

Abstract
Inhibition of the CD40–CD154 pathway controls inflammatory disorders. Unfortunately, administration of anti-CD154 monoclonal antibodies causes thromboembolism. Blockade of signalling downstream of CD40 may represent an approach to treat CD40-driven inflammatory disorders. Blocking tumour necrosis factor receptor-associated factor 6 (TRAF6) signalling downstream of CD40 in MHC II+ cells diminishes inflammation. However, CD40–TRAF6 blockade may cause immunosuppression. We examined the role of CD40–TRAF2,3 and CD40–TRAF6 signalling in the development of pro-inflammatory responses in human non-haematopoietic and monocytic cells. Human aortic endothelial cells, aortic smooth muscle cells, renal proximal tubule epithelial cells, renal mesangial cells and monocytic cells were transduced with retroviral vectors that encode wild-type CD40, CD40 with a mutation that prevents TRAF2,3 recruitment (ΔT2,3), TRAF6 recruitment (ΔT6) or both TRAF2,3 plus TRAF6 recruitment (ΔT2,3,6). Non-haematopoietic cells that expressed CD40 ΔT2,3 exhibited marked inhibition in CD154-induced up-regulation of vascular cell adhesion molecule 1, intercellular adhesion molecule 1 (ICAM-1), monocyte chemotactic protein 1 (MCP-1), tissue factor and matrix metalloproteinase 9. Similar results were obtained with cells that expressed CD40 ΔT6. Although both mutations impaired ICAM-1 up-regulation in monocytic cells, only expression of CD40 ΔT6 reduced MCP-1 and tissue factor up-regulation in these cells. Treatment of endothelial and smooth muscle cells with cell-permeable peptides that block CD40–TRAF2,3 or CD40–TRAF6 signalling impaired pro-inflammatory responses. In contrast, while the CD40–TRAF2,3 blocking peptide did not reduce CD154-induced dendritic cell maturation, the CD40–TRAF6 blocking peptide impaired this response. Hence, preventing CD40–TRAF2,3 or CD40–TRAF6 interaction inhibits pro-inflammatory responses in human non-haematopoietic cells. In contrast to inhibition of CD40–TRAF6 signalling, inhibition of CD40–TRAF2,3 signalling did not impair dendritic cell maturation. Blocking CD40–TRAF2,3 signalling may control CD40–CD154-dependent inflammatory disorders.
Keywords: endothelial cell, epithelial cell, inflammation, monocyte, muscle smooth cell
Introduction
CD40 is a member of the tumour necrosis factor (TNF) receptor superfamily that is constitutively or inducibly expressed on haematopoietic cells and various non-haematopoietic cells.1–4 CD154 (CD40 ligand) is expressed primarily on activated CD4+ T cells and activated platelets, although it can also be present at lower levels on other cell types.1,2,4 CD154 also exists as a soluble protein in plasma that appears to be biologically active.5 CD40–CD154 interaction is central for regulation of cellular and humoral immunity.1,2 This pathway is of clinical relevance not only because it promotes resistance against various pathogens, but also because it drives inflammatory disorders such as atherosclerosis, inflammatory bowel disease, systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis and graft rejection.6 The pathogenic role of CD40–CD154 in these disorders is probably explained by pathology caused by activated T and B cells as well as by pro-inflammatory responses triggered by CD40 ligation in non-haematopoietic cells.
Although under basal conditions CD40 is either not expressed or is expressed weakly in non-haematopoietic cells, CD40 expression is increased in endothelial cells, smooth muscle cells, intestinal epithelial cells and renal parenchymal cells in inflammatory disorders like atherosclerosis, ligation-induced vascular injury, inflammatory bowel disease, glomerulonephritis and graft rejection.7–13 Moreover, T cells and platelets present at these sites express CD154.7,9,10,13–15 CD40 triggers cellular responses relevant to inflammation: increased adhesion molecule expression and chemokine production by endothelial cells,3,16 production of chemokines and pro-inflammatory cytokines by vascular smooth muscle cells and macrophages,4,17–19 enhanced expression of matrix metalloproteinases (MMP) and the pro-coagulant tissue factor by vascular smooth muscle cells, endothelial cells and macrophages20–22 as well as increased chemokine production by renal proximal tubule epithelial cells.23,24 Hence, the CD40–CD154 pathway probably induces responses involved in the initiation of various inflammatory disorders and the development of complications such as plaque rupture and thrombus formation in atherosclerosis.
Animal studies showed that blockade of CD40–CD154 is therapeutically effective in various inflammatory disorders.25–31 Unfortunately, clinical application of neutralizing anti-CD154 monoclonal antibody in humans has been complicated by thromboembolic events.32 Moreover, indiscriminate inhibition of the CD40–CD154 pathway is likely to result in susceptibility to opportunistic infections because CD40–CD154 interaction is crucial for protection against numerous pathogens.1,2
The evidence that CD40−/− mice exhibit diminished atherosclerosis and neointima formation after vascular injury, decreased intestinal inflammation in a model of inflammatory bowel disease, as well as decreased glomerular inflammation in a model of crescentic glomerulonephritis indicates that CD40 can become a therapeutic target for the control of various inflammatory disorders.13,33–36 CD40 signals through adaptor proteins such as TNF receptor-associated factors (TRAF) and janus kinase 3.37,38 TRAF are major mediators of signalling downstream of CD40.37 CD40 has a domain (PxQxT) that directly binds TRAF2 and TRAF339 (TRAF3 inhibits CD40 signalling), a distal domain (SVxE) that binds TRAF2,40 and a site that binds TRAF6 (PxExxAr/Ac).39 Whereas both TRAF2,3 and TRAF6 binding sites can induce similar cellular responses, the effects downstream of these sites can be non-overlapping. The TRAF6 binding site promotes interleukin-12 (IL-12) production by dendritic cells, TNF-α, IL-1β, IL-6 and nitric oxide synthase 2 up-regulation in macrophages, induction of autophagy-dependent anti-microbial activity in macrophages, IL-6 production by B cells and plasma cell formation.19,41–46 In contrast, the TRAF2,3 binding site appears to be dominant for isotype switch.47 Studies using transgenic mice where either wild-type (wt) CD40 or CD40 with mutations in TRAF binding sites were expressed in MHC II+ cells, indicated that the TRAF6 binding site in this cell population plays an important role in promoting atherosclerosis and neointima formation after vascular injury.35,36
CD40–TRAF signalling can be cell specific. While the role of CD40–TRAF binding sites in haematopoietic cells has been studied, much less is known about the role of these sites in the induction of pro-inflammatory responses in non-haematopoietic cells. This is important as CD40 expression in non-haematopoietic cells promotes inflammation after tissue injury.13 Studies in epithelial cells revealed that the TRAF2 binding site plays the dominant role in nuclear factor-κB (NF-κB), p38 mitogen-activated protein kinase and Jun N-terminal kinase activation.48 In contrast, fibroblasts that express chimeras of extracellular CD8 fused to the intracellular domain of wt CD40 or CD40 that expressed either the TRAF2,3 or TRAF6 binding sites suggested that the latter site is sufficient for IL-6 and IL-8 production in response to CD8 cross-linking.49 Various studies used non-haematopoietic cells deficient in TRAFs or that expressed a TRAF dominant negative mutant to examine the role of TRAFs in CD40-dependent chemokine and adhesion molecule up-regulation.13,24,50,51 Although these studies provided useful information, data interpretation may be complicated by the fact that: (i) TRAFs can form heterocomplexes after CD40 ligation and hence, deficiency of a particular TRAF can affect additional TRAF signalling48,52; (ii) TRAFs can signal downstream of various receptors in addition to CD40; and (iii) partial TRAF knock-down or suboptimal expression of TRAF dominant negative mutants may result in insufficient inhibition of TRAF signalling.
By inducing expression of either wt CD40 or CD40 with mutations in the TRAF2,3 and/or TRAF6 binding sites that prevent recruitment of the appropriate TRAFs or by using cell-permeable CD40–TRAF blocking peptides, we dissected the role of CD40–TRAF signalling in the induction of a broad range of cellular responses that have been linked to the pathogenesis and complications of inflammatory disorders. Our studies indicate that blockade of either CD40–TRAF2,3 or CD40–TRAF6 signalling was sufficient to markedly impair various pro-inflammatory responses in non-haematopoietic cells. Whereas the CD40–TRAF6 binding site played the main role in production of monocyte chemotactic protein 1 (MCP-1) and tissue factor up-regulation in human monocytic cells, this site also appeared to play the dominant role in human dendritic cell maturation. Given that CD40–TRAF6 signalling stimulates various responses key to cell-mediated immunity, our findings raise the possibility that blockade of CD40–TRAF2 signalling may improve CD40-induced inflammation with a potential reduced risk of immunosuppression.
Materials and methods
Cells
Human aortic endothelial cells (HAEC) and human aortic smooth muscle cells (HASMC) were obtained from Lonza (Walkersville, MD); human renal proximal tubular epithelial cells (HRPTEC) and human renal mesangial cells (HRMC) were obtained from ScienCell Research Laboratories (Carlsbad, CA). Cells were cultured following the manufacturers’ recommendations. MonoMac6 cells were a gift from Rene de Waal Malefyt (DNAX Research Institute, Palo Alto, CA) and were cultured in RPMI-1640 plus 10% fetal bovine serum (Hyclone, Logan, UT). Human monocyte-derived dendritic cells were generated as described previously.53 Briefly, monocytes were isolated from peripheral blood mononuclear cells using a Monocyte Isolation Kit II (Miltenyi Biotec, Auburn, CA). Monocytes were cultured in RPMI-1640 plus 10% fetal bovine serum containing granulocyte–macrophage colony-stimulating factor (1000 U/ml) plus IL-4 (500 U/ml; both from PeproTech, Rocky Hill, NJ) for 7 days. Cytokines were added every 3 days. The mouse endothelial cell line that expresses the extracellular domain of human CD40 and the intracellular domain of mouse CD40 (hmCD40 mHEVc) was previously described.46
In vitro stimulation
Cells were treated with or without human CD154 (3 μg/ml; a gift from William Fanslow, Amgen, Thousand Oaks, CA or cell-free supernatants containing multimeric CD15454 obtained from Dr Richard Kornbluth, Multimeric Biotherapeutics Inc., La Jolla, CA) for 24 hr at 37° as described.55 Responses induced by both preparations of CD154 were similar. Specificity of CD154 was confirmed by detecting > 95% neutralization in response to co-incubation with anti-human CD154 monoclonal antibody (Ancell Corporation, Bayport, MN). Omission of CD154 or incubation with a non-functional CD154 mutant (T147N; obtained from Dr Richard Kornbluth) was used as control. Endothelial cells were also incubated with interferon-γ (500 IU/ml; PeproTech) plus TNF-α (500 IU/ml; PeproTech) or PMA (50 ng/ml; Sigma Chemical, St Louis, MO).
Retroviral vectors and transductions
The cDNA for wt human CD40 (hCD40), hCD40Δ22 (a mutant that ablates binding to TRAF2 and TRAF3; CD40 ΔTRAF2,3), hCD40EEAA (a mutant that prevents binding to TRAF6; CD40 ΔTRAF6), and hCD40Δ55 (a mutant that ablates binding to TRAF2, TRAF3 and TRAF6; CD40 ΔTRAF2,3,6) have been previously described.56,57 The murine stem cell virus-based bicistronic retroviral vector MIEG3 that encodes enhanced green fluorescence protein (EGFP) and either cDNA for wt human CD40, CD40 ΔTRAF2,3, CD40 ΔTRAF6, or CD40 ΔTRAF2,3,6 were previously described.58 Ecotropic retroviral supernatants were generated as described58 except for the use of the envelope plasmid RD114 (gift from Yasu Takeuchi, University College London, London, UK). Briefly, Phoenix-gp cell line (gift from Gary Nolan, Stanford University, CA) was transfected with MIEG3-based retroviral vectors and plasmids encoding envelope and gag-pol using a calcium phosphate transfection kit (Invitrogen Corporation, Carlsbad, CA). Cells were incubated overnight with retrovirus in the presence of polybrene (8 μg/ml, Sigma Chemical).
Cell-permeable peptides
Peptides that consisted of the TRAF2,3 and TRAF6 binding sites of CD40 were made cell permeable by linking them to the TAT47–57 cell penetrating peptide. The sequences for the CD40–TRAF2,3 and the CD40–TRAF6 blocking peptides were NH2-NTAAPVQETLHG YGRKKRRQRRR-OH and NH2-KQEPQEIDFPDD YGRKKRRQRRR-OH. The TAT47–57 sequence is underlined. Control peptide consisted of either TAT47–57 or TAT47–57 linked to a scrambled peptide. Peptides were manufactured by Proteintech Group (San Diego, CA) and were low in endotoxin and > 98% pure by HPLC.
Flow cytometry
Cells were stained with phycoerythrin-conjugated anti-CD40 (BD Biosciences, San Jose, CA), anti-CD80, anti-CD83, anti-CD86, anti-HLA-DR, anti-intercellular adhesion molecule 1 (ICAM-1), anti-vascular cell adhesion molecule 1 (VCAM-1) (all from eBiosciences, San Diego, CA), anti-tissue factor (American Diagnostica Inc., Stanford, CT) plus donkey anti-mouse–phycoerythrin (eBiosciences) or isotype control monoclonal antibodies. Cells were washed and fixed with 1% paraformaldehyde followed by analysis using an LSR II (BD Biosciences).
ELISA
Supernatants from cell monolayers cultured in 96-well plates were collected at 24 hr. Supernatants were used to measure concentrations of MCP-1 by ELISA (R&D Systems, Minneapolis, MN). MMP-9 proteolytic activity was measured with a Biotrak Activity Assay System (GE Healthcare, Chalfont St Giles, UK).
Luciferase assay
Peptides were tested as previously described using the mouse endothelial cell mHEVc that express a chimera of the extracellular domain of human CD40 and intracytoplasmic domain of mouse CD40 (hmCD40) with either a mutation that prevents recruitment of TRAF2,3 (hmCD40 ΔT2,3) or TRAF6 (hmCD40 ΔT6).46 These cells were also transfected with pGL4.32luc2P/NF-κB-RE/Hygro vector (Promega Corporation, Madison, WI), a plasmid that encodes an NF-κB response element that drives transcription of the luciferase reporter gene luc2P (Photinus pyralis).46 Cells were pre-incubated with peptides for 3 hr followed by stimulation with human CD154. Luciferase activity was assessed using a Steady-Glo luciferase assay system (Promega Corporation) and a luminometer. Bioluminescence signals were normalized to total protein concentration as determined by BCA protein assay (Pierce Protein Research Products, Rockford, IL). The effects of peptides on cell viability were examined using a commercially available alamarBlue cell viability assay (Invitrogen Corporation).
Statistical analysis
All results were expressed as the mean ± SEM. Data were analysed by two-tailed Student's t-test and analysis of variance. Differences were considered statistically significant at P < 0·05.
Results
Role of the CD40–TRAF2,3 and the CD40–TRAF6 binding sites in CD154-induced up-regulation of VCAM-1, ICAM-1, MCP-1 and tissue factor in human aortic endothelial cells
CD40 expression is increased in non-haematopoietic cells in inflammatory diseases and contributes to pro-inflammatory responses in these disorders.7–13 In contrast, CD40 is either not expressed or is expressed weakly in non-haematopoietic cells under basal conditions. To study the role of CD40–TRAF signalling in the induction of pro-inflammatory responses, primary human non-haematopoietic cells were induced to express wt CD40 or CD40 with mutations that prevent TRAF recruitment. This approach has been shown to be well suited for studying the role of TRAF signalling downstream of CD40.35,36,42,47,57,59 Human cells were transduced with retroviral vectors that encode either wt CD40 or CD40 with deletions or point mutations at TRAF binding sites proven to ablate binding to TRAF2,3 (ΔT2,3), TRAF6 (ΔT6) or TRAF2,3,6 (ΔT2,3,6).57,59 Primary HAEC were transduced with these vectors and the percentages of transduced cells (EGFP+) as well as the corrected mean fluorescence intensity (cMFI) for CD40 on EGFP+ cells were similar (Fig. 1a; P > 0·05).
Figure 1.

Role of CD40–tumour necrosis factor (TNF) receptor-associated factor 6 (TRAF) binding sites on vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), monocyte chemoattractant protein 1 (MCP-1) and tissue factor up-regulation in human aortic endothelial cells (HAEC). HAEC were transduced with MIEG3-based retroviral vectors that encode EGFP alone or EGFP and either wild-type (wt) CD40, CD40 ΔT2,3, CD40 ΔT6 or CD40 ΔT2,3,6. (a) Percentages of transduced cells (EGFP+) and corrected mean fluorescence intensity (cMFI) for CD40 on EGFP+ cells. (b) Expression of VCAM-1 and EGFP was analysed by flow cytometry at 24 hr post-incubation with or without CD154. (c, d), Expression of VCAM-1 and ICAM-1 on gated EGFP+ cells expressed as cMFI. (e) HAEC were incubated with interferon-γ (IFN-γ) plus TNF-α and expression of VCAM-1 was assessed by flow cytometry at 24 hr. (f) MCP-1 concentrations determined by ELISA in the supernatants collected at 24 hr. (g, h) Tissue factor expression was assessed by flow cytometry on gated EGFP+ cells at 24 hr after incubation with or without CD154 (g) or PMA (h). Results are shown as mean ± SEM and are representative of three to six experiments. *P < 0·05; **P < 0·01; ***P < 0·001.
We examined the role of CD40–TRAF binding sites on VCAM-1 and ICAM-1 up-regulation in HAEC. Incubation with CD154 caused marked up-regulation of VCAM-1 and ICAM-1 on transduced (EGFP+) HAEC that expressed wt CD40 (Fig. 1b–d). This effect was ablated by co-incubation with a neutralizing anti-CD154 monoclonal antibody (> 95% inhibition; data not shown). In contrast, HAEC transduced with the empty retroviral vector (MIEG3) exhibited a much less pronounced up-regulation of VCAM-1 (mean cMFI Ctr = 22; CD154 = 56) and ICAM-1 (mean cMFI Ctr = 55; CD154 = 104) in response to CD154. These findings are consistent with previous reports on the effect of CD154 on ICAM-1/VCAM-1 expression in HAEC and are probably explained by the low levels of CD40 under basal conditions.16 Hence, our findings indicate that responses observed in transduced HAEC are driven primarily by retrovirus-induced CD40. Next, we examined the effect of mutations in TRAF binding sites on adhesion molecule up-regulation. VCAM-1 and ICAM-1 up-regulation were ablated in cells that express CD40 that does not recruit TRAF2,3 and TRAF6 (Fig. 1c,d). Compared with wt CD40, expression of either CD40 ΔT2,3 or CD40 ΔT6 was sufficient to markedly inhibit VCAM-1 and ICAM-1 up-regulation (Fig. 1c,d). Moreover, the effects of mutations in TRAF binding sites were specific because up-regulation of VCAM-1 in response to interferon-γ/TNF-α was similar regardless of the retroviral vector used to transduce HAEC (Fig. 1e). Hence, up-regulation of VCAM-1 and ICAM-1 on HAEC stimulated through CD40 is TRAF dependent, and a mutation that prevents CD40–TRAF2,3 or CD40–TRAF6 interaction is sufficient to profoundly impair these responses.
CD154 enhanced MCP-1 production by HAEC that expressed wt CD40 (Fig. 1f). Expression of CD40 ΔT2,3,6 ablated MCP-1 production (Fig. 1f). Expression of CD40 ΔT6 markedly inhibited MCP-1 production in response to CD40 stimulation while expression of CD40 ΔT2,3 ablated this response (Fig. 1f). Hence, MCP-1 production induced by stimulation through CD40 is TRAF dependent and a mutation that prevents TRAF2,3 recruitment is sufficient to abrogate this response.
Incubation with CD154 up-regulated tissue factor expression on HAEC (Fig. 1g). This effect was TRAF binding site dependent because it was ablated by expression of CD40 ΔT2,3,6 (Fig. 1g). Expression of CD40 ΔT2,3 or CD40 ΔT6 markedly impaired tissue factor up-regulation (Fig. 1g). The effects of CD40 mutations were specific because up-regulation of tissue factor in response to PMA was similar regardless of the retroviral vector used (Fig. 1h). Hence, mutations in the CD40–TRAF2,3 or CD40–TRAF6 binding sites impaired up-regulation of tissue factor on HAEC stimulated through CD40. Taken together, a mutation that prevents CD40–TRAF2,3 or CD40–TRAF6 interaction is sufficient to profoundly impair pro-inflammatory responses (VCAM-1/ICAM-1 up-regulation and MCP-1 production) and tissue factor up-regulation in CD40-stimulated HAEC.
Both CD40–TRAF2,3 and CD40–TRAF6 binding sites are required for optimal CD154-induced up-regulation of VCAM-1, MCP-1, tissue factor and MMP-9 activity in human aortic smooth muscle cells
We determined whether CD40 stimulation causes VCAM-1 up-regulation in HASMC and determined the role of TRAF binding sites in this response as well as in CD40-driven MCP-1 up-regulation. HASMC were transduced with retroviral vectors. As in HAEC, the percentages of transduced cells (EGFP+) and the cMFI for CD40 were similar for all groups (Fig. 2a; P > 0·5). Incubation with CD154 caused VCAM-1 up-regulation (Fig. 2b). This response was dependent on CD40–TRAF signalling because it was ablated in HASMC that express CD40 ΔT2,3,6 (Fig. 2b). Expression of CD40 ΔT2,3 or CD40 ΔT6 significantly impaired up-regulation of VCAM-1 (Fig. 2b). Hence, blockade of either the TRAF2,3 or TRAF6 binding site is sufficient to significantly diminish VCAM-1 up-regulation in HASMC. CD40 stimulation increased MCP-1 production by HASMC that was TRAF dependent because it was ablated in cells that expressed CD40 ΔT2,3,6 (Fig. 2c). Mutations in either the TRAF2,3 or TRAF6 binding sites were sufficient to remarkably inhibit MCP-1 up-regulation (Fig. 2c).
Figure 2.
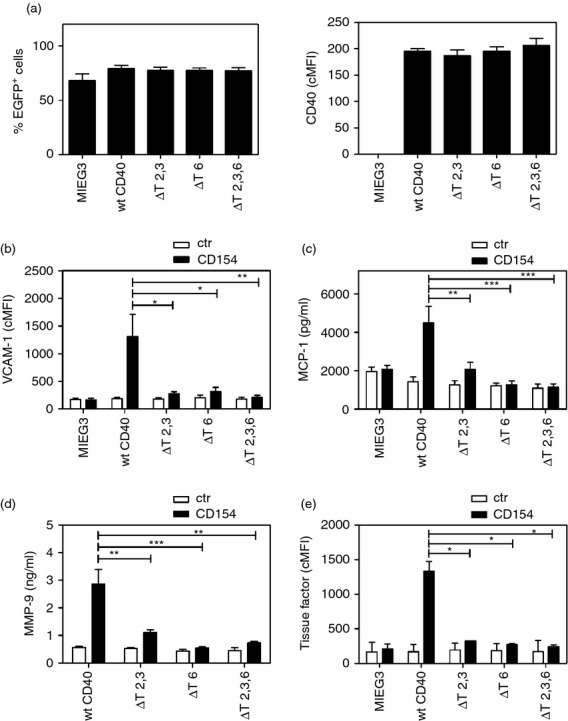
Both CD40––tumour necrosis factor (TNF) receptor-associated factor 2,3 (TRAF2,3) and CD40–TRAF6 binding sites are required for optimal CD154-induced up-regulation of vascular cell adhesion molecule 1 (VCAM-1), monocyte chemoattractant protein 1 (MCP-1), tissue factor and matrix metalloproteinase 9 (MMP-9) activity in human aortic smooth muscle cells (HASMC). Cells were transduced with MIEG3-based retroviral vectors that encode EGFP alone or EGFP and either wild-type (wt) CD40, CD40 ΔT2,3, CD40 ΔT6 or CD40 ΔT2,3,6. (a) Percentages of transduced cells (EGFP+) and corrected mean fluorescence intensity (cMFI) for CD40 on EGFP+ cells. (b–e) HASMC were incubated with or without CD154 for 24 hr. (b) Flow cytometric assessment of VCAM-1 expression on gated EGFP+ cells. (c) MCP-1 concentrations were measured by ELISA. (d) MMP-9 activity was measured using a Biotrak Activity Assay System. (e) Flow cytometric assessment of tissue factor expression on gated EGFP+ cells. Results are shown as mean ± SEM and are representative of three or four experiments. *P < 0·05; **P < 0·01; ***P < 0·001.
We examined the effect of CD40–TRAF signalling in the up-regulation of MMP-9 activity in smooth muscle cells. HASMC that expressed wt CD40 exhibited increased MMP-9 activity after CD40 stimulation (Fig. 2d). This effect was markedly inhibited by expression of CD40 ΔT2,3 or CD40 ΔT6 and ablated in cells that expressed CD40 ΔT2,3,6 (Fig. 2d). Tissue factor up-regulation on HASMC was also TRAF binding site-dependent and expression of CD40 ΔT2,3 or CD40 ΔT6 markedly impaired this response (Fig. 2e). Taken together, a mutation that prevents CD40–TRAF2,3 or CD40–TRAF6 interaction is sufficient to profoundly impair pro-inflammatory responses (VCAM-1 up-regulation and MCP-1 production), MMP-9 activity and tissue factor up-regulation in CD40-stimulated HASMC.
A mutation that prevents CD40–TRAF2,3 or CD40–TRAF6 interaction is sufficient to inhibit pro-inflammatory responses in human renal proximal tubule epithelial cells and mesangial cells
TRAF6 knockdown studies indicated that HRPTEC secrete MCP-1 that was dependent on TRAF6.24 However, these studies did not address the role of other TRAFs.24 Transduction of HRPTEC with retroviral vectors yielded similar levels of CD40 (P > 0·05; Fig. 3a). In agreement with TRAF6 knockdown studies, ablation of CD40–TRAF6 signalling markedly impaired MCP-1 production (Fig. 3b). Importantly, blockade of CD40–TRAF2,3 interaction also caused marked inhibition in MCP-1 production (Fig. 3b). ICAM-1 can promote the development of glomerulonephritis.60,61 ICAM-1 up-regulation in cells within the glomerulus is probably important for the interaction with infiltrating leucocytes and the development of glomerulonephritis.62 Moreover, patients with glomerulonephritis can exhibit ICAM-1 up-regulation in mesangial cells.63 Hence, we determined whether CD40 stimulation up-regulates ICAM-1 in HRMC and ascertained the role of TRAF signalling in such a response. CD40 stimulation up-regulated ICAM-1 on HRMC (Fig. 3c). This effect was markedly inhibited in cells that expressed CD40 ΔT2,3 or CD40 ΔT6 and was ablated in those that expressed CD40 ΔT2,3,6 (Fig. 3c). These effects were unlikely to be explained by differences in CD40 expression as cMFI for CD40 were similar among cells that expressed wt CD40 or CD40 mutants (P > 0·05; Fig. 3a). Hence, blockade of either CD40–TRAF2,3 or CD40–TRAF6 interaction is sufficient to inhibit pro-inflammatory responses not only in vascular wall cells but also in non-haematopoietic cells from the kidney.
Figure 3.
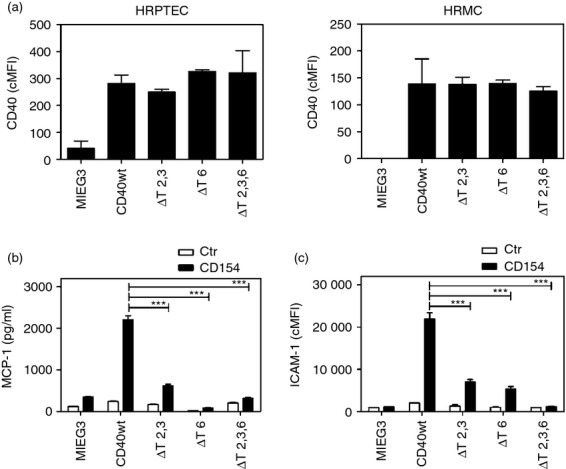
A mutation that prevents CD40–tumour necrosis factor receptor-associated factor 2,3 (TRAF2,3) or CD40–TRAF6 interaction is sufficient to inhibit pro-inflammatory responses in human renal proximal tubule epithelial cells (HRPTEC) and human renal mesangial cells (HRMC). HRPTEC and HRMC were transduced with MIEG3-based retroviral vectors that encode EGFP alone or EGFP and either wild-type (wt) CD40, CD40 ΔT2,3, CD40 ΔT6 or CD40 ΔT2,3,6. (a) Corrected mean fluorescence intensity (cMFI) for CD40 on EGFP+ cells. (b, c) Cells were incubated with or without CD154 for 24 hr. (b) Monocyte chemoattractant protein 1 (MCP-1) production by HRPTEC. MCP-1 concentrations were measured by ELISA. (c) Flow cytometric assessment of intercellular adhesion molecule 1 (ICAM-1) expression on gated EGFP+ HRMC. Results are shown as mean ± SEM and are representative of three experiments. ***P < 0·001.
The CD40–TRAF6 binding site is dominant for the induction of pro-inflammatory responses in human monocytic cells
We examined the effect of CD40–TRAF binding sites in various pro-inflammatory responses and tissue factor up-regulation in human monocytic cells. The human monocytic line MonoMac6 (lacks endogenous CD40) was transduced with MIEG3-based retroviral vectors. EGFP+ cells were sorted and maintained as lines (> 95% EGFP+). The cMFI for CD40 was similar for all MonoMac6 cells transduced with a CD40 encoding retroviral vector (Fig. 4a; P > 0·5). Incubation with CD154 caused up-regulation of ICAM-1, MCP-1 and tissue factor on monocytic cells that expressed wt CD40 (Fig. 4b–d). Expression of CD40 ΔT6 markedly inhibited these responses while expression of CD40 ΔT2,3 had a more moderate inhibitory effect (Fig. 4b–d). Hence, in contrast to non-haematopoietic cells, the CD40–TRAF6 binding site plays the dominant role in the induction of pro-inflammatory and pro-thrombotic responses in human monocytic cells. Table 1 summarizes the inhibitory effects of expression of CD40 mutants on the various cellular responses tested.
Figure 4.
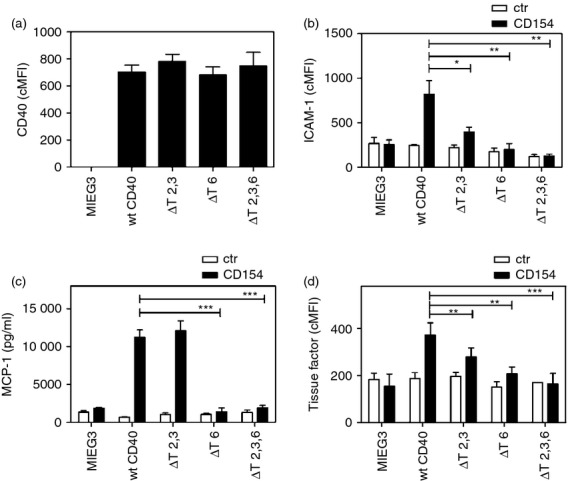
The CD40–tumour necrosis factor receptor-associated factor 6 (TRAF6) binding site is dominant for the induction of pro-inflammatory responses in human monocytic cells. MonoMac6 cells transduced with MIEG3-based retroviral vector that encode EGFP alone or EGFP and either wild-type (wt) CD40, CD40 ΔT2,3, CD40 ΔT6, CD40 ΔT2,3,6. (a) Corrected mean fluorescence intensity (cMFI) for CD40 on EGFP+ cells. (b–d) Cells were incubated with or without CD154 for 24 hr. (b) Flow cytometric assessment of intercellular adhesion molecule 1 (ICAM-1) expression on gated EGFP+ cells. (c) Monocyte chemoattractant protein 1 (MCP-1) concentrations were measured by ELISA. (d) Flow cytometric assessment of tissue factor expression on gated EGFP+ cells. Results are shown as mean ± SEM and are representative of three or four experiments. *P < 0·05; **P < 0·01; ***P < 0·001.
Table 1.
Effects of expression of CD40 with mutations in CD40–tumour necrosis factor (TNF) receptor-associated factor (TRAF) binding sites on up-regulation of various pro-inflammatory responses on human aortic endothelial cells, aortic smooth muscle cells, renal proximal tubule epithelial cells, renal mesangial cells and human monocytic cells
| ΔTRAF2,3 | ΔTRAF6 | ΔTRAF2,3,6 | ||
|---|---|---|---|---|
| Aortic endothelial cells | VCAM-1 | 75·3 ± 8·3 | 83·4 ± 3·2 | 97·1 ± 1·3 |
| ICAM-1 | 74·8 ± 7·1 | 80·2 ± 3·4 | 97·1 ± 0·6 | |
| MCP-1 | 93·6 ± 4·6 | 56·3 ± 6·9 | 95·7 ± 4·3 | |
| Tissue factor | 72·6 ± 9·6 | 76·1 ± 4·0 | 99·0 ± 0·6 | |
| Aortic smooth muscle cells | VCAM-1 | 89·9 ± 2·0 | 87·5 ± 5·4 | 96·6 ± 0·8 |
| MCP-1 | 81·7 ± 6·9 | 92·6 ± 3·9 | 99·3 ± 0·4 | |
| MMP-9 | 72·0 ± 5·0 | 91·2 ± 2·1 | 97·3 ± 2·7 | |
| Tissue factor | 88·4 ± 7·5 | 92·8 ± 7·2 | 94 3 ± 3·3 | |
| Renal epithelial cells | MCP-1 | 84·2 ± 1·1 | 96·9 ± 1 | 95·3 ± 1·1 |
| Mesangial cells | ICAM-1 | 65·4 ± 7·2 | 70·0 ± 3·6 | 98·5 ± 0·8 |
| Monocytic cells | ICAM-1 | 71·2 ± 1·1 | 91·2 ± 5·1 | 97·7 ± 1·8 |
| MCP-1 | 10·9 ± 5·1 | 98·2 ± 1·8 | 99·2 ± 0·8 | |
| Tissue factor | 15·0 ± 7·6 | 93·5 ± 4·2 | 97·7 ± 2·3 |
Data are expressed as mean inhibition compared with cells that expressed wt CD40. Results are averages ± SEM from 3–6 experiments
ICAM-1, intercellular adhesion molecule 1; MCP-1, monocyte chemoattractant protein 1; VCAM-1, vascular cell adhesion molecule 1.
CD40–TRAF blocking peptides inhibit pro-inflammatory responses in human non-haematopoietic cells
Peptides can readily block protein–protein interactions.64 CD40–TRAF signalling can be inhibited by peptides that consist of the amino acid sequence of the TRAF2,3 and TRAF6 binding sites of CD40 fused to a membrane transduction domain to make the peptides cell-permeable.19,46 To examine the effects of pharmacological inhibition of CD40–TRAF signalling, we used peptides that consisted of the amino acid sequence of the TRAF2,3 or the TRAF6 binding sites of CD40 linked to TAT47–57. First, we confirmed that peptides blocked CD40–TRAF signalling using reporter mouse endothelial cells that express a human–mouse CD40 chimera (extracellular human CD40 and intracellular mouse CD40) that signalled either through the TRAF2,3 binding site (hmCD40 ΔT6) or the TRAF6 binding site (hmCD40 ΔT2,3).46 These cells also express an NF-κB response element that drives a luciferase reporter gene.46 Reporter cells were incubated with peptides followed by stimulation with human CD154. Peptides did not affect cell viability (not shown). The CD40–TRAF2,3 blocking peptide inhibited NF-κB activity in cells that express CD40 that signals through the TRAF2,3 binding site but had no significant effect on cells that express CD40 that signals through the TRAF6 binding site (Fig. 5a). The CD40–TRAF6 blocking peptide had the reverse effect (Fig. 5a).
Figure 5.

CD40–tumour necrosis factor (TNF) receptor-associated factor (TRAF) blocking peptides inhibit pro-inflammatory responses in human non-haematopoietic cells. (a) mHEVc endothelial cells that express a nuclear factor-κB (NF-κB) response element that drives transcription of a luciferase reporter plus either hmCD40 ΔT2,3 or hmCD40 ΔT6 were pre-incubated with CD40–TRAF2,3 blocking peptide, CD40–TRAF6 blocking peptide or control peptide (all at 1 μm) or medium alone followed by stimulation with human CD154. Data are expressed as fold-increase in normalized luciferase activity in cells stimulated with CD154 compared with cells treated with respective peptide in the absence of CD154. (b, c) Human aortic endothelial cells (HAEC) were incubated with control peptide (Ctr P), CD40–TRAF2,3 blocking peptide (T2,3 BP) or CD40–TRAF6 blocking peptide (T6 BP) all at 1 μm. After 3 hr, HAEC were stimulated with CD154 (b) or interferon-γ (IFN-γ)/TNF-α (c). Vascular cell adhesion molecule 1 (VCAM-1) expression was examined by flow cytometry after 24 hr. (d) Human aortic smooth muscle cells (HASMC) transduced with the wild-type (wt) CD40 retroviral vector were incubated with peptides (10 μm) followed by stimulation with CD154 for 24 hr. MCP-1 production was examined by ELISA. Results are shown as mean ± SEM and are representative of three experiments. **P < 0·01; ***P < 0·001.
Next, we tested the effects of peptides on adhesion molecule up-regulation induced by CD154. While the control peptide did not affect VCAM-1 up-regulation, CD40–TRAF2,3 and CD40–TRAF6 blocking peptides impaired up-regulation of VCAM-1 in response to CD154 (Fig. 5b). The effects were specific because CD40–TRAF blocking peptides did not affect VCAM-1 up-regulation induced by interferon-γ/TNF-α (Fig. 5c). The CD40–TRAF2,3 and CD40–TRAF6 blocking peptides also inhibited MCP-1 production by CD154-treated HASMC (Fig. 5d). Taken together, a pharmacological approach that impairs either CD40–TRAF2,3 or CD40–TRAF6 signalling inhibits pro-inflammatory responses in vitro.
CD40–TRAF6 signalling appears to play the dominant role in CD154-induced maturation of human dendritic cells
Dendritic cells play a key role in antigen presentation and production of IL-12. It is well established that CD40–TRAF6 but not CD40–TRAF2,3 signalling mediates IL-12 production.43 Although less clear, CD40–TRAF signalling appears to promote co-stimulatory ligand up-regulation in mouse dendritic cells.43 We used CD40–TRAF blocking peptides to examine the role of TRAF signalling in maturation of human dendritic cells. Human monocyte-derived dendritic cells incubated with CD154 up-regulated CD80, CD86 and HLA-DR, as well as acquired expression of the maturation marker CD83 (Fig. 6). While the CD40–TRAF2,3 blocking peptide had no significant effects on up-regulation of these molecules, their up-regulation was impaired by the CD40–TRAF6 blocking peptide (Fig. 6). Hence, CD40-driven maturation of human dendritic cells appears to be mediated mainly by CD40–TRAF6 signalling.
Figure 6.
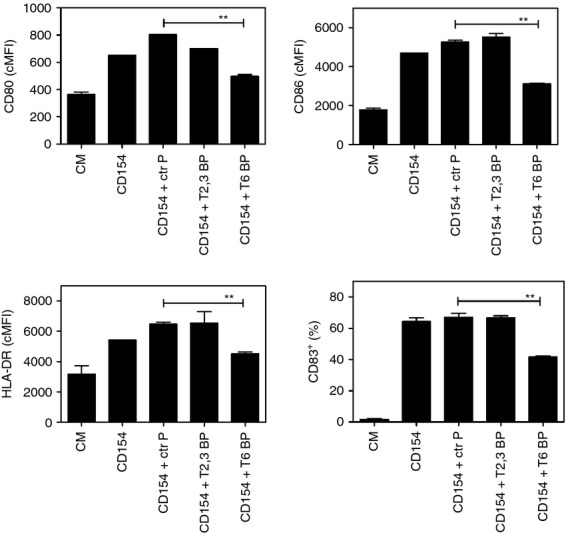
CD40–tumour necrosis factor (TNF) receptor-associated factor 6 (TRAF6) signalling appears to play the dominant role in CD154-induced maturation of human dendritic cells. Human monocyte-derived dendritic cells were incubated with control peptide (Ctr P), CD40–TRAF2,3 blocking peptide (T2,3 BP) or CD40–TRAF6 blocking peptide (T6 BP) all at 10 μm. After 3 hr, dendritic cells were stimulated with CD154. Expression of CD80, CD86, HLA-DR and CD83 was examined by flow cytometry after 24 hr. Results are shown as mean ± SEM and are representative of three experiments. **P < 0·01.
Discussion
The need to identify approaches to control CD40-driven inflammatory disorders by blocking the effects of this molecule stresses the importance of characterizing the cascades downstream of CD40 that mediate pro-inflammatory responses. Little is known about the role of adaptor proteins required for cellular responses triggered by CD40 in non-haematopoietic cells. We report that blockade of either CD40–TRAF2,3 or CD40–TRAF6 was sufficient to markedly impair CD40-induced VCAM-1/ICAM-1 up-regulation and MCP-1 production in HAEC, HASMC, HRPTEC and HRMC, tissue factor up-regulation in HAEC and HASMC, as well as increased MMP-9 activity in HASMC. These findings suggest that cooperation between the two pathways is required for optimal cellular responses. Studies with human monocytic cells also showed that TRAF signalling is needed for ICAM-1 and tissue factor up-regulation, as well as production of MCP-1. However, in contrast to non-haematopoietic cells, the CD40–TRAF6 pathway played the more dominant role in promoting MCP-1 and tissue factor up-regulation. Moreover, maturation of human dendritic cells appeared to be driven mainly by the CD40–TRAF6 pathway.
In contrast to TRAF knock-down or expression of dominant negative TRAFs, the use of cells that express either wt CD40 or CD40 with mutations that affect the TRAF2,3 and/or the TRAF6 binding sites allows the selective study of TRAF signalling in cellular responses triggered by CD40 ligation. Although the effectiveness of these mutations in preventing TRAF recruitment has not been confirmed in primary non-haematopoietic cells, the hCD40EEAA mutant used in this study has been proven to block recruitment of TRAF6 in 293-T cells.57 Some in vitro and in vivo studies examined CD40–TRAF2,3 signalling using CD40 with a point mutation in the proximal TRAF2,3 binding site (PxQxA). Although this mutation impairs TRAF2 recruitment to CD40, TRAF2 recruitment still takes place,40,59 a process dependent on the distal TRAF2 binding site.40 In contrast, the CD40 deletion mutant used in this study has been shown to ablate TRAF2 recruitment in B cells.59 The level of CD40 expression is another important aspect that has to be taken into account in the interpretation of data obtained in experimental systems that use wt CD40 or CD40 mutants.47 Differences in the responses induced by wt CD40 and CD40 mutants may be the result of significant dissimilarities in CD40 expression rather than the effects of mutations in TRAF binding sites. To address this potential caveat, the levels of CD40 expression were assessed in all the experiments reported in this study and no significant differences in CD40 expression were detected among cells that expressed wt CD40 or CD40 mutants. The approach taken in these studies established the critical role of CD40–TRAF signalling in a broad variety of cellular responses linked to inflammatory disorders and show that the role of CD40–TRAF signalling can be cell-type specific. In contrast to monocytic cells, both the CD40–TRAF2,3 and CD40–TRAF6 binding sites are required to optimally promote various pro-inflammatory responses in HAEC, HASMC, HRPTEC and HRMC. Importantly, blockade of CD40–TRAF2,3 interaction was sufficient to markedly impair adhesion molecule up-regulation, chemokine production, tissue factor and MMP-9 activity up-regulation. Of relevance, B cells have also been shown to signal through both the CD40–TRAF2,3 and CD40–TRAF6 binding sites.42,47,57,59
The importance of CD40–TRAF2,3 signalling in promoting adhesion molecule up-regulation and MCP-1 production is supported not only by studies that used cells that expressed CD40 with mutations in TRAF binding sites but also by treatment with CD40–TRAF blocking peptides. These findings are relevant to the pathogenesis of various inflammatory diseases including atherosclerosis, vascular injury, inflammatory bowel disease and glomerulonephritis because animal studies have revealed the pivotal role of VCAM-1, ICAM-1 and MCP-1 in these disorders60,61,65–69 and CD40 expression in the vascular wall is required for neointima formation after arterial injury.13 Blockade of CD40–TRAF2,3 signalling also inhibited increased MMP-9 activity in HASMC. MMP are believed to play a complex role in atherosclerosis and mechanical arterial injury that appears to include promoting thinning of the fibrous cap in atheromas, plaque destabilization and subsequent rupture.70 Finally, blockade of CD40–TRAF2,3 signalling significantly blunted tissue factor up-regulation in HAEC and HASMC, findings that may have implications to thrombosis. These results raise the possibility that the CD40–TRAF2,3 pathway at the level of non-haematopoietic cells may play an important role in the development of inflammatory disorders and complications that can accompany vascular inflammation.
Studies in transgenic mice indicated that CD40–TRAF6 signalling at the level of MHC II+ cells plays an important role in progression of atherosclerosis and neointima formation after vascular injury.35,36 Our studies in human monocytic cells indeed show that the TRAF6 binding site of CD40 plays the dominant role in the up-regulation of molecules that are key for the development of vascular inflammation and probably thrombosis (MCP-1 and tissue factor). Moreover, the CD40–TRAF6 pathway is responsible for production of TNF-α, IL-1 and IL-6 in response to CD40 stimulation in macrophages.19 Blockade of CD40–TRAF6 signalling has been proposed as an approach to control CD40-driven inflammation. However, this signalling pathway also controls various cellular immune responses that are important for protection against infections. Only the CD40–TRAF6 pathway is responsible for the induction of macrophage antimicrobial activities mediated by autophagy and NOS2 up-regulation.45,46,58 The TRAF6 binding site of CD40 is required for CD40-driven production of IL-12 in dendritic cells.43 The reported role of TRAF signalling in CD40-driven up-regulation of co-stimulatory ligands in mouse dendritic cells is less clear. Upon incubation with CD154+ fibroblasts, mouse dendritic cells that express human CD40 up-regulated co-stimulatory ligands even if CD40 could not signal through TRAFs.43 TRAF signalling appeared to promote co-stimulatory ligand up-regulation only in the setting of suboptimal CD40 ligation.43 Using human monocyte-derived dendritic cells we report that only the CD40–TRAF6 pathway appears to promote dendritic cell maturation. Although the role of the CD40–TRAF6 pathway in other subsets of dendritic cells remains to be studied, it is clear that multiple lines of evidence support the notion of a crucial role of this pathway in mechanisms for host protection.
Using CD40–TRAF2,3 and CD40–TRAF6 blocking peptides as a model of pharmacological inhibition of CD40–TRAF signalling, our work indicates that targeting either of these pathways inhibits CD40-induced pro-inflammatory responses in various non-haematopoietic cells. A CD40–TRAF6 blocking peptide has also been reported to inhibit production of TNF-α, IL-1β and IL-6 by human monocytes.19 However, the inhibitory effect of CD40–TRAF6 blocking peptides on CD40-induced dendritic cell maturation (this study) and macrophage anti-microbial activities45,46,58 support the concern that pharmacological inhibition of CD40–TRAF6 signalling may cause significant immunosuppression. Blockade of CD40–TRAF2,3 signalling may lack major effects on cell-mediated immune responses in myeloid cells although it could inhibit humoral responses dependent on CD40–TRAF2,3 signalling in B cells. Given that CD40 expressed on non-haematopoietic cells is pivotal to the development of inflammation, our findings raise the possibility that pharmacological inhibition of CD40–TRAF2 signalling may be useful for the management of CD40-dependent inflammatory disorders with a likely reduced risk of impairing cell-mediated immunity. This could be accomplished by administration of cell-permeable peptides71 or small molecules that impair CD40–TRAF2 interaction.
Acknowledgments
This work was supported by NIH Grant EY019250, grants 5-2008-233 and 1-2009-204 from the Juvenile Diabetes Foundation International and grants 0555327B and 0755336B from the American Heart Association Ohio Valley Affiliate, all to CSS as well as by the Flow Cytometry Core Facility of the Comprehensive Cancer Center of Case Western Reserve University and University Hospitals of Cleveland (P30 CA43703).
Glossary
- cMFI
corrected mean fluorescence intensity
- EGFP
enhanced green fluorescence protein
- HAEC
human aortic endothelial cells
- HASMC
human aortic smooth muscle cells
- HRPTEC
human renal proximal tubule epithelial cells
- HRMC
human renal mesangial cells
- ICAM-1
intercellular adhesion molecule-1
- IL-12
interleukin-12
- MCP-1
monocyte chemoattractant protein-1
- MMP
metalloproteinase
- NF-κB
nuclear factor-κB
- PBMC
peripheral blood mononuclear cells
- TNF
tumour necrosis factor
- TRAF
tumour necrosis factor receptor associated factor
- VCAM-1
vascular cell adhesion molecule 1
- wt
wild-type
Disclosures
MCS and CSS have applied for a patent for the use of CD40–TRAF blocking peptides to modulate inflammatory disorders.
Author's contribution
JACP, JAG, IS and CSS performed the experiments. MCS and CSS designed the study. JACP and CSS wrote the article.
References
- 1.Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–35. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 2.van Kooten C, Banchereau J. CD40–CD40 ligand. J Leukoc Biol. 2000;67:2–17. doi: 10.1002/jlb.67.1.2. [DOI] [PubMed] [Google Scholar]
- 3.Karmann K, Hughes CCW, Schechner J, Fanslow WC, Pober JS. CD40 on human endothelial cells: inducibility by cytokines and functional regulation of adhesion molecule expression. Proc Natl Acad Sci USA. 1995;92:4342–6. doi: 10.1073/pnas.92.10.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mach F, Schonbeck U, Sukhova GK, Bourcier T, Bonnefoy JY, Pober JS, Libby P. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells and macrophages: implications for CD40–CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci USA. 1997;94:1931–6. doi: 10.1073/pnas.94.5.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu H, Zhang X, Mannon RB, Kirk AD. Platelet-derived or soluble CD154 induces vascularized allograft rejection independent of cell-bound CD154. J Clin Invest. 2006;116:769–74. doi: 10.1172/JCI27155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peters AL, Stunz LL, Bishop GA. CD40 and autoimmunity: the dark side of a great activator. Semin Immunol. 2009;21:293–300. doi: 10.1016/j.smim.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yellin MJ, D'Agati V, Parkinson G, et al. Immunohistologic analysis of renal CD40 and CD40L expression in lupus nephritis and other glomerulonephritis. Arthritis Rheum. 1997;40:124–34. doi: 10.1002/art.1780400117. [DOI] [PubMed] [Google Scholar]
- 8.Battaglia E, Biancone L, Resegotti A, Emanuelli G, Ruggero Fronda G, Camussi G. Expression of CD40 and its ligand, CD40L, in intestinal lesions of Crohn's disease. Am J Gastroenterol. 1999;94:3279–84. doi: 10.1111/j.1572-0241.1999.01538.x. [DOI] [PubMed] [Google Scholar]
- 9.Hakkinen T, Karkola K, Yla-Herttualla S. Macrophages, smooth muscle cells, endothelial cells, and T-cells express CD40 and CD40L in fatty streaks and more advanced human atherosclerotic lesions. Virchows Arch. 2000;437:396–405. doi: 10.1007/s004280000239. [DOI] [PubMed] [Google Scholar]
- 10.Bruemmer D, Riggers U, Holzmeister J, et al. Expression of CD40 in vascular smooth muscle cells and macrophages is associated with early development of human atherosclerotic lesions. Am J Cardiol. 2001;87:21–7. doi: 10.1016/s0002-9149(00)01266-2. [DOI] [PubMed] [Google Scholar]
- 11.Ohashi Y, Okazaki H, Sato T, Miura S, Amada N, Yamaguchi M. Function of CD40/CD40L in peritubular capillary renal allograft rejection. Transplant Proc. 2001;33:407–8. doi: 10.1016/s0041-1345(00)02070-4. [DOI] [PubMed] [Google Scholar]
- 12.Borcherding F, Nitschke M, von Smolinski D, Bieber K, van Kooten C, Lehnert H, Fellermann K, Buning J. The CD40–CD40L pathway contributes to the proinflammatory function of intestinal epithelial cells in inflammatory bowel disease. Am J Pathol. 2010;176:1816–27. doi: 10.2353/ajpath.2010.090461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song Z, Jin R, Yu S, Nanda A, Granger DL, Li G. Crucial role of CD40 signaling in vascular wall cells in neointima formation and vascular remodeling after vascular interventions. Atheroscler Thromb Vasc Biol. 2012;32:50–64. doi: 10.1161/ATVBAHA.111.238329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaweco AS, Mitchell BL, Lucas BA, McClatchey KD, van Thiel DH. CD40 expression on graft infiltrates and parenchymal CD154 (CD40L) induction in human chronic renal allograft rejection. Kidney Int. 1999;55:1543–52. doi: 10.1046/j.1523-1755.1999.00379.x. [DOI] [PubMed] [Google Scholar]
- 15.Danese S, de la Motte C, Sturm A, Vogel JD, West GA, Strong SA, Katz JA, Fiocchi C. Platelets trigger a CD40-dependent inflammatory response in the microvasculature of inflammatory bowel disease patients. Gastroenterology. 2003;124:1249–64. doi: 10.1016/s0016-5085(03)00289-0. [DOI] [PubMed] [Google Scholar]
- 16.Davis B, Zou M-H. CD40 ligand-dependent tyrosine nitration of prostacyclin synthase in vivo. Circulation. 2005;112:2184–92. doi: 10.1161/CIRCULATIONAHA.105.553206. [DOI] [PubMed] [Google Scholar]
- 17.Kornbluth RS, Kee K, Richman DD. CD40 ligand (CD154) stimulation of macrophages to produce HIV-1-suppressive β-chemokines. Proc Natl Acad Sci USA. 1998;95:5205–10. doi: 10.1073/pnas.95.9.5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukundan L, Milhorn DM, Matta B, Suttles J. CD40-mediated activation of vascular smooth muscle cell chemokine production through a Src-initiated. MAPK-dependent pathway. Cell Signal. 2004;16:375–84. doi: 10.1016/j.cellsig.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 19.Mukundan L, Bishop GA, Head KZ, Zhang L, Wahl L, Suttles J. TNF receptor-associated factor 6 is an essential mediator of CD40-activated proinflammatory pathways in monocytes and macrophages. J Immunol. 2005;174:1081–90. doi: 10.4049/jimmunol.174.2.1081. [DOI] [PubMed] [Google Scholar]
- 20.Mach F, Schonbeck U, Bonnefoy JY, Pober JS, Libby P. Activation of monocyte/macrophage functions related to acute atheroma complication by ligation of CD40. Induction of collagenase, stromelysin, and tissue factor. Circulation. 1997;96:396–9. doi: 10.1161/01.cir.96.2.396. [DOI] [PubMed] [Google Scholar]
- 21.Schonbeck U, Mach F, Sukhova GK, Murphy C, Bonnefoy JY, Fabunmi RP, Libby P. Regulation of matrix metalloproteinase expression in human vascular smooth muscle cells by T-lymphocytes. A role for CD40 signaling in plaque rupture? Circ Res. 1997;81:448–54. doi: 10.1161/01.res.81.3.448. [DOI] [PubMed] [Google Scholar]
- 22.Schonbeck U, Mach F, Sukhova GK, Herman M, Graber P, Kehry MR, Libby P. CD40 ligation induces tissue factor expression in human vascular smooth muscle cells. Am J Pathol. 2000;156:7–14. doi: 10.1016/S0002-9440(10)64699-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deckers JGM, de Haij S, van der Woude FJ, van der Kooij SW, Daha MR, van Kooten C. IL-4 and IL-13 augment cytokine- and CD40-induced RANTES production by human renal tubular epithelial cells in vitro. J Am Soc Nephrol. 1998;9:1187–93. doi: 10.1681/ASN.V971187. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Nord EP. CD40 ligation stimulates MCP-1 and IL-8 production, TRAF6 recruitment, and MAPK activation in proximal tubule cells. Am J Physiol Renal Physiol. 2002;282:F1020–33. doi: 10.1152/ajprenal.00291.2001. [DOI] [PubMed] [Google Scholar]
- 25.Mohan C, Shi Y, Laman JD, Datta SK. Interaction between CD40 and its ligand gp39 in the development of murine lupus nephritis. J Immunol. 1995;154:1470–80. [PubMed] [Google Scholar]
- 26.Stuber E, Strober W, Neurath M. Blocking the CD40L–CD40 interaction in vivo specifically prevents the priming of T helper 1 cells through the inhibition of interleukin-12 secretion. J Exp Med. 1996;183:693–8. doi: 10.1084/jem.183.2.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kirk AD, Harlan DM, Armstrong NM, et al. CTLA4-Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc Natl Acad Sci USA. 1997;94:8789–94. doi: 10.1073/pnas.94.16.8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mach F, Schonbeck U, Sukhova GK, Atkinson E, Libby P. Reduction of atherosclerosis in mice by inhibition of CD40 signaling. Nature. 1998;394:200–3. doi: 10.1038/28204. [DOI] [PubMed] [Google Scholar]
- 29.Schonbeck U, Sukhova GK, Shimizu K, Mach F, Libby P. Inhibition of CD40 signaling limits evolution of established atherosclerosis in mice. Proc Natl Acad Sci USA. 2000;97:7458–63. doi: 10.1073/pnas.97.13.7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lutgens E, Cleutjens KBJM, Heeneman S, Koteliansky VE, Burkly LC, Daemen MJAP. Both early and delayed anti-CD40L antibody treatment induces a stable plaque phenotype. Proc Natl Acad Sci USA. 2000;97:7464–89. doi: 10.1073/pnas.97.13.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Z, Geboes K, Colpaert S, et al. Prevention of experimental colitis in SCID mice reconstituted with CD45RBhigh CD4+ T cells by blocking the CD40–CD154 interactions. J Immunol. 2000;164:6005–14. doi: 10.4049/jimmunol.164.11.6005. [DOI] [PubMed] [Google Scholar]
- 32.Boumpas DT, Furie R, Manzi S, Ilei GG, Wallace DJ, Balow JE, Vaishnaw A. A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003;48:719–27. doi: 10.1002/art.10856. [DOI] [PubMed] [Google Scholar]
- 33.Ruth A-J, Kitching AR, Semple TJ, Tipping PG, Holdsworth SR. Intrinsic renal cell expression of CD40 directs Th1 effectors inducing experimental crescentic glomerulonephritis. J Am Soc Nephrol. 2003;14:2813–22. doi: 10.1097/01.asn.0000091381.60059.fb. [DOI] [PubMed] [Google Scholar]
- 34.Vowinkel T, Anthoni C, Wood KC, et al. CD40–CD40 ligand mediates the recruitment of leukocytes and platelets in the inflamed murine colon. Gastroenterology. 2007;132:955–65. doi: 10.1053/j.gastro.2006.12.027. [DOI] [PubMed] [Google Scholar]
- 35.Donners MMPC, Beckers L, Lievens D, et al. The CD40–TRAF6 axis is the key regulator of the CD40/CD40L system in neointima formation and arterial remodeling. Blood. 2008;111:4596–604. doi: 10.1182/blood-2007-05-088906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lutgens E, Lievens D, Beckers L, et al. Deficient CD40–TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J Exp Med. 2010;207:391–404. doi: 10.1084/jem.20091293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bishop GA, Hostager BS, Brown KD. Mechanisms of TNF receptor-associated factor (TRAF) regulation in B lymphocytes. J Leukoc Biol. 2002;72:19–23. [PubMed] [Google Scholar]
- 38.Hanissian SH, Geha R. Jak3 is associated with CD40 and is critical for CD40 induction of gene expression in B cells. Immunity. 1997;6:379–88. doi: 10.1016/s1074-7613(00)80281-2. [DOI] [PubMed] [Google Scholar]
- 39.Pullen SS, Miller HG, Everdeen DS, Dang TT, Crute JJ, Kehry MR. CD40-tumor necrosis factor receptor-associated factor (TRAF) interactions: regulation of CD40 signaling through multiple TRAF binding sites and TRAF hetero-oligomerization. Biochemistry. 1998;37:11836–45. doi: 10.1021/bi981067q. [DOI] [PubMed] [Google Scholar]
- 40.Lu LF, Cook WJ, Lin LL, Noelle RJ. CD40 signaling through a newly identified tumor necrosis factor receptor-associated factor 2 (TRAF2) binding site. J Biol Chem. 2003;278:45414–18. doi: 10.1074/jbc.M309601200. [DOI] [PubMed] [Google Scholar]
- 41.Baccam M, Bishop GA. Membrane-bound CD154, but not anti-CD40 mAb, induces NF-κB independent B cell IL-6 production. Eur J Immunol. 1999;29:3855–66. doi: 10.1002/(SICI)1521-4141(199912)29:12<3855::AID-IMMU3855>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 42.Ahonen CL, Manning EM, Erickson LD, O'Connor BP, Lind EF, Pullen SS, Kehry MR, Noelle RJ. The CD40–TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells. Nat Immunol. 2002;3:451–6. doi: 10.1038/ni792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mackey MF, Wang Z, Eichelberg K, Germain RN. Distinct contributions of different CD40 TRAF binding sites to CD154-induced dendritic cell maturation and IL-12 secretion. Eur J Immunol. 2003;33:779–89. doi: 10.1002/eji.200323729. [DOI] [PubMed] [Google Scholar]
- 44.Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest. 2006;116:2366–77. doi: 10.1172/JCI28796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subauste CS, Andrade RM, Wessendarp M. CD40–TRAF6 and autophagy-dependent anti-microbial activity in macrophages. Autophagy. 2007;3:245–8. doi: 10.4161/auto.3717. [DOI] [PubMed] [Google Scholar]
- 46.Portillo J-AC, Muniz-Feliciano L, Subauste MC, Heinzel FP, Subauste CS. CD40 and TNF-α synergize to induce nitric oxide synthase in macrophages. Immunology. 2012;135:140–50. doi: 10.1111/j.1365-2567.2011.03519.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jabara HH, Laouini D, Tsitsikov E, et al. The binding site for TRAF2 and TRAF3 but not for TRAF6 is essential for CD40-mediated immunoglobulin class switching. Immunity. 2002;17:265–76. doi: 10.1016/s1074-7613(02)00394-1. [DOI] [PubMed] [Google Scholar]
- 48.Davies CC, Mak TW, Young LS, Eliopoulos AG. TRAF6 is required for TRAF2-dependent CD40 signal transduction in non-hemopoietic cells. Mol Cel Biol. 2005;25:9806–19. doi: 10.1128/MCB.25.22.9806-9819.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pukerson JM, Smith RS, Pollock SJ, Phipps RP. The TRAF6, but not the TRAF2/3, binding domain of CD40 is required for cytokine production in human lung fibroblasts. Eur J Immunol. 2005;35:2920–8. doi: 10.1002/eji.200526219. [DOI] [PubMed] [Google Scholar]
- 50.Schwabe RF, Schnabl B, Kweon YO, Brenner DA. CD40 activates NF-κB and c-Jun N-terminal kinase and enhances chemokine secretion on activated human hepatic stellate cells. J Immunol. 2001;166:6812–19. doi: 10.4049/jimmunol.166.11.6812. [DOI] [PubMed] [Google Scholar]
- 51.Zirlik A, Bavendiek U, Libby P, et al. TRAF-1, -2, -3, -5, and -6 are induced in atherosclerotic plaques and differentially mediate proinflammatory functions of CD40L in endothelial cells. Atheroscler Thromb Vasc Biol. 2007;27:1101–7. doi: 10.1161/ATVBAHA.107.140566. [DOI] [PubMed] [Google Scholar]
- 52.Ogolla P, Portillo J-AC, White CL, Patel K, Lamb B, Sen GC, Subauste CS. The protein kinase double-stranded RNA-dependent (PKR) enhances protection against disease cause by a non-viral pathogen. PLoS Pathog. 2013;9:e100557. doi: 10.1371/journal.ppat.1003557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Subauste CS, Wessendarp M. Human dendritic cells discriminate between viable and killed Toxoplasma gondii tachyzoites: dendritic cell activation after infection with viable parasites results in CD28 and CD40 ligand signaling that controls IL-12-dependent and -independent T cell production of IFN-γ. J Immunol. 2000;165:1498–505. doi: 10.4049/jimmunol.165.3.1498. [DOI] [PubMed] [Google Scholar]
- 54.Haswell LE, Glennie MJ, Al-Shamkhani A. Analysis of the oligomeric requirement for signaling by CD40 using soluble multimeric forms of its ligand, CD154. Eur J Immunol. 2001;31:3094–100. doi: 10.1002/1521-4141(2001010)31:10<3094::aid-immu3094>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 55.Portillo J-AC, Van Grol J, Zheng L, et al. CD40 mediates retinal inflammation and neuro-vascular degeneration. J Immunol. 2008;181:8719–26. doi: 10.4049/jimmunol.181.12.8719. [DOI] [PubMed] [Google Scholar]
- 56.Hsing Y, Hostager BS, Bishop GA. Characterization of CD40 signaling determinants regulating nuclear factor-κB activation in B lymphocytes. J Immunol. 1997;159:4898–906. [PubMed] [Google Scholar]
- 57.Jalukar SV, Hostager BS, Bishop GA. Characterization of the roles of TNF receptor-associated factor 6 in CD40-mediated B lymphocyte effector functions. J Immunol. 2000;164:623–30. doi: 10.4049/jimmunol.164.2.623. [DOI] [PubMed] [Google Scholar]
- 58.Andrade RM, Wessendarp M, Portillo J-AC, Yang J-Q, Gomez FJ, Durbin JE, Bishop GA, Subauste CS. TRAF6 signaling downstream of CD40 primes macrophages to acquire anti-microbial activity in response to TNF-α. J Immunol. 2005;175:6014–21. doi: 10.4049/jimmunol.175.9.6014. [DOI] [PubMed] [Google Scholar]
- 59.Haxhinasto SA, Hostager BS, Bishop GA. Cutting Edge: molecular mechanisms of synergy between CD40 and the B cell antigen receptor: role for TNF receptor-associated factor 2 in receptor interaction. J Immunol. 2002;169:1145–9. doi: 10.4049/jimmunol.169.3.1145. [DOI] [PubMed] [Google Scholar]
- 60.Nishikawa K, Guo YJ, Miyasaka M, Tamatani T, Collins AB, Sy MS, McCluskey RT, Andres G. Antibodies to intercellular adhesion molecule 1/lymphocyte function-associated antigen 1 prevent crescent formation in rat autoimmune glomerulonephritis. J Exp Med. 1993;177:667–77. doi: 10.1084/jem.177.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Okada S, Shikata K, Matsuda M, et al. Intercellular adhesion molecule-1-deficient mice are resistant against renal injury after induction of diabetes. Diabetes. 2003;52:2586–93. doi: 10.2337/diabetes.52.10.2586. [DOI] [PubMed] [Google Scholar]
- 62.Baran D, Vendeville B, Ogborn M, Katz N. Cell adhesion molecule expression in murine lupus-like nephritis induced by lipopolysaccharide. Nephron. 2000;84:167–76. doi: 10.1159/000045565. [DOI] [PubMed] [Google Scholar]
- 63.Dal Canton A, Fuiano G, Sepe V, Caglioti A, Ferrone S. Mesangial expression of intercellular adhesion molecule-1 in primary glomerulosclerosis. Kidney Int. 1992;41:951–5. doi: 10.1038/ki.1992.145. [DOI] [PubMed] [Google Scholar]
- 64.Prive GG, Melnick A. Specific peptides for the therapeutic targeting of oncogenes. Curr Opin Gen Dev. 2006;16:71–7. doi: 10.1016/j.gde.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 65.Taniguchi T, Tsukada H, Nakamura H, Kodama M, Fukuda K, Saito T, Miyasaka M, Seino Y. Effects of the anti-ICAM-1 monoclonal antibody on dextran sodium sulphate-induced colitis in rats. J Gastroenterol Hepatol. 1998;13:945–9. doi: 10.1111/j.1440-1746.1998.tb00766.x. [DOI] [PubMed] [Google Scholar]
- 66.Huo Y, Hafezi-Moghadam A, Ley K. Role of vascular cell adhesion molecule-1 and fibronectin connecting segment-1 in monocyte rolling and adhesion on early atherosclerotic lesions. Circ Res. 2000;87:153–9. doi: 10.1161/01.res.87.2.153. [DOI] [PubMed] [Google Scholar]
- 67.Nageh MF, Sandberg ET, Marotti KR, Lin AH, Melchior EP, Bullard DC, Beaudet AL. Deficiency of inflammatory cell adhesion molecules protects against atherosclerosis in mice. Atheroscler Thromb Vasc Biol. 1997;17:1517–20. doi: 10.1161/01.atv.17.8.1517. [DOI] [PubMed] [Google Scholar]
- 68.Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, Rollins BJ. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–81. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- 69.Egashira K, Zhao Q, Kataoka C, et al. Importance of monocyte chemoattractant protein-1 pathway in neointimal hyperplasia after periarterial injury in mice and monkeys. Circ Res. 2002;90:1167–72. doi: 10.1161/01.res.0000020561.03244.7e. [DOI] [PubMed] [Google Scholar]
- 70.Newby AC. Matrix metalloproteinase inhibition therapy for vascular diseases. Vascular Pharmacol. 2012;56:232–44. doi: 10.1016/j.vph.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 71.Johnson RM, Harrison SD, Maclean D. Therapeutic applications of cell-penetrating peptides. Methods Mol Biol. 2011;683:535–51. doi: 10.1007/978-1-60761-919-2_38. [DOI] [PubMed] [Google Scholar]