

Abstract
MicroRNAs (miRs) are small noncoding RNAs that regulate mRNA stability and/or translation. Because of their release into the circulation and their remarkable stability, miR levels in plasma and other biological fluids can serve as diagnostic and prognostic disease biomarkers. However, quantifying miRs in the circulation is challenging due to issues with sensitivity and specificity. This Letter describes for the first time the design and characterization of a regenerative, solid-state localized surface plasmon resonance (LSPR) sensor based on highly sensitive nanostructures (gold nanoprisms) that obviates the need for labels or amplification of the miRs. Our direct hybridization approach has enabled the detection of subfemtomolar concentration of miR-X (X = 21 and 10b) in human plasma in pancreatic cancer patients. Our LSPR-based measurements showed that the miR levels measured directly in patient plasma were at least 2-fold higher than following RNA extraction and quantification by reverse transcriptase-polymerase chain reaction. Through LSPR-based measurements we have shown nearly 4-fold higher concentrations of miR-10b than miR-21 in plasma of pancreatic cancer patients. We propose that our highly sensitive and selective detection approach for assaying miRs in plasma can be applied to many cancer types and disease states and should allow a rational approach for testing the utility of miRs as markers for early disease diagnosis and prognosis, which could allow for the design of effective individualized therapeutic approaches.
Keywords: Nanoprisms, localized surface plasmon resonance, label-free detection, microRNAs, pancreatic cancer, patient plasma
Pancreatic ductal adenocarcinoma (PDAC)-related deaths are a major health concern in the United States since the five-year survival rate is only 6%.1 A crucial contributor to this dismal statistic is the absence of a biomarker for early PDAC detection. Moreover, most patients with PDAC do not develop specific symptoms until the disease is quite advanced. Therefore, at clinical presentation, PDAC patients often have locally advanced and/or metastatic disease, which precludes effective therapy in the vast majority of patients. In this context microRNAs (miRs),2 which are small single-stranded, noncoding RNAs often play a major role in cell proliferation, survival, migration, invasion, and metastasis in various cancers,3−5 including PDAC.6,7 Moreover, miRs are released into the circulation, where they exhibit remarkable stability. Therefore, the development of sensitive and specific detection techniques, which precisely and quantitatively measure the concentration of miRs in their native environments such as blood or plasma, may provide a unique opportunity for developing diagnostic and prognostic markers in PDAC.6−8
Microarrays9 and quantitative reverse transcription polymerase chain reaction (qRT-PCR)10,11 assays are routinely used to detect miRs. However, these methods are semiquantitative, require sequence-based amplification and radioactive labeling steps, and suffer from cross-hybridization and invalid internal controls. Other analytical techniques such as electrochemical12,13 and fluorescence-based assays14 are also used to quantify the miRs. However, such techniques require either additional amplification or labeling, or complex electron/energy transfer processes, and cannot be performed in physiological media. A few label-free techniques such as photonic microring resonators,15 nanopores,16,17 and nanoparticle-based biobarcode gel assay18 can detect miRs associated with cancer patients. However, microring resonators suffer from low sensitivity and do not work in physiological media. Although nanopore-based sensors have shown the ability to detect miRs in the circulation of lung cancer patients, the technique requires a complicated fabrication procedure, a high probe concentration, and a specific probe signature.16 The biobarcode gel technique relies on complex sandwich-type capturing methods, uses of the toxic chemical potassium cyanide, and may not be applicable to clinically relevant patient samples.
Plasmonic nanostructures have gained significant attention because of their geometrical feature-dependent localized surface plasmon resonance (LSPR) properties,19−23 which can be further controlled by modulating their local dielectric environment.21,24 Utilizing these properties, several molecular25,26 and biological27−34 sensors have been developed where analyte binding to nanostructure surface-bound receptors results in an increase in refractive index and consequently a LSPR peak shift.35,36 In this context, it has not been possible to detect and quantify sequence specific miRs by their direct hybridization to nanostructure probes followed by monitoring the LSPR properties of nanostructures without using labeling steps. We now report for the first time the fabrication of label-free, solid-state plasmonic biosensors for miR detection. The biosensing involves the direct hybridization of PDAC-relevant miRs in plasma to their complementary single-stranded DNAs (HS-C6-ssDNA) that were functionalized on the surface of gold nanoprisms attached onto a glass substrate. This construct serves as a plasmonic biosensor through monitoring the LSPR dipole peak (λLSPR). We also demonstrate that our sensors are extremely specific in miR detection and that addition of DNA-RNA duplex cleaving enzymes regenerates the sensor, allowing for multiple uses without compromising sensing efficiency.
Fabrication of the Plasmonic Biosensor for miRs Detection
Figure 1 represents the schematic diagram of our solid-state, label-free plasmonic biosensor fabrication for miR detection. Chemically synthesized gold nanoprisms (Figure 1a), which displayed their λLSPR at ∼797 nm upon attachment to solid substrate immersed in PBS buffer, were selected as nanoantennas for our biosensor fabrication because (1) their λLSPR peak position (in the 700–900 nm wavelength range) is particularly suitable for biomolecule detection because of negligible background scattering and adsorption of endogeneous chromophores from physiological media such as blood and plasma;37,38 (2) they have strong electromagnetic (EM) field enhancement at the sharp tips;39,40 (3) they exhibits a strong LSPR response to small changes in their surrounding environment;36,41−46 (4) their atomically smooth surface allows formation of a self-assembled monolayer (SAM)47 of receptors with both a tightly packed lower layer of alkylthiols and a more loosely packed upper layer that provide the required space for duplex formation with complementary miR strands; (5) gold is nontoxic and stable under extreme physiological conditions;48 and (6) the gold–sulfur bond is very stable with thiol-modified receptors making a strong covalent bond with the gold surface. Details describing the synthesis of gold nanoprisms and their attachment onto the silanized glass substrate are provided in Supporting Information. Recently,49 we have shown that a molecular sensor fabricated using an ∼35 nm average edge-length gold nanoprisms displayed an unprecedentedly large 21 nm reversible shift upon a minor 0.6 nm increase in the thickness of the local dielectric environment. Therefore, gold nanoprisms of this size and geometry are unique and should provide extremely high sensitivity if plasmonic biosensors are fabricated using them, which is the scope of this Letter. This Letter provides the first example of LSPR-based miRs sensing in physiological media.
Figure 1.

Design of plasmonic biosensors and detecting miR-X in various physiological media. (a) Chemically synthesized and freshly prepared gold nanoprisms were covalently attached onto a 3-mercaptopropyltriethoxysilane-functionalized glass coverslip (substrate). (b) Surface of gold nanoprisms was chemically modified with a 1.0 μM 1:1 mixture of SH-C6-ssDNA-X and PEG6-SH in PBS buffer (pH 7.4) to prepare the plasmonic biosensor. (c) Incubation of sensor in miR-X solution and formation of DNA duplex. (d) Schematic of the extinction spectrum of the biosensor collected in PBS buffer after modification with a 1.0 μM 1:1 mixture of SH-C6-ssDNA-X and PEG6-SH (blue curve). The extinction spectrum was again collected after incubation in miR-X solution and careful rinsing with PBS buffer to determine the new peak position (red curve). The extent of LSPR dipole peak shift (ΔλLSPR) depends on the concentration of miR-X used during the incubation in (c), which ranged from 100 nM to 50 fM. (e) Plot of ΔλLSPR versus log of miR-X concentrations used to determine the limit of detection. The image is not to scale.
For detection and quantification, our targets were miR-21 and miR-10b, because we have shown by locked nuclei acid–based in situ hybridization that they are overexpressed in pancreatic cells (PCCs) within the tumor mass50,51 and that circulating miR-10b may serve as biomarker for diagnosis of PDAC.8 We designed the sensing strategy based on the hybridization between complementary probes (-C6-ssDNA-X, X = 21 and 10b) attached to gold nanoprisms and their target miRs. The introduction of spacers in-between the DNA probes was included to reduce steric hindrance between the probes and the miRs and therefore to enhance the hybridization and ultimately the sensitivity. As shown in Figure 1b, poly(ethylene glycol)6-thiols (PEG6-SH) were used as spacers because they avoid nonspecific adsorption of extraneous materials onto the nanoprism’s surface and are not reactive toward miRs or other biological constituents present in plasma. Previously, we demonstrated that functionalization of a nanoantenna’s surface with an equal mole ratio of receptor and spacer provided the best sensitivity and lowest limit of detection (LOD).42 Therefore, a 1:1 ratio of HS–C6-ssDNA-X:PEG6-SH was used to prepare the plasmonic biosensors (Figure 1b). All the miRs and oligonucleotides sequences used in these studies are provided in Supporting Information, Table S1.
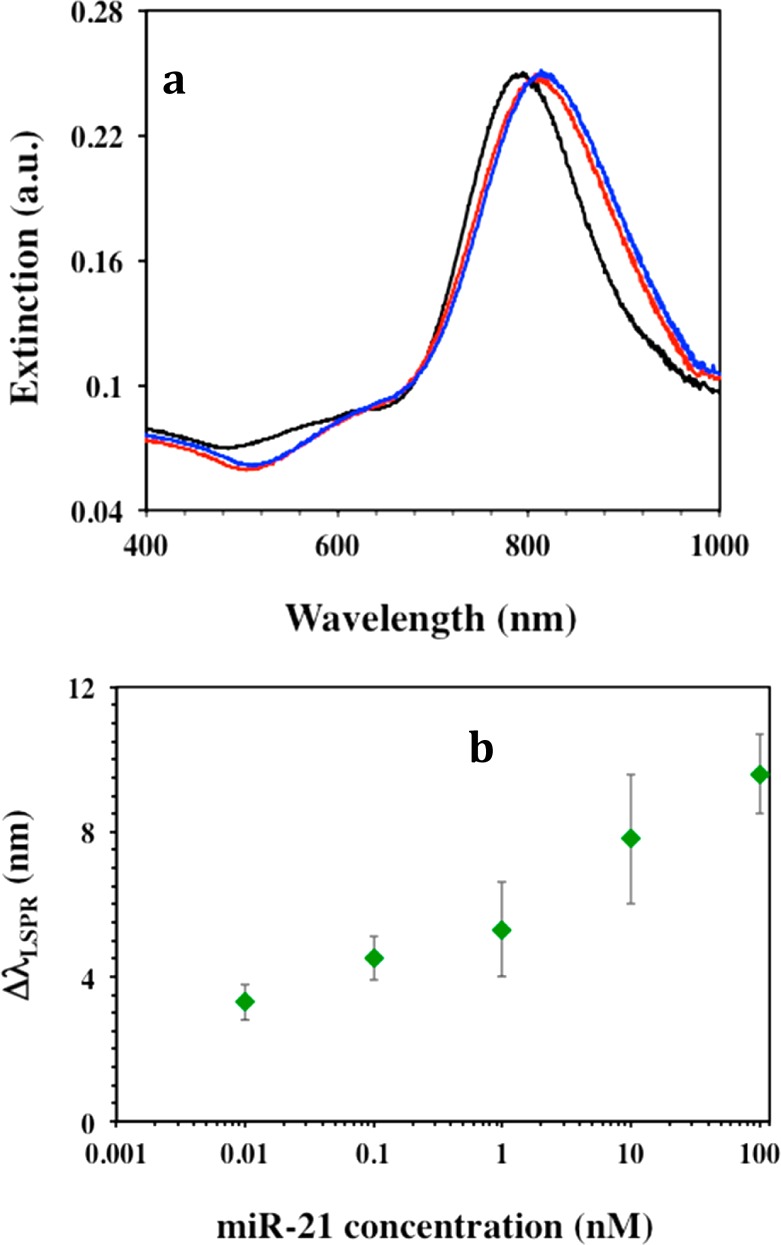
As illustrated in Figure 2a, we used UV–vis spectroscopy to monitor the changes in λLSPR of the gold nanoprisms at different functionalization steps. The functionalization of substrate-bound nanoprisms with 1:1 ratio of HS–C6-ssDNA-21:PEG6-SH resulted in an ∼20.5 ± 3.2 nm red-shift of λLSPR as a result of the increase in local refractive index, which suggested the attachment of both molecular species onto the nanoprism’s surface. These plasmonic biosensors were utilized for miR detection by incubating miR-21 (obtained from Sigma-Aldrich, U.S.A.) with concentration ranging from 100 nM to 50 fM in PBS buffer, 40% bovine plasma, or 40% human plasma. The λLSPR response of gold nanoprisms for each miR-21 concentration was measured where the highest 18.8 ± 1.9 nm λLSPR red shift was observed for 100 nM miR-21 (Figure 2a, blue) in PBS buffer. We hypothesize that the λLSPR red-shift is due to hybridization between ssDNA-21 and miR-21. It was found that the magnitude of the λLSPR shift was concentration dependent, where 50 fM miR-21 caused a 3.7 ± 0.3 nm λLSPR red shift in PBS buffer. Figure 2b illustrates the magnitude of the λLSPR shift (ΔλLSPR) upon DNA/RNA duplex formation for various miR-21 concentrations in the three different media. Evidently higher concentrations of miR-21 induced a larger number of ssDNA-21 strands to convert to DNA/RNA duplexes and consequently a larger change in the local refractive index around the nanoprisms, which results in a larger value of ΔλLSPR.
Figure 2.

MicroRNA detection using label-free plasmonic biosensors. (a) Monitoring LSPR dipole peak (λLSPR) changes by UV–visible absorption spectra of gold nanoprisms during various functionalization steps: before (black, λLSPR = 800 nm) and after functionalized with a 1 μM/1 μM ratio of HS-C6-ssDNA-21/PEG6-SH (red, λLSPR = 821 nm), and after incubation with 100 nM miR-21 solution in PBS buffer (blue, λLSPR = 839 nm). (b) average λLSPR peak shift (ΔλLSPR) of gold nanoprisms functionalized with a 1 μM/1 μM ratio of HS-C6-ssDNA-21/PEG6-SH after incubation in different concentrations of miR-21 in PBS buffer (red triangles), 40% human plasma (black squares), and 40% bovine plasma (blue diamonds). The ΔλLSPR were calculated by taking the difference between the λLSPR peak position of the plasmonic biosensor after and before the hybridization with miR-21 in the various media. (c) Average ΔλLSPR of the plasmonic biosensor functionalized with a 1 μM/1 μM ratio of HS-C6-ssDNA-10b/PEG6-SH after hybridization with different miR-10b concentrations in PBS buffer (red triangles), in 40% human plasma (black squares), and in 40% bovine plasma (blue diamonds). All extinction spectra recorded after miR-X incubation were measured in PBS buffer after rinsing with PBS buffer.
The highest red shift of 18.8 ± 1.9 nm we observed for 100 nM miR-21 incubation with our plasmonic biosensor is significant considering only an ∼5% change in the refractive index upon ssDNA/RNA duplex formation.52 We believe such a high sensitivity of our plasmonic biosensors is because of the unique LSPR properties of our gold nanoprisms and the possibility of electron delocalization as the ssDNA forms the duplex and becomes double-strand. The atomically flat surface, extremely small height (∼8 nm), and sharp tips of our nanoprisms display strong EM field enhancement near their surface and therefore are expected to be extremely sensitive to small changes of their local dielectric environment.35,40 Moreover, transformation of ssDNA into double-strand DNA significantly changes the refractive index because of the high charge density and polarizability of the DNAs.52 The duplex DNA is capable of long-range charge transfer and alters the electron density around the nanoprisms thus influencing their LSPR properties. This interesting phenomenon requires further scientific study, which is currently under our investigation.
Our sensing mechanism is based on the hypothesis that the attachment of complementary target miRs to our plasmonic biosensor will shift the λLSPR to higher wavelength (Figure 1c). The total shift (ΔλLSPR) depended on the miR concentration (Figure 1d) and could be used to determine the limit of detection (LOD) (Figure 1e). The LODs calculated for miR-21 in three different media were found to be in the range of 23–35 fM, which was more than 1000 and 3 fold lower than with the label-free microring resonator (150 fmol)15 and the nanopore based (100 fM)16 miR sensors, respectively. Importantly, these techniques detected miRs in PBS buffer whereas we have demonstrated here for the first time a sensing approach in physiological media. Utilizing our same direct hybridization-based detection approach, plasmonic biosensors were constructed with of −C6-ssDNA-10, while keeping other parameters constant. The LOD for miR-10b in the above media was determined over a concentration range from 100 nM to 50 fM. The average ΔλLSPR and LODs for miR-10b in three diverse media are shown in Figure 2c.
The principle underlying the actions of plasmonic biosensors is based on the successful hybridization between miRs and ssDNA attached to nanoprisms, where a higher number of duplex formations will result in a larger change in the refractive index surrounding the nanoprisms resulting in larger ΔλLSPR and higher sensitivity. Therefore, it would be expected that functionalization of gold nanoprisms with 100% HS–C6-ssDNA-X (without the PEG6-thiol spacer) should reduce the LOD values because of steric hindrance and low attachment of miRs. To investigate this, gold nanoprisms were functionalized with 100% −C6-ssDNA-21 resulting in an ∼15.0 ± 1.8 nm λLSPR red shift (Figure 3a). The sensor was then incubated in different concentrations of miR-21 prepared in 40% human plasma. As illustrated in Figure 3a, an ∼9.6 ± 1.1 nm red shift was observed for a 100 nM miR-21 concentrations and the lowest concentration that can be repeatedly detected was 10 pM from a ΔλLSPR of 3.4 ± 0.5 nm. Figure 3b shows the ΔλLSPR versus concentration plot. Evidently, functionalization of the nanoprism’s surface with 100% −C6-ssDNA-21 resulted in a 200-fold increase in detection limit in comparison to the 1:1 ratio −C6-ssDNA-21/PEG6-SH mixed functionalization (Supporting Information Table S2). These experimental data further highlight our rationale for using spacers that increase the likelihood of hybridization. We believe fully covered gold nanoprisms were obtained when 100% −C6-ssDNA-21 was used for functionalization, which creates steric hindrance and does not allow the maximum number of miR-21 strands to come into close proximity with −C6-ssDNA-21 for hybridization. Therefore, not all the −C6-ssDNA-21 attached on the gold nanoprisms’ surface was hybridized with miR-21 strands resulting in low sensing response. Thus, if we introduce a spacer between the −C6-ssDNA-21, it will allow the maximum -ssDNA-21 strands to be freely available for hybridization without any interference and ultimately enhance the sensitivity of the biosensor. Accordingly, for the remaining of our investigation, we used a 1:1 mixed −C6-ssDNA-X:PEG6-SH to functionalize the gold nanoprisms.
Figure 3.
Determining the optimum detection condition. (a) UV–visible extinction spectra monitoring the LSPR dipole peak (λLSPR) of gold nanoprisms attached to silanized glass substrate before (black, λLSPR = 796 nm) and after (red, λLSPR = 811 nm) functionalization with 1 μM of HS-C6-ssDNA-21 without PEG6-SH spacers and after incubation in 100 nM miR-21 solution in 40% human plasma (blue, λLSPR = 822 nm). (b) Average ΔλLSPR of these HS-C6-ssDNA-21 functionalized gold nanoprisms upon hybridization with different miR-21 concentrations in 40% human plasma. The ΔλLSPR were calculated by taking the difference between the λLSPR peak position of the nanoprisms after and before the incubation with miR-21 in PBS buffer.
In order to confirm the hybridization of miR-X with −C6-ssDNA-X that resulted in the ΔλLSPR, the enzyme RNase H was used to selectively cleave the DNA: RNA duplex and potentially reverse the ΔλLSPR. Initially, the plasmonic biosensor for miR-21 was incubated in a 100 nM solution of miR-21, which resulted in red-shift of λLSPR potentially reflecting hybridization. The biosensor was then immersed in 15 units of RNase H solution for 2 h. Afterward the λLSPR showed a blue shift and reverted back to its original position before miR-21 incubation (Figure 4a). When the 1:1 ratio −C6-ssDNA-21:PEG6-SH mixed functionalized biosensor was incubated with RNase H solution alone overnight, no noticeable change in λLSPR value was observed (Supporting Information Figure S7). These experimental results validate our previous observation that the λLSPR blue shift was due to the cleavage of heteroduplex done by RNase H. The biosensors were rinsed with RNase free water to PBS buffer and again incubated in 100 nM miR-21 solution for rehybridization where an ∼14 nm red shift of the λLSPR was observed. These experiments confirm our working hypothesis that hybridization between the nanoprism’s surface ligands (−C6-ssDNA-X) and the miR-X resulted in changes in the local dielectric environment around the nanoprisms causing wavelength shift. As shown in Figure 4b, the λLSPR responses were identical for several cycles due to hybridization and dehybridization of miR-21 over a period of 6 days. The same experiments were done for the miR-10b biosensor and similar results were observed, underscoring the long-term stability of the sensors and their potential for being developed into cost-effective point of care diagnostic tools.
Figure 4.
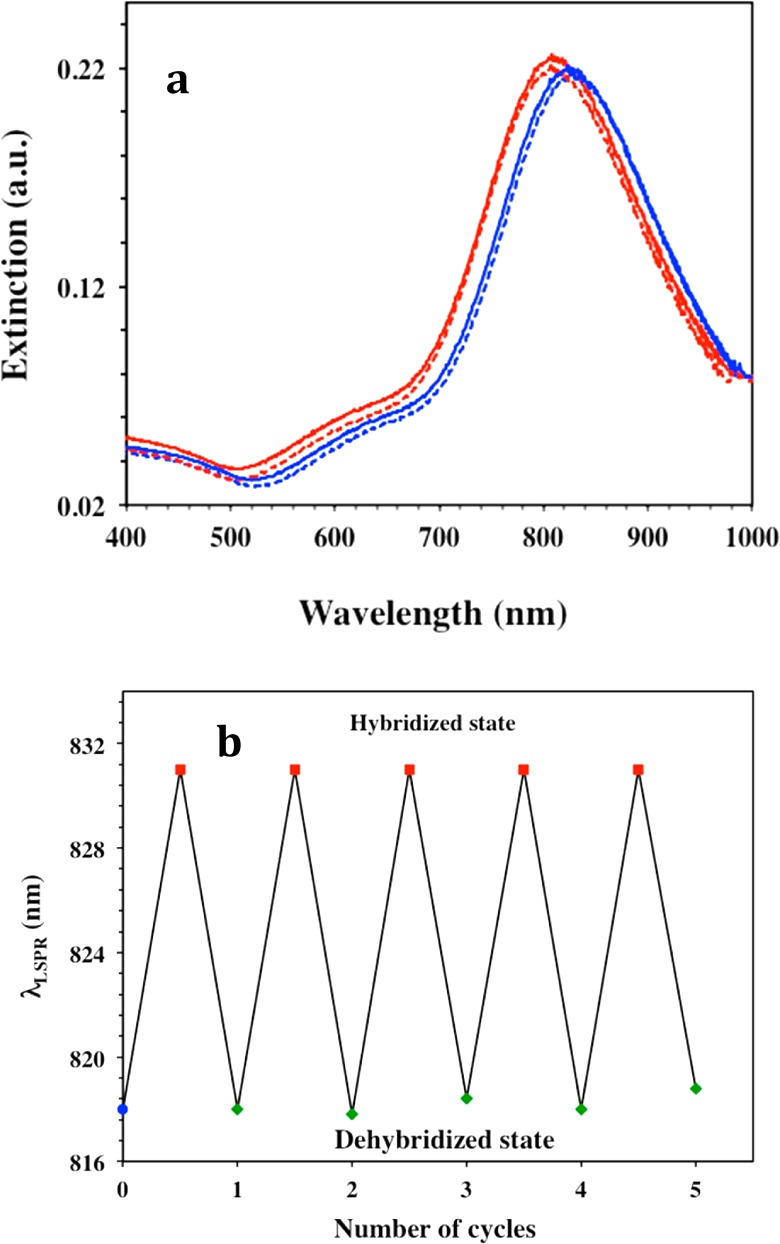
Characterization of the plasmonic biosensors regeneration ability. (a) UV–visible extinction spectra of gold nanoprisms functionalized with a 1 μM/1 μM ratio of HS-C6-ssDNA-21/PEG6-SH (red, λLSPR = 818 nm) attached to silanized glass, after incubation with 100 nM of miR-21 (blue, λLSPR = 832 nm), after treatment with 15 units of RNase H for 2 h (red dotted, λLSPR = 818 nm), and after rehybridized with 100 nM of miR-21 (blue dotted, λLSPR = 832 nm). (b) Changes in LSPR dipole peak position (λLSPR) of gold nanoprisms functionalized with a 1 μM/1 μM ratio of HS-C6-ssDNA-21/PEG6-SH upon hybridization and dehybridization with miR-21 for several cycles. The λLSPR peak shifts back and forth upon sensor regeneration with RNase H by cleaving DNA/RNA duplex and rehybridization after incubation into 100 nM miR-21 in 40% human plasma. After each of the dehybridization steps, the plasmonic biosensors were thoroughly rinsed with PBS buffer to completely remove enzyme RNase H.
The hybridization takes place at the 5′ end of −C6-ssDNA-21 and the 3′ end of miR-21, which evidently increased the refractive index. Additionally such hybridization would also increase the thickness of the local dielectric environment of the nanoprisms. Together, a significantly large ΔλLSPR was generated for both miR-21 and miR-10b. Atomic force microscopy (AFM) analysis was conducted to characterize our plasmonic biosensors and also to verify the change in surface area caused by miR-21 incubation with mixed −C6-ssDNA-21 and PEG6-SH-functioanlized gold nanoprisms. After analyzing 40 different nanoprisms (Figure 5 and Supporting Information Figure S8) – the exact same area of four different sections of the sensor - an average 2.4 × 10–15 m2 increase in surface area was observed by AFM. Thus, attachment of miRs to plasmonic biosensors has increased the thickness of local dielectric environment around the gold nanoprisms and influenced their LSPR properties. Ultrasensitive refractive index-induced LSPR response of nanoprisms allows us to fabricate label-free plasmonic biosensor.
Figure 5.

Surface characterization of the plasmonic biosensors. Atomic force microscopy images of gold nanoprisms bound to silanized glass substrate (a) after functionalization with 1:1 ratio of HS-C6-ssDNA-21/PEG6-SH (b) and after hybridization with100 nM miR-21 in 40% human plasma (c). The measurements were conducted in air. (d) Changes in the surface area of gold nanoprisms after each functionalization steps. Forty nanoprisms were selected to determine the average change in the surface area. Detailed method of surface area calculation is provided in the Supporting Information file.
The successful implementation of plasmonic biosensors for use with real biological samples mandates documentation of their specificity toward target miRs in that patient samples containing multiple miR species. The mix functionalized (−C6-ssDNA-21 and PEG6-SH) biosensors were incubated overnight in 40% human plasma solution containing 100 nM each of miR-16, miR-122, miR-126, and miR-141 because these miRs are commonly present in human plasma. The λLSPR response was measured before and after incubation (Supporting Information Figure S9) and resulted in an ∼2.5 ± 0.3 nm λLSPR red shift, which is within the instrument noise level and/or minor nonspecific adsorption of extraneous materials present in human plasma. In another control experiment, gold nanoprisms attached as before to glass substrate were functionalized with 100% PEG6-SH by incubation in a 1 μM aqueous solution, and after rinsing with large amounts of water, incubated in a 40% human plasma solution of 100 nM miR-21 for 12 h. This procedure resulted in only an ∼0.9 ± 0.7 nm λLSPR red shift (Supporting Information Figure S10), confirming that the plasmonic biosensors we designed are highly specific toward the target miRs.
Detection of miR Levels in in Plasma from Pancreatic Cancer Patients
Pancreatic cancer is the fourth-leading cause of cancer death in the United States with an annual mortality of nearly 40 000 and a dismal five-year survival rate of 6%.1 PDAC is characterized by chemotherapeutic resistance and by the absence of an effective screening procedure for early disease. It is generally accepted that early diagnosis could reduce mortality rates substantially and thus a noninvasive early PDAC test must be developed. Several miRs (such as miR-21, -10b, -103, -155, -196a, 210, and -221) were found to be overexpressed in PDAC.6−8,53 Given their resistance to degradation, plasma miRs have the potential to serve as biomarkers for the noninvasive diagnosis of PDAC. Previously, nanopore sensors were used to detect miRs in lung cancer patients, but to the best of our knowledge no sensors have been developed to date to detect PDAC-related miRs in human plasma.
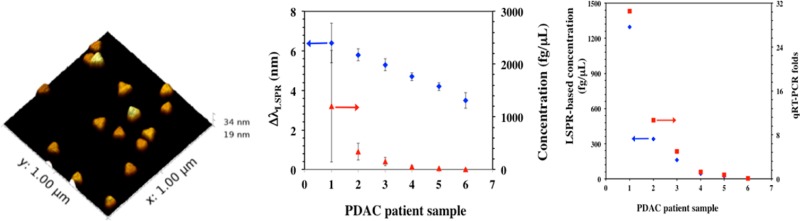
Utilizing our plasmonic biosensors we detected miR-21 and miR-10b in plasma from PDAC patients. Plasma samples were collected from six patients and six normal control subjects. Total plasma RNAs including miRs were extracted from 100 μL of each plasma sample using a TRIZOL kit with a final elution volume of 28 μL. Next, 14 μL volumes were used for miR quantification by the plasmonic biosensor and the remaining 14 μL were used in the qRT-PCR assay. The plasmonic biosensors were fabricated as described before for both miR-21 and miR-10b detection. The extracted human miR-21 or miR-10b samples were diluted in PBS buffer and incubated with the biosensors were for 12 h, followed by rinsing with PBS buffer and measurement of the λLSPR response in PBS buffer. The observed λLSPR shift for each miR-21 and miR-10b sample was converted into the corresponding concentration using the calibration curve derived for miR-21 or 10b under optimized conditions and compared with the value from normal control subjects (Figure 6a,c). The concentrations of miR-21 and miR-10b determined from plasmonic biosensors were also compared with the values obtained from the qRT-PCR assay (Figure 6b and Supporting Information Figure S16). Importantly, for the first time through a label-free technique we have shown that miR-10b concentration is nearly 4-fold higher than the miR-21 level in patient samples. Inasmuch as both miR-21 and miR-10b are overexpressed in PDAC, it is possible that miR-10b is released more efficiently by pancreatic cancer cells than miR-21, allowing it to achieve higher levels in the circulation. It is therefore possible that miR-10b levels are also increased within the pancreatic tumor microenvironment, where it could be acting to enhance PDAC biological aggressiveness.
Figure 6.

Determination of microRNA concentration in PDAC patients and normal control subjects. (a) The average λLSPR peak shifts of gold nanoprisms functionalized with a 1:1 ratio of HS-C6-ssDNA-21/PEG6-SH upon hybridization with miR-21 from the total RNAs extracted from plasma samples of PDAC patients (blue diamonds) and normal control subjects (blue squares). The respective λLSPR peak shifts were converted to concentrations using the calibration curve established for miR-21 in PBS buffer as shown in Figure 2b [PDAC patients (red triangles), and for normal control subjects (red circles)]. (b) Comparison of miR-21 concentration for six PDAC patients determined using plasmonic biosensors (blue diamond) and qRT-PCR (red square). (c) Similar experiments were conducted to detect miR-10b where the λLSPR peak shifts and concentrations for PDAC patients are shown in blue diamonds and red triangles, respectively. The λLSPR peak shifts (blue squares) and concentrations (red circles) for normal controls are shown for comparison. (d) The average λLSPR peak shifts (blue diamonds) and concentration (red triangles) for the miR-21 in plasma samples from PDAC patients without any purification.
We also detected miR-21 levels directly in human plasma samples collected from PDAC patients without RNAs extraction. Thus, human plasma (50 μL/sample) from six pancreatic cancer patients were diluted in PBS buffer followed by incubation with our plasmonic biosensors for 12 h. The λLSPR response of each sample was measured through UV–vis spectroscopy and showed a steady increase in concentration from sample 6 to 1 (Figure 6d). Both plasmonic biosensor and qRT-PCR results indicated that miR-10b levels were higher in PDAC patients compared to normal control subjects and that the levels of miR-21 and miR-10b can be quantified with high accuracy using our gold nanoprism-based plasmonic biosensor without any modification, amplification, or labeling. Importantly, the miR-21 concentration in extracted samples was at least 2-fold lower than in the pure plasma samples. We believe this is due to loss of miRs during the RNA extraction process, which requires multiple steps for RNA purification. Therefore, the most widely used qRT-PCR method to determine the concentration of miRs in patients may not accurately represent the actual concentration. This limitation and imprecise quantification can be avoided by using our newly developed plasmonic biosensors, which can provide a unique opportunity as potential diagnostic and prognostic markers in PDAC, other cancers, and potentially other disease states.
Conclusion
We have designed, fabricated, and characterized a plasmonic biosensor that was able to detect PDAC relevant miRs in human plasma without using RNAs extraction, which opens a new avenue for the direct detection and quantification of miR levels in clinical samples without any form of sample preparation. To our knowledge, this is the first LSPR-based, label-free, direct hybridization method for miR detection, which eliminates all the current drawbacks such as labeling, tagging, amplification, use of highly toxic chemical, and further modification of the sensor. Furthermore, it vastly simplifies the detection approach without requiring detailed knowledge of the electron or energy transfer processes involved as in other more complicated techniques. Additionally, this ultrasensitive, plasmonic-based, direct hybridization-controlled detection approach is applicable to any type of miRs that are relevant to various diseases. It was found that our plasmonic biosensor can be regenerated through several cycles and is stable for several days without compromising its sensitivity and selectivity, which should enable the development of simple, cost-effective tools for the early detection of miRs and thus facilitate the early diagnosis of various cancers. Finally, the large EM-field enhancement at the nanoprism’s sharp tips39 will enhance the Raman scattering intensity of the analytes. In theory, therefore, nanoprisms can be used to design effective substrates for surface-enhanced Raman spectroscopy-based54−57 detection and quantification of multiple miRs simultaneously through integration of their spectral characteristic with the λLSPR shifts.
Materials and Methods
All synthetic DNA probes and microRNAs were purchased from Sigma-Aldrich (U.S.A.). PBS buffer prepared with RNase-free water was used to dilute oligonucleotides and miRs solutions. Patient plasma was obtained from the Indiana University Simon Cancer Center Solid Tissue Bank (Indianapolis, Indiana).
Fabrication of LSPR-Based miR Sensors and Detection
The gold nanoprism-based miR sensors were designed using our published procedure with modification.49 The attachment of gold nanoprisms on silanized glass substrates is described in the Supporting Information file. The substrate-bound nanoprisms were incubated in PBS buffer solution containing 1 μM each of HS–C6-ssDNA-X and PEG6-SH overnight and rinsed with PBS buffer. The initial LSPR peak position of each sensing platform was determined using UV–visible spectroscopy in PBS buffer and then was incubated in the different concentrations of miR solutions, for exmaple, either in PBS buffer, 40% bovine plasma, or 40% human plasma for 12 h at room temperature. The plasmonic biosensors were thoroughly washed with PBS buffer to remove any nonspecifically adsorbed species. The miR bound biosensor was then placed in PBS buffer for 10 min before the LSPR peak position was determined. For UV–vis extinction spectra measurement, one particular solvent was chosen to avoid the solvent dielectric constant effect, which is known to shift the LSPR peak.21,58,59
Total RNA Extraction and Quantification of MicroRNA by qRT-PCR
Total RNA was isolated from plasma samples that were obtained from the Indiana University Simon Cancer Center Solid Tissue Bank (Indianapolis, IN, U.S.A.) using Trizol-LS reagent (Life Technologies, Carlsbad, CA, U.S.A.). cDNA was generated using 10 ng of RNA and miR-10b, miR-21, or miR-425–5p RT primers and a miR reverse transcription kit (Life Technologies) as per the manufacturer’s recommendations. Quantitative PCR (qPCR) was performed using Taqman miR expression assay reagents. Expression levels as determined by qPCR were normalized to miR-425-5p, since this miR was expressed at similar levels in all samples and exhibited <1 cycle threshold (Ct) difference across all samples. After normalization to miR-425-5p (ΔCt), the ΔCt values for miRs in controls were averaged and subtracted from the ΔCt values of each individual sample (ΔΔCt). miR levels were then calculated using the 2–ΔΔCt method.60
Acknowledgments
The authors would like to thank B. Muhoberac and S. Savant for invaluable advice regarding the manuscript and K. Lawrence for providing PEG6-thiol. We also thank the IUSCC Cancer Center at Indiana University School of Medicine for the use of the Tissue Procurement and Distribution Core, which provided patient and control plasma samples for microRNA assays. The research was partially supported by Indiana University-Purdue University Indianapolis startup funds (R.S), Purdue Research Foundation (R.S), the Indiana Economic Development Fund (IEDF) and the National Institute of Health Grant R37-CA-075059 (M.K). The authors also thank Amy Trotch (from Affymetrix) for providing RNase H sample and the Wallace lab (IUPUI) for the use of the AFM instrument. S.M. thanks ACS Project SEED program for financial support.
Supporting Information Available
Experimental detail of nanoprisms synthesis, fabrication of plasmonic biosensors, and additional UV–visible spectra, AFM images, and histograms. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
G.K.J. designed and performed the LSPR sensing experiments and collected and analyzed the LSPR data. S.D-M. designed and performed the qRT-PCR experiments, analyzed the data, and processed patient samples. M.J. and S.M performed AFM characterizations and LSPR measurements, respectively. M.K. analyzed patient samples. R.S. conceived the original idea and designed the LSPR experiments. G.K.J., M.K., and R.S wrote the manuscript and other authors commented on it.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Siegel R.; Ma J.; Zou Z.; Jemal A. Ca: Cancer J. Clin. 2014, 64, 9–29. [DOI] [PubMed] [Google Scholar]
- Bartel D. P. Cell 2004, 116, 281–297. [DOI] [PubMed] [Google Scholar]
- Winter J.; Jung S.; Keller S.; Gregory R. I.; Diederichs S. Nat. Cell Biol. 2009, 11, 228–234. [DOI] [PubMed] [Google Scholar]
- Esquela-Kerscher A.; Slack F. J. Nat. Rev. Cancer 2006, 6, 259–269. [DOI] [PubMed] [Google Scholar]
- He L.; Thomson J. M.; Hemann M. T.; Hernando-Monge E.; Mu D.; Goodson S.; Powers S.; Cordon-Cardo C.; Lowe S. W.; Hannon G. J.; Hammond S. M. Nature 2005, 435, 828–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szafranska A. E.; Doleshal M.; Edmunds H. S.; Gordon S.; Luttges J.; Munding J. B.; Barth R. J.; Gutmann E. J.; Suriawinata A. A.; Marc Pipas J.; Tannapfel A.; Korc M.; Hahn S. A.; Labourier E.; Tsongalis G. J. Clin. Chem. 2008, 54, 1716–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setoyama T.; Zhang X.; Natsugoe S.; Calin G. A. Clin. Cancer Res. 2011, 17, 5527–5529. [DOI] [PubMed] [Google Scholar]
- Ouyang H.; Gore J.; Deitz S.; Korc M. Oncogene 2014, 33, 4664–4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S.; Lee H. J.; Wark A. W.; Corn R. M. J. Am. Chem. Soc. 2006, 128, 14044–14046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Yao B.; Huang H.; Wang Z.; Sun C.; Fan Y.; Chang Q.; Li S.; Wang X.; Xi J. Anal. Chem. 2009, 81, 5446–5451. [DOI] [PubMed] [Google Scholar]
- Chen C.; Ridzon D. A.; Broomer A. J.; Zhou Z.; Lee D. H.; Nguyen J. T.; Barbisin M.; Xu N. L.; Mahuvakar V. R.; Andersen M. R.; Lao K. Q.; Livak K. J.; Guegler K. J. Nucleic Acids Res. 2005, 33, e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B. N.; Mutharasan R. Anal. Chem. 2012, 84, 10426–10436. [DOI] [PubMed] [Google Scholar]
- Ramnani P.; Gao Y.; Ozsoz M.; Mulchandani A. Anal. Chem. 2013, 85, 8061–8064. [DOI] [PubMed] [Google Scholar]
- Cissell K. A.; Rahimi Y.; Shrestha S.; Hunt E. A.; Deo S. K. Anal. Chem. 2008, 80, 2319–2325. [DOI] [PubMed] [Google Scholar]
- Qavi A. J.; Bailey R. C. Angew. Chem., Int. Ed. 2010, 49, 4608–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zheng D.; Tan Q.; Wang M. X.; Gu L.-Q. Nat. Nanotechnol. 2011, 6, 668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanunu M.; Dadosh T.; Ray V.; Jin J.; McReynolds L.; Drndic M. Nat. Nanotechnol. 2010, 5, 807–814. [DOI] [PubMed] [Google Scholar]
- Lee H.; Park J.-E.; Nam J.-M. Nat. Commun. 2014, 5, 1–7. [Google Scholar]
- Jain P. K.; Huang X.; El-Sayed I. H.; El-Sayed M. A. Acc. Chem. Res. 2008, 41, 1578–1586. [DOI] [PubMed] [Google Scholar]
- Halas N. J.; Lal S.; Chang W.-S.; Link S.; Nordlander P. Chem. Rev. 2011, 111, 3913–3961. [DOI] [PubMed] [Google Scholar]
- Mayer K. M.; Hafner J. H. Chem. Rev. 2011, 111, 3828–3857. [DOI] [PubMed] [Google Scholar]
- Jin R.; Cao Y.; Mirkin C. A.; Kelly K. L.; Schatz G. C.; Zheng J. G. Science 2001, 294, 1901–1903. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Xia Y. Science 2002, 298, 2176–2179. [DOI] [PubMed] [Google Scholar]
- Anker J. N.; Hall W. P.; Lyandres O.; Shah N. C.; Zhao J.; Van Duyne R. P. Nat. Mater. 2008, 7, 442–453. [DOI] [PubMed] [Google Scholar]
- Weiss P. S. Acc. Chem. Res. 2008, 41, 1772–1781. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Chen J. I. L.; Ginger D. S. Nano Lett. 2012, 12, 2530–2536. [DOI] [PubMed] [Google Scholar]
- Saha K.; Agasti S. S.; Kim C.; Li X.; Rotello V. M. Chem. Rev. 2012, 112, 2739–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart M. E.; Anderton C. R.; Thompson L. B.; Maria J.; Gray S. K.; Rogers J. A.; Nuzzo R. G. Chem. Rev. 2008, 108, 494–521. [DOI] [PubMed] [Google Scholar]
- Beeram S. R.; Zamborini F. P. J. Am. Chem. Soc. 2009, 131, 11689–11691. [DOI] [PubMed] [Google Scholar]
- Chen S.; Svedendahl M.; Duyne R. P. V.; Käll M. Nano Lett. 2011, 11, 1826–1830. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Munechika K.; Ginger D. S. Nano Lett. 2007, 7, 690–696. [DOI] [PubMed] [Google Scholar]
- Haes A. J.; Hall W. P.; Chang L.; Klein W. L.; Van Duyne R. P. Nano Lett. 2004, 4, 1029–1034. [Google Scholar]
- Stranahan S. M.; Willets K. A. Nano Lett. 2010, 10, 3777–3784. [DOI] [PubMed] [Google Scholar]
- Elghanian R.; Storhoff J. J.; Mucic R. C.; Letsinger R. L.; Mirkin C. A. Science 1997, 277, 1078–1081. [DOI] [PubMed] [Google Scholar]
- Willets K. A.; Van Duyne R. P. Annu. Rev. Phys. Chem. 2007, 58, 267–297. [DOI] [PubMed] [Google Scholar]
- Malinsky M. D.; Kelly K. L.; Schatz G. C.; Van Duyne R. P. J. Am. Chem. Soc. 2001, 123, 1471–1482. [Google Scholar]
- Nusz G. J.; Curry A. C.; Marinakos S. M.; Wax A.; Chilkoti A. ACS Nano 2009, 3, 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson J. B.; Westcott S. L.; Hirsch L. R.; West J. L.; Halas N. J. Appl. Phys. Lett. 2003, 82, 257–259. [Google Scholar]
- Hao E.; Schatz G. C. J. Chem. Phys. 2004, 120, 357–366. [DOI] [PubMed] [Google Scholar]
- Sherry L. J.; Jin R.; Mirkin C. A.; Schatz G. C.; Van Duyne R. P. Nano Lett. 2006, 6, 2060–2065. [DOI] [PubMed] [Google Scholar]
- Chen H.; Shao L.; Woo K. C.; Ming T.; Lin H.-Q.; Wang J. J. Phys. Chem. C 2009, 113, 17691–17697. [Google Scholar]
- Joshi G. K.; McClory P. J.; Muhoberac B. B.; Kumbhar A.; Smith K. A.; Sardar R. J. Phys. Chem. C 2012, 116, 20990–21000. [Google Scholar]
- Joshi G. K.; McClory P. J.; Dolai S.; Sardar R. J. Mater. Chem. 2012, 22, 923–931. [Google Scholar]
- Haes A. J.; Zou S.; Zhao J.; Schatz G. C.; Van Duyne R. P. J. Am. Chem. Soc. 2006, 128, 10905–10914. [DOI] [PubMed] [Google Scholar]
- Hall W. P.; Modica J.; Anker J.; Lin Y.; Mrksich M.; Van Duyne R. P. Nano Lett. 2011, 11, 1098–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland A. D.; Van Duyne R. P. Nano Lett. 2003, 3, 1057–1062. [Google Scholar]
- Bain C. D.; Whitesides G. M. J. Am. Chem. Soc. 1989, 111, 7164–7175. [Google Scholar]
- Murphy C. J.; Gole A. M.; Stone J. W.; Sisco P. N.; Alkilany A. M.; Goldsmith E. C.; Baxter S. C. Acc. Chem. Res. 2008, 41, 1721–1730. [DOI] [PubMed] [Google Scholar]
- Joshi G. K.; Blodgett K. N.; Muhoberac B. B.; Johnson M. A.; Smith K. A.; Sardar R. Nano Lett. 2014, 14, 532–540. [DOI] [PubMed] [Google Scholar]
- Preis M.; Gardner T. B.; Gordon S. R.; Pipas J. M.; Mackenzie T. A.; Klein E. E.; Longnecker D. S.; Gutmann E. J.; Sempere L. F.; Korc M. Clin. Cancer Res. 2011, 17, 5812–5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sempere L. F.; Preis M.; Yezefski T.; Ouyang H.; Suriawinata A. A.; Silahtaroglu A.; Conejo-Garcia J. R.; Kauppinen S.; Wells W.; Korc M. Clin. Cancer Res. 2010, 16, 4246–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhadj S.; Singh G.; Saraf R. F. Langmuir 2004, 20, 5539–5543. [DOI] [PubMed] [Google Scholar]
- Greither T.; Grochola L. F.; Udelnow A.; Lautenschläger C.; Würl P.; Taubert H. Int. J. Cancer 2010, 126, 73–80. [DOI] [PubMed] [Google Scholar]
- Stiles P. L.; Dieringer J. A.; Shah N. C.; Van Duyne R. P. Annu. Rev. Anal. Chem. 2008, 1, 601–626. [DOI] [PubMed] [Google Scholar]
- Moskovits M. J. Raman Spectrosc. 2005, 36, 485–496. [Google Scholar]
- Singh A. K.; Khan S. A.; Fan Z.; Demeritte T.; Senapati D.; Kanchanapally R.; Ray P. C. J. Am. Chem. Soc. 2012, 134, 8662–8669. [DOI] [PubMed] [Google Scholar]
- Jackson J. B.; Halas N. J. Proc. Nat. Acad. Sci. U.S.A. 2004, 101, 17930–17935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otte M. A.; Sepulveda B.; Ni W.; Juste J. P.; Liz-Marzan L. M.; Lechuga L. M. ACS Nano 2009, 4, 349–357. [DOI] [PubMed] [Google Scholar]
- Novo C.; Funston A. M.; Mulvaney P. Nat. Nanotechnol. 2008, 3, 598–602. [DOI] [PubMed] [Google Scholar]
- Livak K. J.; Schmittgen T. D. Methods 2001, 25, 402–408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.