

Abstract
The immune response against hapten is T-cell-dependent, and so requires the uptake, processing and presentation of peptides on MHC class II molecules by antigen-presenting cells to the specific T cell. Some haptens, following conjugation to the available free amines on the surface of the carrier protein, can reduce its immunogenicity. The purpose of this study was to explore the mechanism by which this occurs. Four proteins were tested as carriers and six molecules were used as haptens. The immune response to the carrier proteins was reduced > 100-fold by some of the haptens (termed carrier immunogenicity reducing haptens – CIRH), whereas other haptens did not influence the protein immunogenicity (carrier immunogenicity non-reducing haptens – nCIRH). Conjugation of the protein to a CIRH affected protein degradation by lysosomal cathepsins, leading to the generation of peptides that differ in length and sequence from those derived from the same native protein or that protein modified with nCIRH. Injection of CIRH-conjugated protein into mice induced an increase in the population of regulatory T cells. The results of this study provide a putative mechanism of action for the reduction of immune response to haptenated proteins.
Keywords: carrier immunogenicity, hapten, peptide repertoire, tolerance
Introduction
Haptens are small chemical groups that cannot stimulate antibody responses in their free soluble form, because they cannot cross-link B-cell receptors and do not recruit T-cell help. However, when coupled to a carrier protein, they become immunogenic, as the protein carries multiple hapten groups that can now cross-link B-cell receptors and activate T cells through peptides derived from the carrier protein.1 Understanding the mechanisms of hapten–carrier systems involved in the T-cell-dependent immune response enables the development of bacterial capsule–polysaccharide conjugate-based vaccines.2 Although much knowledge has been accumulated on the immune response against haptens, little is known about the specificity of the immune response to the carrier protein. It is uncertain whether the haptenated and native proteins are degraded into the same peptides, to be presented by the MHC to T cells with and without the conjugated hapten, and as a concequence, whether different populations of T cells are induced following immunization with native or haptenated protein. Although T lymphocytes commonly recognize short peptides presented by MHC molecules, there are reports that T cells may specifically respond to peptides that are phosphorylated,3 glycosylated,4 or that carry chemicals,5 drugs,6 metal ions7 or lipids.8 Ortmann et al.9 identified murine CD8+ T cells specific for trinitrophenol-modified MHC class I-binding peptides as antigens. Shimizu et al.9 suggested that anti-hapten B cells are assisted by T cells specific to the haptenated carrier, rather than a molecule composed solely of a linear sequence of amino acids.
Previous studies have shown that conjugation of some haptens to proteins reduces the antibody response to the carrier.10,11 This phenomenon may have implications for the reduction of therapeutic protein immunogenicity – an important goal in modern medicine12,13 that would allow the use of foreign proteins for therapeutic use. The hapten–carrier system is of interest as a model of epitope modulations or altered self-proteins because it raises questions as to how chemical modification of proteins affects their immunogenicity and their contribution to the induction of allergies and autoimmune diseases.14
The mechanism underlying the reduction in antibody immune response to the carrier by part of the hapten molecule is currently unknown. The protein's modification may alter its immunogenicity by several routes. One possible route is via degradation of the antigen into peptides. Following immunization, antigens internalized by antigen-presenting cells (APC) are digested by proteases into peptides that may be loaded and presented on an MHC class II molecule, which further interacts with the specific T-cell receptor.
The different types of APC are equipped with similar and distinct intracellular acidic proteases, referred to as cathepsins, most of which contain a cysteine as the attacking nucleophile in the catalytic cleft.15 The function of endocytic proteases in antigen processing determines the kinetics and intracellular location of MHC class II peptide loading, leading to the generation or destruction of T-cell epitopes.16 Examples of both situations have been found with various proteins and attached molecules. For example, N-glycosylation of mammalian protein asparagine residues blocks proteolytic cleavage in vitro by eliminating sites of endopeptidase processing;17 the lack of N-glycosylation of the neuronal glutamate receptor subunit 3 in Rasmussens’ encephalitis, a severe form of paediatric epilepsy, exposes a cleavage site, so creating a new autoantigen.18 Hapten conjugation to a protein may affect the protein's interaction with the cathepsins and therefore, may affect the peptide repertoire obtained from a given protein.19
The T-cell receptor is specific for a unique combination of a particular peptide and a particular MHC molecule; the presence, absence or generation of different peptides from the same protein may lead to inactivation or activation of different T-cell sub-populations.15
The aim of the present study was to elucidate the mechanism by which hapten reduces antibody immune response to a carrier. For this purpose, the effect of haptens on protein degradation into peptides, and their presentation by APC followed by induction of specific cells and cytokines were studied.
Materials and methods
Synthesis of adduct haptens
N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (480 mg, 2.5 mmol) was added to a solution containing biotin (400 mg, 1.7 mmol), disthiobiotin (400 mg, 1.88 mmol) or lipoic acid (400 mg, 1.94 mmol) and N-hydroxysuccinimide (250 mg, 2.17 mmol) in dimethylformamide (4 ml). After stirring for 0.5 hr at room temperature, another solution of 2-mannosamine or 2-glucosamine (400 mg, 2.2 mmol) dissolved in 1 ml DMSO was added dropwise and the final solution was stirred overnight at room temperature. Then the solvents were removed and the products were isolated and purified by flash chromatography (silica gel, methanol : dichloromethane, 5 : 95 as solvents). Using conventional analytical methods, the pure products were identified as mannosamine–biotin adduct (MBA), mannosamine–desthiobiotin adduct (MDTBA), mannosamine–lipoic acid adduct (MLA) and glucosamine–biotin adduct (GBA).
Protein–hapten conjugation
MBA, GBA, MDTBA and MLA (1.5 mg each) dissolved in DMSO were added to horse (hs) IgG, BSA, ovalbumin (OVA) or hydrophilic recombinant gp100 (1 mg/ml each) in 25 mm phosphate buffer pH 6 (the molar concentration of the haptens was three orders of magnitude greater than that of the proteins). After 1 hr at room temperature, NaBCNH3 (2 mg in 50 μl double-distilled water) was added to reduce the imine bond. Biotin (2 mg), N-hydroxysuccinimide (1.2 mg) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (2 mg) in DMSO (50 μl) were stirred for 0.5 hr at room temperature and were added to 1 ml of protein solution in PBS (1 mg/ml). The solutions were mixed for 3 hr at room temperature and dialysed with PBS. Excess reagent was removed by filtration through an Amicon ultracentrifugal filter device (MWCO 10 000). Hapten–protein conjugation efficiency was tested by available free amine test and Western blot.13 Free amine test is a quantitative method that determines the reduction in the number of free amines as a result of hapten attachment. The number of free amines in the tested proteins was in the range of 12–20. Following conjugation, the number of free amines was reduced to < 1 for all tested haptens.
Immunization of mice
BALB/c mice (10 weeks old; Harlan Laboratories, Jerusalem, Israel) were injected intramuscularly with 50 μg of carrier or hapten-conjugated carrier at 2-week intervals. To avoid irrelevant effects, immunizations were carried out without the use of adjuvant. Two weeks after each injection, blood was drawn and sera were separated and kept at −20° until analysis. All animal studies have been reviewed and approved by an appropriate institutional and national review committee.
Detection of antibody response
The presence of antibodies against carrier protein or hapten in mouse sera following immunization with native or with haptenated carrier protein was tested by ELISA. Each of the listed steps was followed by washes with 0.05% Tween-20 in PBS (washing buffer). ELISA plates (Nunc) were incubated overnight at 4° with carrier or with an irrelevant protein conjugated to hapten, diluted in carbonate-coating buffer (pH 9.6) to a final concentration of 1 μg/ml. Blocking buffer was added (5% skim milk in washing buffer) for 1 hr at 37°. Twofold serum dilutions were added to the plate and incubated for 1 hr at 37°. Plates were incubated with a secondary antibody, goat anti-mouse IgG conjugated to horseradish peroxidase (Sigma, Rehovot, Israel), diluted 1 : 5000 in PBS, for 1 hr at 37°. A substrate solution, o-phenylenediaminedihydrochloride (Sigma), was added and optical density at 450 nm (OD450) was determined by ELISA reader. Titre values were determined as the reciprocal value of the twofold serial dilution end-point (twice the negative control OD value) of the tested serum.
Detection of cytokines
The level of plasma cytokine [interleukin-10 (IL-10) and IL-4] was measured with murine cytokine detection ELISA kit according to the manufacturer's protocol (PeproTech, Rehovot, Israel).
BSA internalization and co-localization
BSA–MBA or BSA–biotin (50 μg) were added to 1 × 106 RAW 264.7 line cells/well seeded on eight-well Lab-Tek slides (Nunc, Roskilde, Denmark) for 3 hr at 37° and 5% CO2 followed by a washing step with 0.1% BSA in PBS. Following fixation with 4% formaldehyde for 20 min at room temperature, cells were permeabilized using 0.1% saponin, and 2% BSA in PBS for 15 min and were blocked using mouse serum for 1 hr. Detection of lysosome was performed using 1 : 1000-diluted rat anti-CD107a/LAMP1 (Abcam, Cambridge, UK) and 1 : 5000-diluted donkey anti-rat Alexa Fluor 647 conjugate (Jackson ImmunoResearch Laboratories, West Grove, PA). BSA–MBA or BSA–biotin were detected using 1 : 2000-diluted FITC-conjugated avidin (Sigma).The cells were imaged using a Zeiss LSM-710 laser scanning confocal microscope.
Determination of protein-degradation pattern and peptide sequence
For the preparation of proteolytic enzyme, 2 × 106 RAW 264.7 line cells were lysed by sonication (five cycles of 2 s each at 40 W) in 100 μl of 340 mm sodium acetate buffer (pH 5 with acetic acid) containing 0.1% Triton X-100, 4 mm EDTA and 8 mm dithiothreitol at 4°. Cell lysate was centrifuged for 10 min at 25 000 g. Supernatants were mixed with 5 μg BSA–biotin or BSA–MBA for up to 24 hr at 37° and analysed by mass spectrometry (MS).
Mass spectrometric analysis
Peptides were separated by an ultrafiltration step through a 10 000 molecular weight Microcon (Merck Millipore, Carrigtwohill, Ireland) and then desalted and concentrated with Micro-Tip reverse-phase columns (C18, 200 μl; Harvard Apparatus, Holliston, MA). The peptides were eluted with 80% acetonitrile in 0.1% trifluoroacetic acid. The solvent was evaporated to dryness and the peptides were dissolved in 0.1% trifluoroacetic acid. The eluted peptides were analysed by nanoLC–MS/MS using an Orbitrap XL mass spectrometer (Thermo Fisher, San Jose, CA) fitted with a capillary HPLC (Eksigent, Dublin, CA). The peptides were resolved on a C18 trap column (0.3 × 5 mm, LC-Packings) connected online to 75-μm internal diameter fused silica capillaries (Agilent J&W, Santa Clara, CA) self-packed with 3.5-μm Reprosil C18 (Dr Maisch, GmbH, Germany) as described previously.20 The peptides were eluted at flow rates of 0.25 μl/min, with linear gradients of 7–40% acetonitrile in 0.1% formic acid for 90 min, followed by 15 min at 95% acetonitrile in 0.1% formic acid. Spectra were collected in the orbitrap mass analyser using full ion scan mode over the m/z range 400–2000, which was set to 60 000 resolutions. The seven most intense masses from each full mass spectrum, with singly, doubly and triply charged states, were selected for fragmentation by collision-induced disintegration in the linear ion-trap.
The resolved peptides from the C18 column were also analysed with the Q-Exactive mass spectrometer (Thermo Fisher) fitted with a capillary HPLC (EASY-nLC 1000/Thermo Fisher). Q Exactive analysis resolution was set for the full MS to 70 000 at m/z of 200, automatic gain control (AGC) to 3 × 106, maximum ion time (IT) 100 ms and the dynamic exclusion of 20 s. The MS/MS was set to a start at 100 m/z with resolution of 17 500 and target AGC set to 1 × 105 with maximum IT of 50 ms with normalized collision energy set to 25 eV. Analysis was performed using PEAKS 7 (Bioinformatics Solutions Inc., Waterloo, ON, Canada)21 searched against the mouse part of the SWISS-PROT database (http://www.ebi.ac.uk/swissprot, September 2013) including 16 528 mouse proteins and the sequence of the BSA protein. The search was not limited by enzymatic specificity, the peptide tolerance was set to 0.007 Da, and the fragment ion tolerance was set to 0.5 Da for Orbitrap data and to 0.01 Da for Q-exactive data. Oxidized methionine, biotin (229.12 Da) and mannosamine–biotin (339.13 Da) on lysine were searched as variable modifications. False discovery rate for protein and peptides was set to 1%.
BSA degradation by RAW 264.7 cell line
BSA–MBA or BSA–biotin (100 μg) was added to 1 × 106 cell/well seeded in six-well plates for 3 hr at 37° under 5% CO2. Cells were washed in the plate with 1% BSA in PBS buffer. Cells were fixed with 1 ml/well 4% formalin in PBS for 20 min at room temperature. Cells were permeabilized using 0.1% saponin and 2% BSA in PBS for 15 min at room temperature. Cells were washed in the plate with permeabilization buffer and the protein was detected using 1 : 2000-diluted FITC-conjugated avidin (Sigma). Cells were washed and analysed in a BDcalibur flow cytometer.
Analysis of spleen cell population
Forty-eight hours after boost immunization with hsIgG, hsIgG–biotin or hsIgG–MBA, mouse spleens were extracted and single-cell suspensions were prepared from each spleen with gentle MACS dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany) in cold PBS. Following red blood cell lysis with distilled water, cells were washed with 0.1% BSA in PBS and counted. Cells were diluted to 1 × 106 in 100 μl and incubated with mixed, or separate (for compensation), fluorescently conjugated antibodies using a mouse regulatory T (Treg) cell staining kit (eBioscience, San Diego, CA) according to the manufacturer's protocol. Background staining was determined using unreactive isotype-matched control monoclonal antibodies (eBioscience) with gates positioned to exclude non-reactive cells. Flow cytometry was performed using FACSAria (BD) and data were analysed using fcs express 4 software.
Statistics
ELISAs for immunogenicity and Treg cell population percentage were analysed using one-way analysis of variance, non-parametric Kruskal–Wallis test. All the analyses were performed with graphpadprism5 software.
Results
Reduction of the immune response to a carrier by some haptens
The influence of haptens on the humoral immune response raised against a carrier was tested using hsIgG as the carrier. BALB/c mice were immunized twice with 50 μg of hsIgG or hsIgG conjugated to various haptens (Fig. 1a). Two weeks after the second injection, the level of serum antibodies against hsIgG and the hapten was determined by ELISA.
Figure 1.
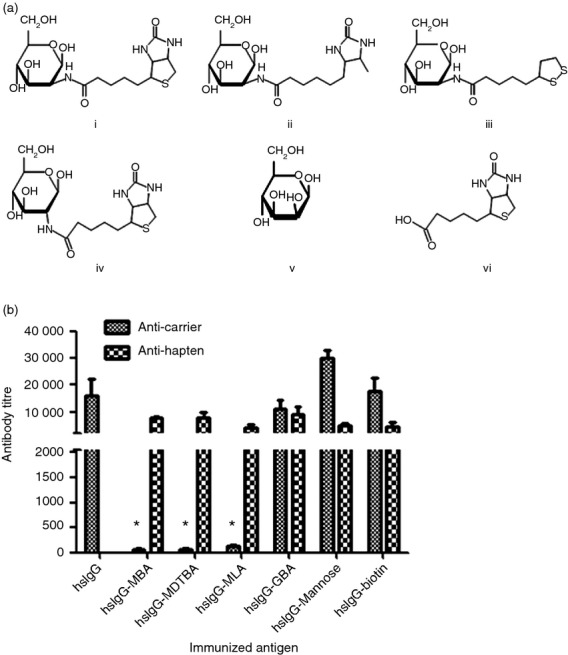
Titres of antibodies against proteins after conjugation of various haptens. (a) Molecular structures of the tested haptens: (i) mannosamine–biotin adduct (MBA), (ii) mannosamine–desthiobiotin adduct (MDTBA), (iii) mannosamine–lipoic acid adduct (MLA), (iv) glucosamine–biotin adduct (GBA), (v) mannose, (vi) biotin. (b) Mice were immunized twice intramuscularly, at 2-week intervals, with horse IgG (hsIgG) or haptenated hsIgG. Anti-carrier (hsIgG) or anti-hapten antibody titre was determined as the reciprocal value of the twofold serial dilution end-point (twice the negative control OD value) of the serum as tested by ELISA (n = 4 to 6). Bar values represent mean antibody levels (±SD). *P < 0.05 for the differences between antibody titre to hsIgG–MDTBA, hsIgG–MLA and hsIgG–MBA, and to hsIgG, hsIgG–biotin, hsIgG–mannose and hsIgG–GBA.
Immunization of mice with hsIgG elicited mean antibody titres of 15 000 (Fig. 1b). Conjugation of hsIgG to MBA, MDTBA or MLA significantly reduced (> 100-fold) the mean titres of anti-carrier antibodies compared with native hsIgG. In contrast, neither the MBA building blocks biotin and mannose, nor GBA reduced hsIgG immunogenicity (Fig. 1b). Hence, certain haptens have the unique ability to reduce antibody responses to the carriers, termed carrier immunogenicity reducing haptens (CIRH), while others do not, termed carrier immunogenicity non-reducing haptens (nCIRH).
All of the haptens elicited an anti-hapten antibody response, regardless of the reduction in antibody response to the carrier (Fig. 1b). No correlation was found between the antibody levels raised against the haptens and those raised against the carriers (Fig. 1b).
Reduction of the antibody response to a carrier by CIRH is not a consequence of the carrier-protein's nature or the antibody's response to the hapten
The phenomenon of a reduction in antibody titre following protein conjugation to a specific CIRH was tested with various carriers that were conjugated to MBA as the CIRH.
BALB/c mice were immunized with 50 μg of native or haptenated gp100, BSA or OVA twice at 2-week intervals. Two weeks after the second injection, serum antibody titre levels against the carrier and against the hapten were determined by ELISA. Immunization with gp100, BSA or OVA elicited mean antibody titre levels of 12 400, 684 and 168, respectively. Conjugation of MBA significantly reduced the mean antibody titres to 600, 190 and undetectable, respectively (Fig. 2). Mean antibody titres of 14 700, 2200 and 2000 were found against the hapten following immunization with gp100–MBA, BSA–MBA and OVA–MBA, respectively. In general, conjugation of hapten to an immunogenic carrier (gp100) elicited a higher hapten-specific antibody titre than its conjugation to a less immunogenic carrier (BSA or OVA). The reduction in the immune response to the carrier was not correlated to the anti-protein or anti-hapten responses.
Figure 2.

Antibody titres against various carriers following conjugation to mannosamine–biotin adduct (MBA). Mice were immunized twice, at 2-week intervals, with native or haptenated gp100, BSA or ovalbumin (OVA). Anti-carrier or anti-hapten antibody titre was determined as the reciprocal value of the twofold serial dilution end-point (twice the negative control OD value) of the serum as tested by ELISA (n = 4 to 6). Bar values represent mean antibody titre levels (±SD).*P < 0.05 and **P < 0.01 for the differences between anti-carrier antibody titre levels following immunization with native protein or MBA-conjugated carrier.
Reduction of antibody response by CIRH is conjugated-carrier specific and not systemic
The CIRH was found to reduce the antibody response to its covalently attached carrier. To determine the need for the CIRH to be conjugated to the carrier to reduce the carrier's immunogenicity, mice were immunized with hsIgG mixed with free MBA or with MBA conjugated to BSA.
Immunization with hsIgG elicited mean antibody titres of 753 and 15 237 following the first and second injections, respectively (Table 1). Immunization with hsIgG in a mix with MBA elicited a mean antibody titre similar to the native hsIgG. No reduction in anti-hsIgG antibody titre was observed following injection of hsIgG mixed with free MBA or MBA conjugated to BSA. Hence, the immune-response-reduction effect of CIRH is specific to the conjugated carrier, rather than systemic.
Table 1.
Antibody titre against horse serum IgG, mixed with mannosamine–biotin
| First injection antibody titre |
Second injection antibody titre |
|||
|---|---|---|---|---|
| Immunizing antigen | Mean | SD | Mean | SD |
| hsIgG | 753 | 217 | 15 237 | 2254 |
| hsIgG + MBA (not conjugated) | 659 | 260 | 14 600 | 3010 |
| hsIgG + BSA–MBA | 720 | 310 | 13 100 | 3940 |
| hsIgG–MBA (conjugated) | < 32a | 77a | 15 | |
Mice were immunized twice, at 2-week intervals, with horse IgG (hsIgG), hsIgG mixed with mannosamine–biotin (MBA) or haptenated hsIgG. Anti-carrier (hsIgG) antibody titre was determined as the reciprocal value of the twofold serial dilution end-point (twice the negative control OD value) of the serum as tested by ELISA (n = 4 to 6). Titres < 32 are equal to non-immune serum.
P < 0.05 for the differences between anti-carrier antibody titre levels following immunization with MBA-conjugated carrier and other treatments.
Only a protein carrier can elicit an immune response against the tested haptens
Immunization of mice with MBA conjugated to self or non-self proteins elicited an anti-hapten antibody immune response (Table 2). To test the necessity of protein as an hapten carrier to elicit an immune response, mice were immunized with MBA conjugated to polystyrene microparticles, and no antibody response was triggered (Table 2). In mice immunized with free MBA, no antibody against MBA was found.
Table 2.
Antibody titres against hapten conjugated to self-protein or microparticles
| Anti-hapten antibody titre |
||
|---|---|---|
| Immunizing antigen | Mean | SD |
| hsIgG–MBA | 7780 | 902 |
| Mouse IgG–MBA | 2809 | 825 |
| Microparticles–MBA | < 32a | |
| Unconjugated MBA | < 32a | |
Mice were immunized twice with horse IgG conjugated to mannosamine–biotin (hsIgG–MBA), mouse IgG–MBA or microparticles–MBA. Anti-hapten antibody titre was determined as the reciprocal value of the twofold serial dilution end-point (twice the negative control OD value) of the serum as tested by ELISA (n = 4 to 6). Titres < 32 are equal to non-immune serum.
P < 0.05 for the differences between anti-MBA antibody titre levels following immunization with MBA-conjugated proteins and other treatments.
Protein–CIRH boost injection to mice pre-immunized with native protein reduces recall antibody response
The ability of CIRH to suppress the secondary anti-carrier humoral immune response in mice immunized with the native protein was tested in mice that were immunized twice with native hsIgG, followed by a third injection of hsIgG–MBA. The two injections with hsIgG elicited specific anti-carrier antibodies with a mean titre of 15 200. The third injection of hsIgG increased the mean antibody titre to 44 800. However, if the third injection was with hsIgG–MBA, the mean antibody titre did not increase, remaining at 14 600 (Fig. 3).
Figure 3.
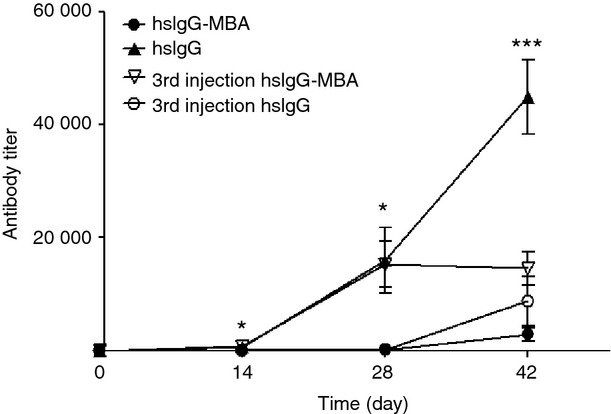
Antibody titre of immune response against carrier following third injection of native horse serum IgG (hsIgG) or hsIgG conjugated to mannosamine–biotin (hsIgG–MBA) into mice pre-immunized with hsIgG or hsIgG–MBA. Mice were immunized three times with hsIgG or hsIgG–MBA. Antibody titres were determined as the reciprocal value of the twofold serial dilution end-point (twice the negative control OD value) of the serum as tested by ELISA (n = 6). Symbol values represent antibody mean titres (±SD). Black circle: hsIgG–MBA, all three injections; black triangle: hsIgG, all three injections; inverted, white triangle: third injection, hsIgG–MBA following two injections of hsIgG; white circle: third injection, hsIgG following two injections of hsIgG–MBA. *P < 0.05 and ***P < 0.001 for the differences between antibody titre levels against hsIgG and antibody titre levels against hsIgG–MBA at the same time-point.
In mice immunized twice with hsIgG–MBA the anti-carrier-specific mean antibody titre level was 90. Following a third injection with hsIgG–MBA, mean antibody titre increased to 2800. A third injection with native hsIgG induced a mean antibody titre of 8700, not significantly different from the second immunization with native carrier protein.
Haptenated proteins internalize normally to the lysosome
Binding of CIRH to the free amine group alters the isoelectric point of the carrier protein (e.g. for hsIgG, from 6.5 to 4.9). This might affect the protein's solubility and its internalization and transport to the lysosome by APC. Any alteration in this pathway might affect the immune response. To test the effect of hapten binding on the carrier's endocytic route, a mouse macrophage cell line was incubated for 3 hr with BSA–biotin (nCIRH) or BSA–MBA (CIRH). Cells were then washed, fixed and permeabilized. BSA conjugated to biotin or MBA was detected with avidin (red) and lysosome was detected with anti-LAMP1 (green). BSA–biotin and BSA–MBA were co-localized to the cell lysosome (Fig. 4). No difference between nCIRH and CIRH was found in terms of the carrier–hapten lysosome co-localization. Hence, internalization and lysosome localization were not affected by conjugation of the protein to CIRH and cannot explain the reduction in antibody titre.
Figure 4.

Localization of haptenated protein in macrophage cellular compartment. Confocal microscopy analysis of antigen-presenting cells. RAW cells were seeded on eight-well Lab-Tek plates and incubated with 50 μg BSA conjugated to mannosamine–biotin (BSA–MBA) or BSA–biotin for 3 hr at 37°. After washing, fixation and permeabilization steps, intracellular staining for lysosome was performed with anti-LAMP1 (green) and carrier staining with avidin (red). Original magnification, 63 × Oil. Bars, 10 μm.
CIRH affects the degradation kinetics and pattern, and the amount of peptide derived from the carrier protein
To determine degradation patterns of hapten-conjugated proteins by cathepsins, BSA–biotin or BSA–MBA were incubated with macrophage lysosome extract or with live macrophages. Degradation products at 24 hr were analysed by MS. The haptens were found to be conjugated to the peptides following the enzymatic degradation, and peptides of different lengths and sequences were derived from the same protein in its native form or modified with CIRH or nCIRH (Fig. 5a). The number of peptides derived from degradation of BSA–MBA was significantly less than those of BSA–biotin or native BSA (51, 113, 238, respectively), as detected by MS (data summarized in table of Fig. 5a). Those peptides cover 34, 60 and 82%, respectively, of the BSA sequence. The reduction effect of CIRH on protein degradation rate was supported by flow cytometry analysis with live cells in which BSA–biotin or BSA–MBA was incubated with live macrophages for 3 hr in a pulse and chase manner. During this period, the proteins were in saturation. Next, the cells were washed and the presence of haptenated BSA in the live cells was monitored over 18 hr at different incubation times by fixation and permeabilization of the cells, and intercellular staining of the protein with avidin conjugated to fluorescent dye. A reduction in fluorescence intensity at 3, 6 and 18 hr indicates a significant reduction in the degradation rate of BSA–MBA compared with BSA–biotin (Fig. 5b).
Figure 5.
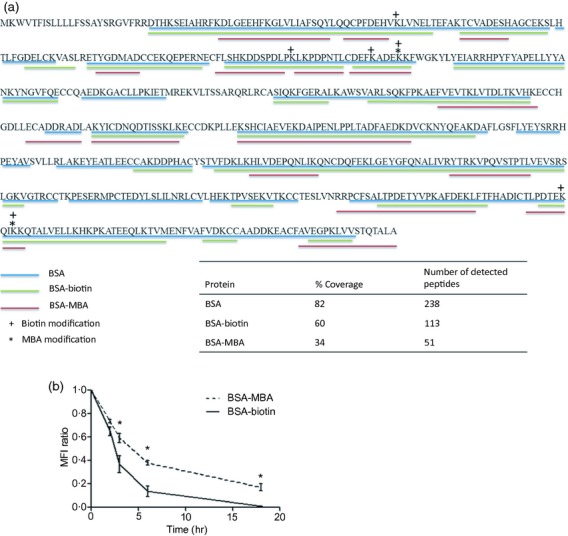
Degradation of haptenated BSA by lysosomal proteases and in live cells. BSA–biotin or BSA–mannosamine–biotin (MBA) were incubated for 24 hr with macrophage (RAW 264.7) cell line lysate in sodium acetate buffer. (a) Degradation products of BSA, BSA–biotin or BSA–MBA were analysed by LC–MS/MS for peptide sequence. (b) Macrophages were incubated with haptenated carriers, BSA–biotin or BSA–MBA, for 3 hr. The level of the haptenated carriers was monitored at various time points up to 18 hr. The cells were fixed, permeabilized and intercellularly stained with avidin–FITC. Fluorescence analysis was performed by flow cytometry. The results are shown as calculation of geometric mean fluorescence intensity (MFI) ratio between time 0 and four time-points over 18 hr (±SD of three independent experiments). *P < 0.05 for the differences in the degradation rates between BSA–MBA and BSA–biotin.
Elevation in Treg cell population in hsIgG–MBA-immunized mice
To test the effect of CIRH on the Treg cell population, mice were immunized with hsIgG, hsIgG–MBA (CIRH), hsIgG–biotin (nCIRH) or saline. Forty-eight hours after the second immunization, splenocytes were isolated and stained for CD4+, CD25+ and FoxP3+. It was found that in mice immunized with hsIgG, hsIgG–biotin or saline, the percentage of FoxP3+ out of CD4+ cells was 12.6, 12.16 and 11.4, respectively. Immunization with hsIgG–MBA induced a significant elevation in Treg cells to a mean of 13.9 (Fig. 6a,b). No differences were found in the CD3+ or CD19+ cell population (data not shown). The experiment was repeated twice, with similar results.
Figure 6.

T regulatory (Treg) cell population by flow cytometry analysis. Splenocytes were isolated and stained for CD4+ and CD25+ followed by intracellular staining for FoxP3+. CD25+, FoxP3+ cell frequencies out of CD4+ within the indicated gates are shown. Mice were immunized with horse serum (hs) IgG (i), hsIgG–biotin (ii), hsIgG– mannosamine–biotin (MBA) (iii), and saline (iv) (n = 6) and data represent two independent experiments (a). Bar values represent FoxP3+ percentage mean of gated CD4+ (±SD) (b). **P < 0.01 for the differences between hsIgG–MBA- and hsIgG-, hsIgG–biotin- or saline-immunized mice.
CIRH affects levels of plasma cytokines IL-4 and IL-10
To test the effect of CIRH on the cytokines levels, mice were immunized with hsIgG, hsIgG–MBA (CIRH), hsIgG–biotin (nCIRH) or saline. Twenty-four hours after the second immunization, plasma cytokine levels were determined by ELISA. The mean plasma IL-4 levels in hsIgG-immunized, hsIgG–biotin-immunized and saline-immunized mouse groups were 222.28, 162.79 and 220.89 pg/ml, respectively; IL-4 levels in the hsIgG–MBA group were significantly decreased to 43.7 pg/ml. The IL-10 level in hsIgG–MBA-immunized mice was 2022 pg/ml, significantly higher than in the hsIgG-injected and hsIgG–biotin-injected mice (863.99 and 1349.71, respectively), but similar to the saline control (Table 3).
Table 3.
Cytokine levels in immunized mice
| hsIgG |
hsIgG–biotin |
hsIgG–MBA |
Saline |
|||||
|---|---|---|---|---|---|---|---|---|
| Plasma cytokine | Mean | SD | Mean | SD | Mean | SD | Mean | SD |
| IL-4 (pg/ml) | 222.28 | 91.14 | 180.79 | 50.8 | 43.7a | 26.01 | 220.89 | 47.7 |
| IL-10 (pg/ml) | 863.99 | 335.81 | 1349.71 | 284.62 | 2022.81a | 327.15 | 2423.81a | 20.79 |
Mice were immunized twice with horse serum (hs) IgG, hsIgG–biotin, hsIgG–mannosamine–biotin (MBA) or saline. Twenty-four hours after the second immunization, mouse plasma was tested by ELISA. Plasma cytokine levels are presented as pg/ml (± SD).
P < 0.05 between different treatments in the same tested cytokine.
Discussion
One of the important hallmarks of the immune system is its ability to react differently by distinguishing between different pathogenic stimuli. To achieve an appropriate balance between effector and regulatory responses over the course of an infection, several layers of regulation are required. One of these is likely provided by the infection-driven conversion of Foxp3+ Treg cells.22 Aside from evidence that natural Treg cells arise and mature in the thymus, there is clear evidence that they can develop in the periphery under certain conditions.
Activation of Treg cells may be achieved by APC, which can orchestrate the immune response via regulation of peptide-loaded MHC molecules for T-cell receptors.23 In APC, antigen-processing is mediated by the cathepsins, which recognize a certain motif in the antigen and degrade the protein to peptides.24 Protein modification may affect their normal recognition and cleavage by cathepsin and thereby influence the peptide–MHC class II repertoire.
The aim of this study was to explore the mechanism by which hapten reduces the antibodies immune response to its carrier. This was done by comparing the effect of various haptens on the immunogenicity of various protein carrier conjugates and by analysing the effect of hapten on protein internalization and degradation to peptide by APC. The effect of haptens on protein immunogenicity has been tested previously.10–12 In this study, mice were immunized with hsIgG as a potent immunogenic carrier conjugated to MBA, MDTBA, MLA, GBA, mannose or biotin.
One possible explanation for the reduction in antibody immune response is the masking effect, which can be achieved by conjugation of molecules to a protein's surface.25 Immunization with hsIgG conjugated to MBA, MDTBA or MLA significantly reduced the antibody immune response against hsIgG. In contrast, conjugation of mannose or biotin, which are the building blocks of MBA, did not reduce the immune response to the carrier (Fig. 1a,b). Interestingly, conjugation of GBA, a stereoisomer of MBA that differs only in the stereochemistry at the C-2 position, did not reduce the immune response in a similar manner (Fig. 1a,b). Considering that the hapten-to-carrier ratio and the degree of binding to available free amines are identical for all haptens and that there are relatively minor differences in the molecular weights of the various haptens, these results suggest that the reduction effect on the antibody response is not due to a passive physical masking effect of the carrier epitopes by the haptens, but rather that an active mechanism is involved in this reduction by CIRH. Moreover, polyclonal antibody raised in mice against the native protein can effectively bind the haptenated protein in vitro as tested by ELISA (data not shown). Another possible explanation for the reduction in immune response might be an immunodominant effect of the hapten.
Immunodominance is described as epitopes in an antigen that are preferentially recognized by B and T cells, and hence the immune response focuses on only a few of the many potential epitopes on the antigen.26 However, the haptenated carriers induced an antibody response against the hapten, regardless of its type (CIRH or nCIRH) (Fig. 1b) and regardless of the nature of the carrier (Fig. 2). The antibody response against the haptens was not correlated with the reduction in immune response against the carrier (Fig. 1b). Hence, the reduction in the antibody response to the carrier cannot be explained by an immunodominant effect of the hapten.
The immune-response-reducing ability of CIRH was demonstrated on various known carriers, including BSA, OVA and gp100, a member of a family of melanoma/melanocyte differentiation antigens strongly expressed in most melanomas and known as a potent immunogenic protein27 (Fig. 2). Hence, the decrease in carrier immunogenicity by conjugation of CIRH to the carrier protein is a general phenomenon that is not dependent on the nature of the protein.
To determine whether the reduction effect by CIRH is systemic or specific to the conjugated protein, mice were immunized with hsIgG mixed with, but not conjugated to MBA, or with MBA conjugated to BSA. No reduction in the anti-hsIgG antibody immune response was observed following immunization of mice with native hsIgG without chemically conjugating the CIRH to the carrier (Table 1). Hence, the reduction effect of CIRH only applies to the conjugated protein and it does not reflect systemic suppression of the immune response.
To determine whether carrier-specific memory B cells can be stimulated by haptenated proteins, hsIgG–MBA was injected into mice pre-immunized with hsIgG. Even though antibody induced by the native protein can bind the haptenated protein in vitro, no induction of antibody response was observed in vivo (Fig. 3). This result suggests that there is no further activation of B cells, because of suppression or skew of the immune response from the B cells.
Triple immunization of mice with hsIgG–MBA induced a mean titre of hsIgG-specific antibodies of 2800, a 16-fold reduction as compared to the antibody titre induced following triple immunization with native hsIgG. Immunization with native hsIgG in mice pre-immunized twice with hsIgG–MBA elevated the antibody titre against hsIgG to 8700, significantly higher than that to hsIgG–MBA (Fig. 3). This suggests that immunization with haptenated carrier induces memory cells, but not anti-carrier plasma B cells.
To examine whether the B-cell response to the hapten is T-cell-dependent, mice were immunized with free MBA, or MBA conjugated to polystyrene beads to imitate the multivalent effect. Hence there were no protein-derived peptides for the activation of T cells by APC through the MHC molecule. No induction of antibody was found following immunization with the free MBA or with beads conjugated to MBA (Table 2). This result indicates that the B-cell response to hapten is T-cell-dependent, in agreement with previous studies using other haptens.11
To initiate a T-cell-dependent immune response, antigens must first be internalized by APC, transported to the lysosome and digested by proteases into peptides that can be loaded and presented by the MHC class II molecule. Chemical modification of proteins may alter their cleavage sites, resulting in the generation of different peptides. To test the effect of the conjugation of CIRH versus nCIRH on carrier internalization, BSA–MBA or BSA–biotin were incubated with the macrophage cell line for 3 hr. Following incubation, the proteins were analysed for their intracellular location by confocal microscopy. The conjugation of nCIRH and CIRH to BSA did not affect BSA internalization and localization to the lysosome (Fig. 4). To determine the effect of the conjugation of CIRH in comparison to nCIRH on the carrier's degradation to peptides, BSA–MBA or BSA–biotin were incubated with macrophage lysosome extract. Degradation of the proteins and the peptides derived from the degraded proteins were analysed by MS. The kinetics of the degradation in the cells was slower in BSA–MBA than in BSA–biotin as tested by incubating the haptenated proteins with live macrophages for 3 hr, washing the cells, and monitoring the presence of the haptenated BSA in the cells by flow cytometry using fluorescent dye for the protein (Fig. 5b). Analysis by MS showed the generation of peptides of different lengths and sequences from BSA–MBA (CIRH) compared with BSA–biotin (nCIRH) or native BSA (Fig. 5a). In addition, on part of the peptide, the hapten modification was found to be attached (Fig. 5a), meaning that part of the peptides may be presented on the MHC molecule with the hapten. The relatively low frequency of MBA compared with biotin modification on the detected peptides may be explained by the low number of peptides obtained following degradation of BSA-MBA (Fig. 5a). This alteration in protein processing by APC may be a key point in understanding the impact of modification on the generation of protein-derived peptides for presentation on MHC molecules to activate the corresponding T cells. As any given TCR is specific for a unique combination of a particular peptide and a particular MHC molecule, the presence, absence or new peptides generated from the same protein may lead to inactivation or activation of different T-cell subpopulations. Examination of the lymphocyte population in mice following boost immunization with hsIgG–MBA revealed significant elevation in the Treg cell population compared with mice that were immunized with native hsIgG, hsIgG–biotin or saline (Fig. 6a,b). A possible model for the involvement of Treg cells in reduction of antibody response against the carrier is that part of the haptens (the intrinsic properties of which could not be generalized in this study) may activate a pathway in APC or specifically B cells, which induces the transformation of part of T helper cells into Treg cells. As the reduced response by CIRH is not systemic, these Treg cells may be antigen-specific. In a previous study it was found that the immune response elicited by haptens is TLR-independent, and CD4+ T-cell responses to haptenated antigens are therefore largely MyD88-independent, differing from the response to protein.11 This may explain how those Treg cells are involved in the suppression of the response to the carrier but not to the hapten, which triggers the antibody production via an alternative pathway. In further analysis of plasma cytokines, lower levels of IL-4 were found in hsIgG–MBA-immunized mice than in those immunized with native hsIgG, hsIgG–biotin or saline. Plasma IL-10 levels were lower in mice immunized with native hsIgG and hsIgG–biotin than in saline-immunized and hsIgG–MBA-immunized mice. No differences were found in the cytokine levels between native hsIgG-immunized and hsIgG–biotin-immunized mice (Table 3). To analyse the cytokine levels, blood was drawn 24 hr after boost injection. Because each cytokine has an optimal point for sampling and a relatively short protein half-life, the differences in the cytokine levels between the groups, rather than the absolute levels, were considered. The differences in the plasma cytokine levels emphasize the different activation of the immune response following immunization of mice with conjugated CIRH.
In previous studies, reduction of the immune response against proteins following their conjugation to haptens was presented as a general phenomenon, without discriminating CIRH from nCIRH.10,11 In this study, different types of haptens were shown to have different impacts on the immune response. This research provides insight into the potential effect of protein modification on activation or silencing of the immune response. Post-translational modification, directed and spontaneous, of self-antigens can explain the loss of immune tolerance to self-antigens by creating new antigens that alter the processing and presentation of peptides.19 For instance, post-translation-modified type II collagen has been shown to contribute to the development of arthritis.28
In conclusion, some haptens reduced carrier-protein immunogenicity, but all of the tested haptens induced anti-hapten antibodies, regardless of their ability to reduce the immune response against the carrier. In the current study, the effect of haptenation on protein degradation for presentation on MHC by APC and the subsequent alteration in the immune response were explored. Further analysis of this mechanism could provide a better understanding of the interaction and impact of protein modification on the immune response, particularly its role in autoimmune diseases and allergy.
Acknowledgments
TG, EA, JP had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Experimental design: TG, EA, JP, EDH and JV. Molecule synthesis and protein conjugation: IR, SH and JV. Acquisition of data: TG, IR, ML. MS analysis: EB and AA. Analysis and interpretation of the data: TG, IR, EA and JP. Drafting of the manuscript: TG, EA and JP.
Glossary
- APC
antigen-presenting cell
- CIRH
carrier immunogenicity reducing haptens
- GBA
glucosamine–biotin adduct
- hs
horse serum
- MBA
mannosamine–biotin adduct
- MDTBA
mannosamine–desthiobiotin adduct
- MLA
mannosamine-lipoic acid adduct
- nCIRH
carrier immunogenicity non-reducing haptens
- OVA
ovalbumin
- Treg
regulatory T
Disclosures
The authors have no conflicts of interest to disclose.
References
- 1.Murphy K, Travers P, Walport M, Janeway C. Immunization. In: Lawrence E, editor. Janeway's Immunobiology. 8th edn. New York: Garland Science; 2012. pp. 718–9. [Google Scholar]
- 2.Pollard AJ, Perrett KP, Beverley PC. Maintaining protection against invasive bacteria with protein–polysaccharide conjugate vaccines. Nat Rev Immunol. 2009;9:213–20. doi: 10.1038/nri2494. [DOI] [PubMed] [Google Scholar]
- 3.Zarling AL, Ficarro SB, White FM, Shabanowitz J, Hunt DF, Engelhard VH. Phosphorylated peptides are naturally processed and presented by major histocompatibility complex class I molecules in vivo. J Exp Med. 2000;192:1755–62. doi: 10.1084/jem.192.12.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdel-Motal UM, Berg L, Rosen A, et al. Immunization with glycosylated Kb-binding peptides generates carbohydrate-specific, unrestricted cytotoxic T cells. Eur J Immunol. 1996;26:544–51. doi: 10.1002/eji.1830260307. [DOI] [PubMed] [Google Scholar]
- 5.Weltzien HU, Moulon C, Martin S, Padovan E, Hartmann U, Kohler J. T cell immune responses to haptens. Structural models for allergic and autoimmune reactions. Toxicology. 1996;107:141–51. doi: 10.1016/0300-483x(95)03253-c. [DOI] [PubMed] [Google Scholar]
- 6.Engler OB, Strasser I, Naisbitt DJ, Cerny A, Pichler WJ. A chemically inert drug can stimulate T cells in vitro by their T cell receptor in non-sensitised individuals. Toxicology. 2004;197:47–56. doi: 10.1016/j.tox.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 7.Thierse HJ, Gamerdinger K, Junkes C, Guerreiro N, Weltzien HU. T cell receptor (TCR) interaction with haptens: metal ions as non-classical haptens. Toxicology. 2005;209:101–7. doi: 10.1016/j.tox.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 8.Ly D, Kasmar AG, Cheng TY, et al. CD1c tetramers detect ex vivo T cell responses to processed phosphomycoketide antigens. J Exp Med. 2013;210:729–41. doi: 10.1084/jem.20120624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ortmann B, Martin S, von Bonin A, Schiltz E, Hoschutzky H, Weltzien HU. Synthetic peptides anchor T cell-specific TNP epitopes to MHC antigens. J Immunol. 1992;148:1445–50. [PubMed] [Google Scholar]
- 10.Shimizu T, Osaka Y, Banri-Koike C, et al. T cells specific to hapten carrier but not to carrier alone assist in the production of anti-hapten and anti-carrier antibodies. Int Immunol. 2007;19:1157–64. doi: 10.1093/intimm/dxm080. [DOI] [PubMed] [Google Scholar]
- 11.Palm NW, Medzhitov R. Immunostimulatory activity of haptenated proteins. Proc Natl Acad Sci USA. 2009;106:4782–7. doi: 10.1073/pnas.0809403105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gefen T, Pitcovski J, Vaya J, et al. Coated cross-species antibodies by mannosamine-biotin adduct confer protection against snake venom without eliciting humoral immune response. Vaccine. 2010;28:8197–202. doi: 10.1016/j.vaccine.2010.09.032. [DOI] [PubMed] [Google Scholar]
- 13.Vaya J, Aizenshtein E, Khatib S, Gefen T, Fassler M, Musa R, Krispel S, Pitcovski J. Mannosamine-biotin as a novel masking agent for coating IgG for immune response silencing and augmentation of antibody–antigen interaction. Vaccine. 2009;27:6869–76. doi: 10.1016/j.vaccine.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 14.Holmdahl M, Ahlfors SR, Holmdahl R, Hansson C. Structure-immune response relationships of hapten-modified collagen II peptides in a T-cell model of allergic contact dermatitis. Chem Res Toxicol. 2008;21:1514–23. doi: 10.1021/tx8001077. [DOI] [PubMed] [Google Scholar]
- 15.Honey K, Rudensky AY. Lysosomal cysteine proteases regulate antigen presentation. Nat Rev Immunol. 2003;3:472–82. doi: 10.1038/nri1110. [DOI] [PubMed] [Google Scholar]
- 16.Beck H, Schwarz G, Schroter CJ, et al. Cathepsin S and an asparagine-specific endoprotease dominate the proteolytic processing of human myelin basic protein in vitro. Eur J Immunol. 2001;31:3726–36. doi: 10.1002/1521-4141(200112)31:12<3726::aid-immu3726>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 17.Manoury B, Hewitt EW, Morrice N, Dando PM, Barrett AJ, Watts C. An asparaginyl endopeptidase processes a microbial antigen for class II MHC presentation. Nature. 1998;396:695–9. doi: 10.1038/25379. [DOI] [PubMed] [Google Scholar]
- 18.Gahring L, Carlson NG, Meyer EL, Rogers SW. Granzyme B proteolysis of a neuronal glutamate receptor generates an autoantigen and is modulated by glycosylation. J Immunol. 2001;166:1433–8. doi: 10.4049/jimmunol.166.3.1433. [DOI] [PubMed] [Google Scholar]
- 19.Doyle HA, Mamula MJ. Autoantigenesis: the evolution of protein modifications in autoimmune disease. Curr Opin Immunol. 2012;24:112–8. doi: 10.1016/j.coi.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishihama Y, Rappsilber J, Andersen JS, Mann M. Microcolumns with self-assembled particle frits for proteomics. J Chromatogr A. 2002;979:233–9. doi: 10.1016/s0021-9673(02)01402-4. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, Xin L, Shan B, et al. PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol Cell Proteomics. 2012;11:M111.010587. doi: 10.1074/mcp.M111.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grainger JR, Hall JA, Bouladoux N, Oldenhove G, Belkaid Y. Microbe-dendritic cell dialog controls regulatory T-cell fate. Immunol Rev. 2010;234:305–16. doi: 10.1111/j.0105-2896.2009.00880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce TH2 and tolerogenic responses. Nat Immunol. 2010;11:647–55. doi: 10.1038/ni.1894. [DOI] [PubMed] [Google Scholar]
- 24.Bryant P, Ploegh H. Class II MHC peptide loading by the professionals. Curr Opin Immunol. 2004;16:96–102. doi: 10.1016/j.coi.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 25.Gefen T, Vaya J, Khatib S, Harkevich N, Artoul F, Heller ED, Pitcovski J, Aizenshtein E. The impact of PEGylation on protein immunogenicity. Int Immunopharmacol. 2013;15:254–9. doi: 10.1016/j.intimp.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 26.Frank SA. Immunology and Evolution of Infectious Disease. New Jersey: Princeton University Press; 2002. Immunodominance within Hosts; pp. 73–89. [PubMed] [Google Scholar]
- 27.Eisenberg G, Machlenkin A, Frankenburg S, Mansura A, Pitcovski J, Yefenof E, Peretz T, Lotem M. Transcutaneous immunization with hydrophilic recombinant gp100 protein induces antigen-specific cellular immune response. Cell Immunol. 2010;266:98–103. doi: 10.1016/j.cellimm.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Duivenvoorde LM, Han WG, Bakker AM, et al. Immunomodulatory dendritic cells inhibit Th1 responses and arthritis via different mechanisms. J Immunol. 2007;179:1506–15. doi: 10.4049/jimmunol.179.3.1506. [DOI] [PubMed] [Google Scholar]