

Abstract
IgE is known to enhance some antibody responses to specific antigens, but whether this contributes to allergic asthma remains unclear. We have previously found that repeated antigen challenges in mice sensitized with antigen-specific IgE monoclonal antibody (mAb) exacerbated airway inflammation and remodelling accompanied by increased levels of endogenous antigen-specific IgE and IgG1. Here, we investigated whether IgE/antigen-mediated enhancement of endogenous IgE production contributes to the exacerbation of airway inflammation and remodelling. BALB/c mice passively sensitized with ovalbumin (OVA) -specific IgE mAb were challenged with OVA intratracheally seven times; anti-IgE mAb was intraperitoneally administered 1 day before the fourth challenge. Treatment with anti-IgE mAb inhibited the increased level of endogenous OVA-specific IgE in serum, but not OVA-specific IgG1, and a biphasic increase in airway resistance at the fourth challenge. Furthermore, a biphasic increase in airway resistance, airway hyper-responsiveness to methacholine, OVA-specific IgE and IgG1 production, and infiltrations by neutrophils and eosinophils in the lungs at the seventh challenge were suppressed by treatment; airway remodelling, such as goblet cell hyperplasia and sub-epithelial fibrosis, was also reduced. In addition, the production of interleukin-17A, interleukin-33 and CXCL1 in the lungs related to these IgE-mediated responses was decreased by treatment. Collectively, we found that the mechanism leading to the exacerbation of allergic asthma is closely related to IgE/antigen-mediated enhancement of IgE production, suggesting that this may create a vicious circle leading to the chronic status in asthmatic patients having levels of antigen-specific IgE ready to form complexes with antigen.
Keywords: airway hyper-responsiveness, asthma, biphasic increase in airway resistance, IgE, inflammation, remodelling
Introduction
Patients with allergic asthma have elevated levels of antigen-specific IgE, and the disease is characterized by antigen-induced early-phase and late-phase asthmatic responses, airway hyper-responsiveness (AHR) and airway inflammation.1,2 IgE plays a critical role in the early-phase asthmatic response. IgE sensitizes mast cells by binding to their high-affinity IgE receptor (FcεRI); subsequently, activation of the cells triggers the secretion of a chemical mediator within minutes after exposure to inhaled antigen,3,4 resulting in the induction of acute airway obstruction. In contrast, the late-phase asthmatic response begins several hours after exposure, and airway narrowing is largely resolved by 24 hr, at which time AHR to methacholine was observed. Furthermore, the main pathological changes are the migration of neutrophils, eosinophils and lymphocytes from the bloodstream into the lung associated with these responses.3,5,6 Additionally, airway remodelling, including goblet cell hyperplasia and sub-epithelial fibrosis, is regarded as one of the characteristic features of chronic asthma.4,7–10
IgE is a unique antibody capable of enhancing the antibody responses to specific antigens. An antigen administered intravenously to mice along with a specific IgE up-regulated the production of antigen-specific IgE and IgG, peaking as early as 6 days after priming;11,12 furthermore, marked proliferation of antigen-specific CD4+ T cells was induced, peaking at 3 days.13 Hence, these data suggest that, when an antigen-specific IgE is present, the IgE complexes with the antigen to produce antigen-specific antibodies such as IgE and IgG via the activation of CD4+ T cells. However, the actual relationship between IgE-mediated antibody responses and allergic asthma remains unclear.
In a previous study, we reported that BALB/c mice passively sensitized with an intraperitoneal injection of ovalbumin (OVA) -specific IgE monoclonal antibody (mAb) (OE-1) showed a biphasic increase in airway resistance, AHR, airway inflammation, and remodelling after seven repeated intratracheal OVA challenges.14,15 A key feature of this IgE-sensitized model is that the exacerbation of airway inflammation and remodelling was observed during the fourth to seventh antigen challenges; furthermore, the level of antigen-specific IgE was significantly increased over the injection level of OE-1 alone at the fourth and seventh antigen challenges.14 However, it remains unclear whether IgE/antigen-mediated enhancement of endogenous IgE production contributes to the exacerbation of airway inflammation and remodelling during the fourth to seventh challenges in IgE-sensitized mice.
In this study, we first examined the role of endogenous antigen-specific IgE produced after immunization with antigen-specific IgE mAb (OE-1) and antigen in the development of a biphasic increase in airway resistance at the fourth challenge by the depletion of endogenous IgE using anti-IgE mAb. Furthermore, the infiltration by inflammatory cells such as neutrophils and eosinophils into the lungs, and airway remodelling, including goblet cell hyperplasia and sub-epithelial fibrosis, at the seventh challenge, were evaluated when anti-IgE mAb was administered at the fourth challenge. Additionally, we have reported that interleukin-17A (IL-17A), IL-33 and glutamic acid-leucine-arginine (ELR)+ CXC chemokines such as CXCL1 contributed to the development of these IgE-mediated responses;15–17 therefore, the effect of anti-IgE mAb on cytokine and chemokine production at the seventh challenge was evaluated.
Materials and methods
Animals
Male 7-week-old BALB/c mice were obtained from Japan SLC (Hamamatsu, Japan). These mice were maintained in a temperature-controlled environment with free access to standard rodent chow and water. All of the experimental procedures were approved by the Experimental Animal Research Committee at Kobe Pharmaceutical University.
OVA-specific IgE mAb
Ovalbumin-specific IgE mAb (OE-1) was derived from a B-cell hybridoma producing murine IgE, as described previously.14 The hybridoma was grown in CELLine CL1000 with BD-Cell-MAb medium (BD Biosciences, San Diego, CA) supplemented with 20% heat-inactivated fetal bovine serum (FBS), 1% l-glutamine, and 1% penicillin-streptomycin. The level of OE-1 in culture supernatants of hybridoma was assessed by ELISA.14 OE-1 was added to plates coated with anti-IgE antibody followed by adding biotin-labelled anti-mouse IgE antibody. Furthermore, alkaline phosphate anti-biotin was added, the plate was developed with p-nitrophenyl phosphate, and measurements were made at 405 nm using a microplate reader. OE-1 levels were calculated by comparison with mouse IgE standards (Southern Biotech, Birmingham, AL).
Passive sensitization with OVA-specific IgE mAb
Passive sensitization with OE-1 was performed with the protocol described previously.15 As shown in Fig. 3(a), BALB/c mice were passively sensitized with intraperitoneal injections of a supernatant containing OE-1 (100 μg/0·3 ml/mouse) of hybridoma on days 0, 1 and 2. Both the sensitized and non-sensitized mice were challenged on days 1, 2, 3, 8, 9, 10 and 15 under anaesthesia with isoflurane (Wako Pure Chemicals, Osaka, Japan) with 1% OVA (grade V; Sigma-Aldrich, St Louis, MO) in a volume of 20 μl by intratracheal administration as reported previously.18
Figure 3.

Effect of anti-IgE monoclonal antibody (mAb) on IgE-mediated airway inflammation at the seventh challenge. (a) Experimental protocol for treatment with anti-IgE mAb. Anti-IgE mAb was intraperitoneally administered on day 7 [OE-1 (7th) + anti-IgE mAb]. Negative and positive controls were non-sensitized–challenged [NS-C (7th)] and OE-1-sensitized, rat IgG1 mAb-treated [OE-1 (7th) + rat IgG1] mice, respectively. (b) Effect of anti-IgE mAb on the increased levels of OVA-specific IgE and IgG1 in serum 24 hr after the seventh challenge (Day 16). (c) Effect of anti-IgE mAb on increase in the inflammatory cell numbers in BALF 24 hr after the seventh challenge. (d) Effect of anti-IgE mAb on the biphasic increase in airway resistance after the seventh challenge. (e) Effect of anti-IgE mAb on the development of AHR 24 hr after the seventh challenge. Each value is the mean ± SEM of four to seven animals from two separate experiments. *P < 0·05 and **P < 0·01 compared with OE-1 (7th) + rat IgG1 group.
Treatment with anti-IgE mAb and anti-FcεRI mAb
As shown in Figs 1(a) and 3(a), on day 7, a dose (50 μg/mouse) of anti-IgE mAb (R35-92, rat IgG1; BD Biosciences) was administered intraperitoneally to mice sensitized with OE-1, and control mice were given the same amount of rat IgG1. It has been reported that the dose is enough to deplete the increased level of IgE in the serum of actively sensitized mice.19
Figure 1.
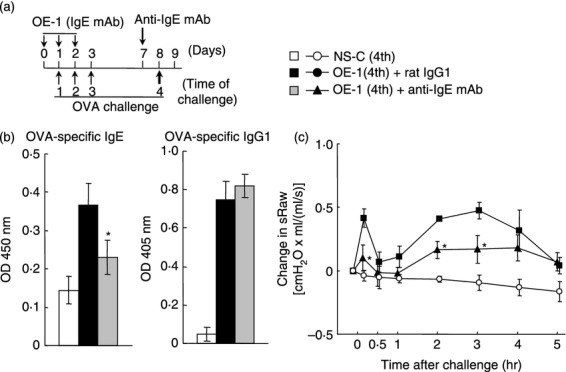
Effect of anti-IgE monoclonal antibody (mAb) on IgE-mediated biphasic increase in airway resistance at the fourth challenge. (a) Experimental protocol for treatment with anti-IgE mAb. Anti-IgE mAb was intraperitoneally administered on day 7 [OE-1 (4th) + anti-IgE mAb]. Negative and positive controls were non-sensitized–challenged [NS-C (4th)] and OE-1-sensitized, rat IgG1 mAb-treated [OE-1 (4th) + rat IgG1] mice, respectively. (b) Effect of anti-IgE mAb on the increased levels of ovalbumin (OVA) -specific IgE and IgG1 in serum 24 hr after the fourth challenge (Day 9). (c) Effect of anti-IgE mAb on the biphasic increase in airway resistance after the fourth challenge. Each value is the mean ± SEM of three to five animals from one experiment. *P < 0·05 compared with OE-1 (4th) + rat IgG1 group.
In addition, on days – 2, – 1 and 0, a dose (10 μg/mouse) of anti-FcεRIα mAb (Mar-1, hamster IgG; BioLegend, San Diego, CA) or hamster IgG was administered intraperitoneally twice a day to mice sensitized with OE-1 ( Fig. 2a). It has been reported that mast cells and basophils in the lungs of a murine model of asthma were depleted by treatment with Mar-1 by about 70%, and the depletion was sustained for 7 days.20,21
Figure 2.
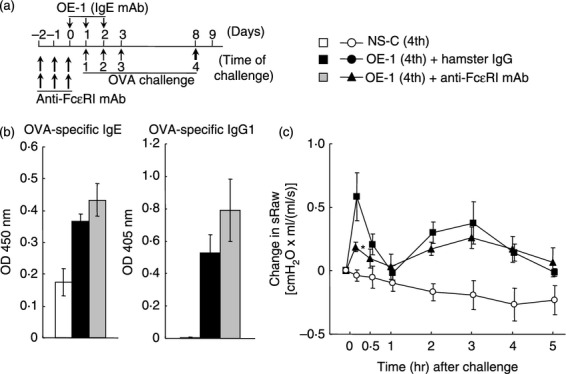
Effect of anti-FcεRI monoclonal antibody (mAb) on IgE-mediated biphasic increase in airway resistance at the fourth challenge. (a) Experimental protocol for treatment with anti- FcεRI mAb. Anti-FcεRI mAb was intraperitoneally administered twice a day on days –2, –1 and 0 [OE-1 (4th) + anti-FcεRI mAb]. Negative and positive controls were non-sensitized–challenged [NS-C (4th)] and OE-1-sensitized, hamster IgG-treated [OE-1 (4th) + hamster IgG] mice, respectively. (b) Effect of anti-FcεRI mAb on the increased levels of ovalbumin (OVA) -specific IgE and IgG1 in serum 24 hr after the fourth challenge (Day 9). (c) Effect of anti-FcεRI mAb on the biphasic increase in airway resistance after the fourth challenge. Each value is the mean ± SEM of three to five animals from one experiment. *P < 0·05 compared with OE-1 (4th) + rat IgG1 group.
Measurement of OVA-specific IgE and IgG1 in serum
Levels of OVA-specific IgE and IgG1 antibodies in serum were measured by ELISA, as described previously.14 Serum samples were collected 24 hr after the fourth (day 9) or seventh (day 16) antigen challenge in IgE-sensitized mice. OVA-specific IgE antibody was detected using plates coated with anti-mouse IgE antibody and adding biotin-labelled OVA. Streptavidin–horseradish peroxidase was added, the plate was developed with 3,3′,5,5′-tetramethylbenzidine, and measurements were made at 450 nm using a microplate reader after stopping the reaction with sulphuric acid. Values for serum OVA-specific IgE (1 : 5) are expressed as absorbance units.
Ovalbumin-specific IgG1 was detected using plates coated with OVA and adding alkaline phosphate-conjugated anti-mouse IgG1. The plates were developed with p-nitrophenyl phosphate and read at 405 nm using a microplate reader. Values for serum OVA-specific IgG1 (1 : 1000) are expressed as absorbance units.
Measurement of airway resistance
Airway resistance and AHR were determined as described previously.16 To evaluate the degree of early-phase and late-phase increases in airway resistance, we measured specific airway resistance [sRaw; cmH2O × ml/(ml/s)] before and 10 min to 5 hr after the fourth or seventh challenge using a two-chambered, double-flow plethysmograph system (Pulmos-I; M.I.P.S., Osaka, Japan) according to the method of Pennock et al.22
Airway hyper-responsiveness was assessed 24 hr after the seventh challenge. Briefly, increasingly higher doses of methacholine (3·125, 6·25, and 12·5 μg/ml) in solution were consecutively administered via the intratracheal route to non-sensitized and sensitized mice under isoflurane-induced anaesthesia at 30-min intervals. sRaw was measured 2 min after the respective instillations of the three doses of methacholine.
Analysis of cells recovered by bronchoalveolar lavage
To evaluate airway cellular inflammation, we examined the accumulation of inflammatory cells in bronchoalveolar lavage fluid (BALF) as described previously.18 IgE-sensitized mice were killed 24 hr after the seventh antigen challenge. The trachea was cannulated, and the left bronchi were tied for histological examination. The right air lumen was washed twice with 0·5 ml Hanks’ balanced salt solution (HBSS) containing 2% heat-inactivated FBS. The cell pellet was suspended with a defined volume (200 μl/sample) of HBSS containing 2% heat-inactivated FBS. The total leucocyte count in the lavage fluid was determined by staining with Turk's solution. For differential cell counts, BAL cells were stained with Diff-Quik solution (Sysmex International Reagents, Kobe, Japan). A minimum of 200 cells were counted under a microscope and, based on their morphological criteria, classified as macrophages, lymphocytes, neutrophils or eosinophils.
Histological study
Left lungs were fixed in 10% neutral-buffered formalin, then dissected, embedded in paraffin, and cut into 4-μm thick sections. These sections were stained with haematoxylin & eosin (H&E), periodic acid-Schiff (PAS) and Masson's trichrome. Each section was evaluated for inflammation (H&E), goblet cell hyperplasia (PAS) and sub-epithelial fibrosis (Masson) on a scale of 0–4 with increments of 0·5 by a blinded observer as described previously.18
Measurement of cytokines in lung tissue supernatants
The right lobe (after BAL) was homogenized in 1 ml T-PER tissue protein extraction reagent (Thermo Scientific, Rockford, IL) containing a Complete Mini Protease Inhibitor Cocktail tablet (Roche, Mannheim, Germany; 1 tablet/10 ml T-PER stock reagent) as described previously.15 Lung homogenates were centrifuged at 9000 g for 10 min at 4°. The levels of IL-4, IL-5, IL-13, IL-33, CXCL1 (R&D Systems, Minneapolis, MN) and IL-17 (BioLegend) in supernatants of lung homogenates were measured using quantitative colorimetric sandwich ELISA kits.
Statistical analyses
Data are shown as the mean ± SEM. Statistical analyses between the two groups were performed using Student's t-test. A probability value of P < 0·05 was considered significant.
Results
Effect of anti-IgE mAb on IgE-mediated biphasic increase in airway resistance at the fourth challenge
We have previously found that the level of antigen-specific IgE at the fourth and seventh antigen challenges in IgE-sensitized mice was higher than that in non-sensitized—challenged mice; additionally, the levels significantly increased over the level of injection of OE-1 alone without antigen challenges.14 Furthermore, the fourth and seventh challenges caused a biphasic increase in airway resistance.14,16,17 First, we examined whether the depletion of endogenous IgE using anti-IgE mAb reduced the biphasic increase in airway resistance at the fourth challenge. OVA-specific IgE and IgG1 in the serum of mice sensitized with OE-1 were significantly increased 24 hr after the fourth challenge (day 9) compared with non-sensitized–challenged mice; treatment with anti-IgE mAb significantly inhibited OVA-specific IgE in serum, but not OVA-specific IgG1 (Fig. 1b). Furthermore, the fourth challenge induced early-phase and late-phase increased airway resistance in IgE-sensitized mice; the biphasic increase in airway resistance was suppressed by treatment with anti-IgE mAb (Fig. 1c).
Effect of anti-FcεRI mAb on IgE-mediated biphasic increase in airway resistance at the fourth challenge
Cross-linking of mast cells and basophils binding IgE antibodies by antigen results in immediate activation of these cells to release chemical mediators that cause tissue injury.23–25 Furthermore, mast cells and basophils have the ability to produce cytokines and chemokines that can mediate chronic allergic inflammation.26–28 Therefore, we investigated the role of these cells in IgE-sensitized mice using treatment with anti-FcεRI mAb, Mar-1, for the depletion of mast cells and basophils. Treatment with anti-FcεRI mAb did not inhibit the production of OVA-specific IgE and IgG1 at the fourth challenge (Fig. 2b); meanwhile, treatment inhibited the early-phase increase in airway resistance, but not the late-phase increase in airway resistance (Fig. 2c).
Effect of anti-IgE mAb on IgE-mediated airway inflammation and airway remodelling at the seventh challenge
We have reported that the infiltration of inflammatory cells such as macrophages, lymphocytes and neutrophils, but not eosinophils, in BALF was observed 24 hr after the fourth challenge; additionally, infiltration by eosinophils was recognized 24 hr after the seventh challenge.15 Therefore, we investigated the effect of treatment with anti-IgE mAb before the fourth challenge on the infiltration of inflammatory cells in BALF at the seventh challenge. As a result, infiltration by neutrophils and eosinophils, but not macrophages and lymphocytes, in BALF at the seventh challenge was inhibited by treatment with anti-IgE mAb; furthermore, the production of antigen-specific IgE and IgG1 at the seventh challenge was reduced (Fig. 3b, c). Meanwhile, consistent with the data shown in Fig. 1, the biphasic increase in airway resistance at the seventh challenge was inhibited by treatment with anti-IgE mAb at the fourth challenge (Fig. 3d). Moreover, we have previously reported that not only a biphasic increase in airway resistance but also AHR was observed in IgE-sensitized mice;14,16 treatment with anti-IgE mAb at the fourth challenge suppressed the development of AHR at the seventh challenge (Fig. 3d, e).
In IgE-sensitized mice, airway remodelling was exacerbated during the fourth to seventh challenges.15 Therefore, we examined the effect of treatment with anti-IgE mAb before the fourth challenge on histological changes such as airway inflammation and remodelling at the seventh challenge in the lungs of IgE-sensitized mice. Consistent with previous data,14,15 airway inflammation (H&E) and remodelling such as goblet cell hyperplasia (PAS) and sub-epithelial fibrosis (Masson) at the seventh challenge in IgE-sensitized mice were significantly observed compared with non-sensitized–challenged mice (Fig. 4). Treatment with anti-IgE mAb at the fourth challenge significantly inhibited airway inflammation and remodelling such as goblet cell hyperplasia and sub-epithelial fibrosis at the seventh challenge in IgE-sensitized mice (Fig. 4).
Figure 4.

Effect of anti-IgE monoclonal antibody (mAb) on IgE-mediated airway remodellling at the seventh challenge. Anti-IgE mAb was intraperitoneally administered on day 7 [OE-1 (7th) + anti-IgE mAb]. Negative and positive controls were non-sensitized–challenged [NS-C (7th)] and OE-1-sensitized, rat IgG1 mAb-treated [OE-1 (7th) + rat IgG1] mice, respectively. (a–c) Effect of anti-IgE mAb on inflammation (haematoxylin & eosin), goblet cell hyperplasia (periodic acid Schiff), and sub-epithelial fibrosis (Masson's trichrome) at the seventh challenge. Scale bar, 100 μm. Histological appearances were scored as measures of inflammation (a (iv)), goblet cell hyperplasia (b (iv)), and sub-epithelial fibrosis (c (iv)). Each value is the mean ± SEM of four to seven animals from two separate experiments. *P < 0·05 compared with OE-1 (7th) + rat IgG1 group.
Effect of anti-IgE mAb on IgE-mediated cytokine and chemokine production at the seventh challenge
We have reported that a biphasic increase in airway resistance and AHR was induced via neutrophilic inflammation associated with ELR+ chemokines such as CXCL1 induced by IL-33 and IL-17A in IgE-sensitized mice;15–17 furthermore, airway remodelling was related to the expression of IL-33.15 Therefore, we examined the effect of anti-IgE mAb on cytokine and chemokine production. The levels of IL-4, IL-5, IL-13, IL-17A, IL-33 and CXCL1 in the lung 24 hr after the seventh challenge in IgE-sensitized mice were greater than in non-sensitized–challenged mice (Fig. 5). Treatment with anti-IgE mAb at the fourth challenge inhibited the production of IL-17A, IL-33 and CXCL1 at the seventh challenge in IgE-sensitized mice; furthermore, T helper type 2 cytokines such as IL-5 and IL-13, but not IL-4, were reduced (Fig. 5).
Figure 5.
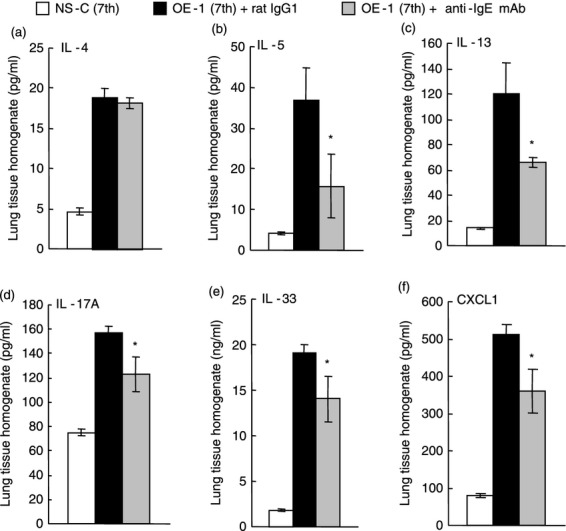
Effect of anti-IgE monoclonal antibody (mAb) on IgE-mediated cytokine production at the seventh challenge. Anti-IgE mAb was intraperitoneally administered on day 7 [OE-1 (7th) + anti-IgE mAb]. Negative and positive controls were non-sensitized–challenged [NS-C (7th)] and OE-1-sensitized, rat IgG1 mAb-treated [OE-1 (7th) + rat IgG1] mice, respectively. Interleukin-4 (IL-4) (a), IL-5 (b), IL-13 (c), IL-17A (d), IL-33 (e), and CXCL1 (f) from the lung homogenate obtained 24 hr after the seventh challenge were measured. Each value is the mean ± SEM of four to seven animals from two separate experiments. *P < 0·05 compared with OE-1 (7th) + rat IgG1 group.
Discussion
The present study shows that treatment with anti-IgE mAb at the fourth challenge decreased the increased endogenous production of antigen-specific IgE in serum, but not antigen-specific IgG1, at the fourth challenge in IgE-sensitized mice; furthermore, not only early-phase but also late-phase increases in airway resistance at the fourth challenge were suppressed. Therefore, we concluded that treatment with anti-IgE mAb specifically inhibited the increased level of antigen-specific IgE, resulting in the suppression of the biphasic increase in airway resistance at the fourth challenge. Furthermore, at the seventh challenge, the increased levels of antigen-specific IgE as well as IgG1 were reduced when anti-IgE mAb was administered at the fourth challenge; additionally, the biphasic increase in airway resistance and AHR was suppressed. On the basis of these results, we found that inhibition of the late-phase increase in airway resistance at the seventh challenge by anti-IgE mAb was stronger than that at the fourth challenge in IgE-sensitized mice. It has been reported that inflammatory cells such as macrophages and mast cells have FcγRs, which are receptors for IgG1;29,30 therefore, the strong suppression of the late-phase increase in airway resistance at the seventh challenge may contribute to the decreased levels of IgG1, which bind to inflammatory cells. Hence, IgE/antigen-mediated enhancement of endogenous IgE production contributes to the development of a biphasic increase in airway resistance and AHR in mice.
We have reported that the depletion of neutrophilic inflammation by anti-Gr-1 mAb inhibited the late-phase increase in airway resistance and AHR in IgE-sensitized mice.16 Therefore, we examined the effect of treatment with anti-IgE mAb before the fourth challenge on the infiltration by neutrophils at the seventh challenge in the lungs of IgE-sensitized mice, resulting in the suppression of not only the late-phase increase in airway resistance and AHR but also the infiltration by neutrophils. These data prompted us to examine the inhibitory mechanisms of neutrophil accumulation in the lungs by treatment with anti-IgE mAb. In our previous studies, IL-33 and IL-17A contributed to the development of the late-phase increase in airway resistance and AHR in IgE-sensitized mice;15–17 additionally, IL-33 cooperated with IL-17A to exacerbate AHR by enhancing neutrophilic inflammation via the production of ELR+ chemokines such as CXCL1.17 In addition to these data, we confirmed that treatment with anti-IgE mAb at the fourth challenge inhibited the production of IL-17A, IL-33 and CXCL1 at the seventh challenge in the lungs of mice sensitized with IgE, suggesting that anti-IgE mAb inhibited the development of a late-phase increase in airway resistance and AHR by suppressing neutrophilic inflammation associated with reduced production of IL-17A, IL-33 and CXCL1.
It has been reported that mast cells and basophils rapidly produce various mediators, such as histamine, associated with the development of an early-phase increase in airway resistance.3,23–25 In our present study, the depletion of mast cells and basophils by treatment with anti-FcεRI mAb suppressed the early-phase increase in airway resistance in IgE-sensitized mice, showing that mast cells and/or basophils contributed to the development of an early-phase increase in airway resistance. However, the late-phase increase in airway resistance was not inhibited by anti-FcεRI mAb at the fourth challenge, although suppression of a late-phase increase in airway resistance by anti-IgE mAb was observed; additionally, the production of antigen-specific IgE and IgG1 was not decreased by treatment with anti-FcεRI mAb. These results suggest that the late-phase increase in airway resistance and the production of IgE and IgG1 at the fourth challenge were independent of mast cells and basophils.
In IgE-sensitized mice, airway inflammation including eosinophils was exacerbated during the fourth to seventh antigen challenges;15 furthermore, treatment with anti-IgE mAb before the fourth challenge decreased the infiltration of eosinophils at the seventh challenge. We have reported that IL-33 induced eosinophil accumulation via T helper type 2 cytokines such as IL-5 and IL-13 produced by activation of CD4+ T cells in IgE-sensitized mice.15 In the present study, treatment with anti-IgE mAb inhibited the production of IL-33, IL-5 and IL-13 in the lungs at the seventh challenge in IgE-sensitized mice. Hence, IgE/antigen-enhanced endogenous IgE production induces the infiltration of eosinophils through the production of IL-33 and T helper type 2 cytokines.
Furthermore, treatment with anti-IgE mAb at the fourth challenge suppressed the development of airway remodelling such as goblet cell hyperplasia and sub-epithelial fibrosis, which are characteristic features of severe and chronic allergic asthma, at the seventh challenge in IgE-sensitized mice, suggesting the importance of IgE/antigen-mediated endogenous IgE production in the exacerbation of airway remodelling in allergic asthma. Additionally, we have reported that IL-33 produced during the fourth to seventh challenges contributed to the exacerbation of airway remodelling in IgE-sensitized mice;15 treatment with anti-IgE mAb inhibited IL-33 production in the lung at the seventh challenge. Hence, it is suggested that IgE/antigen-enhanced endogenous IgE production contributes to the exacerbation of airway remodelling through the production of IL-33.
In conclusion, we demonstrated that IgE/antigen-mediated enhancement of endogenous IgE production contributed to the exacerbation of airway inflammation and remodelling in mice. It has been reported that therapy with anti-IgE is effective for some poorly controlled patients with asthma,31–33 indicating the importance of IgE in the chronic and severe phases of asthma. On the basis of these results, IgE/antigen-mediated enhancement of IgE production may create a vicious circle leading to a chronic or severe status in patients with allergic asthma who have levels of antigen-specific IgE ready to form complexes with antigen.
Acknowledgments
Part of this work was supported by JSPS KAKENHI Grant Number 25460112 (to NM).
Glossary
- AHR
airway hyperresponsiveness
- BALF
bronchoalveolar lavage fluid
- FBS
fetal bovine serum
- H&E
haematoxylin & eosin
- HBSS
Hanks’ balanced salt solution
- IL-17
interleukin-17
- mAb
monoclonal antibody
- OVA
ovalbumin
- PAS
periodic acid-Schiff
- sRaw
specific airway resistance
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Cohn L, Elias JA, Chupp GL. Asthma: mechanisms of disease persistence and progression. Annu Rev Immunol. 2004;22:789–815. doi: 10.1146/annurev.immunol.22.012703.104716. [DOI] [PubMed] [Google Scholar]
- 2.Robinson DS, Hamid Q, Ying S, et al. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 3.Pauwels R. The relationship between airway inflammation and bronchial hyperresponsiveness. Clin Exp Allergy. 1989;19:395–8. doi: 10.1111/j.1365-2222.1989.tb02404.x. [DOI] [PubMed] [Google Scholar]
- 4.Aikawa T, Shimura S, Sasaki H, Ebina M, Takishima T. Marked goblet cell hyperplasia with mucus accumulation in the airways of patients who died of severe acute asthma attack. Chest. 1992;101:916–21. doi: 10.1378/chest.101.4.916. [DOI] [PubMed] [Google Scholar]
- 5.Bousquet J, Chanez P, Lacoste JY, et al. Eosinophilic inflammation in asthma. N Engl J Med. 1990;323:1033–9. doi: 10.1056/NEJM199010113231505. [DOI] [PubMed] [Google Scholar]
- 6.De Monchy JG, Kauffman HF, Venge P, Koeter GH, Jansen HM, Sluiter HJ, De Vries K. Bronchoalveolar eosinophilia during allergen-induced late asthmatic reactions. Am Rev Respir Dis. 1985;131:373–6. doi: 10.1164/arrd.1985.131.3.373. [DOI] [PubMed] [Google Scholar]
- 7.Ebina M, Takahashi T, Chiba T, Motomiya M. Cellular hypertrophy and hyperplasia of airway smooth muscles underlying bronchial asthma. A 3-D morphometric study. Am Rev Respir Dis. 1993;148:720–6. doi: 10.1164/ajrccm/148.3.720. [DOI] [PubMed] [Google Scholar]
- 8.Hossain S, Heard BE. Hyperplasia of bronchial muscle in chronic bronchitis. J Pathol. 1970;101:171–84. doi: 10.1002/path.1711010212. [DOI] [PubMed] [Google Scholar]
- 9.Jeffery PK, Godfrey RW, Adelroth E, Nelson F, Rogers A, Johansson SA. Effects of treatment on airway inflammation and thickening of basement membrane reticular collagen in asthma. A quantitative light and electron microscopic study. Am Rev Respir Dis. 1992;145:890–9. doi: 10.1164/ajrccm/145.4_Pt_1.890. [DOI] [PubMed] [Google Scholar]
- 10.Roche WR, Beasley R, Williams JH, Holgate ST. Subepithelial fibrosis in the bronchi of asthmatics. Lancet. 1989;1:520–4. doi: 10.1016/s0140-6736(89)90067-6. [DOI] [PubMed] [Google Scholar]
- 11.Heyman B, Tianmin L, Gustavsson S. In vivo enhancement of the specific antibody response via the low-affinity receptor for IgE. Eur J Immunol. 1993;23:1739–42. doi: 10.1002/eji.1830230754. [DOI] [PubMed] [Google Scholar]
- 12.Gustavsson S, Hjulström S, Liu T, Heyman B. CD23/IgE-mediated regulation of the specific antibody response in vivo. J Immunol. 1994;152:4793–800. [PubMed] [Google Scholar]
- 13.Getahun A, Hjelm F, Heyman B. IgE enhances antibody and T cell responses in vivo via CD23+ B cells. J Immunol. 2005;175:1473–82. doi: 10.4049/jimmunol.175.3.1473. [DOI] [PubMed] [Google Scholar]
- 14.Mizutani N, Goshima H, Nabe T, Yoshino S. Establishment and characterization of a murine model for allergic asthma using allergen-specific IgE monoclonal antibody to study pathological roles of IgE. Immunol Lett. 2012;141:235–45. doi: 10.1016/j.imlet.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 15.Mizutani N, Nabe T, Yoshino S. Interleukin-33 and alveolar macrophages contribute to the mechanisms underlying the exacerbation of IgE-mediated airway inflammation and remodelling in mice. Immunology. 2013;139:205–18. doi: 10.1111/imm.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizutani N, Goshima H, Nabe T, Yoshino S. Complement C3a-induced IL-17 plays a critical role in an IgE-mediated late-phase asthmatic response and airway hyperresponsiveness via neutrophilic inflammation in mice. J Immunol. 2012;188:5694–705. doi: 10.4049/jimmunol.1103176. [DOI] [PubMed] [Google Scholar]
- 17.Mizutani N, Nabe T, Yoshino S. IL-17A promotes the exacerbation of IL-33-induced airway hyperresponsiveness by enhancing neutrophilic inflammation via CXCR2 signaling in mice. J Immunol. 2014;192:1372–84. doi: 10.4049/jimmunol.1301538. [DOI] [PubMed] [Google Scholar]
- 18.Mizutani N, Nabe T, Yoshino S. Complement C3a regulates late asthmatic response and airway hyperresponsiveness in mice. J Immunol. 2009;183:4039–46. doi: 10.4049/jimmunol.0901468. [DOI] [PubMed] [Google Scholar]
- 19.Haak-Frendscho M, Robbins K, Lyon R, Shields R, Hooley J, Schoenhoff M, Jardieu P. Administration of an anti-IgE antibody inhibits CD23 expression and IgE production in vivo. Immunology. 1994;82:306–13. [PMC free article] [PubMed] [Google Scholar]
- 20.Nabe T, Matsuya K, Akamizu K, et al. Roles of basophils and mast cells infiltrating the lung by multiple antigen challenges in asthmatic responses of mice. Br J Pharmacol. 2013;169:462–76. doi: 10.1111/bph.12154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Denzel A, Maus UA, Rodriguez Gomez M, et al. Basophils enhance immunological memory responses. Nat Immunol. 2008;9:733–42. doi: 10.1038/ni.1621. [DOI] [PubMed] [Google Scholar]
- 22.Pennock BE, Cox CP, Rogers RM, Cain WA, Wells JH. A noninvasive technique for measurement of changes in specific airway resistance. J Appl Physiol Respir Environ Exerc Physiol. 1979;46:399–406. doi: 10.1152/jappl.1979.46.2.399. [DOI] [PubMed] [Google Scholar]
- 23.Oettgen HC, Geha RS. IgE in asthma and atopy: cellular and molecular connections. J Clin Invest. 1999;104:829–35. doi: 10.1172/JCI8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galli SJ. Mast cells and basophils. Curr Opin Hematol. 2000;7:32–9. doi: 10.1097/00062752-200001000-00007. [DOI] [PubMed] [Google Scholar]
- 25.Galli SJ, Kalesnikoff J, Grimbaldeston MA, Piliponsky AM, Williams CM, Tsai M. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu Rev Immunol. 2005;23:749–86. doi: 10.1146/annurev.immunol.21.120601.141025. [DOI] [PubMed] [Google Scholar]
- 26.Falcone FH, Haas H, Gibbs BF. The human basophil: a new appreciation of its role in immune responses. Blood. 2000;96:4028–38. [PubMed] [Google Scholar]
- 27.Schroeder JT, MacGlashan DW, Jr, Lichtenstein LM. Human basophils: mediator release and cytokine production. Adv Immunol. 2001;77:93–122. doi: 10.1016/s0065-2776(01)77015-0. [DOI] [PubMed] [Google Scholar]
- 28.Voehringer D, Shinkai K, Locksley RM. Type 2 immunity reflects orchestrated recruitment of cells committed to IL-4 production. Immunity. 2004;20:267–77. doi: 10.1016/s1074-7613(04)00026-3. [DOI] [PubMed] [Google Scholar]
- 29.Guilliams M, Bruhns P, Saeys Y, Hammad H, Lambrecht BN. The function of Fcγ receptors in dendritic cells and macrophages. Nat Rev Immunol. 2014;14:94–108. doi: 10.1038/nri3582. [DOI] [PubMed] [Google Scholar]
- 30.Tkaczyk C, Okayama Y, Woolhiser MR, Hagaman DD, Gilfillan AM, Metcalfe DD. Activation of human mast cells through the high affinity IgG receptor. Mol Immunol. 2002;38:1289–93. doi: 10.1016/s0161-5890(02)00077-9. [DOI] [PubMed] [Google Scholar]
- 31.Busse W, Corren J, Lanier BQ, McAlary M, Fowler-Taylor A, Cioppa GD, van As A, Gupta N. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol. 2001;108:184–90. doi: 10.1067/mai.2001.117880. [DOI] [PubMed] [Google Scholar]
- 32.Holgate S, Casale T, Wenzel S, Bousquet J, Deniz Y, Reisner C. The anti-inflammatory effects of omalizumab confirm the central role of IgE in allergic inflammation. J Allergy Clin Immunol. 2005;115:459–65. doi: 10.1016/j.jaci.2004.11.053. [DOI] [PubMed] [Google Scholar]
- 33.Holgate ST, Chuchalin AG, Hebert J, et al. Efficacy and safety of a recombinant anti-immunoglobulin E antibody (omalizumab) in severe allergic asthma. Clin Exp Allergy. 2004;34:632–8. doi: 10.1111/j.1365-2222.2004.1916.x. [DOI] [PubMed] [Google Scholar]