

Abstract
MicroRNAs have been shown to be important regulators of immune homeostasis as patients with aberrant microRNA expression appeared to be more susceptible to autoimmune diseases. We recently found that miR-146a was up-regulated in activated B cells in response to rat acetylcholine receptor (AChR) α-subunit 97-116 peptide, and this up-regulation was significantly attenuated by AntagomiR-146a. Our data also demonstrated that silencing miR-146a with its inhibitor AntagomiR-146a effectively ameliorated clinical myasthenic symptoms in mice with ongoing experimental autoimmune myasthenia gravis. Furthermore, multiple defects were observed after miR-146a was knocked down in B cells, including decreased anti-R97-116 antibody production and class switching, reduced numbers of plasma cells, memory B cells and B-1 cells, and weakened activation of B cells. Previously, miR-146a has been identified as a nuclear factor-κB-dependent gene and predicted to base pair with the tumour necrosis factor receptor-associated factor 6 (TRAF6) and interleukin-1 receptor-associated kinase 1 (IRAK1) genes to regulate the immune response. However, our study proved that miR-146a inhibition had no effect on the expression of TRAF6 and IRAK1 in B cells. This result suggests that the function of miR-146a in B cells does not involve these two target molecules. We conclude that silencing miR-146a exerts its therapeutic effects by influencing the B-cell functions that contribute to the autoimmune pathogenesis of myasthenia gravis.
Keywords: experimental autoimmune myasthenia gravis, interleukin-1 receptor-associated kinase-1, miR-146a, RNA interference, tumour necrosis factor receptor-associated factor-6
Introduction
Myasthenia gravis (MG) is a prototypical autoimmune disease characterized by fluctuating weakness and excessive fatigability of voluntary muscles resulting from a blockage of the nerve impulse transmission from nerve endings to muscles.1 Clinical and experimental manifestations indicate that MG is a complex disorder involving a combination of immunopathogenesis, immunogenetics and other unknown factors.2 Evidence demonstrates that secretion of auto-antibodies against the acetylcholine receptor (AChR) in the neuromuscular junction contributes mainly to the pathogenesis of MG and its animal model, experimental autoimmune myasthenia gravis (EAMG).3,4 Serving as the precursors of plasma cells, activated B cells play a critical role in the production of pathogenic auto-antibodies and in the occurrence and development of MG. Importantly, B cells from MG patients are highly activated.5 However, the precise origin of the autoimmune response and its accurate aetiology associated with abnormal B cells remain unclear.
The emergence of microRNAs (miRNAs) provides a new perspective from which to better obtain a complete interpretation of the immune response. MicroRNAs, a family of small endogenous non-coding RNAs, have recently been identified as important regulators of gene expression. They function by repressing specific target genes at the post-transcriptional level. When miRNAs are aberrantly expressed they can contribute to pathological conditions involving the immune system, such as cancer and autoimmunity.6 A member of the miRNA family, miR-146a is regarded as a significant brake on the regulation of autoimmunity.
Located on human chromosome 5, miR-146a is predominantly expressed in immune cells, especially in monocytes, neutrophils, macrophages, dendritic cells and activated T cells, to fine-tune the immune and inflammatory responses.7,8 In human T lymphocytes, miR-146a is scarcely expressed in naive T cells, whereas it is abundantly expressed in memory T cells and is induced upon T-cell receptor stimulation.9 Recently, Boldin et al.8 reported that expression of mature miR-146a was found to be relatively high in peripheral CD19+ B cells as compared with bone marrow-derived CD19+ B cells, but the functional roles of miR-146a in these B cells remained unexplored. In this study, these functional roles are investigated.
Antagomirs are a novel class of synthetic single-stranded RNA analogues complementary to miRNAs that have been chemically modified and cholesterol-conjugated to silence the expression of endogenous miRNAs. The silencing of endogenous miRNAs by this novel method is specific, efficient and long-lasting; indicating that antagomirs are powerful tools to silence specific miRNAs in vivo and may represent a therapeutic strategy for silencing miRNAs in diseases.10
A growing body of evidence suggests that deregulation of miR-146a expression and function plays a pivotal role in the pathogenesis of human autoimmune diseases, including systemic lupus erythematosus (SLE),11 rheumatoid arthritis (RA),12 Sjögren's syndrome (SS),13 multiple sclerosis14 and type 2 diabetes.15 Our previous study has also proved that the expression of miR-146a in peripheral blood mononuclear cells of MG patients was significantly up-regulated compared with healthy controls. In vitro, we further found that transfection with an miR-146a inhibitor dramatically decreased expression of miR-146a and weakened activation of B cells.16 However, the full picture of whether and how miR-146a is involved in humoral immune responses in EAMG in vivo, as it is in vitro, has not yet been painted. Based on our in vitro results, we proposed a model suggesting that aberrant miR-146a expression in B cells is associated with the development of EAMG and AntagomiR-146a can down-regulate the abnormal miR-146a, so ameliorating EAMG.
To test this notion experimentally and to further understand the biological role of miR-146a in B cells, we detected the expression level of miR-146a in B cells following stimulation of the rat AChR α-subunit. Then, AntagomiR-146a was used as a loss-of-function approach to interfere with miR-146a expression to explore its roles in B cells. Our data indicate that miR-146a was up-regulated in activated B cells. Second we describe for the first time the functional defects of B cells after miR-146a was knocked down. Furthermore, silencing of miR-146a by its inhibitor AntagomiR-146a does effectively ameliorate clinical myasthenic symptoms in mice with ongoing EAMG. Our study suggests that miR-146a may be a promising target for the clinical therapy of MG.
Materials and methods
Mice and antigen
Female C57BL/6 mice, 6–8 weeks of age, were purchased from Laboratory Animal Co. Ltd. of Slack King (Longping Sci-tech Park, Changsha, Hunan, China) with the license no. SCXK (Hunan) 2009-0004. Animals were maintained in specific pathogen-free conditions in the animal facility of the Division of Animal Care at Central South University. All animal experiments were performed in accordance with the Principles of Laboratory Animal Care and Use Committee of Xiangya Hospital.
The antigen used to induce EAMG was a synthetic peptide corresponding to region α97-116 of the rat AChR α-subunit (R97-116). The peptides R97-116 (DGDF AIVK FTKV LLDY TGHI) were synthesized by GL Biochem Ltd., Shanghai, China (Lot number: P120409–MJ148487). Peptides were purified by reverse-phase HPLC, and their synthesis was confirmed by mass spectroscopy.
Synthesis of AntagomiR-146a
The single-stranded modified RNA inhibitor AntagomiR-146a (5′-asascccaug gaauucaguucsuscsas-Chol-3′) and its negative control (5′-ususguacuacacaaaag uascsusgs-Chol-3′) were synthesized by GenePharma (Shanghai, China). The lower case letters represent 2′-O-methyl-modified nucleotides, subscript ‘s’ represents a phosphorothioate linkage, and ‘Chol’ represents cholesterol linked through a hydroxyprolinol linkage.
Induction and clinical evaluation of EAMG
Mice were immunized by subcutaneous injection in the hind footpads with 50 μg R97-116 peptide in 200 μl complete Freund's adjuvant (CFA; Sigma, St Louis, MO) supplemented with 1 mg of H37Ra (Difco Laboratories, Franklin Lakes, NJ) on day 0 and mice were boosted on days 30 and 60 with the same peptide in CFA.17 The control mice, designated as the CFA group, received the same emulsion, except PBS was used instead of the peptide.
Clinical scoring was based on the presence of tremor, hunched posture, muscle strength and fatigability. Disease severity was expressed as follows: grade 0, mouse with normal muscle strength and activities; grade 0·5, normal at rest, weak paw grips, or backward movement after 20–30 paw grips (paw exercise) on cage top grid; grade 1, normal at rest, with muscle weakness characteristically shown by hunchback posture, and weak grip or backward movement after exercise; grade 1·5, similar to grade 1 with tremulous movements and chin on the floor and temporary difficulty in raising head; grade 2, showing grade 1·5 symptoms and signs after a minute of walking/slow running on the table top without paw exercise; grade 2·5, grade 2 weakness obvious soon after placing the mouse on the table top; grade 3, dehydrated and moribund with or without closure or secretions of the eyes with grade 2·5 weakness; grade 3·5, imminent death (death within a few hours); grade 4, dead.
Cell culture in vitro
To explore the expression of miR-146a in B cells, sensitized spleen cells were harvested from the mice after the first immunization with R97-116 peptide. B cells were then purified with immunomagnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany) from a portion of spleen cells and were collected for the quantitative PCR analysis. The other cells were subsequently cultured in RPMI-1640 (Invitrogen, Carlsbad, CA) complete medium supplemented with 10% fetal bovine serum (Invitrogen), 10 mm HEPES, 50 U/ml-50 μg/ml penicillin-streptomycin and 50 μm β-mercaptoethanol (Gibco, Grand Island, NY) in the presence of R97-116 peptide (50 μg/ml). AntagomiR-146a (100 nm) was added and AntagomiR negative control was used as the control. After a culture of 24 hr, the B cells were separated for quantitative PCR and further culture. The isolated B cells were seeded into 96-well flat-bottomed plates (1 × 106/ml), and T cells were seeded at a 1 : 5 ratio of T cells : B cells. After a culture of 72 hr, the supernatants were collected for the total IgG detection. Spleen cells from the CFA group were used as controls.
Treatment protocols
To explore the expression and function of miR-146a in EAMG B cells, AntagomiR-146a was used to treat the EAMG mice and perform the loss-of-function studies. The EAMG mice (grade ≥ 1·5) were randomly divided into three groups: (i) AntagomiR-146a group: AntagomiR-146a at 20 mg/kg dose dissolved in 200 μl PBS solution per day was used for treatment. (ii) NC group: AntagomiR negative control (NC) at the same dose dissolved in 200 μl PBS solution per day was used for treatment. (iii) PBS solution group: 200 μl PBS solution per day was used for treatment. The EAMG mice were injected with respective treatment drugs via the caudal vein on three consecutive days (defined successively as day 0, 1, 2). Clinical assessment and myasthenic scoring of the mice in each group were recorded every other day. At the same time the serum specimens were collected to detect the secretion level of anti-R97-116 total IgG and its subtypes by ELISA. At day 10 after the first treatment on day 0, the mice were killed and spleen cells were collected. Frequency of B-cell subsets was detected by flow cytometry, whereas the expression of miR-146a and its possible target genes tumour necrosis factor receptor-associated factor 6 (TRAF-6) and interleukin-1 receptor-associated kinase 1 (IRAK-1) in mouse B cells were detected by quantitative PCR.
ELISA assay for anti-peptide antibodies
A standard ELISA technique was used to detect peptide R97-116-specific antibodies. Briefly, 96-well microtitre plates were coated overnight with 5 μg of peptide at 4° and blocked with 200 μl 1% BSA at room temperature for 0·5 hr. Next, 100 μl of serum (diluted 1/1000) was added to the wells and incubated for 1 hr at room temperature. Plates were washed with 0·05% Tween-20 in PBS. Then, 100 μl of rabbit anti-rat IgG (horseradish peroxidase-conjugated, Immunology Consultants’ Laboratory, Portland, OR) and isotype control (horseradish peroxidase-conjugated; BD Pharmingen, San Diego, CA) were added and incubated for 1 hr at room temperature. Plates were washed with 0·05% Tween-20 in PBS and developed with o-phenylenediamine substrate. The optical density was measured at 450 nm using an automated microplate ELISA reader. Each serum was tested in duplicate and assessed at two different dilutions. The results are expressed as optical density at 450 nm/μl of serum.
Isolation of B cells
Spleens were aseptically removed from three groups of mice and processed into a single-cell suspension. Red blood cells were lysed with cold distilled water. After the erythrocyte lysis, spleen cells were resuspended at 107 cells/ml in PBS with 5% fetal bovine serum. Subsequently, mouse B cells were purified using CD19 beads (Miltenyi) according to the manufacturer's instructions.
Flow cytometry
Spleen cell suspensions were stained for flow cytometry with the indicated antibodies. Phycoerythrin (PE) -conjugated anti-mouse CD5, 488-conjugated anti-mouse CD44, 488-conjugated anti-mouse CD40, PE-conjugated anti-mouse CD27, allophycocyanin-conjugated anti-mouse CD80, PE/Cy7-conjugated anti-mouse CD86 were purchased from Biolegend, San Diego, CA. FITC-conjugated anti-mouse CD19 and Peridinin chlorophyll protein-conjugated anti-mouse CD45R were purchased from Miltenyi. Carboxyfluorescein anti-mouse CD138 were purchased from R&D Systems (Minneapolis, MN). Briefly, anti-CD19 and anti-CD5 were used to discriminate the B-1 cells; anti-CD45R, anti-CD27 and anti-CD44 were used to discriminate the memory B cells; anti-CD45R, anti-CD27 and anti-CD138 were used to discriminate the plasma cells. Anti-CD40, anti-CD80 and anti-CD86 were used to discriminate the surface activation molecules on B cells. Cell analysis was performed on a Beckman Counter (MoFlo™ XDF, Brea, CA).
MicroRNA and mRNA qPCR
To analyse the expression of miR-146a and its possible targets TRAF-6 and IRAK-1 in B cells, quantitative PCR analyses were carried out as follows: total RNA was extracted from B cells using TRIzol reagent (Invitrogen). Then, cDNA synthesis of miR-146a was performed with All-in-One™ miRNA First-strand cDNA Synthesis Kit (GeneCopoeia, Guangzhou, China) as per the manufacturer's instructions. The cDNA synthesis of TRAF-6 and IRAK-1 were performed with All-in-One™ First-strand cDNA Synthesis Kit (GeneCopoeia, Guangzhou, China) as per the manufacturer's instructions. A total reaction volume of 20 μl contained 10 μl qPCR Mix, 5·6 μl RNase-free water, 2 μl forward/reverse primer (for the miRNA detection both a 2 μl universal adaptor PCR primer and a 2 μl miR-146a qPCR primer were used), 2 μl cDNA template and 0·4 μl ROX. Amplification and detection were performed as follows: 95° for 10 min and then 40 cycles of 95° for 10 seconds, 60° for 20 seconds and 72° for 20 seconds, followed by one cycle of 95° for 15 seconds, 60° for 1 min and 95° for 15 seconds. The results were automatically analysed by the Applied Biosystem StepOne™ (Applied Biosystems, Carlsbad, CA) PCR system and the 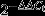 method was used to analyse the mRNA expression (ΔCt represents the difference of threshold cycle value between the target gene and the inner control; ΔΔCt represents the difference of ΔCt between different groups). Housekeeping genes U6 and β-actin were used as endogenous controls to normalize for differences in the amount of total RNA in each sample.
method was used to analyse the mRNA expression (ΔCt represents the difference of threshold cycle value between the target gene and the inner control; ΔΔCt represents the difference of ΔCt between different groups). Housekeeping genes U6 and β-actin were used as endogenous controls to normalize for differences in the amount of total RNA in each sample.
Statistical analysis
The data are presented as the mean ± SEM. Statistical analysis was performed using the one-way classification analysis of variance followed by a Student–Newman–Keuls (SNK) test. A P value <0·05 was considered significant.
Results
miR-146a was up-regulated in B cells following activation
To establish the functional role of miR-146a in the immune system, we first surveyed its expression in mouse splenic B cells after first immunization by the R97-116 peptide. Expression of mature miR-146a was found to be relatively high in B cells stimulated by R97-116 and this up-regulation was significantly attenuated by AntagomiR-146a (Fig. 1). This result suggests that miR-146a may play an important role in the response of B cells to pathological antigens.
Figure 1.
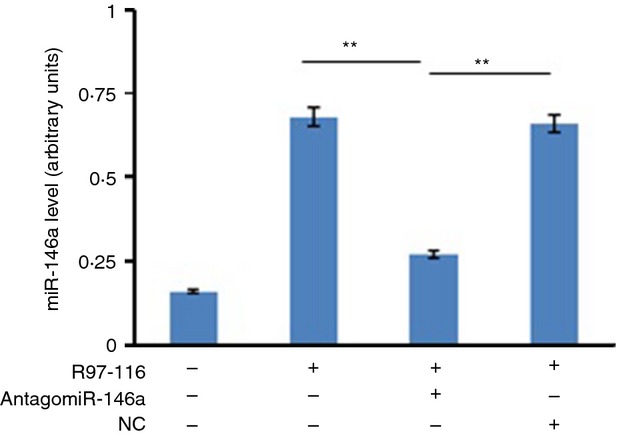
miR-146a was up-regulated in B cells following activation. The miR-146a mRNA was determined by quantitative PCR analysis in sensitized B cells. These B cells were cultured with a second immunization by R97-116 peptide in the absence or presence of AntagomiR-146a and/or AntagomiR Negative Control (NC). Non-activated B cells from the complete Freund's adjuvant (CFA) group were used as negative controls. The data were from three independent experiments and are shown as means ± SEM, with n = 3. The results showed that miR-146a expression was decreased significantly in R97-116-stimulated plus AntagomiR-146a-inhibited B cells (R97-116+ AntagomiR-146a subgroup) when compared with R97-116-stimulated but plus NC-inhibited B cells (R97-116+ NC subgroup) and normal R97-116-stimulated B cells (Positive control) (**P < 0·01).
B cells with knockdown of miR-146a showed decreased total IgG in vitro
Previous experiments have demonstrated that AntagomiR-146a could significantly inhibit the up-regulated miR-146a in B cells following activation. To explore its follow-up effects, ELISA was used to detect the secretion of total IgG antibodies. In this study, we observed a significant decrease of total IgG in the group cultured with AntagomiR-146a which has been confirmed that miR-146a was knocked down in B cells (Fig. 2).
Figure 2.
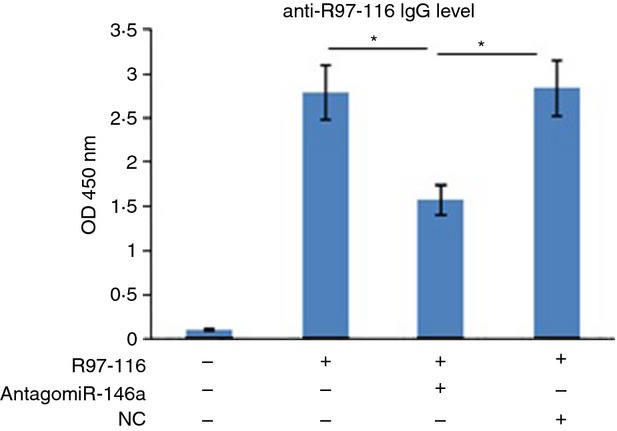
B cells with miR-146a knockdown secreted less total IgG in vitro. Samples were the same cultured B cells as the previous experiments. Supernatants from these cultures were harvested 72 hr after co-culture with T cells and assayed by ELISA for total IgG. The data were from three independent experiments and shown as means ± SEM, with n = 3. The results showed that the secretion levels of total IgG in R97-116+ AntagomiR-146a subgroup was significantly lower than in the R97-116+ NC subgroup and Positive control. (*P < 0·05).
Treatment with AntagomiR-146a ameliorated clinical myasthenic symptoms in mice with ongoing EAMG
It is well accepted that anti-AChR antibodies serve as the critical components in the pathogenesis of MG/EAMG. We presumed that AntagomiR-146a might benefit EAMG because of the evidence that AntagomiR-146a might attenuate the production of anti-R97-116 antibodies after stimulation with R97-116. To confirm these, EMAG mice were treated with AntagomiR-146a, AntagomiR NC, or PBS solution. Clinical assessment and myasthenic score of the mice in each group were recorded every other day. As expected, we observed a significant amelioration of the clinical severity of the EAMG mice treated with AntagomiR-146a (Fig. 3).
Figure 3.
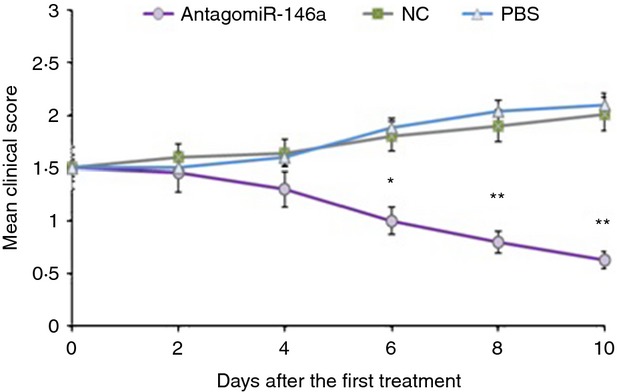
Treatment with AntagomiR-146a ameliorated clinical myasthenic symptoms in mice with ongoing experimental autoimmune myasthenia gravis (EAMG). Each symbol represents the mean clinical score (MCS) of mice in the AntagomiR-146a group (n = 10), the NC group (n = 10) and the PBS group (n = 10) at various times after treatment with respective treatment drugs via the caudal vein for 3 days continuously. Differences of the MCS were statistically significant between three groups since the sixth days after enrolment began. The MCS of AntagomiR-146a group was significantly lower than NC and PBS groups. At the end of the experiment, the MCS of the AntagomiR-146a group was 0·63 ± 0·33, the NC group was 2·01 ± 0·41, and the PBS group was 2·14 ± 0·55 (*P < 0·05; **P < 0·01).
Silence of miR-146a inhibited the production of anti-R97-116 antibodies in vivo
To confirm the role of miR-146a in EAMG, we detected anti-R97-116 total IgG and its subtypes in vivo by ELISA. We observed a significant decrease of total IgG, IgG1 and IgG2b in the AntagomiR-146a group (Fig. 4), which was consistent with the results in vitro. Therefore, AntagomiR-146a greatly influenced the expression and class switching of serum anti-R97-116 antibodies in the serum.
Figure 4.

miR-146a knockdown inhibited the production of anti-R97-116 antibodies in vivo. The secretion levels of total IgG, IgG1 and IgG2b were significantly lower in the AntagomiR-146a group compared with the NC group and PBS group. There were no significant differences of IgG2a, IgG2c and IgM in all groups. The complete Freund's adjuvant (CFA) group, which served as the control and exhibited no secretion of antigen-specific antibody, demonstrated the success of experimental autoimmune myasthenia gravis induction in the three experimental groups (*P < 0·05; **P < 0·01).
AntagomiR-146a could effectively silence miR-146a of B cells in vivo
To verify whether systemic administration of AntagomiR-146a in vivo effectively targeted B cells and exerted its biological effects, we first isolated B cells on the 10th day after AntagomiR-146a treatment. The sorting purity was > 95% (Fig. 5a). Then, quantitative PCR was performed to detect the expression of miR-146a. As expected, expression of miR-146a in the B cells was significantly reduced in the AntagomiR-146a group compared with the NC group and PBS group. The CFA group served as controls and had little expression of miR-146a, which also proved that miR-146a was up-regulated in B cells only when the B cells were stimulated by antigens and in response to activation (Fig. 5b).
Figure 5.
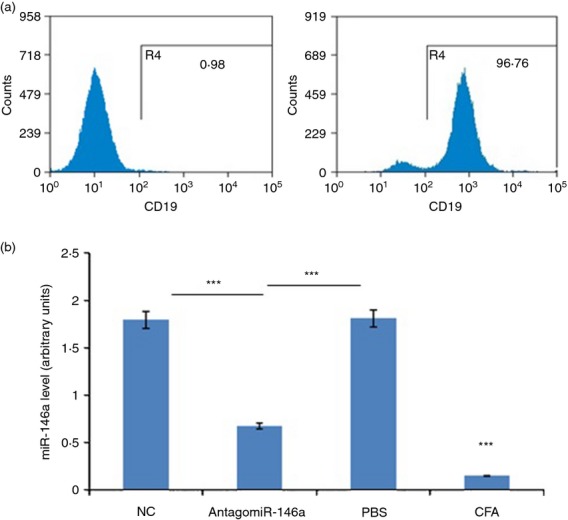
AntagomiR-146a could effectively silence miR-146a of B cells in vivo. At day 10 after the treatment, the mice were killed to collect spleen cells and B cells were isolated. (a) The high purity of sorted B cells, the left was negative control. (b) The expression of miR-146a in B cells was significantly reduced in the AntagomiR-146a group (0·6747 ± 0·0645) compared with the NC group (1·7971 ± 0·1031) and PBS group (1·8140 ± 0·1021). The complete Freund's adjuvant (CFA) group demonstrated low expression of miR-146a (0·1487 ± 0·0836) (***P < 0·001).
AntagomiR-146a influenced the number of plasma cells, memory B cells and B-1 cells in vivo
To further elucidate the mechanisms of miR-146a in B cells, we investigated the follow-up effects on B-cell subsets attributed to silencing miR-146a in B cells. We found that plasma cells, memory B cells and B-1 cells were significantly reduced in the AntagomiR-146a group compared with the NC group and PBS group (Fig. 6).
Figure 6.
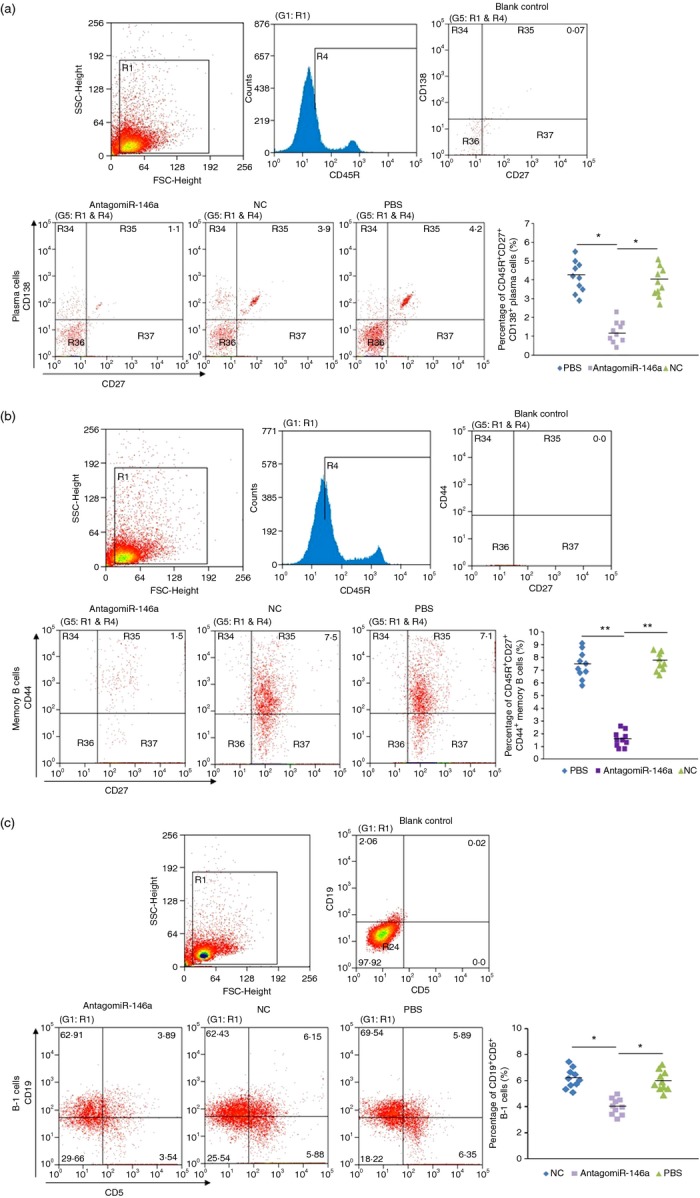
AntagomiR-146a influenced the amount of plasma cells, memory B cells and B-1 cells in vivo. (a) Flow cytometry analysis of CD45R+ CD27+ CD138+ plasma cells in spleen cells of three group mice(n = 10/group). In the AntagomiR-146a group it was 1·24 ± 0·57%, the NC group was 3·87 ± 0·78% and the PBS group was 4·15 ± 0·83%. (b) Flow cytometry analysis of CD45R+ CD27+ CD44+ memory cells. In the AntagomiR-146a group it was 1·57 ± 0·61%, the NC group was 7·62 ± 0·68% and the PBS group was 7·41 ± 1·07%. (c) Flow cytometry analysis of CD19+ CD5+ B-1 cells. In the AntagomiR-146a group it was 4·01 ± 0·63%, the NC group was 6·16 ± 0·76% and the PBS group was 5·97 ± 0·77%. In the dot plots at right, each symbol represents an individual mouse, and the horizontal bars represent the median values. The blank control was used to set the cross mark into four quadrants by the mean of making its probability of the upper right quadrant approximately 0% (*P < 0·05; **P < 0·01).
AntagomiR-146a decreased the expression of CD40, CD80 and CD86 on B cells
To investigate whether anti-mir-146a treatment affected the activation of B cells, the B cells were stained for a variety of differentiation and activation markers. Our data demonstrated that the expression of CD40, CD80 and CD86 were significantly decreased in AntagomiR-146a group compared with the NC group and the PBS group (Fig. 7).
Figure 7.

AntagomiR-146a decreased the expression of CD40, CD80 and CD86 on B cells. (a) Flow cytometry analysis of CD40 on B-cell surface. In the AntagomiR-146a group it was 25·72 ± 3·16%, the NC group was 78·57 ± 2·07% and the PBS group was 79·62 ± 1·43%. (b) Flow cytometry analysis of CD80. In the AntagomiR-146a group it was 10·37 ± 2·33%, the NC group was 30·68 ± 1·98% and the PBS group 32·75 ± 1·65%. (c) Flow cytometry analysis of CD86. In the AntagomiR-146a group it was 22·92 ± 1·38%, the NC group was 67·62 ± 3·72% and the PBS group was 70·15 ± 2·41%. The blank control was used to set the gate by the mean of making the region (R4/R3/R2) approximately 1% (*P < 0·05).
TRAF-6 and IRAK-1 were not the targets of miR-146a-induced immunomodulation in splenic B cells of EAMG
To explore the possible mechanism of miR-146a in B cells from EAMG, we analysed the expression of TRAF-6 and IRAK-1, which have been found to be the target genes in many other immune cells and to participate in several autoimmune diseases. In our study, however, no significant difference in TRAF-6 and IRAK-1 expression was observed in mouse B cells between three groups (Fig. 8). TRAF-6 and IRAK-1 were not the targets of the miR146a-induced immunomodulation in B cells of EAMG. Abnormal expression of miR-146a might act on other potential targets to control the mechanisms of EAMG/MG.
Figure 8.
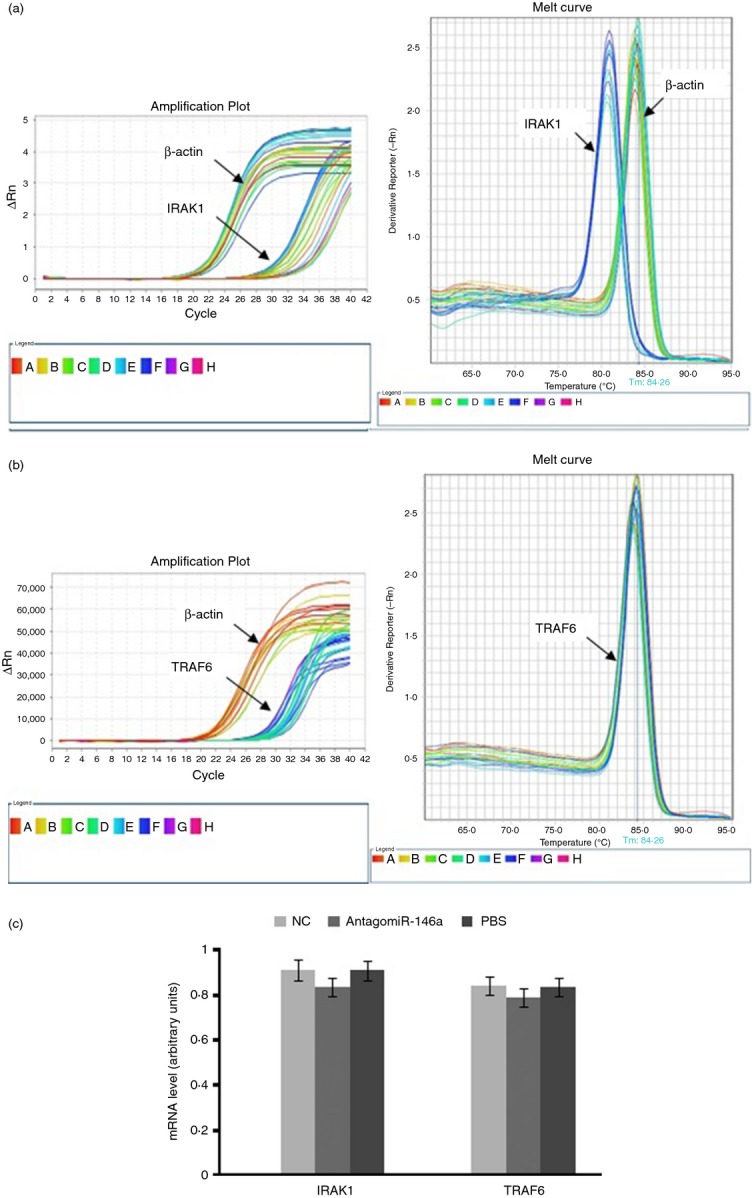
Tumour necrosis factor receptor-associated factor 6 (TRAF6) and interleukin-1 receptor-associated kinase 1 (IRAK1) were not the targets for miR-146a-induced immunomodulation in B cells of experimental autoimmune myasthenia gravis (EAMG). Quantitative PCR measurements of TRAF-6 and IRAK-1 mRNA were taken to analyse the possible targets of miR-146a in B cells. (a) Amplification plot and melt curve for IRAK1 and β-actin. (b) Amplification plot and melt curve for TRAF6. (c) Data analysis showed that the expression differences of TRAF6 (P = 0·296) and IRAK1 (P = 0·463) in mouse B cells from three groups were not statistically significant.
Discussion
In this study, we analysed the effect of miR-146a on the autoimmune response in an EAMG model for MG and the following effects after miR-146a was silenced by AntagomiR-146a in B cells. Our data indicated that miR-146a was up-regulated in activated B cells responding to the AChR antigen. Consistent with the result reported in MG patients in our previous study,16 the current study also implied that miR-146a was up-regulated in EAMG. For the first time, multiple functional defects were observed in B-cell responses after miR-146a was knocked down, including decreased numbers of B-1 cells, memory B cells, and plasma cells, decreased expression of CD40, CD80 and CD86, impaired expression and class switching of serum anti-R97-116 antibodies, which accounted for an amelioration of myasthenic symptoms.
The B-cell compartment has been divided into CD5− conventional B cells, termed B-2, and CD5+ distinctive B cells, termed B-1. Those B cells expressing the T-cell marker CD5 have long been suspected of playing a role in the development of autoimmunity diseases such as SLE, RA and SS.18,19 However, whether the B-1 cells are increased in MG is controversial. Yi et al.20 reported that levels of CD5+ B lymphocytes do not differ between patients with myasthenia gravis and healthy individuals. In contrast to this finding, another study reported a high frequency of CD5+ B cells (> 30%) in the peripheral blood of 57% of MG patients. The administration of immunosuppressive therapy, the age at onset of MG, the presence or absence of detectable serum AChR antibody, clinical extent of MG, and the duration of disease may all affect the frequency of CD5+ B cells in the blood.21 Consistent with the latter observation, our data also imply that the frequency of CD19+ CD5+ B cells in spleen cells is approximately 6% and that the proportion of this cell subset is reduced after AntagomiR-146a treatment. Although CD5+ B cells do not generate an immune memory, they can present antigen to T cells, inducing T-cell responses and secrete interleukin-10 to participate in autoimmune reactions. Therefore, we speculate that the effective treatment of EAMG mice with AntagomiR-146a may correlate with inhibited antigen presentation and inflammatory cytokine release followed by the decreased number of B-1 cells.
Recent data suggest that B-1 cells do not generate pathogenic autoantibodies because they tend to produce low-affinity autoantibodies.18 Antibodies against the AChR were mostly produced by CD5− B cells, also as B-2 cells (hereinafter referred to as B cells).22,23 Anti-AChR autoimmune responses depend on B cells encountering antigen, interacting with helper T cells through co-stimulatory molecules like CD40, CD80 and CD86, proliferating and differentiating into plasma cells and memory B cells. Evidence that B cells from MG patients present signs of activation have been clearly demonstrated: (i) Long-lived plasma cells producing anti-AChR antibodies were present in MG patients because immune-deficient mice transplanted with thymic fragments from these MG patients demonstrated anti-AChR antibodies several weeks after transplantation.24 (ii) There are a large number of pathological self-reactive B cells located in the germinal centres and ectopic germinal centres in the thymus of MG patients and essentially associated with the presence of anti-AChR antibodies.25–27 (iii) B cells from MG patients display increased expression levels of activation makers such as CD71, CD23, CD40, CD80 and CD86.28–30 In this study, we observed significantly high expression of CD40, CD80 and CD86, and also detected the presence of long-lived plasma cells and memory B cells in EAMG mice. These results are consistent with the previous findings that B cells from MG patients are highly activated, and emphasize that the miR-146a contributes to this activation because AntagomiR-146a leads to decreased plasma cells and memory B cells, and expression levels of activation markers in splenic cells.
To gain further insight into the mechanism of miR-146a in B cells of EAMG, we shall answer the following two questions.
What is inducing miR-146a in B cells?
The analysis of the miR-146a promoter provided evidence that miR-146a is a nuclear factor-κB (NF-κB) -dependent gene.31 NF-κB is a transcription regulator that controls diverse biological processes including cell survival, proliferation and immunomodulation. Previous studies have demonstrated that NF-κB can be activated by B-cell activating factor receptor (BAFF-R), which binds BAFF exclusively. Signalling downstream of the BAFF-R leads to B-cell survival through activation of NF-κB.32,33 Studies have proven that patients with MG have higher expressions of BAFF-R in the serum,34–36 and augmented percentages of CD19+ BAFF-R+ B cells.37 Therefore, we propose a model in which the aberrant miR-146a expression in B cells of EAMG/MG is driven by NF-κB and the latter is induced by active BAFF/BAFF-R signal. Furthermore, evidence demonstrates that NF-κB can also be activated by Toll-like receptor 4 (TLR4), which is up-regulated in peripheral mononuclear cells from MG patients and is expressed on B cells.38–40 Our data suggest another putative model in which the aberrant miR-146a expression is induced by the TLR4/NF-κB signal pathway.
What are the relevant targets of miR-146a in B cells?
Many direct targets of miR-146a in immune cells have been identified. TRAF-6 and IRAK-1 are pivotally important ones and associate tightly with autoimmune diseases.31 Both of these target genes encode key adaptor molecules downstream of TLRs and cytokine receptors in the NF-κB activation pathway.41,42 Evidence has shown that miR-146a can inhibit the expression of IRAK-1 and TRAF-6, impair NF-κB activity43and suppress the expression of NF-κB target genes such as interleukin-6 (IL-6), IL-8, IL-1β and tumour necrosis factor-α.12,44 Although both IRAK-1 and TRAF-6 can be under the control of miR-146a the rate of their simultaneous expressions can be independent of each other, it can be different in different diseases. In the case of SS, for example, miR-146a and its target gene TRAF-6 were significantly over-expressed, whereas the expression of IRAK-1 was significantly decreased.44 In contrast, in SLE, the expression of miR-146a was decreased, whereas the expression of IRAK-1 gene was elevated and the expression of TRAF-6 gene was unchanged.45 However, in the peripheral blood mononuclear cells from RA patients, the over-expression of miR-146a did not correlate with a change in expression of TRAF-6 and IRAK-1.12 Although we are now presenting the data on the over-expression of miR-146a in B cells upon activation by AChR antigen, and on the possible network of interactions between miR-146a, TRAF-6 and IRAK-1 in B cells of EAMG, our study demonstrated that an miR-146a inhibitor had no effect on the expression of TRAF-6 and IRAK-1 in B cells. The function of miR-146a in B cells does not involve these two target molecules; we speculate that there may be other target genes upon which miR-146a acts in EAMG/MG.
Given the ameliorating effect of AntagomiR-146a in EAMG and the functional defects in the B-cell compartment after miR-146a is silenced, we conclude that silencing miR-146a may be an effective therapeutic approach in the treatment of MG.
Acknowledgments
This work was supported by two grants from the National Natural Science Foundation of China (No. 30971033 and No. 81271325). In this study, the corresponding author Jing Li is fully responsible for the project. Junmei Zhang contributed mainly to the performance of the experiments, the data analysis and thesis writing. Ge Jia assisted with the statistical analyses. Qun Liu, Jue Hu, Mei Yan and BaiFeng Yang helped with the EAMG animal model, cell culture and collection of experimental data. Huan Yang and Wenbin Zhou assisted in editing this manuscript. We thank the associate researcher YanHong Zhou (Institute for Cancer Research, Central South University) for providing help on flow cytometry.
Glossary
- EAMG
experimental autoimmune myasthenia gravis
- MG
myasthenia gravis
- TRAF-6
TNF receptor-associated factor 6
- IRAK-1
IL-1 receptor-associated kinase 1
- R97-116
rat AChR α-subunit 97-116 peptide
- miRNAs
microRNAs
- miR-146a
microRNA-146a
- NC
AntagomiR negative control
- AChR
acetylcholine receptor
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Drachman DB. Myasthenia gravis. N Engl J Med. 1994;330:1797–810. doi: 10.1056/NEJM199406233302507. [DOI] [PubMed] [Google Scholar]
- 2.Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol. 2009;8:475–90. doi: 10.1016/S1474-4422(09)70063-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindstrom JM, Seybold ME, Lennon VA, Whittingham S, Duane DD. Antibody to acetylcholine receptor in myasthenia gravis. Prevalence, clinical correlates, and diagnostic value. Neurology. 1976;26:1054–9. doi: 10.1212/wnl.26.11.1054. [DOI] [PubMed] [Google Scholar]
- 4.Yang H, Goluszko E, David C, et al. Mapping myasthenia gravis-associated T cell epitopes on human acetylcholine receptors in HLA transgenic mice. J Clin Invest. 2002;109:1111–20. doi: 10.1172/JCI14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berrih-Aknin S, Ragheb S, Le PR, Lisak RP. Ectopic germinal centers, BAFF and anti-B-cell therapy in myasthenia gravis. Autoimmun Rev. 2013;12:885–93. doi: 10.1016/j.autrev.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 6.O'Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 2010;10:111–22. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- 7.Williams AE, Perry MM, Moschos SA, Larner-Svensson HM, Lindsay MA. Role of miRNA-146a in the regulation of the innate immune response and cancer. Biochem Soc Trans. 2008;36(Pt 6):1211–5. doi: 10.1042/BST0361211. [DOI] [PubMed] [Google Scholar]
- 8.Boldin MP, Taganov KD, Rao DS, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med. 2011;208:1189–201. doi: 10.1084/jem.20101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Curtale G, Citarella F, Carissimi C, et al. An emerging player in the adaptive immune response: microRNA-146a is a modulator of IL-2 expression and activation-induced cell death in T lymphocytes. Blood. 2010;115:265–73. doi: 10.1182/blood-2009-06-225987. [DOI] [PubMed] [Google Scholar]
- 10.Krutzfeldt J, Rajewsky N, Braich R, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–9. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 11.Luo X, Yang W, Ye DQ, et al. A functional variant in microRNA-146a promoter modulates its expression and confers disease risk for systemic lupus erythematosus. PLoS Genet. 2011;7:e1002128. doi: 10.1371/journal.pgen.1002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pauley KM, Satoh M, Chan AL, Bubb MR, Reeves WH, Chan EK. Upregulated miR-146a expression in peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Res Ther. 2008;10:R101. doi: 10.1186/ar2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pauley KM, Stewart CM, Gauna AE, et al. Altered miR-146a expression in Sjögren's syndrome and its functional role in innate immunity. Eur J Immunol. 2011;41:2029–39. doi: 10.1002/eji.201040757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waschbisch A, Atiya M, Linker RA, Potapov S, Schwab S, Derfuss T. Glatiramer acetate treatment normalizes deregulated microRNA expression in relapsing remitting multiple sclerosis. PLoS ONE. 2011;6:e24604. doi: 10.1371/journal.pone.0024604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balasubramanyam M, Aravind S, Gokulakrishnan K, et al. Impaired miR-146a expression links subclinical inflammation and insulin resistance in Type 2 diabetes. Mol Cell Biochem. 2011;351:197–205. doi: 10.1007/s11010-011-0727-3. [DOI] [PubMed] [Google Scholar]
- 16.Lu J, Yan M, Wang Y, et al. Altered expression of miR-146a in myasthenia gravis. Neurosci Lett. 2013;555:85–90. doi: 10.1016/j.neulet.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 17.Baggi F, Annoni A, Ubiali F, et al. Breakdown of tolerance to a self-peptide of acetylcholine receptor α-subunit induces experimental myasthenia gravis in rats. J Immunol. 2004;172:2697–703. doi: 10.4049/jimmunol.172.4.2697. [DOI] [PubMed] [Google Scholar]
- 18.Youinou P, Renaudineau Y. The paradox of CD5-expressing B cells in systemic lupus erythematosus. Autoimmun Rev. 2007;7:149–54. doi: 10.1016/j.autrev.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 19.Le PL, Devauchelle V, Pers JO, Jamin C, Youinou P. The mosaic of B-cell subsets (with special emphasis on primary Sjögren's syndrome) Autoimmun Rev. 2007;6:149–54. doi: 10.1016/j.autrev.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 20.Yi Q, Ahlberg R, Pirskanen R, Lefvert AK. Levels of CD5+ B lymphocytes do not differ between patients with myasthenia gravis and healthy individuals. Neurology. 1992;42:1081–4. doi: 10.1212/wnl.42.5.1081. [DOI] [PubMed] [Google Scholar]
- 21.Ragheb S, Lisak RP. Effect of clinical status and treatment on the frequency of CD5+ B cells in patients with myasthenia gravis. Neurology. 1992;42:1076–80. doi: 10.1212/wnl.42.5.1076. [DOI] [PubMed] [Google Scholar]
- 22.Araga S, Kishimoto M, Adachi A, Nakayasu H, Takenaka T, Takahashi K. The CD5+ B cells and myasthenia gravis. Autoimmunity. 1995;20:129–34. doi: 10.3109/08916939509001937. [DOI] [PubMed] [Google Scholar]
- 23.Heidenreich F, Jovin T. Synthesis of anti-acetylcholine receptor antibodies by CD5– B cells from peripheral blood of myasthenia gravis patients. J Neurol. 1996;243:57–62. doi: 10.1007/BF00878532. [DOI] [PubMed] [Google Scholar]
- 24.Yoshikawa H, Lennon VA. ACh receptor protein drives primary and memory autoantibody responses in chimeric human-SCID mice. Clin Immunol. 2002;104:128–37. doi: 10.1006/clim.2002.5251. [DOI] [PubMed] [Google Scholar]
- 25.Guigou V, Emilie D, Berrih-Aknin S, Fumoux F, Fougereau M, Schiff C. Individual germinal centres of myasthenia gravis human thymuses contain polyclonal activated B cells that express all the Vh and Vk families. Clin Exp Immunol. 1991;83:262–6. doi: 10.1111/j.1365-2249.1991.tb05625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sims GP, Shiono H, Willcox N, Stott DI. Somatic hypermutation and selection of B cells in thymic germinal centers responding to acetylcholine receptor in myasthenia gravis. J Immunol. 2001;167:1935–44. doi: 10.4049/jimmunol.167.4.1935. [DOI] [PubMed] [Google Scholar]
- 27.Zuckerman NS, Howard WA, Bismuth J, et al. Ectopic GC in the thymus of myasthenia gravis patients show characteristics of normal GC. Eur J Immunol. 2010;40:1150–61. doi: 10.1002/eji.200939914. [DOI] [PubMed] [Google Scholar]
- 28.Ragheb S, Bealmear B, Lisak R. Cell-surface expression of lymphocyte activation markers in myasthenia gravis. Autoimmunity. 1999;31:55–66. doi: 10.3109/08916939908993860. [DOI] [PubMed] [Google Scholar]
- 29.Murai H, Hara H, Hatae T, Kobayashi T, Watanabe T. Expression of CD23 in the germinal center of thymus from myasthenia gravis patients. J Neuroimmunol. 1997;76:61–9. doi: 10.1016/s0165-5728(97)00030-1. [DOI] [PubMed] [Google Scholar]
- 30.Teleshova N, Matusevicius D, Kivisakk P, Mustafa M, Pirskanen R, Link H. Altered expression of costimulatory molecules in myasthenia gravis. Muscle Nerve. 2000;23:946–53. doi: 10.1002/(sici)1097-4598(200006)23:6<946::aid-mus16>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 31.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–6. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bossen C, Schneider P. BAFF, APRIL and their receptors: structure, function and signaling. Semin Immunol. 2006;18:263–75. doi: 10.1016/j.smim.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 33.Sun SC. Non-canonical NF-κB signaling pathway. Cell Res. 2011;21:71–85. doi: 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scuderi F, Alboini PE, Bartoccioni E, Evoli A. BAFF serum levels in myasthenia gravis: effects of therapy. J Neurol. 2011;258:2284–5. doi: 10.1007/s00415-011-6092-z. [DOI] [PubMed] [Google Scholar]
- 35.Kim JY, Yang Y, Moon JS, et al. Serum BAFF expression in patients with myasthenia gravis. J Neuroimmunol. 2008;199:151–4. doi: 10.1016/j.jneuroim.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 36.Ragheb S, Lisak R, Lewis R, Van Stavern G, Gonzales F, Simon K. A potential role for B-cell activating factor in the pathogenesis of autoimmune myasthenia gravis. Arch Neurol. 2008;65:1358–62. doi: 10.1001/archneur.65.10.1358. [DOI] [PubMed] [Google Scholar]
- 37.Li X, Xiao BG, Xi JY, Lu CZ, Lu JH. Decrease of CD4+CD25highFoxp3+ regulatory T cells and elevation of CD19+BAFF-R+ B cells and soluble ICAM-1 in myasthenia gravis. Clin Immunol. 2008;126:180–8. doi: 10.1016/j.clim.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 38.Hayashi EA, Granato A, Paiva LS, Bertho AL, Bellio M, Nobrega A. TLR4 promotes B cell maturation: independence and cooperation with B lymphocyte-activating factor. J Immunol. 2010;184:4662–72. doi: 10.4049/jimmunol.0903253. [DOI] [PubMed] [Google Scholar]
- 39.Barr TA, Brown S, Ryan G, Zhao J, Gray D. TLR-mediated stimulation of APC: distinct cytokine responses of B cells and dendritic cells. Eur J Immunol. 2007;37:3040–53. doi: 10.1002/eji.200636483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang YZ, Yan M, Tian FF, et al. Possible involvement of toll-like receptors in the pathogenesis of myasthenia gravis. Inflammation. 2013;36:121–30. doi: 10.1007/s10753-012-9526-6. [DOI] [PubMed] [Google Scholar]
- 41.Yang WL, Wang J, Chan CH, et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science. 2009;325:1134–8. doi: 10.1126/science.1175065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gottipati S, Rao NL, Fung-Leung WP. IRAK1: a critical signaling mediator of innate immunity. Cell Signal. 2008;20:269–76. doi: 10.1016/j.cellsig.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 43.Bhaumik D, Scott GK, Schokrpur S, Patil CK, Campisi J, Benz CC. Expression of microRNA-146 suppresses NF-κB activity with reduction of metastatic potential in breast cancer cells. Oncogene. 2008;27:5643–7. doi: 10.1038/onc.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zilahi E, Tarr T, Papp G, Griger Z, Sipka S, Zeher M. Increased microRNA-146a/b, TRAF6 gene and decreased IRAK1 gene expressions in the peripheral mononuclear cells of patients with Sjögren's syndrome. Immunol Lett. 2012;141:165–8. doi: 10.1016/j.imlet.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 45.Tang Y, Luo X, Cui H, et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009;60:1065–75. doi: 10.1002/art.24436. [DOI] [PubMed] [Google Scholar]