

Abstract
Objectives
The mechanism by which atheroma plaque becomes unstable is not completely understood to date but analysis of differentially expressed genes in stable versus unstable plaques may provide clues. This will be crucial toward disclosing the mechanistic basis of plaque instability, and may help to identify prognostic biomarkers for ischaemic events. The objective of our study was to identify differences in expression levels of 59 selected genes between symptomatic patients (unstable plaques) and asymptomatic patients (stable plaques).
Methods
80 carotid plaques obtained by carotid endarterectomy and classified as symptomatic (>70% stenosis) or asymptomatic (>80% stenosis) were used in this study. The expression levels of 59 genes were quantified by qPCR on RNA extracted from the carotid plaques obtained by endarterectomy and analyzed by means of various bioinformatic tools.
Results
Several genes associated with autophagy pathways displayed differential expression levels between asymptomatic and symptomatic (i.e. MAP1LC3B, RAB24, EVA1A). In particular, mRNA levels of MAP1LC3B, an autophagic marker, showed a 5−fold decrease in symptomatic samples, which was confirmed in protein blots. Immune system−related factors and endoplasmic reticulum-associated markers (i.e. ERP27, ITPR1, ERO1LB, TIMP1, IL12B) emerged as differently expressed genes between asymptomatic and symptomatic patients.
Conclusions
Carotid atherosclerotic plaques in which MAP1LC3B is underexpressed would not be able to benefit from MAP1LC3B−associated autophagy. This may lead to accumulation of dead cells at lesion site with subsequent plaque destabilization leading to cerebrovascular events. Identified biomarkers and network interactions may represent novel targets for development of treatments against plaque destabilization and thus for the prevention of cerebrovascular events.
Introduction
Atherosclerosis in the carotid artery is the second leading cause of death and the third cause of disability-adjusted life-years worldwide [1], [2]. Carotid atherosclerosis is a disorder with an important inflammatory component and is considered a risk factor for developing a cerebrovascular accident. A high stenosis grade is a risk factor for a cerebrovascular event but, since it is known that a percentage of patients with high stenosis will present asymptomatic plaques [3], stenosis alone is not sufficient for identification of patients at risk. In contrast, plaques from symptomatic patients are more likely to become unstable and predisposed to rupture [4]. The rupture and destabilization of the plaque in the carotid artery can lead to an ischemic attack [5]. However, the precise mechanisms by which atheroma plaque becomes unstable [6] are still unknown.
Several clinical and pathological studies have revealed specific gene expression biomarkers associated with plaque rupture among symptomatic patients. For instance, matrix metalloproteinase−1 (MMP1) and MMP12, and CD163 and HO−1 have been identified as potential indicators of carotid plaque instability [7], [8]. In addition, ADAMDEC1, MMP9 and legumain genes have been described as over−expressed genes in unstable areas of carotid plaques when compared with stable areas of the same plaque [9]. More recently, IL17A has also been associated with vulnerability of the atheroma plaque [10], while a microarray-based study comparing gene expression levels between symptomatic and asymptomatic patients identified ten genes with significant differences between the two groups [11]. Thus, even if various genes have been suggested to play a role in plaque destabilization, further studies are needed to gain a more comprehensive understanding of the process.
The aim of this study was to perform an extended candidate gene expression analysis in a collection of 80 atheroma sample collection both to identify novel biomarkers and to validate previously reported associated markers. We analyzed 59 genes including 9 genes reported before to be involved in atherogenesis [8], [9], [12], [13], [14], [15], [16], 10 cytokine genes [17], [18], [19], in addition to 40 genes related with endoplasmic reticulum pathways and cellular stress [20], [21]. Our study provides further insight into the mechanism of plaque destabilization associated with cerebrovascular events.
Materials and Methods
Patients and endarterectomy
Patients were recruited from the department of Neurology, Basurto Hospital (Bilbao, Spain) to undergo carotid endarterectomy (CEA). CEA was performed in patients who presented a degree of stenosis higher than 70% with previous history of transient ischemic attack or ipsilateral stroke (symptomatic) or higher than 80% without any presence of cerebrovascular events (asymptomatic). Quantification of degree (%) of stenosis was performed with carotid cervical Eco-Doppler ultrasound and angioresonance imaging vs angio CT according to established criteria [1]. Demographic and clinical data for these patients are summarized in Table 1. This study was approved by the local ethical committee (Ethical Committee of Clinical Research, Basurto Hospital) and all carotid atheroma plaques were collected from patients who had signed written informed consent. This research was performed in agreement with the principles outlined in the Declaration of Helsinki. The CEA plaques were paraffin-embedded and frozen at -80°C until further use.
Table 1. Demographic characteristics of patients included in the study.
| Patient characteristics | All | Symptomatic | Asymptomatic |
| Number, n | 80 | 45 | 35 |
| Age, years ± SD | 68±8 | 68±8 | 67±8 |
| Sex M/F, n | 70/9 | 39/5 | 31/4 |
| Risks Factors (%) | |||
| Contralateral occlusion | 19 | 18 | 20 |
| Hipertensive | 59.5 | 56.8 | 63 |
| DM (diabetes mellitus) | 27.8 | 27 | 28,5 |
| Cholesterol | 45.5 | 41 | 51 |
| Cardiopaty | 28 | 23 | 34 |
| Isquemic cardiopaty | 19 | 13.6 | 26 |
| ATF (atrial fibrillation) | 5 | 4.5 | 5.7 |
| Intermittent claudication | 34 | 25 | 46 |
| Tobacco | 36.7 | 34 | 40 |
| Medications (%) | |||
| Statin | 21.5 | 18.8 | 26 |
| Anticoagulant | 6.3 | 6.8 | 6 |
RNA extraction and reverse transcription
Frozen carotid atheroma plaque samples were immersed in Ambion RNAlater-ICE (Life technologies, UK) and placed overnight at −20°C. Plaques were homogenized following the manufacture's instructions of TRIzol (Life technologies) and the RNA was extracted with the Ambion RiboPure Kit (Life technologies). The purity of RNA samples was estimated with the Nanodrop (Thermo scientific) using the ratio of absorbance values at 260 nm and 280 nm. 250 µg of extracted RNA were retrotranscribed with the High Capacity cDNA Reverse Transcription Kits from ABI (Life technologies) on the Veriti fast thermal cycler (Life technologies) following the manual instructions. The integrity of RNA (18S and 28S rRNA) was verified by 1% agarose gel electrophoresis.
Selection of genes
Genes selected for this study are candidates for involvement in the carotid atherosclerotic processes associated with symptomatology. Literature was scrutinized to identify potential novel pathways involved in the instability of the plaque on the basis of which a total of 59 candidate genes were selected. Nine of these genes were known to be involved in atherogenesis (VCAM1, CD163, MMP9, TIMP1, COL3A1, THBS1, EDN1, ELANE, ELN) [8], [9], [12], [13], [14], [15], [16], another 10 genes were related to the immune system (IL12B, IL12A, TNF, IL10, IL17A, IL18, IL23A, IL6, TGFB1 and IL1A) [17], [18], [19] and the remaining 40 were selected for their involvement in endoplasmic reticulum (ER)−related pathways or cellular stress (i.e. CALR, DDIT3, ERO1LB, etc.) [20], [21] (S1 Table).
Real-time qPCR
SYBR green technology was used to perform Real Time qPCR. Validated specific primers for genes of interest and house keeping genes (β−actin and GAPDH) were purchased from Qiagen (QuantiTect Primer assays) (S1 Table). For each sample we performed SYBR green real-time qPCR in quadruplicates using the PowerSYBR Green Master Mix on the ABI7500fast detection system (Life Technologies) according to manufacture's instructions. The amplification protocol included a melting curve dissociation step to confirm the inexistence of nonspecific amplification products. The normalization of the gene expression data was performed using the geometric mean of the two house-keeping genes (β−actin and GAPDH). The geometric mean of 2 or more selected housekeeping genes has been validated as a normalization method for qPCR data [22]. The analysis was performed using the comparative Ct method (2−ΔΔCt) and the fold change was calculated from normalized Ct values. The statistical significance of fold change differences between the symptomatic and asymptomatic groups was calculated with the non-parametric Mann-Whitney U test and the level of significance was set at P<0.05. PCR amplification efficiency was found close to 100% in all cases.
Bioinformatics enrichment and correlation analysis
Enrichment clustering analysis was performed using the GeneCodis 3.0 program (http://genecodis.cnb.csic.es/), which enables identification of combinations of significant annotations associated with the analyzed gene list. A statistical discrete probability distribution function test (hypergeometric distribution) was used in the enrichment clustering analysis and the P values were adjusted for multiple tests using the false discovery rate method of Benjamini and Hochberg with the cut-off threshold for significance set at 0.001. Spearmann's correlation test was performed using GrapPad version 5.0 (GraphPad Software, La Jolla, CA) to facilitate the identification of interrelated markers and P<0.05 was considered significant.
Protein isolation and western blot
0.02 g of carotid atheroma plaque was washed with PBS and cut at 300 µm with McIllwain Tissue Chopper (The Mickle Laboratory Engineering Co. LTD.) and the resulting mixture was diluted in 100 µl RIPA buffer (150 mM NaCl; 50 mM Tris-Cl, pH 7.5; 1% NP-40; 0.5% deoxycholate; 0.1% sodium dodecyl sulphate) containing protease inhibitors. Samples were homogenized for 1 h and 30 min on a rotator at 4°C followed by centrifugation for 15 min at 14800 rpm. The supernatants were collected and 10 µl of sample was subjected to 15% SDS-PAGE. Proteins were electrophoretically transferred to a PVDF membrane and blocked overnight. Then, membranes were incubated with rabbit anti-LC3B (D11) antibody (Cell Signalling) or mouse anti-GAPDH (6C5) (Millipore) followed by incubation with anti-rabbit or anti-mouse horseradish peroxidase (HRP) conjugate secondary antibody. Bound antibodies were detected with SuperSignal substrate (Thermo Scientific) on a Chemidoc detection system (BioRad). Signals were quantified by densitometric scanning with the Chemidoc software and densitometric values were normalized against GAPDH. Statistical significance was determined by using the non parametric Mann-Whitney U test.
Results
Gene expression profile of symptomatology within carotid plaques
A total of 35 asymptomatic and 45 symptomatic plaques obtained after CEA were tested for differential expression using the comparative Ct method. The demographic and clinical characteristics of the studied group are shown on Table 1.
Quantitative RT-PCR test data analysis based on the comparative Ct method revealed differential expression levels higher than 1.4 fold change (FC) for 25 of the 59 genes scrutinized upon comparison of symptomatic versus asymptomatic (S vs A) and asymptomatic versus symptomatic (A vs S) (Table 2 and Table 3). From the 25 identified differentially expressed genes (FC≥1.4), 15 showed a significant FC (P<0.05) (i.e. TIMP1, ITPR1, CD163, ERP29, EVA1A, PARK2, MMP9, HSP1A1, SEC63, ERO1LB, RAB24, LMAN1, IL12B, ERP27, MAP1LC3B). From this list, MAP1LC3B was uncovered as the gene showing the highest fold difference between the asymptomatic and symptomatic plaques (FC = 5) with a significance level of P<0.0001. In this study we included also genes that have been reported previously to be differentially expressed in carotid plaques upon comparison of symptomatic versus asymptomatic samples (S vs A). This confirmed that CD163 is upregulated in symptomatic plaques (FC = 1.81, P = 0.044) [8]. In addition, we confirmed HMOX1 and MMP9 in our group of samples to be overexpressed (S vs A) with trends towards significance (FC = 2.07, P = 0.065 and FC = 1.4, P = 0.0505 respectively) [8], [9].
Table 2. Ranking of gene expression markers according to the highest fold change in symptomatic (S) compared with asymptomatic (A) samples quantified by real time RT−PCR.
| Gene name | FC (S vs A) | P-value |
| TIMP metallopeptidase inhibitor 1 (TIMP1) | 3.45 | 0.032* |
| Inositol 1,4,5-triphosphate repector, type 1 (ITPR1) | 2.83 | 0.037* |
| Heme oxygenase (decycling) 1 (HMOX1) | 2.07 | 0.065 |
| CD163 molecule/Hemoglobin scavenger receptor (CD163) | 1.81 | 0.044* |
| Mitogen-activated protein kinase 1 (MAPK1) | 1.75 | 0.057 |
| Endoplasmic reticulum protein 29 (ERP29) | 1.68 | 0.031* |
| Mesencephalic astrocyte-derived neurotrophic factor (MANF) | 1.68 | 0.48 |
| Eva-1 homolog A (C. Elegans) (EVA1A) | 1.57 | 0.033* |
| Transforming growth factor, beta 1 (TGFB1) | 1.57 | 0.13 |
| Parkinson protein 2, E3 ubiquitin protein ligase (PARK2) | 1.52 | 0.043* |
| Protein disulfide isomerase family A, member 6 (PDIA6) | 1.47 | 0.093 |
| Collagen, type III, alpha 1 (COL3A1) | 1.42 | 0.063 |
| Matrix metallopeptidase 9 (gelatinase B) (MMP9) | 1.4 | 0.05* |
The statistical significance was analyzed with the non-parametrical statistical test Mann-Whitney U test (* P≤0.05 and ** P≤0.0001).
Table 3. Ranking of gene expression markers according to highest fold change in asymptomatic (A) compared with symptomatic (S) samples quantified by Real Time RT−PCR.
| Gene name | FC (A vs S) | P-value |
| Microtubule-associated protein 1 light chain 3 beta (MAP1LC3B) | 5 | <0.0001** |
| Endoplasmic reticulum protein 27 (ERP27) | 4.07 | 0.047* |
| Interleukin 12B (IL12B) | 2.5 | 0.028* |
| Lectin, mannose-binding, 1 (LMAN1) | 2 | 0.04* |
| RAB24, member RAS oncogene family (RAB24) | 1.75 | 0.031* |
| ERO1-like beta (S.cerevisiae) (ERO1LB) | 1.67 | 0.034* |
| v-raf-1 murine leukemia viral oncogene homolog 1 (RAF1) | 1.67 | 0.074 |
| Protein disulfide isomerase family A, member 4 (PDIA4) | 1.52 | 0.062 |
| SEC63 homolog (S. cerevisiae) (SEC63) | 1.46 | 0.05* |
| Heat shock 70 kDa protein 1A (HSPA1A) | 1.46 | 0.024* |
| Stress-associated endoplasmic reticulum protein 1 (SERP1) | 1.4 | 0.32 |
| DnaJ (Hsp40) homolog, subfamily B, member 9 (DNAJB9) | 1.4 | 0.067 |
The statistical significance was analyzed with the non-parametrical statistical test Mann-Whitney U test (* P≤0.05 and ** P≤0.0001).
In order to identify functional relationships among the differentially expressed genes between the symptomatic and asymptomatic patients, we applied the software GeneCodis 3.0 for modular enrichment analysis that facilitated extraction of regulatory patterns with potential functional/biological significance. Twenty-four annotation groups (with at least 3 genes in each) obtained by including in the analysis the categories of Gene Ontology (biological process, cellular component and molecular function) and KEGG pathways (Kyoto Encyclopedia of Genes and Genomes) are shown in Table 4. Only the statistically corrected significant annotations are shown, with the corrected P−values obtained by hypergeometric analysis corrected by false discovery rate method. Every annotation group and the implicated genes are described with reference to their involved GO categories or pathways. The molecular enrichment analysis based specifically on KEGG pathways and GO molecular function revealed 9 groups of genes (formed of at least 3 genes) with significant concurrent annotations associated with 16 differentially expressed genes (Table 4). The significant concurrent annotations indicated pathways such as protein binding/protein folding chaperone (GO: 0005515), protein processing in the ER (KEGG: 04141), unfolded ER protein binding (GO: 0051082), infectious diseases (e.g. KEGG: 05140, 05145, 05142 and 05152), metal binding (GO: 0046872), pathways related with cancer (e.g. KEGG: 052220, 05200), or vascular smooth muscle contraction (KEGG: 04270).
Table 4. Molecular enrichment associations – GO: Molecular Functions and KEGG pathways.
| Genes groups | Corrected P value | Concurrent Annotations |
| PARK2 (FC +1.52) | 3.15×10−4 | Metal ion binding (GO: 0046872) |
| LMAN1 (FC −2) | Protein binding (GO: 0005515) | |
| RAF1 (FC −1.67) | ||
| TIMP1 (FC +3.45) | ||
| MMP9 (FC +1.4) | ||
| MAPK1 (FC +1.75) | 9.95×10−16 | Protein binding (GO: 0005515) |
| RAF1 (FC −1.67) | Pathways in cancer (Kegg: 05200) | |
| TGFB1 (FC +1.57) | ||
| MMP9 (FC +1.4) | ||
| MAPK1 (FC +1.75) | 0.001 | Protein binding (GO: 0005515) |
| RAF1 (FC −1.67) | MAPK signalling pathway (Kegg: 04010) | |
| TGFB1 (FC +1.57) | Pathways in cancer (Kegg: 05200) | |
| Tuberculosis (Kegg: 05152) | ||
| Renal cell carcinoma (Kegg: 05211) | ||
| Chronic myeloid leukemia (Kegg: 05220) | ||
| Colorectal cancer (Kegg: 05210) | ||
| Pancreatic cancer (Kegg: 05212) | ||
| MAPK1 (FC +1.75) | 0.001 | Protein binding (GO: 0005515) |
| ITPR1 (FC +2.83) | Gap junction (Kegg: 04540) | |
| RAF1 (FC −1.67) | Long-term depression (Kegg: 04730) | |
| Vascular smooth muscle contraction (Kegg: 04270) | ||
| GnHR signalling pathway (Kegg: 04912) | ||
| Long-term potentiation (Kegg: 04720) | ||
| MAPK1 (FC +1.75) | 0.001 | Protein binding (GO: 0005515) |
| RAF1 (FC −1.67) | Pathways in cancer (Kegg: 05200) | |
| MMP9 (FC +1.4) | Bladder cancer (Kegg: 05219) | |
| LMAN1 (FC −2) | 0.001 | Protein binding (GO: 0005515) |
| DNAJB9 (FC −1.38) | Unfolded protein binding (GO: 0051082) | |
| HSPA1A (FC −1.46) | ||
| LMAN1 (FC −2) | 0.001 | Protein processing in ER (Kegg: 04141) |
| SEC63 (FC −1.46) | Unfolded protein binding (GO: 0051082) | |
| ERO1LB (FC −1.67) | ||
| ERP29 (FC +1.68) | 0.001 | Protein processing in ER (Kegg: 04141) |
| PDIA6 (FC +1.47) | Protein disulfide isomerase activity (GO: 0003756) | |
| PDIA4 (FC −1.52) | ||
| MAPK1 (FC +1.75) | 0.001 | Leishmaniasis (Kegg: 05140) |
| IL12B (FC −2.5) | Toxoplasmosis (Kegg: 05145) | |
| TGFB1 (FC +1.57) | Chagas disease (American trypanosomiasis) (Kegg: 05142) | |
| Tuberculosis (Kegg: 05152) |
Confirmation of gene expression pattern in an additional set of samples
In the course of the study, an additional set of 32 atheroma samples (10 asymptomatic and 22 symptomatic) were obtained by CEA from Basurto Hospital and we followed the procedure as before. Clinical data relative to this set of patients was similar to the patients who were included in the first analysis. We validated in this set a selection of genes, that had shown a significant (P<0.05) fold difference between the symptomatic and asymptomatic plaques and have biological functions of putative relevance to the plaque instability process. The following selected genes were tested in this cohort: TIMP1, ITPR1, EVA1A1, COL3A1, ERO1LB, RAB24, LMAN1 and MAP1LC3B. The gene expression levels were analyzed by qPCR with SYBR green technology and we used the Mann-Whitney U test to calculate the P values. Results combining the original and validation sets of samples are shown in Table 5. The FC and P values for the genes tested were maintained exception made for LMAN1, whose significance was lost (Table 5). MAP1LC3B was confirmed as the gene showing the lowest FC of 6.13.
Table 5. Validation of selected markers in an extended sample set.a .
| Gene Symbol | FC (n = 112) | P-value |
| TIMP1 (S vs A) | 5.1 | 0.03* |
| ITPR1 (S vs A) | 2.2 | 0.0357* |
| EVA1A (S vs A) | 2.03 | 0.041* |
| COL3A1 (S vs A) | 1.65 | 0.0039** |
| MAP1LC3B (A vs S) | 6.13 | <0.0001**** |
| EROL1B (A vs S) | 2.15 | 0.028* |
| RAB24 (A vs S) | 1.84 | 0.0026** |
| LMAN1 (A vs S) | 1.3 | n.s. (0.12) |
The statistical significance was analyzed with the non-parametrical statistical test Mann-Whitney U test (* P≤0.05 and **** P≤0.0001). FC; +, overexpressed and –, underexpressed).
qPCR data analysis was performed in the combined set of the first (80 samples) and second cohorts (32 samples).
MAP1LC3B protein expression analysis in carotid atherosclerotic plaques
Protein was extracted from 5 and 4 plaques from asymptomatic and symptomatic patients, respectively, and analyzed for MAP1LC3B levels by western blot. The MAP1LC3B antibody used reacts stronger with the band called LC3B II, which is indicative of autophagosome formation. Levels of LC3B II were significantly lower in symptomatic versus asymptomatic (Fig. 1A, B) suggesting that MAP1LC3B may play a functional role in preventing plaque destabilization.
Figure 1. Differences in MAP1LC3B protein expression between symptomatic and asymptomatic.
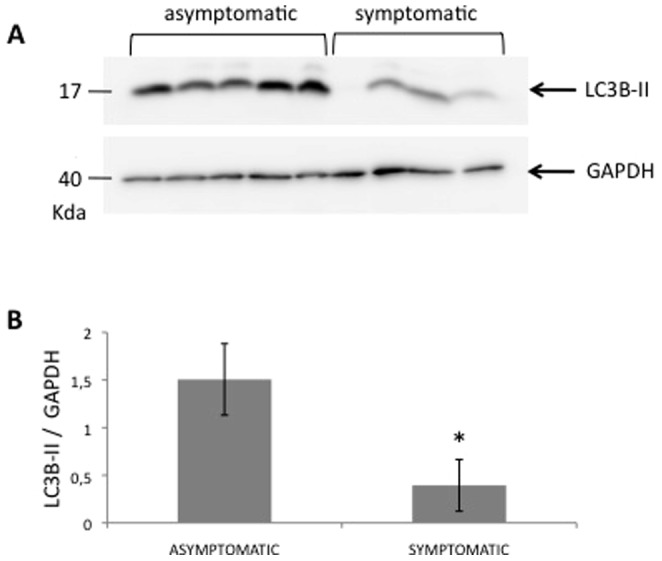
(A) MAP1LC3B and GAPDH carotid atheroma plaque protein levels were analyzed by Western blot and signal was detected on a ChemiDoc (XRS) detection system (BioRad). The blot shows the results from 5 asymptomatic and 4 symptomatic samples. (B) Densitometric analysis of western blot of LC3-II relative to GAPDH. *P = 0.015 symptomatic vs asymptomatic (Mann-Whitney U test).
Correlation analysis: asymptomatic gene expression versus symptomatic gene expression
Correlation analysis between each of the 59 of genes tested in this study was performed using Spearman's rank correlation test using the GraphPad Prism software version 5.0. VCAM1 was correlated with TGFB1 (r = 0.9), MANF (r = 0.86), THBS1 (r = 0.83) and TNF (r = 0.7). On the other hand, ELANE was found to be correlated with IL10 and ELN (r value of 0.9 and 0.71 respectively) while ELN was as well correlated with TGFB1 (r = 0.7). Thus, the correlation analysis identified a pattern of genes that cluster together significantly. Fig. 2 shows correlation values between genes with significant (P<0.001) r value higher than 0.7.
Figure 2. Correlation networks.

The correlation has been computed as the normalised conditional mutual information. Only correlations above 0.7 are shown in this figure and with a significance of P<0.001.
Discussion
The accumulation of atheroma plaque in the carotid artery can lead to stroke. The mechanisms by which a patient with an atherosclerotic plaque in the carotid artery develops ischemic stroke are not completely understood. However, the composition and the vulnerability of the atheroma plaque are important factors in the development of stroke [23], [24], [25]. In this study we adopted a gene expression analysis on carotid atheroma plaques extracted from symptomatic and asymptomatic patients of a series of genes, selected on the bases of literature search, so as to identify genes and/or cellular pathways that would aid in differentiating between the two groups studied and in understanding the mechanism/s that may be involved in this process. We were able to validate the gene expression patterns of previously reported genes (i.e. MMP9, CD163 and HMOX1) [7], [8]. Importantly, we identified a group of previously unreported genes, which appeared differently expressed between the symptomatic and asymptomatic groups (TIMP1, ITPR1, ERP27 or MAP1LC3B). These novel genes are primarily related with inflammation, autophagy, and ER related pathways.
MAP1LC3B emerged as the gene showing the most significant difference in FC between the two groups, with higher expression among asymptomatic patients. This gene has not been identified in previous human carotid plaque studies related with symptomatology. MAP1LC3B is involved in the recruitment of lipid droplets (cholesterol), which may promote autophagy [26]. MAP1LC3B−associated autophagy may be needed to clean up dead cells at the site of atherosclerotic lesions suggesting that autophagy induction could be beneficial in atherosclerosis [27], [28]. In addition, macrophage autophagy has been shown to play a protective role in advanced atherosclerosis [29]. Under hypoxic conditions, known to occur at the lesion site, the UPR (unfolded protein response) is activated as a protective mechanism by regulating the expression of MAP1LC3B [30], [31]. The high level of expression of MAP1LC3B in asymptomatic human carotid atherosclerotic plaques suggests a possible role for preventing the destabilization of the atherosclerotic plaque, probably by promoting basal autophagy activity at the lesion site [28], [32]. Besides, a proteomics study has identified MAP1LC3B as a protein indirectly related with plaque instability [33]. In addition, our data indicates that the nuclear protein high mobility group box 1 (HMGB1; FC = 1.3 (A vs S), P = 0.02), another factor involved in authophagy, may play a role in stimulating beneficial autophagy at the site of lesion. Although HMGB1 has been suggested to be involved in the progression of atherosclerotic plaque, both harmful and beneficial effects of HMGB1 have been documented [34], [35]. In particular, it has been described that HMGB1 regulates autophagy promoting programmed cell survival [36]. In addition, in our cohort we identified RAB24 (FC = 1.75 (A vs S), P = 0.031), a protein considered to play a role in autophagy that colocalizes with MAP1LC3 in autophagosomes [37], to be underexpressed in symptomatic samples. On the other hand, eva-1 homolog A (C. Elegans) (EVA1A; FC = 1.57 (S vs A), P = 0.033) regulates apoptosis and autophagic cell death [38] and it has been described that high levels of EVA1A in rats with middle artery occlusion induced cellular damage conducting to cell death by lysosomal activation [39]. Therefore, EVA1A may play a role in symptomatic plaques by promoting plaque instability caused by autophagic cell death. Calcium homeostasis is also known to play a role in the cellular damage produced by ischemia [40]. Inositol 1,4,5-trisphosphate receptor type 1 (ITPR1; FC = 2.83 (S vs A), P = 0.037) is a channel involved in the influx of calcium from the ER into the cytosol [41]. Calcium release from the ER into the cytosol in basal conditions inhibits autophagy via AMP-activated protein kinase (AMPK) while during stress conditions the calcium signaling stimulates autophagy and apoptosis leading to cellular death [42]. Our results are in concordance with the hypothesis that induction of autophagy may be beneficial for plaque stabilization [43]. While autophagy is needed initially as a repair mechanism at the site of lesion in carotid atherosclerosis to eliminate damaged intracellular material, later on persisting cellular stress induces a type of cell death stimulated by autophagy. For that reason, targeting the later type of autophagy to prevent apoptosis/cell death would be the aim for avoiding the plaque disruption causing fatal symptoms to patients with carotid atherosclerosis.
ER stress-induced apoptosis is known be involved in vascular calcification [20] with its subsequent instability leading to cerebrovascular events [44]. Several differentially expressed genes identified in this study are associated with ER stress pathways (i.e. ERP29, PDIA4, PDIA6, MAP1LC3B, etc), mainly but not only associated with oxidative folding. In our recent study, ER stress induced by a non-coxib celecoxib analogue resulted in increased levels of MAP1LC3B [45], suggesting that the gene is regulated by unfolded protein response (UPR) pathways. The functional enrichment analysis performed, pointed as well to the ER as being associated with symptomatology. Protein binding/protein folding chaperone, protein processing in the ER, metal binding, cancer related pathways, infectious diseases and vascular smooth muscle contraction were biological functions that appeared to be significant in carotid atherosclerosis when we analyzed the 25 identified genes as differently expressed between the two groups.
In the early stage of the cellular stress development, Heat Shock 70 kD Protein−1A (HSPA1A) expression has been shown to exert protective effects by defending against apoptosis and by exerting an anti-inflammatory role [46]. Low levels of expression of HSPA1A (FC = 1.46, P = 0.024), as we observed in our symptomatic cohort, could indicate the initiation of inflammatory stage and cell death. Inflammation is accepted as one of the contributors of atherosclerosis with both the innate and acquired branches of the immune system playing a role in the process [47], [48]. However, our study is indicative for a protective effect displayed by several inflammation biomarkers associated with symptomatology of carotid disease. We identified several factors that seem to point to a beneficial effect of inflammation in asymptomatic patients. In particular, the cytokine subunits belonging to the IL12/IL23 family, IL12B/p40 (FC = 2.5 (A vs S), P = 0.028) and IL23A/p19 (FC = 1.18 (A vs S), P = 0.09), showed higher levels of expression in asymptomatic plaques. IL12B/IL23A forms the heterodimeric IL−23 cytokine that act as an inducer of the Th17 response. The Th17 response may be anti-atherogenic giving protection to patients in whom this response is induced by IL−23 [49], [17]. However, whilst the role of Th17 response in atherosclerosis has not yet been clarified entirely due to contradictory findings [18], [19], some authors have described its protective role in atherosclerosis [49], [50]. Similarly, our results would suggest a role for IL−23−induced Th17 response in carotid plaque stabilization.
In addition, in order to complement the gene expression analysis we attempted to correlate the expression of a gene to the expression of the other gene/s analysed in the carotid plaque samples. The interaction between genes in a network may indicate physical interaction or indirect regulation and it may possible to identify a subgroup of genes that regulate/interact with each other. This information could provide knowledge to develop new concepts for how the instability of plaque occurs. Here we identified groups of genes correlated with differently expressed genes (i.e. VCAM1, TGFB1, THSB1, MANF, ELN, ELANE, IL10, TNF). In this group of genes we observed correlation between the cytokine IL10 and ELANE, an elastin protease known to degrade elastic fibers as elastin; indicating that elastin degradation and immune response process are common interacting regulatory mechanisms in atherosclerosis. Similarly, VCAM1 correlation to TNF and TGFB1 pointed out to an inflammatory pathway for these genes.
In conclusion, this study has identified several biomarkers with altered expression between symptomatic and as symptomatic samples, which are involved in inflammation, ER-related pathways and autophagy. Although the gene expression analysis performed with the technology used here or with similar technologies such as microarrays, have identified several markers associated with symptomatology, these technologies are limited to pre-selected genes. The application to unstable carotid atherosclerosis of the new emerging RNAseq techniques complemented with network analysis would cover the full range of expressed genes allowing to detect genes expressed at low levels and/or splice variants. This would provide unbiased information for identification of mechanistic bases of carotid plaque destabilization.
Supporting Information
List of genes included in the study. Probe gene sets with corresponding NM_number and probe ID (QIAGEN Ltd). FC, fold change (+, upregulated S vs A; -, downregulated S vs A).
(DOC)
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by grants from the Basque Government (Proyectos de Investigación Sanitaria; ref. 2008111024) (http://www.osasun.ejgv.euskadi.net) and from the Basque Government (Grupos de Investigación del Sistema Universitario Vasco; ref. IT512-10 (http://www.hezkuntza.ejgv.euskadi.net). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, et al. (2012) Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. The Lancet 380:2095–2128 10.1016/S0140-6736(12)61728-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Murray CJL, Vos T, Lozano R, Naghavi M, Flaxman AD, et al. (2012) Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. The Lancet 380:2197–2223 10.1016/S0140-6736(12)61689-4 [DOI] [PubMed] [Google Scholar]
- 3. Walker MD, Marler JR, Goldstein M, Grady PA, Toole JF, et al. (1995) Endarterectomy for asymptomatic carotid artery stenosis. JAMA 1995 273:1421–1428 10.1001/jama.1995.03520420037035 [DOI] [Google Scholar]
- 4. Golledge J, Greenhalgh RM, Davies AH (2000) The Symptomatic Carotid Plaque. Stroke 31:774–781 10.1161/01.STR.31.3.774 [DOI] [PubMed] [Google Scholar]
- 5. NASCET C (1991) Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. New Engl J Med 325:445–453 10.1056/NEJM199108153250701 [DOI] [PubMed] [Google Scholar]
- 6. Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, et al. (2003) From Vulnerable Plaque to Vulnerable Patient: A Call for New Definitions and Risk Assessment Strategies: Part I. Circulation 108:1664–1672 10.1161/01.CIR.0000087480.94275.97 [DOI] [PubMed] [Google Scholar]
- 7. Morgan AR, Rerkasem K, Gallagher PJ, Zhang B, Morris GE, et al. (2004) Differences in Matrix Metalloproteinase-1 and Matrix Metalloproteinase-12 Transcript Levels Among Carotid Atherosclerotic Plaques With Different Histopathological Characteristics. Stroke 35:1310–1315 10.1161/01.STR.0000126822.01756.99 [DOI] [PubMed] [Google Scholar]
- 8. Ijäs P, Nuotio K, Saksi J, Soinne L, Saimanen E, et al. (2007) Microarray analysis reveals overexpression of CD163 and HO-1 in symptomatic carotid plaques. Arterioscl Throm Vasc Biol 27:154–160 10.1161/01.ATV.0000251991.64617.e7 [DOI] [PubMed] [Google Scholar]
- 9. Papaspyridonos M, Smith A, Burnand KG, Taylor P, Padayachee S, et al. (2006) Novel Candidate Genes in Unstable Areas of Human Atherosclerotic Plaques. Arterioscl Throm Vasc Biol 26:1837–1844 10.1161/01.ATV.0000229695.68416.76 [DOI] [PubMed] [Google Scholar]
- 10. Erbel C, Dengler T, Wangler S, Lasitschka F, Bea F, et al. (2011) Expression of IL-17A in human atherosclerotic lesions is associated with increased inflammation and plaque vulnerability. Basic Res Cardiol 106:125–134 10.1007/s00395-010-0135-y [DOI] [PubMed] [Google Scholar]
- 11. Saksi J, Ijäs P, Nuotio K, Sonninen R, Soinne L, et al. (2011) Gene expression differences between stroke-associated and asymptomatic carotid plaques. J Mol Med 89:1015–1026 10.1007/s00109-011-0773-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ley K, Huo Y (2001) VCAM-1 is critical in atherosclerosis. J Clin Invest 107:1209–1210 10.1172/JCI13005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kowala MC (1997) The role of endothelin in the pathogenesis of atherosclerosis. Adv Pharmacol 37:299–318. [DOI] [PubMed] [Google Scholar]
- 14. Rekhter MD (2014) Collagen synthesis in atherosclerosis: too much and not enough. Cardiovas Res 41:376–384 10.1016/S0008-6363(98)00321-6 [DOI] [PubMed] [Google Scholar]
- 15. Robert L, Robert AM, Jacotot B (1998) Elastin–elastase–atherosclerosis revisited. Atherosclerosis 140:281–295 10.1016/S0021-9150898900171-3 [DOI] [PubMed] [Google Scholar]
- 16. Stenina OI, Plow EF (2008) Counterbalancing Forces: What Is Thrombospondin-1 Doing in Atherosclerotic Lesions? Circ Res 103:1053–1055 10.1161/CIRCRESAHA.108.188870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ait-Oufella H, Taleb S, Mallat Z, Tedgui A (2011) Recent Advances on the Role of Cytokines in Atherosclerosis. Arterioscl Throm Vasc Biol 31:969–979 10.1161/ATVBAHA.110.207415 [DOI] [PubMed] [Google Scholar]
- 18. Robertson A-KL, Hansson GK (2006) T Cells in Atherogenesis: For Better or For Worse? Arterioscl Throm Vasc Biol 26:2421–2432 10.1161/01.ATV.0000245830.29764.84 [DOI] [PubMed] [Google Scholar]
- 19. Lichtman AH, Binder CJ, Tsimikas S, Witztum JL (2013) Adaptive immunity in atherogenesis: new insights and therapeutic approaches. J Clin Invest 123:27–36 10.1172/JCI63108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duan X, Zhou Y, Teng X, Tang C, Qi Y (2009) Endoplasmic reticulum stress-mediated apoptosis is activated in vascular calcification. Biochem Bioph Res Commun 387:694–699 10.1016/j.bbrc.2009.07.085 [DOI] [PubMed] [Google Scholar]
- 21. Tabas I (2010) The Role of Endoplasmic Reticulum Stress in the Progression of Atherosclerosis. Circ Res 107:839–850 10.1161/CIRCRESAHA.110.224766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, et al. (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:research0034 10.1186/gb-2002-3-7-research0034.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hennerici MG (2004) The unstable plaque. Cerebrovas Dis 17:17–22 10.1159/000075300 [DOI] [PubMed] [Google Scholar]
- 24. Virmani R, Burke AP, Farb A, Kolodgie FD (2006) Pathology of the Vulnerable Plaque. J Am Coll Cardiol 47:C13–C18 10.1016/j.jacc.2005.10.065 [DOI] [PubMed] [Google Scholar]
- 25. Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R (2010) Concept of Vulnerable/Unstable Plaque. Arterioscl Throm Vasc Biol 30:1282–1292 10.1161/ATVBAHA.108.179739 [DOI] [PubMed] [Google Scholar]
- 26. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, et al. (2009) Autophagy regulates lipid metabolism. Nature 458:1131–1135 10.1038/nature07976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, et al. (2011) Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci USA 108:17396–17401 10.1073/pnas.1113421108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martinet W, Meyer I, Verheye S, Schrijvers D, Timmermans J P, et al. (2012) Drug-induced macrophage autophagy in atherosclerosis: for better or worse? Basic Res Cardiol 108:1–11 10.1007/s00395-012-0321-1 [DOI] [PubMed] [Google Scholar]
- 29. Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, et al. (2012) Macrophage Autophagy Plays a Protective Role in Advanced Atherosclerosis. Cell Metab 15:545–553 10.1016/j.cmet.2012.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rouschop KMA, van den Beucken T, Dubois L, Niessen H, Bussink J, et al. (2010) The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 120:127–141 10.1172/JCI40027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Björnheden T, Levin M, Evaldsson M, Wiklund O (1999) Evidence of hypoxic areas within the arterial wall in vivo. Arterioscl Throm Vasc Biol 19:870–876 10.1161/01.ATV.19.4.870 [DOI] [PubMed] [Google Scholar]
- 32. Schrijvers DM, De Meyer GRY, Martinet W (2011) Autophagy in Atherosclerosis: A Potential Drug Target for Plaque Stabilization. Arterioscl Throm Vasc Biol 31:2787–2791 10.1161/ATVBAHA.111.224899 [DOI] [PubMed] [Google Scholar]
- 33. Malaud E, Merle D, Piquer D, Molina L, Salvetat N, et al. (2014) Local carotid atherosclerotic plaque proteins for the identification of circulating biomarkers in coronary patients. Atherosclerosis 233:551–558 10.1016/j.atherosclerosis.2013.12.019 [DOI] [PubMed] [Google Scholar]
- 34. de Souza AWS, Westra J, Limburg PC, Bijl M, Kallenberg CGM (2012) HMGB1 in vascular diseases: Its role in vascular inflammation and atherosclerosis. Autoimmun Rev 11:909–917 10.1016/j.autrev.2012.03.007 [DOI] [PubMed] [Google Scholar]
- 35. Hayakawa K, Qiu J, Lo EH (2010) Biphasic actions of HMGB1 signaling in inflammation and recovery after stroke. Ann NY Acad Sci 1207:50–57 10.1111/j.1749-6632.2010.05728.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, et al. (2010) Endogenous HMGB1 regulates autophagy. J Cell Biol 190:881–892 10.1083/jcb.200911078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Munafó DB, Colombo MI (2002) Induction of Autophagy Causes Dramatic Changes in the Subcellular Distribution of GFP-Rab24. Traffic 3:472–482 10.1034/j.1600-0854.2002.30704.x [DOI] [PubMed] [Google Scholar]
- 38. Wang L, Yu C, Lu Y, He P, Guo J, et al. (2007) TMEM166, a novel transmembrane protein, regulates cell autophagy and apoptosis. Apoptosis 12:1489–1502 10.1007/s10495-007-0073-9 [DOI] [PubMed] [Google Scholar]
- 39. Li L, Khatibi NH, Hu Q, Yan J, Chen C, et al. (2012) Transmembrane protein 166 regulates autophagic and apoptotic activities following focal cerebral ischemic injury in rats. Exp Neurol 234:181–190 10.1016/j.expneurol.2011.12.038 [DOI] [PubMed] [Google Scholar]
- 40. Paschen W (1996) Disturbances of calcium homeostasis within the endoplasmic reticulum may contribute to the development of ischemic-cell damage. Med Hypotheses 47:283–288 10.1016/S0306-9877(96)90068-7 [DOI] [PubMed] [Google Scholar]
- 41. Taylor CW, Genazzani AA, Morris SA (1999) Expression of inositol trisphosphate receptors. Cell Calcium 26:237–251 10.1054/ceca.1999.0090 [DOI] [PubMed] [Google Scholar]
- 42. Decuypere JP, Bultynck G, Parys JB (2011) A dual role for Ca2+ in autophagy regulation. Cell Calcium 50:242–250 10.1016/j.ceca.2011.04.001 [DOI] [PubMed] [Google Scholar]
- 43. Wei K, Wang P, Miao CY (2012) A Double-Edged Sword with Therapeutic Potential: An Updated Role of Autophagy in Ischemic Cerebral Injury. CNS Neurosci Ther 18:879–886 10.1111/cns.12005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Virmani R, Burke AP, Farb A, Kolodgie FD (2006) Pathology of the Vulnerable Plaque. J Am Coll Cardiol 47:C13–C18 10.1016/j.jacc.2005.10.065 [DOI] [PubMed] [Google Scholar]
- 45. Di Penta A, Chiba A, Alloza I, Wyssenbach A, Yamamura T, Villoslada P, et al. (2013) A Trifluoromethyl Analogue of Celecoxib Exerts Beneficial Effects in Neuroinflammation. PLoS ONE 8:e83119 10.1371/journal.pone.0083119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mehta TA, Greenman J, Ettelaie C, Venkatasubramaniam A, Chetter IC, et al. (2005) Heat Shock Proteins in Vascular Disease—A Review. Eur J Vasc Endovasc Surg 29:395–402 10.1016/j.ejvs.2005.01.005 [DOI] [PubMed] [Google Scholar]
- 47. Hansson GK, Hermansson A (2011) The immune system in atherosclerosis. Nat Immunol 12:204–212 10.1038/ni.2001 [DOI] [PubMed] [Google Scholar]
- 48. Chen S, Crother TR, Arditi M (2010) Emerging Role of IL-17 in Atherosclerosis. J Innate Immun 2:325–333 10.1159/000314626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Taleb S, Romain M, Ramkhelawon B, Uyttenhove C, Pasterkamp G, et al. (2009) Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J Exp Med 206:2067–2077 10.1084/jem.20090545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Danzaki K, Matsui Y, Ikesue M, Ohta D, Ito K, et al. (2012) Interleukin-17A Deficiency Accelerates Unstable Atherosclerotic Plaque Formation in Apolipoprotein E-Deficient Mice. Arterioscl Throm Vasc Biol 32:273–280 10.1161/ATVBAHA.111.229997 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of genes included in the study. Probe gene sets with corresponding NM_number and probe ID (QIAGEN Ltd). FC, fold change (+, upregulated S vs A; -, downregulated S vs A).
(DOC)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.