010] channels is displayed in Figure 1e.
Figure 1.

Structural characterization of the LiFePO4 NCs. (a) SEM image of several nanoplatelets. (b) XRD pattern consistent with olivine-type LiFePO4 (triphylite, PDF #01-072-7845). (c) HRTEM image of a single platelet close to [010] zone axis projection. (d) Enlarged view from a [010] orientation (left) after averaging several regions of the platelet, along with simulated image (center) and sketch of atomic columns in projection (right); (e) Atomistic sketch of a LiFePO4 platelet. FeO6 octahedra are shown in brown, PO4 tetrahedra in orange, and Li ions in green.
In spite of the high theoretical capacity of LiFePO4, the pristine NCs were characterized by a poor capacity and cycle life, as discussed later in more detail (see also Figure S10 in SI). This could be due to the presence of oleylamine (Olam) as surfactant molecules that embed the surface of the NCs. These molecules are most likely involved in side reactions with the electrolyte, developing a resistive component during charge/discharge. In addition, the as-synthesized NCs (in the presence of oleylamine as ligand) are hydrophobic and soluble only in nonpolar solvents such as toluene or chloroform, in contrast to glucose (a well-known carbon source for carbon coating)6 that is instead hydrophilic. Therefore, the NCs should be hydrophilic as well in order to be dispersible in an aqueous glucose solution for carbon coating. To this end, we removed the hydrophobic surfactants by treating the LiFePO4 NCs with LiPF6 in a two-phase chloroform/water system. The choice of this particular chemical was dictated by the fact that it is also used as electrolyte in the cell preparation. Furthermore, LiPF6 reacts with water to produce hydrofluoric acid (LiPF6 + H2O => LiF + POF3 + 2HF)19 which can act as an etchant on the surface of the NCs (this does not occur inside the battery where the environment is anhydrous). The procedure consisted in mixing a solution containing NCs in chloroform with an aqueous solution of LiPF6, followed by vigorous shaking. After few minutes, the LiFePO4 NCs were transferred to the aqueous phase, which indicates that the hydrophobic surfactant molecules were displaced by hydrophilic PF6– anions and the NCs became dispersible in water. The excess LiPF6 was removed from the NCs by repeated centrifugation and suspension in ultrapure water. At the moment, the LiPF6 solution cannot be replaced by the direct use of an aqueous HF solution: we observed that a 1.5 M HF solution dissolved completely the NCs, while a more diluted 0.75 M HF solution could not easily extract the NCs from chloroform. Therefore, we conclude that an in situ HF generation via aqueous LiPF6 solution is the most effective and reproducible route to extract the platelet NCs from chloroform.
We performed 1H and 19F liquid phase NMR spectroscopy on the “as-synthesized” NCs (capped by Olam) in deuterated chloroform (CDCl3) and on the “etched” NCs (free of organic surfactants) in D2O. 1H NRM analysis of the as-synthesized NCs in CDCl3 showed only signals which could be assigned to Olam (Figure 2a), and no F species were detected in the 19F NMR spectrum (see Figure S4 of the SI). In the 1H NMR spectrum of the etched NCs sample, we found only trace signals of hydrocarbons (Figure 2a), while its 19F-NMR spectrum revealed the characteristic signals of PF6– at −70.5 and −71.8 ppm (Figure S4 of the SI). In the FTIR spectrum of a film of etched NCs deposited on a silicon substrate, a strong P–F stretching signal at 838 cm–1 was observed, along with trace signals from C–H stretching modes (Figure 2b). We concluded from these data that the surfactant molecules at the surface of the NCs were displaced by PF6– anions. The NCs, before and after the LiFP6 treatment, as well as after carbon coating (performed on etched NCs according to a standard procedure that used glucose as a carbon source, see carbon coating of etched NCs@C in the Methods section), were analyzed via HRTEM. The LiFP6 treated NCs appeared to have rougher edges than the pristine NCs, an indication that they were slightly etched (compare Figure 2c left with Figure 2c at the center; in addition see Figure S7 in the SI).
Figure 2.

Spectroscopic characterization of the NCs before and after LiPF6 etching and HRTEM analysis (including carbon coating). (a) Liquid-phase 1H NMR spectra of a solution of LiFePO4 NCs before (black line) and after LiPF6 treatment (orange line, 10× magnified); (b) FTIR spectra of NC films on silicon substrates: as synthesized NCs, coated with Olam (black line); etched NCs (red line); (c) HRTEM images of: (left) as-synthesized NCs; (center) LiPF6 etched NCs; (right) LiPF6 etched NCs after carbon coating. (d) Scheme depicting how etching of the LiFePO4 NC surface can lead to an intimate contact between the surface and the carbon layer during the carbon coating step.
After carbon coating of the etched NCs,6 a ∼2 nm thick carbon layer was evident on the rough NCs surface (Figure 2c, right) with a total preservation of platelet morphology. In addition, Brunauer–Emmett–Teller (BET) analysis was performed to quantify the variation in surface area due to etching (Table T1 in the SI). The as-synthesized NCs were characterized by a surface area of 57.0 m2/g, while the etched NCs exhibited a surface area of 70.5 m2/g. Considering the ligand contribution to the weight of as-synthesized NCs (around 10% by a thermogravimetric analysis, see Figure S6 in the SI) the increment was due to the roughening of the NCs. BET analysis was performed on the carbon coated samples too. After carbon coating, the etched NCs@C released a surface area of 52.1 m2/g. Such a decrease in surface area with respect to the uncoated sample is most likely due to the thickening of the NCs by the carbon layer.
As a control experiment, we performed carbon coating of the as-synthesized NCs, that is, coated with Olam ligands, with the aim of comparing the electrochemical performance of the two samples (as-synthesized NCs@C and etched NCs@C) and thus identify any beneficial effect of the etching step. This time, however, the only carbon source for the carbon coating process was represented by the Olam ligands at the surface of the NCs. Carbon coating was therefore attempted by simply annealing the as-synthesized NCs under forming gas (95% Ar, 5% H2) at 500 °C for 12 h (see Methods section for additional details). Henceforth, this sample will be named “as-synthesized NCs@C”. The sample, after pyrolysis of the ligands, exhibited a surface area of 22.8 m2/g by BET analysis (see Table T1 in SI) and consisted of particles having roughly spherical shapes with a size distribution between 0.05 and 1 μm (as observed by TEM and SEM analyses, see Figure S9c–d in the SI). The coalescence of starting NCs (which formed particles up to ∼1 μm in size) justified the significant reduction in BET surface area (from 57.0 m2/g of the starting as-synthesized NCs to 22.8 m2/g of as-synthesized NCs@C after pyrolysis of the ligands) and XRD analysis indicated narrowing of all peaks. Overall the loss of platelet morphology is confirmed by the narrowing of the peak from the (020) reflection (Figure S9b in the SI). In addition, we further observed the formation of Li3PO4 impurities after this treatment (see Figure S9a in the SI). Considering the loss of the original platelet morphology and the formation of impurities, the results of this control experiment indicate that the Olam ligands alone are not a suitable source of carbon for a potential carbon coating step on colloidal platelet NCs.
The electrochemical characteristics of the NCs were investigated in coin-type (2032) cells containing an electrode with NCs and a pure Li metal foil as counter electrode. We tested the following four samples: (i) as-synthesized NCs, passivated with Olam, with no LiPF6 treatment and no carbon coating; (ii) as-synthesized NCs, with no LiPF6 treatment, but with carbon coating, using only oleylamine as a carbon source (as-synthesized NCs@C); (iii) NCs treated with LiPF6 but with no carbon coating (etched NCs); (iv) NCs treated with LiPF6 followed by carbon coating, using glucose as a carbon source (etched NCs@C). Electrochemical impedance spectra (EIS, in the form of Nyquist plots) of the NCs electrodes at the fully lithiated state (LiFePO4) are reported in Figure 3a. Here, the intercept on the real axis (Z real) corresponds to the electrolyte resistance (Re), and the semicircle in the middle of the high-frequency region (100 to 1 kHz) is due to the charge-transfer resistance (Rct ∼90–200 Ω) for the Li transfer across the NCs electrode/electrolyte interface. The Rct value for etched NCs@C that we found was actually similar to the Rct value of previously reported electrodes made of carbon-coated LiFePO4 polyaniline (acid-doped) composites20 and comparable to that of ultrathin LiFePO4 nanosheets.10
Figure 3.

Impedance, cyclic voltammetry, and rate capability data. (a) Electrochemical impedance spectra (EIS) in Nyquist plot of the various NC electrodes; (b) cyclic voltammetry (CV) of the etched NCs@C electrode performed at scan rates ranging from 50 to 100 μV/s; (c) cycling characteristics of the etched NCs@C electrode at 1, 5 and 10 C and (d) 24, 61, and 122 C (20.7 A/g).
Electrodes coated with the as-synthesized NCs exhibited a charge transfer resistance (Figure 3a, black curve) that was higher than that of the as-synthesized NCs@C (red curve), due to the improvement of electronic conductivity after carbon coating (Figure 3a). The same as-synthesized NCs exhibited a resistance more than twice that of the etched NCs (orange curve), indicating that the LiPF6 treatment of the latter sample indeed lowered the electrode resistance, possibly due to the improvement of ionic conductivity at the particles surface (Figure 3a). It is therefore evident that, while carbon coating decreases the resistance of the pristine NCs, the LiPF6 etching process can further reduce the electrode resistance. On the other hand, the etched NCs@C exhibited a resistance (green curve) that was slightly higher than that of the etched NC; that is, carbon coating in this case increased the resistance to a small extent. We attribute this slight increase of resistance to a decrease in surface area of the etched NCs@C (52.1 m2/g) with respect to the etched NCs (70.5 m2/g). On the other hand, carbon coating is necessary to avoid degradation of the NCs upon cycling.
As next step we performed cyclic voltammetry (CV) on the cell containing etched NCs@C. At low CV scan rates (50–100 μV/s), the perfectly symmetric (in comparison with as synthesized NCs@C reported in Figure S13 of the SI) and sharp red-ox peak at ∼3.45 V vs Li+/Li associated with Li extraction/insertion processes that switch the Fe2+/Fe3+ redox center in the NCs (Figure 3b) are indicative of fast Li ion kinetics. The calculated Li ion diffusion from the etched NCs@C was indeed higher (∼1.3 × 10–13 cm2 s–1) than that of the as synthesized NCs@C (∼6.9 × 10–14 cm2 s–1). Additional details are reported in Figure S13 of the SI.
To understand the various factors contributing to electrode performance, cells assembled from various samples were cycled. We briefly discuss first the performance of cells built from both the “as-synthesized” NCs and from the etched NCs (in both cases with no carbon coating). The cell built with as-synthesized NCs exhibited only moderate performance at low rates: as an example, at 0.5 C the capacity reached only ∼120 mAh/g, and it faded to 40 mAh/g at 1 C (see Figure S10 in SI). Such capacity was lower than that of the cell built with etched NCs (∼155 mAh/g at 0.5 C and ∼110 mAh/g at 1 C, see Figure S11 in SI). On the other hand, both cells were plagued by limited rate capability. Factors contributing to the low performance may include the lack of a strong mechanical support (which would be provided by carbon coating) and the possibility that the NCs, even if highly conductive during the early cycles (as in the case of the etched NCs), can quickly react with the electrolytes and lead to the formation resistive surface layers, that is, they lack cycling stability. This issue too is circumvented by carbon coating.
We finally focus the discussion on the cells prepared with etched NCs@C. The electrodes were cycled between 2 and 4 V at room temperature to assess their cycling characteristics and rate performance. Figure 3c reports the cycling behavior at different C rates. At 1 C rate, a capacity of ∼155 mAh g–1 was observed. At 5 and 10 C rates, the capacities were ∼135 mAh/g and ∼125 mAh/g, respectively. These values are higher than those of the cells from as-synthesized NCs@C, which released a capacity of 110 mAh/g at 5 C and 90 mAh/g at 10 C (see Figure S12 in the SI). Along the cycles, just after 10 C, the rate was reverted to 1 C to measure again the capacity, and full capacity retention (as that of the original capacity) was observed at 1 C rate for the etched NCs@C. Therefore, the poor performance of as synthesized NCs@C was mainly due to coalescence phenomena (particle size effect on the performance)21 and to Li3PO4 impurity formation (impurity effect on the performance)22 during the carbon-coating process, as discussed earlier.
To evaluate the rate capability of the etched NCs@C electrodes, they were cycled at ultrahigh rates of ∼24 C, 61 C, and even 122 C. The cells were still able to sustain the applied rates and delivered capacities of ∼95 mAh/g, ∼70 mAh/g, and ∼30 mAh/g, respectively. Even if a slight capacity fading between the cycles was noted, the overall capacity values were still found to be at close range for rates up to 24 C (at 24 C the first capacity was 95 mAh/g that faded to 85 mAh/g after 10 cycles). These points to enhanced electronic/ionic conductivities sustaining charge/discharge processes up to high rates, without much loss in capacity. Higher capacity fading (but still at acceptable range) was evident due to the extremely high loads (10.3 A/g and 20.7 A/g) during the charge/discharge cycle. Therefore, considering the fact that the good rate capability of etched NCs@C is due to the preservation of NC morphology after carbon coating with glucose (comparing the etched NC@C with the as-synthesized NCs@C), the LiPF6 etching that makes the NCs hydrophilic and soluble in an aqueous glucose solution is definitively a crucial step to reach high performance.
The rate capability of the etched NCs@C electrodes was higher than that of several LiFePO4-based electrodes reported in other publications: LiFePO4 microspheres,23 LiFePO4 nanocrystals,24 LiFePO4/C nanoplates,25 hierarchical LiFePO4,26 single-crystalline LiFePO4 nanosheets,11 carbon-coated LiFePO4 nanowires,27 LiFePO4@C,28 and graphene–LiFePO4.29 Our deduction is therefore that etching the NCs improves considerably their electrical accessibility. This appears as the overriding factor30 in determining the rate performance.
In conclusion, we have developed a promising pathway for preparing high-power electrodes, in which the LiFePO4 NCs surface is etched by LiFP6, followed by conventional carbon coating. The LiPF6 etched LiFePO4@carbon NC electrodes, at 1 C, 5 and 10 C, achieved ∼155 mAh/g, ∼135 mAh/g, and ∼125 mAh/g, respectively, with significant capacity retention. In addition, the LiPF6 etched LiFePO4@C electrodes exhibited a remarkable rate capability, for example, at 61 C (10.3 A/g), a capacity of ∼70 mAh/g was obtained, while at 122 C (20.7 A/g), the capacity was ∼30 mAh/g. Overall, the rate capability achieved in this work is very attractive for optimizing high-power electrodes. We believe that the surface treatment of the type reported here can be extended to other electrode materials prepared by colloidal synthesis and more in general to other nanomaterials where a “naked” surface (free from ligands) may play a role, and to other olivine type materials in particular. This is important for several applications, as evidenced by the many approaches which have been developed recently to stabilize colloidal NCs with carbon-free ligands.31,32
Methods
Synthesis of Platelet-Shaped LiFePO4 NCs
In a typical up-scalable synthesis of LiFePO4 nanoplatelets, 2.25 g (16.8 mmol) of lithium iodide (LiI), 1.65 g (12.5 mmol) of ammonium phosphate dibasic, 1.575 g (12.5 mmol) of iron(II) chloride, 125 mL (0.38 mmol) of oleylamine (Olam), and 125 mL of 1-octadecene were mixed in a 500 mL three-neck flask connected to a standard Schlenk line. The solution was kept under vacuum at 120 °C for 1 h, after which it was heated at 250 °C under N2 for 3 h. It was then transferred to a nitrogen-filled glovebox and the NCs were precipitated and cleaned by repeated additions of chloroform and ethanol followed by centrifugation at 8000 rpm.
LiPF6 Treatment
The as-synthesized NCs (400 mg) were dissolved in 10 mL of chloroform and 500 mg of LiPF6 was dissolved in 10 mL of water. The two solutions were mixed, and the final 20 mL mixture was vigorously shaken. After few minutes, the LiFePO4 nanoplatelets were transferred into the aqueous phase. The aqueous phase was then collected and centrifuged at 8000 rpm. To the precipitate, 40 mL of ultrapure water was added to remove the excess of LiPF6 from the NCs. The NCs were then redispersed in 5 mL of ultrapure water with a resistivity of 18 MΩ cm–1. This last step was repeated three times.
Carbon Coating of Etched NCs (Etched NCs@C)
The etched LiFePO4 nanoplatelets (500 mg) were dissolved in aqueous glucose solution (20 wt % in glucose: 100 mg in 2 mL of water). The annealing was carried out in a quartz combustion boat containing the dried mixture (NCs + glucose). The boat was placed in the middle of the furnace while flowing 50 sccm of argon (Ar) gas and was rapidly warmed up to 450 °C (50 °C/min) and then slowly heated to 500 °C (10 °C/min) under forming gas (95% Ar, 5% H2). The slow part of the heating process was chosen to avoid temperature overshoots, and the heating procedure took about 35 min. After that, the sample was kept at constant temperature (500 °C) in 50 sccm of forming gas (95% Ar, 5% H2) for 12 h.
Carbon Coating of As-Synthesized NCs (As-Synthesized NCs@C)
In this approach, the as-synthesized NCs were carbon-coated using only the Olam ligand molecules at their surface as carbon source. Annealing was carried out in a quartz combustion boat containing the as-synthesized NCs dried powder. The boat was placed in the middle of the furnace while flowing 50 sccm of argon (Ar) gas and was rapidly warmed up to 450 °C (50 °C/min) and then slowly heated to 500 °C (10 °C/min) under forming gas (95% Ar, 5% H2). The slow part of the heating process was chosen to avoid temperature overshoots, and the heating procedure took about 35 min. After that, the sample was kept at constant temperature (500 °C) in 50 sccm of forming gas (95% Ar, 5% H2) for 12 h.
X-ray Diffraction (XRD)
XRD patterns were recorded on a Rigaku SmartLab X-ray powder diffractometer equipped with a 9 kW CuKα rotating anode, operating at 40 kV and 150 mA. A Göbel mirror was used to convert the divergent X-ray beam into a parallel beam and to suppress the Cu Kβ radiation (1.392 Å). The diffraction patterns were collected at room temperature and over an angular range of 10–80°, with a step size of 0.05°. A graphite (002) flat crystal was used to remove the X-ray fluorescence. XRD data analysis was carried out using PDXL 2.1 software from Rigaku.
Brunauer–Emmett–Teller (BET) Measurements
Specific surface area measurements were carried out by nitrogen physisorption at 77 K in a Quantachrome equipment, model autosorb iQ. The specific surface areas were calculated using the multipoint BET (Brunauer–Emmett–Teller) model, considering 11 equally spaced points in the P/P0 range of 0.05–0.30. Prior to measurements, the samples (50–200 mg in form of powder) were degassed for 1 h at 30 °C under vacuum to eliminate weakly adsorbed species.
Thermogravimetric Analysis (TGA)
The as-synthesized NCs (dried powder) were analyzed by TGA to quantify the fraction of organic component. We performed the thermal analysis using a TGA Q500 instrument (under N2, equilibration time 5 min at 30 °C and then 10 °C/min for heating).
X-ray Photoelectron Spectroscopy (XPS)
XPS was performed on a Kratos Axis Ultra DLD spectrometer, using a monochromatic Al Kα source (15 kV, 20 mA). Wide scans were acquired at analyzer pass energy of 160 eV. High-resolution narrow scans were performed at a constant pass energy of 20 eV and steps of 0.1 eV. The photoelectrons were detected at a takeoff angle Φ = 0° with respect to the surface normal. The pressure in the analysis chamber was maintained below 5 × 10–9 Torr for data acquisition. The data were converted to VAMAS format and processed using Casa XPS software, version 2.3.15. The binding energy (BE) scale was internally referenced to the C 1s peak (BE for C–C = 284.8 eV).
Transmission Electron Microscopy (TEM)
Conventional TEM imaging was performed using a JEOL JEM-1011 microscope working at an acceleration voltage of 100 kV. High-resolution TEM (HRTEM) and scanning TEM (STEM) measurements were acquired with a JEOL JEM-2200FS microscope equipped with a Schottky gun working at an acceleration voltage of 200 kV, a CEOS spherical aberration corrector in the objective lens allowing a spatial resolution of 0.9 Å, and an in-column energy Ω-filter.
High-Resolution SEM (HRSEM)
Analyses of selected samples were carried out using a JEOL JSM 7500FA, equipped with a cold field emission gun and operating in secondary electron (SE) imaging mode (ultimate resolution of 1 nm at 15 kV of accelerating voltage). EDS analysis was performed using an Oxford Instruments X-Max 80, LN2-free silicon drift detector (SDD), with 80 mm2 active area and 129 eV of energy resolution at 5.9 keV (Mn Kα). The extended Pouchou and Pichoir (XPP) matrix correction algorithm included in the Oxford Aztec software was used to analyze the data.
Electron Energy Loss Spectroscopy (EELS)
To quantify the elemental composition of the LiFePO4 NCs (and in particular for what concerns Li), we carried out energy loss spectroscopy analysis in the TEM (EELS). The spectra were collected on the JEOL JEM-2200FS with the Ω-filter in spectroscopy mode, from a region of the sample containing tens of nanocrystals, after careful calibration of the illumination and collection angles (α ∼1.5 mrad and β ∼5.0 mrad, respectively). For better accuracy we used the model-based fitting method33 and parametrized atomic Hartree–Slater cross sections (from Gatan Inc.). Atomic models in the figures were done with VESTA.34
Nuclear Magnetic Resonance (NMR) and Fourier Transform Infrared (FT-IR) Spectroscopy
The sample bearing oleylamine (PF6–) ligands was resuspended in 500 μL of CDCl3 (D2O). For both samples their 19F NMR spectra were recorded on a Bruker Ultrashield Plus FT-NMR 600 MHz Advance III and their 1H NMR NMR on a Bruker DRX 400 spectrometer with predefined pulse programs and sample rotation. The FTIR spectra were recorded on a Bruker Vertex 70v FTIR spectrometer in ATR mode.
Battery Fabrication and Electrochemical Measurements
All of the electrochemical measurements (EC) were carried out using a PARSTAT2273 potentiostat/galvanostat and a MPG-2 (biologic) multichannel battery testing unit. All of the steps in the battery fabrication were carried out in an Ar filled glovebox. The NC samples were thoroughly dried at 70 °C overnight to obtain a powder. The NCs powder was mixed with carbon black and polyvinylidene difluoride (PVDF) in N-methyl-2-pyrrolidone (degassed), such that the weight fraction of NCs in the resulting mixture was 75%. The vial containing the mixture was stirred for about 2 h in order to obtain an homogeneous slurry. The NCs slurry was then applied onto the current collector, dried at 120 °C overnight, and then pressed, which resulted in a uniform and compact coating. Coin type 2032 cells were assembled with the coated current collector serving as one electrode, while a pure Li metal disk served both as reference and counter electrode. A solution of 1 M lithium hexafluorophosphate in 1:1:1 (v/v) propylene carbonate, ethylene carbonate, and diethyl carbonate was used as electrolyte, and polypropylene layers were employed to separate the electrodes. For electrochemical impedance spectroscopy (EIS) measurements, the cells were allowed to equilibrate to a stable open circuit voltage. Impedance spectra of the cells were recorded at room temperature between 100 kHz and 10 mHz, with an AC amplitude of 20 mV. Cyclic voltammetry (CV) measurements were carried out at 50 and 100 μV s–1 scan rates in the potential range from 2.5 to 4.0 V.
Acknowledgments
This work was supported in part by the European Union through the FP7 consolidator ERC Grant TRANS-NANO (Contract No. 614897) and CNECT-ICT-604391 (Graphene Flagship). We would like to acknowledge Dr. Karol Miszta, Dr. Alessandro Genovese, Dr. Alberto Casu, Dr. Tania Montanari, Dr. Milena Arciniegas, Maria Elena Materia, and Giammarino Pugliese for helpful discussions.
Supporting Information Available
Chemical compound information. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
A.P., G.B., and S.M. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Padhi A. K.; Nanjundaswamy K. S.; Goodenough J. B. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 1997, 144, 1188–1194. [Google Scholar]
- Herle P. S.; Ellis B.; Coombs N.; Nazar L. F. Nano-network electronic conduction in iron and nickel olivine phosphates. Nat. Mater. 2004, 3, 147–152. [DOI] [PubMed] [Google Scholar]
- Xu Y. N.; Chung S. Y.; Bloking J. T.; Chiang Y. M.; Ching W. Y. Electronic structure and electrical conductivity of undoped LiFePO4. Electrochem. Solid State Lett. 2004, 7, A131–A134. [Google Scholar]
- Whittingham M. S.; Song Y. N.; Lutta S.; Zavalij P. Y.; Chernova N. A. Some transition metal (oxy)phosphates and vanadium oxides for lithium batteries. J. Mater. Chem. 2005, 15, 3362–3379. [Google Scholar]
- Prosini P. P.; Lisi M.; Zane D.; Pasquali M. Determination of the chemical diffusion coefficient of lithium in LiFePO4. Solid State Ionics 2002, 148, 45–51. [Google Scholar]
- Wang J.; Sun X. Understanding and recent development of carbon coating on LiFePO4 cathode materials for lithium-ion batteries. Energy Environ. Sci. 2012, 5, 5163–5185. [Google Scholar]
- Jayaprakash N.; Kalaiselvi N.; Periasamy P. Synthesis and characterization of LiMXFe1-XPO4 (M = Cu, Sn; X = 0.02) cathodes - A study on the effect of cation substitution in LiFePO4 material. Int. J. Electrochem. Sci. 2008, 3, 476–488. [Google Scholar]
- Chung S. Y.; Bloking J. T.; Chiang Y. M. Electronically conductive phospho-olivines as lithium storage electrodes. Nat. Mater. 2002, 1, 123–128. [DOI] [PubMed] [Google Scholar]
- Delacourt C.; Poizot P.; Levasseur S.; Masquelier C. Size effects on carbon-free LiFePO4 powders. Electrochem. Solid State Lett. 2006, 9, A352–A355. [Google Scholar]
- Rui X.; Zhao X.; Lu Z.; Tan H.; Sim D.; Hng H. H.; Yazami R.; Lim T. M.; Yan Q. Olivine-Type Nanosheets for Lithium Ion Battery Cathodes. ACS Nano 2013, 7, 5637–5646. [DOI] [PubMed] [Google Scholar]
- Zhao Y. P.; Liu B.; Yu G. Single-Crystalline LiFePO4 Nanosheets for High-Rate Li-Ion Batteries. Nano Lett. 2014, 14, 2849–2853. [DOI] [PubMed] [Google Scholar]
- Wang L.; Zhou F.; Meng Y. S.; Ceder G.. First-principles study of surface properties of LiFePO(4): Surface energy, structure, Wulff shape, and surface redox potential, Phys. Rev. B 2007, 76, article number 165435. [Google Scholar]
- Fedorkova A.; Orinakova R.; Orinak A.; Wiemhoefer H.-D.; Kaniansky D.; Winter M. Surface treatment of LiFePO4 cathode material with PPy/PEG conductive layer. J. Solid State Electrochem. 2010, 14, 2173–2178. [Google Scholar]
- Song G.-M.; Wu Y.; Liu G.; Xu Q. Influence of AlF3 coating on the electrochemical properties of LiFePO4/graphite Li-ion batteries. J. Alloys Comp. 2009, 487, 214–217. [Google Scholar]
- Sun Y. K.; Cho S. W.; Lee S. W.; Yoon C. S.; Amine K. AlF3-coating to improve high voltage cycling performance of Li Ni1/3Co1/3Mn1/3 O-2 cathode materials for lithium secondary batteries. J. Electrochem. Soc. 2007, 154, A168–A172. [Google Scholar]
- An J.; Liu C.; Guo R.; Li Y.; Xu W. Ti3SiC2 Modified LiFePO4/C Cathode Materials with Improved Electrochemical Performance. J. Electrochem. Soc. 2012, 159, A2038–A2042. [Google Scholar]
- Park K.-S.; Xiao P.; Kim S.-Y.; Dylla A.; Choi Y.-M.; Henkelman G.; Stevenson K. J.; Goodenough J. B. Enhanced Charge-Transfer Kinetics by Anion Surface Modification of LiFePO4. Chem. Mater. 2012, 24, 3212–3218. [Google Scholar]
- Paolella A.; Bertoni G.; Dilena E.; Marras S.; Ansaldo A.; Manna L.; George C. Redox Centers Evolution in Phospho-Olivine Type (LiFe0.5Mn0.5 PO4) Nanoplatelets with Uniform Cation Distribution. Nano Lett. 2014, 14, 1477–1483. [DOI] [PubMed] [Google Scholar]
- Barlow C. G. Reaction of water with hexafluorophosphates and with Li bis(perfluoroethylsulfonyl)imide salt. Electrochem. Solid State Lett. 1999, 2, 362–364. [Google Scholar]
- Su C.; Lu G.; Xu L.; Zhang C. Preparation of LiFePO4/Carbon/PANI-CSA Composite and Its Properties as High-Capacity Cathodes for Lithium Ion Batteries. J. Electrochem. Soc. 2012, 159, A305–A309. [Google Scholar]
- Bruce P. G.; Scrosati B.; Tarascon J.-M. Nanomaterials for Rechargeable Lithium Batteries. Angew. Chem., Int. Ed. 2008, 47, 2930–2946. [DOI] [PubMed] [Google Scholar]
- Hu C.; Yi H.; Fang H.; Yang B.; Yao Y.; Ma W.; Dai Y. Suppressing Li3PO4 impurity formation in LiFePO4/Fe2P by a nonstoichiometry synthesis and its effect on electrochemical properties. Mater. Lett. 2011, 65, 1323–1326. [Google Scholar]
- Sun C.; Rajasekhara S.; Goodenough J. B.; Zhou F. Monodisperse Porous LiFePO4 Microspheres for a High Power Li-Ion Battery Cathode. J. Am. Chem. Soc. 2011, 133, 2132–2135. [DOI] [PubMed] [Google Scholar]
- Rangappa D.; Sone K.; Ichihara M.; Kudo T.; Honma I. Rapid one-pot synthesis of LiMPO4 (M = Fe, Mn) colloidal nanocrystals by supercritical ethanol process. Chem. Commun. 2010, 46, 7548–7550. [DOI] [PubMed] [Google Scholar]
- Saravanan K.; Balaya P.; Reddy M. V.; Chowdari B. V. R.; Vittal J. J. Morphology controlled synthesis of LiFePO4/C nanoplates for Li-ion batteries. Energy Environ. Sci. 2010, 3, 457–464. [Google Scholar]
- Guo B.; Ruan H.; Zheng C.; Fei H.; Wei M. Hierarchical LiFePO4 with a controllable growth of the (010) facet for lithium-ion batteries. Sci. Rep. 2013, 3, 2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C.; Yu Y.; Gu L.; Weichert K.; Maier J. Electrospinning of Highly Electroactive Carbon-Coated Single-Crystalline LiFePO4 Nanowires. Angew. Chem. Int. Ed. 2011, 50, 6278–6282. [DOI] [PubMed] [Google Scholar]
- Lee Y. C.; Han D.-W.; Park M.; Jo M. R.; Kang S. H.; Lee J. K.; Kang Y.-M. Tailored Surface Structure of LiFePO4/C Nanofibers by Phosphidation and Their Electrochemical Superiority for Lithium Rechargeable Batteries. ACS Appl. Mater. Interfaces 2014, 6, 9435–9441. [DOI] [PubMed] [Google Scholar]
- Kim W.; Ryu W.; Han D.; Lim S.; Eom J.; Kwon H. Fabrication of Graphene Embedded LiFePO4 Using a Catalyst Assisted Self Assembly Method as a Cathode Material for High Power Lithium-Ion Batteries. ACS Appl. Mater. Interfaces 2014, 6, 4731–4736. [DOI] [PubMed] [Google Scholar]
- Zhang W.-J. Comparison of the Rate Capacities of LiFePO4 Cathode Materials. J. Electrochem. Soc. 2010, 157, A1040–A1046. [Google Scholar]
- Kovalenko M. V.; Scheele M.; Talapin D. V. Colloidal Nanocrystals with Molecular Metal Chalcogenide Surface Ligands. Science 2009, 324, 1417–1420. [DOI] [PubMed] [Google Scholar]
- Rosen E. L.; Buonsanti R.; Llordes A.; Sawvel A. M.; Milliron D. J.; Helms B. A. Exceptionally Mild Reactive Stripping of Native Ligands from Nanocrystal Surfaces by Using Meerwein’s Salt. Angew. Chem. Int. Ed. 2012, 51, 684–689. [DOI] [PubMed] [Google Scholar]
- Bertoni G.; Verbeeck J. Accuracy and precision in model based EELS quantification. Ultramicroscopy 2008, 108, 782–790. [DOI] [PubMed] [Google Scholar]
- Momma K.; Izumi F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 12721276. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.