

Abstract
The antiproliferative activities of new substituted tetrahydroisoquinolines (THIQs) are described. Their cytotoxicities against Ishikawa human endometrial cell line were determined after 72 h drug expose employing Celtiter-Glo assay at concentrations ranging from 0.01 to 100,000 nM. The antiproliferative activities of the compounds understudy were compared to tamoxifen (TAM). In-vitro results indicated that most of the compounds showed better activity than TAM. The most active compounds obtained in this study were 1, 2, 3 and 22 whose IC50 values are 1.41, 0.91, 0.74 and 0.36 μM respectively. This study helped us to evaluate the risk of developing endometrial cancer in the design of non-steroid estrogen receptor modulators with no agonistic effects on uterus. In-silico pharmacophore hypotheses were generated using GALAHAD and PHASE and the best models with a probable bioactive conformation(s) for these compounds were proposed. These conformations and the alignments of the molecular structures give us an insight in designing compounds with better biological activity.
Keywords: Antiproliferative agents, Ishikawa Cell lines, Tetrahydroisoquinolines, Pharmacophore Models
INTRODUCTION
Breast cancer is the second leading cause of cancer-related deaths in women and is the most common cancer among women, excluding non-melanoma skin cancers. An estimated 232,340 new cases of invasive breast cancer are expected among American women. [1]. The nuclear receptor, estrogen receptor (ER) and progesterone receptor (PR) and their associated steroid hormones are known to play important roles in the growth of breast tumors and the status of these hormones is employed as diagnostic indicators for endocrine responsiveness and tumor recurrence. Estrogens are involved in the stimulation of cancer cell proliferation. Unopposed or increased estrogen exposure is also associated with an increase risk for endometrial cancer. One way of blocking the estrogen action on tumor cells is preventing the binding of estrogen (17β-estradiol) to estrogen receptor (ER) by designing novel inhibitors (Antagonists) of ER [2]. Tamoxifen has been the leading drug to treat breast cancer for more than two decades and has proven to be an effective treatment for (ER)-positive breast cancer, particularly in the post-menopausal women [3–5]. It is licensed as a chemo preventive agent following findings of a 49% reduction in invasive breast cancer in treated women [6]. However, it is not without adverse side effects. Tamoxifen behaves as ER antagonist in the breast tissue and as ER agonist in bone, and has prophylactic use in breast cancer [7]. Its agonistic effect on the uterus is said to be associated with increased risk of developing endometrial cancer [8]. Thus, alternative chemical entities, preferably non-steroid estrogen receptor modulators are sought with no agonistic effects on uterus.
Tetrahydroisoquinoline natural product analogs have been shown to exhibit biological activity, rendering potential pharmaceutical agents [9]. The tetrahydroisoquinoline family of alkaloids include potent cytotoxic agents that display a range of biological properties such as antitumor and antimicrobial activities studied thoroughly over the past 25 years starting with the isolation of naphthyndinomycin in 1974 [10]. Ecteinascidin-743 (ET-743) is a marine tetrahydroisoquinoline alkaloid isolated from the tunicate Ekteinascidia turbinate with a potent cytotoxic activity against a variety of tumor cell lines in vitro and against several rodent tumors and human xenografts in vivo [11].
Tetrahydroisoqinoline derivatives were identified as subtype selective estrogen receptor antagonists/agonists hence, potential therapeutic agents for breast cancer [12, 13]. Structure activity relationship studies (SAR) of ER-α selective tetrahydroisoquinolines were reported by Renaud et al. [14]. Tetrahydroisoquinolines incorporating conformationally restricted side chains as the replacement of the aminoethoxy residue, typical of SERMs were reported exhibiting binding affinity to ER-α and antagonistic properties [15]. More recently, new steroidomimetic tetrahydroquinolines were reported which act as microtubule disruptors [16].
However, in the development of anti-estrogenic drugs, it is critical that the molecules under study do not cause estrogenic stimulation of the uterus, which could lead to both increase in uterine bleeding and an increased risk of developing uterine cancer [17]. Ishikawa cell line, a well-differentiated human endometrial adenocarcinoma cell line expresses functional estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ) isoforms. Therefore testing the new potential anti-proliferative moieties on Ishikawa cell line in the initial stages of the design is a good idea to understand the risk associated in developing endometrial cancer and its treatment [18]. Also the compounds which act as antiproliferative agents against endometrial cancer can be used in combination with Tamoxifen (TAM) in the treatment of breast cancer, particularly in post-menopausal women.
We report in this study, the in-vitro antiproliferative activity of new tetrahydroisoquinolines (THIQs) against Ishikawa human endometrial adenocarcinoma cell lines. Pharmacophore hypotheses using Genetic Algorithm with Linear Assignment of Hypermolecular Alignment of Datasets (GALAHAD) and Pharmacophore Alignment and Scoring Engine (PHASE) were generated and evaluated. This analysis would set a stage for further optimization of the lead compounds.
METHODS
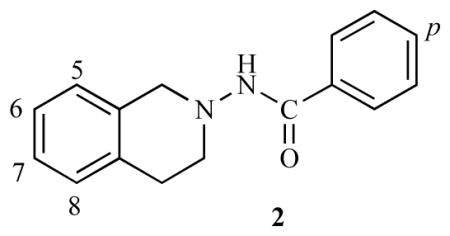
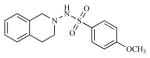
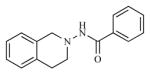
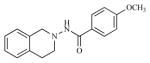
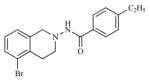
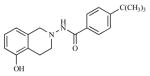
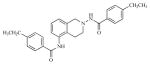
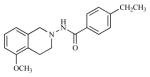
We have used a set of 23 substituted tetrahydroisoquinoline analogs (Table 1) whose pIC50 values are in the range of 4.47–6.44 against Ishikawa cell lines. The in vitro cancer cell antiproliferative activity was tested in triplicates to obtain high-quality reliable IC50 data for the modeling. Observed activities (IC50) were converted to negative logarithm (pIC50) before pharmacophore generation.
Table 1.
Activity profile of the compounds under study.
| |||
|---|---|---|---|
| ID | Compound | IC50 (μM, Ishikawa) | pIC50 (Ishikawa) |
| 1 |
|
1.41 | 5.8500 |
| 2 |
|
0.91 | 6.0402 |
| 3 |
|
0.74 | 6.1286 |
| 4 |
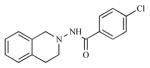
|
2.13 | 5.6722 |
| 5 |
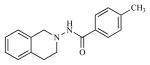
|
1.65 | 5.7819 |
| 6 |
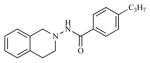
|
2.54 | 5.5939 |
| 7 |
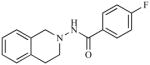
|
30.60 | 4.5127 |
| 8 |
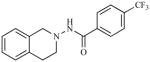
|
1.56 | 5.8066 |
| 9 |
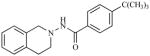
|
23.09 | 4.6366 |
| 10 |
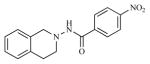
|
28.23 | 4.5494 |
| 11 |
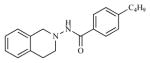
|
3.11 | 5.5068 |
| 12 |
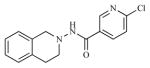
|
19.53 | 4.7092 |
| 13 |
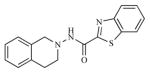
|
29.09 | 4.5372 |
| 14 |

|
26.68 | 4.5739 |
| 15 |
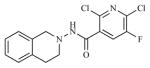
|
29.24 | 4.5338 |
| 16 |
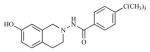
|
2.93 | 5.5337 |
| 17 |
|
3.19 | 5.4956 |
| 18 |
|
33.78 | 4.4712 |
| 19 |
|
22.11 | 4.6554 |
| 20 |
|
20.41 | 4.6900 |
| 21 |
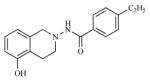
|
33.49 | 4.4750 |
| 22 |
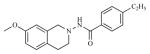
|
0.36 | 6.4435 |
| 23 |
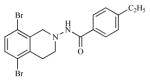
|
1.92 | 5.7153 |
| 24 |
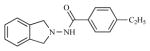
|
>100 | >100 |
| TAM |
|
22.92 | 4.64 |
Antiproliferative Activity Studies
The Ishikawa human endometrial cancer cell line was purchased from Sigma. The cell lines were cultured in Phenol Red-Free RPMI-1640 medium (HyClone, 500 mL) supplemented with 10 % fetal bovine serum (FBS) procured from Atlanta Biologicals. They were maintained in exponential growth phase by sub-culturing twice weekly in 150-cm2 flasks at 37 °C, 95 % air with 5 % CO2. The media was removed from the flasks, the cells washed with phosphate buffer solution (PBS) (HyClone) and then detached using 5 ml of TryplExpress solution (Invitrogen) (incubation 5–10 min) followed by addition of growth media. Cells were centrifuged (1,500 rpm) for 5 min. and re-suspended in growth media at 105cell/mL. The cell lines were placed in 20, 96 well plates at a density of 5000 cells/well in total volume of 50 μL in phenol-red free medium and incubated overnight. Compounds were weighed and dissolved in DMSO (10 μM) and tested at different concentrations ranging from 0.01 to 100,000 nM. Tamoxifen (Sigma) (TAM, 10 μM) was used as a positive control. Estradiol (Sigma) (25 μL of 40 nM) was added to all appropriate wells in the plate. 25μL media was added to all wells that did not receive estradiol. 25 μL of stocks (contain compounds, DMSO and phenol-red free medium) were added to cells and medium already on plate. 50μL media was added to media wells, 50 μL mix (contain 32 mL DMSO + 768 mL phenol-red free medium) was added to all vehicle control wells and 10 μM TAM was also added to appropriate wells. Drug exposed cells were incubated or 72 h, after which the plates were removed for Cell-Titer-GLo assay (Promega) from 37 °C, 5% CO2 incubator and equilibrated at room temperature for 30 min. 100 μL of CellTiter-Glo assay reagent was added to each well and cell-lysis was induced on an orbital shaker for 2 min. followed by a further 10 min incubation at room temperature. Luminescence results were read on TriLux Luminometer. The luminescent signal is proportional to the number of active cells present in culture. Dead cells do not affect cell counts because they do not contribute to ATP content. As a consequence, the number of metabolically active cells can be directly derived from the luminescent signal using a specific calibration curve. The results expressed as IC50 (inhibitory concentration of 50 %) were the averages of three data points for each concentration and were calculated using GraphPad Prism 4.0.
Molecular Modeling
All computational studies were performed using Tripos SYBYL-X 1.3 and Schrodinger software packages based on a Windows XP workstation. The structures used in this manuscript were drawed with Concord and energy minimized using Tripos force field and Gasteigner-Huckel charges (method: Powell, termination: gradient 0.05 kcl/mol A° and max. iterations 1,00,000) as implemented in SYBYL for GALAHAD and OPLS-2005 force field for PHASE. Confgen as implemented in Schrodinger was used as the low energy bioactive conformation generator for PHASE.
RESULTS AND DISCUSSION
Twenty three substituted tetrahydro isoquinolines (THIQs) have been synthesized [19] following the good activity profile (antiproliferative activity) of the unsubstituted compound 2 on human endometrial Ishikawa cell lines. An effort to evaluate the effect of various substitutions on the phenyl ring of the THIQs towards activity is undertaken. In this regard, high-quality biological testing results were collected for the newly synthesized compounds along with Tamoxifen (Table 1). The most active compounds are 1, 2, 3 and 22 (IC50 = 1.41, 0.91, 0.74 and 0.36 μM respectively). These results strongly indicate that the antiproliferative activity of most of our compounds on Ishikawa cell lines were far better than the standard Tamoxifen, indicating that the compounds in the present study have low associated risk in developing endometrial cancer and its treatment. As far as the structure activity relationship is concerned, ethyl group with the right steric bulk proved to be the optimum substitution at the -para position of the aromatic ring. Similarly, -methoxy substitutions on the seventh position of the THIQ aromatic ring resulted in decrease in activity compared with the unsubstituted compound 2. Substitutions on the other positions of the THIQ aromatic ring did not resulted in better activity. Fifth position on the tetrahydroisoquinoline ring proved to be the wrong site for substitution leading to considerable loss in activity. Similarly, hetero atoms incorporated inside the aromatic ring as in compounds 12, 13, 14 and 15 led to loss in activity.
Pharmacophore modeling is used to propose the 3D spatial arrangement of chemical features that are essential for biological activity. Briefly, a pharmacophore model consists of a group of features located relatively close to each other in 3D space, surrounded by a sphere of tolerance, which encode location dependent chemical characteristics that account for activity. The sphere represents the 3D area that should be occupied by specific chemical functional groups for optimal activity. Pharmacophore models would then be developed based on a set of ligands superposed to maximize steric overlap and minimizing strain energy. GALAHAD [20–22] is a module in Sybyl [23] modeling environment which was used initially to generate the pharmacophore hypothesis in this study. GALAHAD identifies a set of molecular conformations with an optimal combination of low strain energy, steric overlap, and pharmacophoric similarity. The biological activity data were used in conjunction with GALAHAD to develop a pharmacophore based on hypotheses generated by compound molecular structures and properties. The model outlines features that can be used for virtual screening and/or can be used as a guide to rationally design and synthesize potentially high activity compounds. It uses a generalized multi-dimensional cost function that takes into account the hydrophobic, ionic, hydrogen bonding and steric attributes of the test compounds when generating various possible alignments, and consequently, pharmacophores [24].
Alignment and Pharmacophore Generation
The alignment was performed in two stages. In the first stage, top six active molecules whose pKI are in the range 6.44–5.78 were aligned flexibly by GALAHAD, independent of template. The GALAHAD produces a set of hypotheses following the flexible alignment. Specificity, Number of Hits, Features, Pareto Ranking, Energy, Sterics, Hydrogen bonds and Molecular Query for the characteristics of the models were generated. In addition, information about how the individual compound used to generate the pharmacophore compares to the query tuplet (multiplet) is also displayed in the GALAHAD results [25]. The top models generated were examined and the best model would be selected based on three criteria: 1. Number of “hits” should equal to the number of active molecules which in this case is six. 2. The model needs to have reasonable energies (within the same order of magnitude as compared to other competing models). 3. The model should have the maximum pharmcophore features. The model which satisfies all of the above criteria was then selected and the associated pharmacophore was used as a template to align the remaining molecules in the data set using GALAHAD’s “Align to template” procedure.
The most active molecules were used to generate the initial pharmacophore and the GALAHAD results were shown in Table 2. Here, all the ligands were aligned with each other with a population size of 45 and maximum generation value of 70 and required the number of hits to be the number of active molecules. Each row is a probable alignment based on the overlap of pharmacophoric features of the six molecules that can be used as a template to align the remaining molecules in the second step. Similarly, each of the columns is a model property that can help isolate the ideal hypothesis that can be used to align the remaining molecules. Small values of energy and high values of steric and pharmacophoric concordance are desired for the best model. Among the top 12 models that were produced by GALAHAD, all the models showed five features and the number of ‘hits’ were equal to the active molecules viz., six. The PARETO scores were ‘0’ indicating models were not superior to each other based on the energy, sterics, H-bond and MOL_Qry. The high energy alignment models, MODEL_009 and MODEL_012 were discarded. All the models except MODEL_007 have energies in the same range. The MODEL_007 with relatively high energy was discarded. Model with low specificity (MODEL_010) was also discarded. Among the final 8 models, MODEL_002 with high steric values was selected as the template to align the remaining molecules in the data set. In this step, a hypermolecule that contains pharmacophoric information from multiple molecules in the dataset is generated. Here an extention of the LAMDA methodology [26] to aggregate features into a single hypermolecule by sequentially processing structurally similar compounds was used.
Table 2.
Galahad models and scores.
| Model | Specificity | N_Hits | Feats | Pareto | Energy | Sterics | hbond | MOL_QRY |
|---|---|---|---|---|---|---|---|---|
| MODEL_001 | 3.9180 | 6 | 5 | 0 | 4.55 | 614.7 | 74.2 | 17.32 |
| MODEL_002 | 3.9180 | 6 | 5 | 0 | 4.61 | 638.6 | 74.2 | 17.32 |
| MODEL_003 | 3.9160 | 6 | 5 | 0 | 3.50 | 446.2 | 74.2 | 17.32 |
| MODEL_004 | 3.9160 | 6 | 5 | 0 | 3.30 | 358.5 | 74.2 | 17.32 |
| MODEL_005 | 3.9180 | 6 | 5 | 0 | 4.21 | 595.9 | 74.2 | 17.32 |
| MODEL_006 | 3.9180 | 6 | 5 | 0 | 4.14 | 452.2 | 73.1 | 17.32 |
| MODEL_007 | 3.9160 | 6 | 5 | 0 | 11.66 | 645.5 | 74.2 | 15.32 |
| MODEL_008 | 3.9180 | 6 | 5 | 0 | 4.07 | 456.4 | 69.5 | 15.66 |
| MODEL_009 | 2.9130 | 6 | 5 | 0 | 6.880658 | 675.8 | 70.1 | 15.66 |
| MODEL_010 | 2.9100 | 6 | 5 | 0 | 3.86 | 478.2 | 59.1 | 15.08 |
| MODEL_011 | 3.9110 | 6 | 5 | 0 | 3.39 | 428.0 | 58.2 | 11.75 |
| MODEL_012 | 3.8900 | 6 | 5 | 0 | 214338908 | 688.8 | 60.9 | 12.29 |
The optimized pharmacophore model from this procedure is shown in Fig. (1). This includes two hydrophobic features centered on the benzene rings (HY_3, HY_4), one acceptor atom center on the oxygen atom (AA_2), one donor nitrogen atom (DA_1) and one positive nitrogen atom embedded in the THIQ ring (NP_5). The model shows the importance of all these five features for the antiproliferative activity. This model was helpful in predicting the probable bioactive conformation of the molecules understudy and would lead to design most active molecules.
Fig. (1).
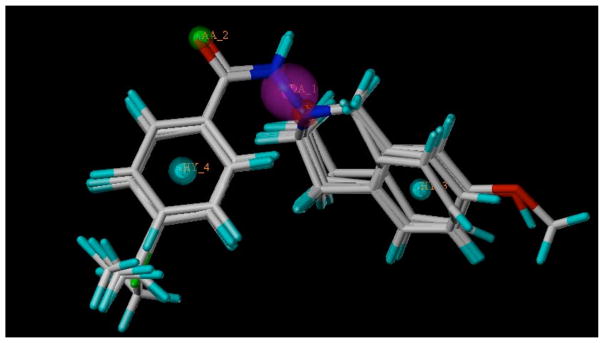
Galahad Alignment.
Next, we attempted to generate a PHASE pharmacophore model. The 3D structures of molecules constructed earlier for GALAHAD using Sybyl-X 1.3 software were imported into Maestro project table as implemented in Schrodinger modeling suite [27]. Given the set of defined active and inactive molecules in the dataset, PHASE utilizes fine-grained conformational sampling and a range of scoring functions to identify a common pharmacophore hypothesis, which display the characteristics of 3-D chemical structures that are supposed to be critical for binding [28, 29]. The hypothesis discloses a set of aligned conformations that suggest the relative manner in which the molecules most likely bind to the receptor. The various conformer generators in PHASE ensure that only relevant low energy bioactive conformations are generated. The generated hypotheses together with the aligned conformations of the dataset can be then combined with compound’s activity data to create a 3D-QSAR model that identifies overall aspects of molecular structures responsible for biological activity.
The pharmacophore models were generated using the “Develop Common Pharmacophore Hypotheses” module of PHASE. In the process, multiple conformers for each molecule were generated followed by energy minimization based on OPLS-2005 force field. The conformational space was explored by ConfGen [30, 31], with 500 conformers per rotatable bond and 1000 maximum of conformers per structure. A distance dependent dielectric was applied for solvation treatment. The pharmacophore models were generated using the same active compounds in the dataset which were used for generating GALAHAD pharmacophore models. These are defined as “active ligands” for pharmacophore generation. A set of pharmacophoric sites based on features defined in PHASE were assigned to the molecules. These features include hydrogen bond acceptor (A), hydrogen bond donor (D), hydrophobic group (H), and aromatic rings (R). Pharmacophores with five features that match to all active ligands were generated by using a tree-based partitioning technique with maximum tree depth of five. Box size of pharmacophore was adjusted to 2 Å. Active and inactive molecules were then scored for a given pharmacophore using default weights of scoring parameters. The variants are defined with maximum number of sites to be five, minimum number of sites to be four and should match all six of the actives. This gave four variations of which the variants ‘ADHRR’ gave three maximum hypotheses and ‘AADRR’ gave two maximum hypotheses. The generated pharmacophore hypotheses were clustered and scored with default parameters. The quality of the alignment was measured by a survival score (S). Scoring of a pharmacophore with respect to the activity of the ligand was conducted using default parameters for site, vector and volume terms. The four final models generated were AADRR.2, AADRR.6, ADHRR.4 and ADHRR.6. All four hypotheses were closely examined and scores reported in Table 3. The best models were ADHRR.4 and ADHRR.6 shown in Fig. (2). The pharmcophore site distances for the models ADHRR.4 and ADHRR.6 are shown in Table 4. In the second step, unaligned molecules in the dataset were aligned using pharmacophore features for models under study. Thereafter, all the four models were selected for further generation of pharmacophore based 3D-QSAR models (part of the PHASE module) with the grid spacing at 1.00 Å. Since actives and inactives were defined in generating pharmacophore models, uniform sampling was opted over activity coordinates for QSAR model building campaign. The maximum PLS factors were set to five. The random seed was set to a non-zero integer i.e., 6 in this case, and random training was set to 50 %, so that the results are reproducible. The modest Q2 (0.364) obtained for the model ADHRR.4 is not surprising as the dataset consists of only limited compounds which were structurally related. Compound 24 (Table 1) was synthesized later with one carbon less in the tetrahydroisoquinoline ring than the parent compound 2 (0.91 μM). As expected, compound 24 turned out to be inactive in our preliminary studies supporting the importance of hydrophobic group (H6) for the activity as shown in pharmacophore models ADHRR.4 and ADHRR.6. This inactivity may also be the result of the different conformations adopted by the less flexible five membered ring.
Table 3.
Phase hypothesis scores.
| Model | Survival Active | Survival Inactive | Post-hoc | Site | Vector | Volume | Selectivity |
|---|---|---|---|---|---|---|---|
| AADRR.2 | 3.958 | 1.106 | 3.958 | 1.00 | 1.000 | 0.960 | 1.367 |
| AADRR.6 | 3.958 | 1.043 | 3.958 | 1.00 | 1.000 | 0.960 | 1.387 |
| ADHRR.6 | 3.880 | 1.036 | 3.880 | 0.94 | 1.000 | 0.939 | 1.816 |
| ADHRR.4 | 3.853 | 1.095 | 3.853 | 0.92 | 1.000 | 0.934 | 1.821 |
Fig. (2).

PHASE Alignment Model: ADHRR.4 and Model: ADHRR.6.
Table 4.
Pharmacophore site distances for Models ADHRR.4 and ADHRR.6.
| Site 1 | Site 2 | Distance |
|---|---|---|
| A2 | D5 | 2.477 |
| A2 | H6 | 5.399 |
| A2 | R8 | 7.138 |
| A2 | R9 | 3.677 |
| D5 | H6 | 3.625 |
| D5 | R8 | 5.469 |
| D5 | R9 | 4.751 |
| H6 | R8 | 2.639 |
| H6 | R9 | 5.102 |
| R8 | R9 | 6.384 |
| A2 | D5 | 3.169 |
| A2 | H6 | 4.432 |
| A2 | R8 | 6.073 |
| A2 | R9 | 3.699 |
| D5 | H6 | 3.708 |
| D5 | R8 | 5.458 |
| D5 | R9 | 3.594 |
| H6 | R8 | 2.755 |
| H6 | R9 | 6.931 |
| R8 | R9 | 8.721 |
CONCLUSION
The pharmacophore models generated in this study provides rational to synthesize better ligands that optimize the perceived features and also assist in building a more reliable 3D-QSAR models and ligand based Virtual Screening. Pharmacophore model ADHRR.4 appears to be closer to the reality in the way the ligands orient themselves in the active site of the receptor. The pharmacophore features revealed in the models can provide necessary interactions with the binding pockets which would be crucial for activity. Based on these models, lead optimization efforts are under way to obtain compounds with enhanced activity.
Supplementary Material
Acknowledgments
We are grateful to the National Center for Research Resources and the National Institute of Minority Health and Health Disparities of the National Institutes of Health through Grant Number 8 G12MD007582-28. We are also grateful to Dr. Michael J. Roberts, Cell Biology and Immunology, Southern Research Institute, Birmingham, AL, for testing cytotoxicity on Ishikawa cell lines.
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
A general reaction scheme giving details of the synthesis of Tetrahydroisoquinolines (THIQs) and the experimental data of a representative compound, N-(3,4-dihydroisoquinolin-2(1H)-yl)-4-methoxybenzamide 3 are enclosed as supplementary data
References
- 1.Cancer Facts & Figures. American Cancer Society; Atlanta: 2013. [Google Scholar]
- 2.Johnston S. Fulvestrant and the sequential endocrine cascade for advanced breast cancer. British Journal of Cancer. 2004;90(Suppl 1):S15–S18. doi: 10.1038/sj.bjc.6601632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuiper GGJM, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors α and β. Endocrinology. 1997;138(3):863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 4.Jordan VC. The strategic use of antiestrogens to control the development and growth of breast cancer. Cancer. 1992;70(4):977–982. [PubMed] [Google Scholar]
- 5.Seeger H, Huober J, Wallwiener D, Mueck AO. Inhibition of human breast cancer cell proliferation with estradiol metabolites is as effective as with tamoxifen. Horm Metab Res. 2004;36(5):277–280. doi: 10.1055/s-2004-814480. [DOI] [PubMed] [Google Scholar]
- 6.Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N, Atkins J, Daly M, Wieand S, Tan-Chiu E, Ford L, Wolmark N. Tamoxifen for prevention of breast cancer: Report of the national surgical adjuvant breast and bowel project P-1 Study. J Natl Cancer Inst. 1998;90(18):1371–1388. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 7.Phillips DH, Venitt S. Safety of prophylactic tamoxifen. Lancet. 1993;341(8858):1485–1486. doi: 10.1016/0140-6736(93)90934-9. [DOI] [PubMed] [Google Scholar]
- 8.Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2(3):205–213. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- 9.Kahsai AW, Cui J, Kaniskan HU, Garner PP, Fenteany G. Analogs of tetrahydroisoquinoline natural products that inhibit cell migration and target galectin-3 outside of its carbohydrate-binding site. J Biol Chem. 2008;283(36):24534–24545. doi: 10.1074/jbc.M800006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buckingham JB, Buckingham J. Dictionary of natural products. Vol. 9. Chapman and Hall; USA: 1996. [Google Scholar]
- 11.Erba E, Bergamaschi D, Bassano L, Damia G, Ronzoni S, Faircloth G, D’Incalci M. Ecteinascidin-743 (ET-743), a natural marine compound, with a unique mechanism of action. Eur J Cancer. 2001;37(1):97–105. doi: 10.1016/s0959-8049(00)00357-9. [DOI] [PubMed] [Google Scholar]
- 12.Chesworth R, Zawistoski MP, Lefker BA, Cameron KO, Day RF, Mangano FM, Rosati RL, Colella S, Petersen DN, Brault A, Lu B, Pan LC, Perry P, Ng O, Castleberry TA, Owen TA, Brown TA, Thompson DD, DaSilva-Jardine P. Tetrahydroisoquinolines as subtype selective estrogen agonists/antagonists. Bioorg Med Chem Lett. 2004;14(11):2729–2733. doi: 10.1016/j.bmcl.2004.03.077. [DOI] [PubMed] [Google Scholar]
- 13.Lin HR, Safo MK, Abraham DJ. Identification of a series of tetrahydroisoquinoline derivatives as potential therapeutic agents for breast cancer. Bioorg Med Chem Lett. 2007;17(9):2581–2589. doi: 10.1016/j.bmcl.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Renaud J, Bischoff SF, Buhl T, Floersheim P, Fournier B, Halleux C, Kallen J, Keller H, Schlaeppi JM, Stark W. Estrogen Receptor Modulators: Identification and structure-activity relationships of potent ERα-selective tetrahydroisoquinoline ligands. J Med Chem. 2003;46(14):2945–2957. doi: 10.1021/jm030086h. [DOI] [PubMed] [Google Scholar]
- 15.Renaud J, Bischoff SF, Buhl T, Floersheim P, Fournier B, Geiser M, Halleux C, Kallen J, Keller H, Ramage P. Selective estrogen receptor modulators with conformationally restricted side chains. Synthesis and structure-activity relationship of ERalpha-selective tetrahydroisoquinoline ligands. J Med Chem. 2005;48(2):364–379. doi: 10.1021/jm040858p. [DOI] [PubMed] [Google Scholar]
- 16.Leese MP, Jourdan F, Dohle W, Kimberley MR, Thomas MP, Bai R, Hamel E, Ferrandis E, Potter BVL. Steroidomimetic tetrahydroisoquinolines for the design of new microtubule disruptors. ACS Med Chem Lett. 2012;3(1):5–9. doi: 10.1021/ml200232c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rorke EA, Kendra KL, Katzenellenbogen BS. Relationships among uterine growth, ornithine decarboxylase activity and poly-amine levels: studies with estradiol and antiestrogens. Molecular and Cellular Endocrinology. 1984;38(1):31–38. doi: 10.1016/0303-7207(84)90142-4. [DOI] [PubMed] [Google Scholar]
- 18.Wan Z, Musa MA, Joseph P, Cooperwood JS. Synthesis and Biological Activity of 3-N-Substituted Estrogen Derivatives as Breast Cancer Agents. Mini Rev Med Chem. 2013;13(9):1381–1388. doi: 10.2174/1389557511313090012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Redda KK, Gangapuram M, Ardley TW. Synthesis of substituted 1,2,3,4-tetrahydroisoquinolines as anticancer agents. Proceedings of the 101st Annual Meeting of the American Association for Cancer Research; Washington, DC., USA. Apr 17–21, 2010; p. 735. [Google Scholar]
- 20.GALAHAD™. Distributed by Tripos Inc; 1699 S. Hanley Rd., St. Louis, MO, USA: 2009. [Google Scholar]
- 21.Clark RD, Abrahamian E, Strizhev A. A computer-implemented method for aligning flexible molecules by performing ensemble alignment in the internal coordinate space followed by rigid body alignment in Cartesian space. 20,070,143,030. U S Patent. 2007 Jun 21;
- 22.Richmond NJ, Abrams CA, Wolohan PRN, Abrahamian E, Willett P, Clark RD. GALAHAD: 1. Pharmacophore identification by hypermolecular alignment of ligands in 3D. J Comput-Aided Mol Des. 2006;20(9):567–587. doi: 10.1007/s10822-006-9082-y. [DOI] [PubMed] [Google Scholar]
- 23.SYBYL-X 1.3. Tripos International; 1699 S. Hanley Rd., St. Louis, MO, 63144, USA: 2011. [Google Scholar]
- 24.Tripos Bookshelf for SYBYL-X 1.3. Tripos Internatioanl; St. Louis, MO, 63144, USA: 2011. [Google Scholar]
- 25.Abrahamian E, Fox PC, Naerum L, Christensen IT, Thogersen H, Clark RD. Efficient generation storage and manipulation of fully flexible pharmacophore multiplets and their use in 3-D similarity searching. J Chem Inf Comput Sci. 2003;43(2):458–468. doi: 10.1021/ci025595r. [DOI] [PubMed] [Google Scholar]
- 26.Richmond NJ, Willett P, Clark RD. Alignment of three-dimensional molecules using an image recognition algorithm. J Mol Graph Model. 2004;23(2):199–209. doi: 10.1016/j.jmgm.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 27.Maestro, version 9.3. Schrödinger, LLC; New York, NY, USA: 2012. [Google Scholar]
- 28.Phase, version 3.4. Schrödinger, LLC; New York, NY, USA: 2012. [Google Scholar]
- 29.Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA. PHASE: A New Engine for Pharmacophore Perception, 3D QSAR Model Development, and 3D Database Screening. 1. Methodology and Preliminary Results. J Comput Aided Mol Des. 2006;20(10–11):647–671. doi: 10.1007/s10822-006-9087-6. [DOI] [PubMed] [Google Scholar]
- 30.ConfGen, version 2.3. Schrödinger, LLC; New York, NY, USA: 2012. [Google Scholar]
- 31.Watts KS, Dalal P, Murphy RB, Sherman W, Friesner RA, Shelley JC. ConfGen: A Conformational Search Method for Efficient Generation of Bioactive Conformers. J Chem Inf Model. 2010;50(4):534–546. doi: 10.1021/ci100015j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.