

Abstract
The extracellular matrix regulates tissue development and homeostasis, and its dysregulation contributes to neoplastic progression. The extracellular matrix serves not only as the scaffold upon which tissues are organized but provides critical biochemical and biomechanical cues that direct cell growth, survival, migration and differentiation and modulate vascular development and immune function. Thus, while genetic modifications in tumor cells undoubtedly initiate and drive malignancy, cancer progresses within a dynamically evolving extracellular matrix that modulates virtually every behavioral facet of the tumor cells and cancer-associated stromal cells. Hanahan and Weinberg defined the hallmarks of cancer to encompass key biological capabilities that are acquired and essential for the development, growth and dissemination of all human cancers. These capabilities include sustained proliferation, evasion of growth suppression, death resistance, replicative immortality, induced angiogenesis, initiation of invasion, dysregulation of cellular energetics, avoidance of immune destruction and chronic inflammation. Here, we argue that biophysical and biochemical cues from the tumor-associated extracellular matrix influence each of these cancer hallmarks and are therefore critical for malignancy. We suggest that the success of cancer prevention and therapy programs requires an intimate understanding of the reciprocal feedback between the evolving extracellular matrix, the tumor cells and its cancer-associated cellular stroma.
Keywords: ECM, hallmarks of cancer, mechanotransduction
Introduction
The extracellular matrix (ECM) regulates the development and maintains tissue homeostasis 1. The ECM is composed of a complex network of macromolecules that assemble into three-dimensional supramolecular structures with distinct biochemical and biomechanical properties that regulate cell growth, survival, motility and differentiation by ligating specific receptors such as integrins, syndecans and discoidin receptors 2,3. The ECM also provides the structural foundation for tissue function and mechanical integrity, regulates the availability of growth factors and cytokines and maintains the hydration and pH of the local microenvironment. A critical aspect of the ECM is that it is dynamically remodeled and specifically tailored to the structure/function of each organ, and its composition, biomechanics and anisotropy are exquisitely tuned to reflect the physiological state of the tissue 4,5.
Tumors often display desmoplasia, and this fibrotic state is characterized by increased deposition, an altered organization and enhanced post-translational modifications of ECM proteins 6. The cancer-associated ECM is not only an integral feature of a tumor but also actively contributes to its histopathology and behavior 7,8. For instance, patients with pancreatic cancer show a marked stromal desmoplasia that often associates with tumor progression and poor disease outcome 9. Similarly, expression of matrix remodeling genes such as MMPs and collagen cross-linkers is predictive of a poor prognosis for breast cancer patients 10,11. Fibrosis can also predispose a tissue to malignancy; patients with cirrhosis of the liver or cystic fibrosis, conditions that are characterized by abnormal accumulation of collagen, have an increased risk of developing cancer 12,13. Moreover, increased mammographic density, which associates with increased collagen deposition, correlates with an elevated risk of developing breast cancer 14. Indeed, MMPs and high mechanical stress are predictive of tumor formation in breast cancer patients 15.
Originally described by Hanahan and Weinberg 16, the hallmarks of cancer encompass fundamental biological capabilities acquired during the development of human cancers including sustained proliferation, evasion of growth suppression, death resistance, replicative immortality, induced angiogenesis, and initiation of invasion and metastasis. In 2011, Hanahan and Weinberg 17 revisited the hallmarks, adding two emerging features: dysregulated cellular metabolism and the evasion of immune destruction. Importantly, the ECM regulates many of the same cellular responses that characterize the cancer hallmarks (Fig1). This overlap suggests that the biochemical and biophysical properties of the ECM should be considered when examining tumor behavior and therapeutic interventions 18. In this review, we discuss how the composition and the mechanical properties of the ECM influence the acquisition and maintenance of each of the original and emerging cancer hallmarks.
Figure 1.

Influences of ECM on the hallmarks of cancer
From tumor initiation to metastasis, the ECM influences each of the classically defined and emerging hallmarks of cancer as first described by Hanahan and Weinberg in 2000 and amended in 2011. ECM molecules bind to cell surface receptors, which activates intracellular signaling pathways. ECM adhesion-induced signals through ERK and PI3K promote self-sufficient growth 22. FAK signaling inhibits growth suppressors p15 and p21 and limits the induction of apoptosis through p53 42. ECM components and biophysical properties promote EMT induction and enhance pro-migratory pathways, particularly TGF-β and RhoA/Rac signaling 96. ECM stiffness also enhances angiogenesis and increases VEGF signaling in endothelial cells 78. At each phase of tumorigenesis, the ECM adapts to reinforce the progression of the disease through promotion of the hallmarks.
Sustaining proliferative signaling
Cellular transformation and tumor progression require escape from proliferative suppression. Proliferation is initiated by the ligation of growth factor receptors whose activation promotes intracellular signaling that facilitates cell cycle progression. Cell cycle progression in turn is tightly controlled by the G1/S cell cycle checkpoint. G1/S cell cycle transition requires cellular adhesion to the ECM. Adhesion to the ECM permits growth factor-dependent activation of Ras, which through Erk signaling promotes G1/S transition 19–21. Adhesion-dependent Fak phosphorylation stimulates Ras and PI3K signaling to activate Erk, promoting its nuclear translocation and cyclin D1 induction to sequester the growth suppressors CDK1 and CDK4 22. Although Erk mediates cell cycle progression in fibroblasts, recent work suggests that the Rac GTPases mediate adhesion-dependent cyclin D1 expression in epithelial cells. Interestingly, many malignantly transformed cells secrete their own ECM ligands and, in doing so, are able to escape proliferative suppression to grow and survive in hostile environments 23,24. Tumor cells which acquire the ability to synthesize their own ECM proteins are shown to be highly metastatic 25. Indeed, transformation by oncogenes such as Ras, which stimulates Erk signaling to promote anchorage independence for growth and survival, simultaneously induces expression of several ECM proteins 26. In addition, a malignant tissue is typically stiffer than its normal counterpart, and this altered biomechanical property is largely mediated by a highly cross-linked and oriented collagenous ECM 6,8 (see 27–29 for review). This stiffened ECM associates with tumor aggression and correlates with an increased propensity toward metastasis and poorer patient outcome 6,30. Consistently, cells interacting with a stiffer ECM proliferate more in response to growth factors and express genes that positively correlate with a proliferative signature 7,31. Indeed, in response to a stiffened matrix, cells elevate Fak phosphorylation and stimulate Erk, PI3K and Rac, which accelerates cell cycle progression through increased expression of cyclin D1 7,32–34. Thus, the ECM and its receptors regulate cell proliferation, and corruption of these interactions modulates tumor progression.
Evading growth suppressors
Cellular quiescence must be overcome to establish a neoplastic lesion. Many tumor suppressors limit cell cycle transitions by blocking progression from G1 to S phase of the cell cycle 35,36. Activated p53 and Smad phosphorylation by TGF-β induces proteins such as p21 and p27 that inhibit the activity of cyclin-dependent kinases that are critical for cell cycle progression, thus suppressing cell growth 37–40. However, cell ligation to an ECM can temper the activity of many of these tumor suppressor pathways, thereby overriding this growth suppression mechanism 41. For instance, cell–ECM interactions regulate TGF-β signaling by inducing p130Cas to prevent Smad3 phosphorylation and reduce p15 and p21 expressions to subvert cell cycle arrest 42. ECM adhesion also directly and indirectly inhibits the function of tumor suppressors such as BRCA1, thereby compromising cell cycle checkpoint control 43. In this manner, an increase in tumor cell adhesion to the ECM can circumvent many of the normal growth suppression pathways to foster malignant transformation. Indeed, the quiescence or dormant state observed in some extravasated metastatic cells may be due, at least in part, to their inability to actively engage the ECM and activate integrin and growth factor-dependent signaling at the secondary site 44,45. Thus, in the absence of integrin-mediated adhesion to the ECM, Src and Erk signaling is not activated, and tumor suppressor levels of cell cycle inhibitors such as p27 remain high, preventing cell proliferation 46,47. Consistently, lung tumor metastasis is enhanced by ECM pre-conditioning, stiffening with lysyl oxidase and enhanced fibronectin deposition, which may foster tumor cell growth and survival by overriding the activity of these tumor suppressors. Indeed, a stiffened ECM reduces the expression of genes that typically inhibit cell cycle progression 27,48,49. Matrix stiffness also induces the expression of microRNAs that lower expression of the tumor suppressor PTEN, thereby enhancing PI3K/Akt activity to promote cell growth and survival (Fig2) 50. In this respect, the HIPPO pathway components YAP and TAZ regulate cell proliferation and apoptosis to control organ development, and the activity of these transcription factors is exquisitely sensitive to mechanical cues from the ECM. Moreover, overexpression or mechanical activation of YAP permits tumor cells to overcome growth suppression by contact inhibition and to achieve uncontrolled proliferation 51,52. These findings argue that tumor cells that are able to leverage interactions with the ECM should have a distinct growth advantage.
Figure 2.

Effects of matrix rigidity on tumor progression
Recent work from Mouw et al 50 shows how matrix rigidity impacts on tumor progression. (A) Culturing MCF10a cells on stiff polyacrylamide gels in vitro promotes FAK phosphorylation and suppresses the levels of the tumor suppressor PTEN. In vivo inhibition of collagen cross-linking (LOX-i) in the polyoma middle T (PyMT) mouse model of breast cancer results in the opposite phenotype, with PTEN levels being increased feeding into a suppression of Akt activity. (B) These data suggest that the stiffening of the ECM works through focal adhesions to inhibit tumor suppressors and promote tumor progression.
Resisting cell death
Malignant transformation is accompanied by enhanced cell survival. Cell death is mediated by the cleavage of cell death-associated caspases and the mitochondrial release of pro-apoptotic proteins such as cytochrome c, with tight regulation by a coterie of pro- and anti-apoptotic molecules including those from the Bcl2 family 53. Oncogenic transformation is frequently accompanied by the acquisition of anchorage-independent survival (suppression of anoikis) as occurs when the receptor tyrosine kinase Erbb2 is overexpressed and Bim is inhibited through increased Erk activation 54. Similarly, cell adhesion to an ECM inactivates pro-apoptotic molecules such as Bax and induces expression of several anti-apoptotic genes including Bcl2 to promote cell survival 55–57. Likewise, laminin ligation of α6β4 integrin permits EGFR activation of Rac to promote anchorage-independent survival by stimulating NF-κB 58.
Intriguingly, ECM ligation can also enhance a cell's ability to resist apoptosis induction. Indeed, breast tumor stiffness, a feature associated with elevated integrin signaling, associates positively with reduced chemotherapeutic responsiveness 59. Although the molecular mechanisms underlying this phenotype remain poorly understood, prior studies suggest that activation of β1 integrin and FAK via ECM ligation can suppress p53-induced apoptosis through Mdm2-mediated ubiquitination and p53 degradation in response to DNA damaging agents 60–62. Moreover, tissue polarity, mediated through laminin ligation of α6β4 integrin and hemidesmosome formation, permits NF-κB activation and death resistance of mammary tumor cells in response to a plethora of chemotherapeutic and immune receptor death stimuli 63.
Enabling replicative immortality
The unrestrained growth observed in many tumors associates with replicative immortality. Normal cells demonstrate limited replicative ability due primarily to shortening of telomeres, which are the regions of noncoding nucleotide sequence at the end of each chromosome. Because conventional DNA polymerases are unable to replicate the entire DNA strand, the telomere at the end of each chromosome progressively shortens after each cell division. Cancer cells overcome this limitation by expressing the enzyme telomerase, which elongates the telomeres, and thereby, overcoming replicative senescence. Interestingly, patients with idiopathic pulmonary fibrosis, which is characterized by increased deposition of ECM proteins and tissue stiffening, exhibit elevated levels of telomerase, suggesting increased ECM adhesion may influence the replicative behavior of cells 64,65. Indeed, epithelial cells expressing high levels of ITGB1 were enriched for telomerase activity 66.
Inducing angiogenesis
Tumors stimulate neo-vascularization to provide the oxygen and nutrients required for their growth and survival 67. Angiogenesis is stimulated by growth factors such as VEGF and FGF and involves the proliferation and migration of endothelial cells into the nutrient-deprived tissue, specifically regions adjacent to the tumor followed by their assembly into patent blood vessels 68,69. The ECM surrounding the tumor acts as a reservoir for pro- and anti-angiogenic factors, provides a conduit for the migration of endothelial cells, and fosters the growth and survival of newly recruited endothelial cells 70–75. A stiffened tumor-associated ECM also favors angiogenesis by promoting endothelial cell migration and by inducing GATA2 and TFII-I transcriptional programs to enhance expression of VEGFR2 receptors that support endothelial cell growth and survival 76–80. Nevertheless, a highly rigid ECM can also compromise vascular integrity and activate MMPs that degrade the ECM by releasing anti-angiogenic factors. These data indicate that the ECM can both promote and inhibit angiogenesis 76,77,81.
Activating invasion and metastasis
Malignant transformation is defined as the invasion of transformed cells into the adjacent parenchyma and requires the acquisition of a motile, invasive phenotype 82. Actin-rich protrusions, termed invadopodia, which require integrin-mediated adhesion and focal adhesion formation, direct tumor cell invasion through localized MMP-mediated matrix degradation 83,84. ECM stiffness promotes invadopodia formation and enhances tumor cell invasion by driving focal adhesion assembly 85. Once the physical barriers surrounding a benign tumor are compromised, tumor cell migration is driven through elevated activity of Rho and Rac GTPases, which stimulate actin assembly and turnover and actomyosin-dependent cell tension 86. Thereafter, the nature of the migratory phenotype is dictated by the activity of the dominant Rho-family GTPase; a mesenchymal migratory phenotype is largely dictated by the activity of Rac GTPases while elevated RhoA GTPase activity favors ameboid migration 87. Consistently, the oncogene Ras stimulates Rho activity to promote an ameboid migratory phenotype, whereas the tumor suppressor p53 inhibits tumor cell migration by reducing RhoA activity 88,89. Once within the parenchyma, tumor cell metastasis depends upon efficient navigation through the tissue to the vasculature, its successful intravasation into the circulatory system (blood or lymphatic vessels), where the tumor cell can disseminate throughout the body, eventual extravasation into a secondary tissue site and colonization (survival and growth) within the secondary site to form a viable tumor colony (Fig3) 90. In this regard, the metastatic potential of a transformed cell is favored by an epithelial to mesenchymal transition (EMT), which is fostered by exposure to TGF-β secreted by infiltrating immune cells or via localized degradation of the ECM 91–95. A stiffened ECM promotes TGF-β-induced EMT and induces a basal-like tumor cell phenotype to stimulate cancer metastasis 96; conversely, inhibiting collagen crosslinking and reducing matrix stiffening prevents tumor metastasis 10,95,97. Thus, ECM stiffness promotes malignant transformation and metastasis by fostering integrin-dependent cell adhesion and migration and regulating tumor plasticity.
Figure 3.
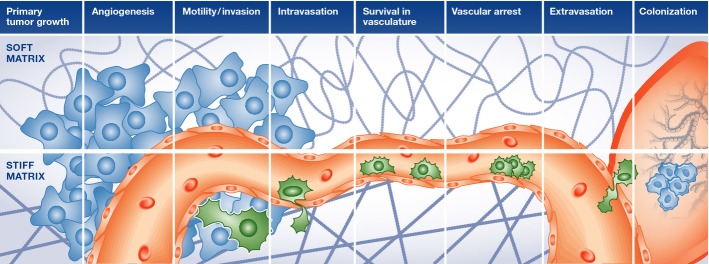
Influences of ECM on the metastatic cascade
Tumor cell dissemination and establishment of metastatic lesions are controlled by several stringent processes that include induction of an invasive phenotype, migration through the tissue parenchyma, intravasation into the bloodstream, survival in the circulation, followed by extravasation and growth and survival at a secondary organ site. Adhesion to the ECM regulates each of these stages of tumor metastasis.
Emerging hallmark: avoiding immune destruction
Immune surveillance by the adaptive immune response is a key physiological mechanism that prevents tumor formation. Adaptive immunity relies on the ability of cytotoxic T cells to recognize foreign or mutated antigens displayed on transformed cells and to thereafter induce their demise through T-cell-mediated cell death. The ECM can both support and compromise the adaptive tumor immune response. The pro-immunogenic activity of the ECM is mediated in part through the provision of migratory ‘highways’ onto which T cells can invade into the tissue 98 in response to chemoattractant ECM fragments released via MMP-mediated cleavage, as has been demonstrated for monocytes migrating into inflamed lung tissue following the release of digested elastin by MMP12 and elastase 99. The ECM can also directly inhibit T-cell proliferation through type I collagen ligation of LAIR receptors, and T-cell activation from a naïve state 100. However, T-cell activity can also be impeded by the ECM through the impairment of antigen presentation by APCs 101. In this regard, a stiffened ECM can compromise T-cell activation by CD3 and CD28, possibly by impairing IL-2 production, which is necessary for T-cell proliferation and Th1-cell differentiation 102. As such, a stiffened tumor-associated ECM could suppress anti-tumorigenic T-cell function.
Emerging hallmark: deregulating cellular energetics
The final emerging cancer hallmark is metabolic reprogramming of tumor cells. Termed the Warburg effect, tumor cells shift from predominantly aerobic glycolysis toward anaerobic glycolysis. While the overall yield of usable ATP from anaerobic glycolysis is less efficient, producing only 2 ATP compared to aerobic glycolysis which produces 36 ATP, this glycolytic switch permits the tumor cells to use glucose for other regulatory processes such as protein synthesis and cell division 103. Effective partitioning of glucose consumption is essential for the rapid cell division required by a highly proliferative tumor cell, so that a shift toward anaerobic glycolysis provides the cells with a selective growth advantage compared with cells using aerobic glycolysis. Even in the presence of high oxygen tension, tumor cells exploit the anaerobic metabolic process to facilitate rapid cell division 104.
At its most basic level, the ECM is essential for the uptake of extracellular nutrients and production of functional ATP. Focal adhesion signaling mediates the transmission of ECM signals into the tumor cells which, in turn, promotes the activation of the PI3K pathway which increases glycolysis 105. Facilitating the shift in metabolic processes, PI3K signaling increases the expression of GLUT1 and GLUT4 along with additional cell surface transport proteins to increase the cellular influx of glucose 106. Additionally, FAK cooperation with oncogenic drivers such as Ras and Myc supports the conversion of glutamate to glutamine, which promotes cell survival through enhanced protein biosynthesis and feeds into the TCA cycle to maintain high tumor cell proliferation 107. Intriguingly, tumor cells interacting with a stiffened ECM show a marked upregulation of growth factor-dependent PI3K/Akt signaling, which by virtue of its ability to increase aerobic glycolysis suggests that tissue tension may also directly regulate tumor cell metabolism 108.
Enabling characteristic: genomic mutation and instability
Cancer cells display genomic alterations due to mutations and chromosomal rearrangements as well as loss of tumor suppressors and compromised DNA repair mechanisms. These various acquired genetic alterations promote malignant transformation and facilitate tumor progression. Data suggest that an aberrant ECM may promote genetic instability and can even compromise DNA repair pathways necessary to prevent malignant transformation. For instance, inherited mutations in collagen components, such as the alpha 5 and 6 chains of collagen IV or collagen VII, increase the probability of developing smooth muscle tumors or skin cancer 109,110. Consistently, ectopic expression of stromelysin or MMP3 in the mouse mammary gland induced mammary epithelial cell proliferation and precocious branching morphogenesis and promoted malignant transformation and genomic instability, even in the absence of oncogene expression 111,112.
Enabling characteristic: tumor-promoting inflammation
Tumors are characterized by tissue inflammation, and a chronically inflamed tissue has a heightened risk for malignant transformation. Intriguingly, a chronically inflamed tissue is frequently fibrotic and shows increased collagen and fibronectin deposition 113,114. Furthermore, a fibrotic ECM and ECM receptor ligation can profoundly influence the recruitment of cellular components of the innate immune system. For instance, in the absence of expression of the laminin-binding receptor, α6β1 integrin, neutrophil recruitment into tissues is severely compromised 115. Similarly, macrophages require expression of the collagen receptor DDR1 to infiltrate atheroschlerotic plaques 116, whereas the ECM protein SPARC significantly inhibits macrophage infiltration 98. The composition of the tissue ECM can also dramatically modify the activation state of the recruited innate immune cells. Thus, a collagen-rich ECM promotes macrophage proliferation and activation 117 and favors a pro-tumorigenic M2 polarization phenotype, whereas a fibronectin-rich ECM promotes the M1 or anti-tumorigenic potential of macrophages 118,119. Indeed, the tumor-associated stiffened ECM is often enriched for type I collagen, and a rigid matrix promotes the M2 polarization of macrophages possibly by diminishing expression of the M1 macrophage regulator TNF-α in response to LPS 120.
Translation to the clinic
Conventional therapeutics typically target rapidly proliferating tumors and/or inhibit the activity of specific signaling pathways that drive malignancy, such as HER2, EGFR or RAF 121,122. Such treatments often enjoy enormous initial success, yet are plagued by the emergence of resistant tumors. Cell adhesion to the ECM and the mechanical feature of the ECM can profoundly regulate many of the classic and emerging cancer hallmarks. As such, the ECM and its adhesion receptors constitute tractable therapeutic targets that might prove useful for preventing or treating cancer or at the very least might prove useful as a combinatorial treatment with classic chemotherapies or with targeted therapies. Indeed, the biochemical and biophysical features of the ECM are not only essential for tumor progression, but can also regulate the efficacy of conventional therapies 123,124. Thus, abrogation of ECM remodeling or deposition into the stroma of tumors in preclinical models of cancer progression has successfully retarded cancer progression 125–128. The success of interventions targeting signaling molecules and remodeling enzymes, such as Shh, FAK and hyaluronidase inhibitors, has led to the development of clinical trials which utilize these interventions to neoadjuvantly enhance patient therapy (NCT01938443, NCT01130142, NCT01959139). Numerous FAK inhibitors have been developed and tested in phase I clinical trials 129. Interestingly, preliminary studies in these patients show that these drugs show promise in slowing tumor growth as well as the metastatic nature of late stage cancers (clinicaltrials.gov, GSK2256098). Unfortunately, Shh inhibitors were ineffective in slowing the disease progression in pancreatic cancer patients 130. This is surprising given the preclinical success of inhibition of the Shh pathway on pancreatic cancer progression, desmoplasia and mortality. Yet, such data do not negate therapeutically targeting pancreatic fibrosis as this discrepancy could be due to the systemic effects on various cell types within the tumor or genetic differences between preclinical models and patient disease. These results indicate that there is still significant work to be done to determine the genetic contexts and microenvironmental alterations, which would allow ECM targeting interventions to provide a therapeutic benefit to patients.
Conclusions and future perspectives
The hallmarks of cancer are driven by oncogenic mutations and influenced by biochemical and biomechanical properties of the extracellular matrix surrounding the developing tumor. During tumor progression, tumors develop from heterogeneous cell populations containing many different oncogenic mutations. These heterogeneous tumor populations are driven through very different oncogenic mutations and interact with the tumor microenvironment in different ways 131. Heterogeneous development is also reflected in the tumor-associated ECM with a large degree of variability seen in ECM deposition and stiffening in a single tumor 132,133. Hetero-geneity within the ECM could explain why therapeutics targeting this feature of tumor development have not had significant success in clinical trials 130. How ECM heterogeneity influences tumor development and therapeutic efficacy is one of the many unanswered questions yet to be addressed (see Sidebar A). A recent study abrogating stromal fibroblasts from pancreatic tumors suggests that the influences of this cell population on ECM composition and mechanical stiffening are inhibitory to tumor progression 134. However, this is contrary to other studies, particularly in the breast, where increased deposition of ECM components results in enhanced tumor progression 7. These data, which present opposing results for the role of the ECM in tumor progression, suggest that the influence of the ECM on the hallmarks of cancer cannot be broadly applied to all cancer types. These potentially conflicting results indicate that we must broaden our focus to encompass the various dynamic changes, outlined above, which occur to the biochemical and biophysical properties of the ECM during tumor progression. Given the essential need for matrix stiffness to drive many tumor-promoting effects of the ECM, it is essential to determine whether this ECM property is a correlative phenotype to tumor progression or a causative factor driving tumor initiation. While there are many unanswered questions with regard to how the ECM and its biophysical properties influence tumor progression, the potential for this component to be an efficacious target in treating cancer patients remains exceedingly high.
Sidebar A: In need of answers
-
What are the cellular sources inducing an altered ECM deposition and remodeling during cancer progression? And how do these various sources influence tumor progression?
In vitro and in vivo data show that the ECM can be deposited and altered by either epithelial or stromal cells in a tumor. However, the relative contribution of these actions to tumor progression is unknown. To answer these questions, novel transgenic models must be developed to specifically target either the expression of ECM proteins or remodeling capabilities from various cell populations in mouse models of tumor development. Additionally, modulating a tumor cells' response to an altered ECM at various time points in tumor progression would allow for the delineation of contribution of these ECM changes to the various steps in tumor cell progression to metastasis.
-
Are the enhanced biomechanical properties of the ECM observed in tumors a causative or correlative factor to tumor development?
Diseases associated with biochemical ECM changes are correlated with increased propensity for the development of cancers. The contribution of biomechanical processes to this development has yet to be addressed. Careful analysis of the mechanical properties of diseased tissue, like fatty liver or cystic fibrosis, would bring to answer this question.
-
Is the influence of the ECM on tumor progression consistent through various cancer origins and subtypes?
Oncogenes have been shown to influence the intracellular signaling mechanisms, which control a cells' response to the ECM. As different tumors and subtypes within a given tumor display oncogenic drivers, the response to ECM cues is very different, thus dictating how intervening in this interaction could influence tumor progression. Determining what situations are applicable for what types of inhibitors through in vitro and in vivo analysis of oncogene-directed tumor–ECM interactions is critical to our understanding of when to target this aspect of the tumor microenvironment.
-
At which point in a tumor cells' response to the altered ECM biochemical and biophysical properties it is most efficacious to therapeutically intervene?
Cellular adhesion to an ECM involves numerous extracellular and intracellular factors. Each of these factors represents an independent opportunity to intervene in the tumor cells' response to ECM changes. Determining whether intervention should be done through inhibiting collagen itself, the cell membrane mediators of tumor–ECM interactions or intracellular signaling in response to ECM adhesion is an essential question. In vivo examination of each of these components of a cells' response to the ECM through the use of transgenic knockouts as well as chemical inhibitors would provide invaluable information about efficacy of interventions during tumor progression.
Acknowledgments
We apologize to all colleagues whose work cannot be cited owing to space limitations. This work was supported by NIH NCI T32CA108462 (MWP), DOD BCRP Grant W81XWH-07-1-0538 (JKM), DOD BCRP Grants W81XWH-05-1-0330 and W81XWH-13-1-0216 (VMW), NIH NCI Grants R01 CA138818, U54 CA143836, U54 CA163155, R01 CA085492, R01 GM066047, U01 CA151925 and U01 ES019458, R33 CA183685 (VMW), AACR PANCAN A121989 (VMW), and Susan G. Komen Grant KG110560PP (VMW).
Glossary
- Anoikis
Form of programmed cell death, which is induced by anchorage-dependent cells detaching from the surrounding ECM
- Akt
v-akt murine thymoma viral oncogene homolog
- APC
Antigen-presenting cell
- Bax
BCL2-associated X protein
- Bcl2
B-cell CLL/lymphoma 2
- Bim
BCL2-like 11
- BRCA1
Breast cancer 1
- CD3
Cluster of differentiation 3
- CD28
Cluster of differentiation 28
- CDK
Cyclin-dependent kinase
- Chemoattractant
A cytokine that induces the movement of a cell toward a higher concentration of the chemical signal
- DDR1
Discoidin domain receptor tyrosine kinase 1
- Desmoplasia
The growth of fibrous or connective tissue
- EGFR
Epidermal growth factor receptor
- EMT
Epithelial to mesenchymal transition
- Erbb2
v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2
- Erk
Extracellular regulated MAP kinase
- Fak
Focal adhesion kinase
- FGF
Fibroblast growth factor
- Extravasation
The movement of a cell out of the circulatory system
- G1/S transition
A restriction point between the G1 phase and the S phase of the cell cycle, which must meet a specific set of requirements to be overcome. Progression through this point signifies a point of no return for cell cycle progression
- GATA2
GATA binding protein 2
- GLUT1
Glucose transporter 1
- Hemidesmosome
A small bud-like structure attaching an epithelial cell to the basal lamina
- HER2
Human epidermal growth factor receptor 2
- IL-2
Interleukin 2
- Intravasation
The invasion of cancer cells through the basement membrane and into the blood or lymphatic vessel
- ITGB1
Integrin, beta 1
- LAIR
Leukocyte-associated immunoglobulin-like receptor
- LPS
Lipopolysaccharide
- Mdm2
Transformed mouse 3T3 cell double minute 2, E3 ubiquitin ligase and proto-oncogene
- Mechanotransduction
The means by which a cell converts mechanical stimulus from the ECM into downstream signaling changes
- MMP
Matrix metalloproteinase
- Myc
Myelocytomatosis oncogene
- Neoadjuvant
The administration of a therapeutic agent before the standard treatment regiment, usually to enhance the efficacy of the conventional intervention
- NF-κB
Nuclear factor kappa B
- p130Cas
RAB3 GTPase-activating protein subunit 1
- p21
Cyclin-dependent kinase inhibitor 1A
- p27
Cyclin-dependent kinase inhibitor 1B
- p53
Transformation-related protein 53
- PI3K
Phosphatidylinositol 3-kinase
- Posttranslational modifications
a step in protein biosynthesis in which the chemical or structural nature of the amino acids comprising a protein is altered after the translation of the protein
- PTEN
Phosphatase and tensin homolog
- Rac
Ras-related C3 botulinum toxin substrate 1
- RAF
Raf-1 proto-oncogene
- Ras
Rat sarcoma viral oncogene homolog
- Rho
Rhodopsin
- Shh
Sonic hedgehog
- Smad
Mothers against DPP homologs
- SPARC
Secreted protein acidic and rich in cysteine
- Src
Rous sarcoma oncogene
- TAZ
Transcriptional co-activator with PDZ binding motif
- TCA cycle
Tricarboxylic acid cycle, also Krebs cycle
- TFII-I
General transcription factor II-I
- TGF-β
Transforming growth factor β
- Th1 cell
Type 1 helper T cell
- TNF-α
Tumor necrosis factor α
- Tumor-associated ECM
Extracellular matrix that has been modified over the course of tumor progression to have altered composition, density and mechanical properties
- VEGF
Vascular endothelial growth factor
- VEGFR2
Vascular endothelial growth factor receptor 2
- YAP
Yes-associated protein
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Mammoto T, Ingber DE. Mechanical control of tissue and organ development. Development. 2010;137:1407–1420. doi: 10.1242/dev.024166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitinger B, Hohenester E. Mammalian collagen receptors. Matrix Biol. 2007;26:146–155. doi: 10.1016/j.matbio.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Xian X, Gopal S, Couchman JR. Syndecans as receptors and organizers of the extracellular matrix. Cell Tissue Res. 2010;339:31–46. doi: 10.1007/s00441-009-0829-3. [DOI] [PubMed] [Google Scholar]
- Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol. 2010;22:697–706. doi: 10.1016/j.ceb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, White JG, Keely PJ. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventhal KR, Yu H, Kass L, Lakin JN, Egebald M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandol S, Edderkaoui M, Gukovsky I, Lugea A, Gukovskaya A. Desmoplasia of pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol. 2009;7:S44–S47. doi: 10.1016/j.cgh.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erler JT, Bennewith KL, Nicolau M, Dornhofer N, Kong C, Le QT, Chi JT, Jeffrey SS, Giaccia AJ. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- Slattery ML, John E, Torres-Mejia G, Stern M, Lundgreen A, Hines L, Giuliano A, Baumgartner K, Herrick J, Wolff RK. Matrix metalloproteinase genes are associated with breast cancer risk and survival: the Breast Cancer Health Disparities Study. PLoS One. 2013;8:e63165. doi: 10.1371/journal.pone.0063165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen HT, Friis S, Olsen JH, Thulstrup AM, Mellemkjaer L, Linet M, Trichopoulos D, Vilstrup H, Olsen J. Risk of liver and other types of cancer in patients with cirrhosis: a nationwide cohort study in Denmark. Hepatology. 1998;28:921–925. doi: 10.1002/hep.510280404. [DOI] [PubMed] [Google Scholar]
- Neglia JP, FitzSimmons SC, Maisonneuve P, Schoni MH, Schoni-Affolter F, Corey M, Lowenfels AB. The risk of cancer among patients with cystic fibrosis. Cystic Fibrosis and Cancer Study Group. N Engl J Med. 1995;332:494–499. doi: 10.1056/NEJM199502233320803. [DOI] [PubMed] [Google Scholar]
- Boyd NF, Guo H, Martin LJ, Sun L, Stone J, Fishell E, Jong RA, Hislop G, Chiarelli A, Minkin S, et al. Mammographic density and the risk and detection of breast cancer. N Engl J Med. 2007;356:227–236. doi: 10.1056/NEJMoa062790. [DOI] [PubMed] [Google Scholar]
- Boghaert E, Gleghorn JP, Lee K, Gjorevski N, Radisky DC, Nelson CM. Host epithelial geometry regulates breast cancer cell invasiveness. Proc Natl Acad Sci USA. 2012;109:19632–19637. doi: 10.1073/pnas.1118872109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Weigelt B, Ghajar CM, Bissell MJ. The need for complex 3D culture models to unravel novel pathways and identify accurate biomarkers in breast cancer. Adv Drug Deliv Rev. 2014;69–70:42–51. doi: 10.1016/j.addr.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA, Assoian RK. Integrins and cell proliferation: regulation of cyclin-dependent kinases via cytoplasmic signaling pathways. J Cell Sci. 2001;114:2553–2560. doi: 10.1242/jcs.114.14.2553. [DOI] [PubMed] [Google Scholar]
- Xiong J, Balcioglu HE, Danen EH. Integrin signaling in control of tumor growth and progression. Int J Biochem Cell Biol. 2013;45:1012–1015. doi: 10.1016/j.biocel.2013.02.005. [DOI] [PubMed] [Google Scholar]
- Keely PJ. Mechanisms by which the extracellular matrix and integrin signaling act to regulate the switch between tumor suppression and tumor promotion. J Mammary Gland Biol Neoplasia. 2011;16:205–219. doi: 10.1007/s10911-011-9226-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva Y, Gillen KM, Gerald W, Beggs HE, Reichardt LF, Giancotti FG. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J Clin Invest. 2009;119:252–266. doi: 10.1172/JCI37160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics. 2012;11:M111.014647. doi: 10.1074/mcp.M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahir N, Weaver VM. Death in the third dimension: apoptosis regulation and tissue architecture. Curr Opin Genet Dev. 2004;14:71–80. doi: 10.1016/j.gde.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Lamar JM, Carr SA, Hynes RO. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. Elife. 2014;3:e01308. doi: 10.7554/eLife.01308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabiraman PP, Rao PV. Mechanistic basis of Rho GTPase-induced extracellular matrix synthesis in trabecular meshwork cells. Am J Physiol Cell Physiol. 2010;298:C749–C763. doi: 10.1152/ajpcell.00317.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilghman RW, Cowan CR, Mih JD, Koryakina Y, Gioeli D, Slack-Davis JK, Blackman BR, Tschumperlin DJ, Parsons JT. Matrix rigidity regulates cancer cell growth and cellular phenotype. PLoS One. 2010;5:e12905. doi: 10.1371/journal.pone.0012905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J, Moscona A. Role of cell shape in growth control. Nature. 1978;273:345–349. doi: 10.1038/273345a0. [DOI] [PubMed] [Google Scholar]
- Chen CS. Mechanotransduction - a field pulling together? J Cell Sci. 2008;121:3285–3292. doi: 10.1242/jcs.023507. [DOI] [PubMed] [Google Scholar]
- Schwartz MA. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb Perspect Biol. 2010;2:a005066. doi: 10.1101/cshperspect.a005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells RG. The role of matrix stiffness in regulating cell behavior. Hepatology. 2008;47:1394–1400. doi: 10.1002/hep.22193. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Keely PJ. Mechanical signaling through the cytoskeleton regulates cell proliferation by coordinated focal adhesion and Rho GTPase signaling. J Cell Sci. 2011;124:1195–1205. doi: 10.1242/jcs.067009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambard JC, Lefloch R, Pouyssegur J, Lenormand P. ERK implication in cell cycle regulation. Biochim Biophys Acta. 2007;1773:1299–1310. doi: 10.1016/j.bbamcr.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Bae YH, Mui KL, Hsu BY, Liu SL, Cretu A, Razinia Z, Xu T, Pure E, Assoian RK. A FAK-Cas-Rac-lamellipodin signaling module transduces extracellular matrix stiffness into mechanosensitive cell cycling. Sci Signal. 2014;7:ra57. doi: 10.1126/scisignal.2004838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins K, Jacks T, Pavletich NP. The cell cycle and cancer. Proc Natl Acad Sci USA. 1997;94:2776–2778. doi: 10.1073/pnas.94.7.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. TGFbeta in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal ML, Agarwal A, Taylor WR, Stark GR. p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc Natl Acad Sci USA. 1995;92:8493–8497. doi: 10.1073/pnas.92.18.8493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Bae SC. TGF-beta-dependent cell growth arrest and apoptosis. J Biochem Mol Biol. 2002;35:47–53. doi: 10.5483/bmbrep.2002.35.1.047. [DOI] [PubMed] [Google Scholar]
- Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt HC, Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012;28:128–136. doi: 10.1016/j.tig.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard C, Goldbeter A. The balance between cell cycle arrest and cell proliferation: control by the extracellular matrix and by contact inhibition. Interface Focus. 2014;4:20130075. doi: 10.1098/rsfs.2013.0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Seok Kang Y, Soo Kim J, Shin NY, Hanks SK, Song WK. The integrin-coupled signaling adaptor p130Cas suppresses Smad3 function in transforming growth factor-beta signaling. Mol Biol Cell. 2008;19:2135–2146. doi: 10.1091/mbc.E07-10-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell FC, Martin F. Laminin-rich extracellular matrix association with mammary epithelial cells suppresses Brca1 expression. Cell Death Differ. 2000;7:360–367. doi: 10.1038/sj.cdd.4400647. [DOI] [PubMed] [Google Scholar]
- Barkan D, Green JE, Chambers AF. Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. Eur J Cancer. 2010;46:1181–1188. doi: 10.1016/j.ejca.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibue T, Weinberg RA. Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci USA. 2009;106:10290–10295. doi: 10.1073/pnas.0904227106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontier SM, Muller WJ. Integrins in breast cancer dormancy. APMIS. 2008;116:677–684. doi: 10.1111/j.1600-0463.2008.01026.x. [DOI] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK) Cancer Res. 2003;63:1684–1695. [PubMed] [Google Scholar]
- Schrader J, Gordon-Walker TT, Aucott RL, van Deemter M, Quass A, Walsh S, Benten D, Forbes SJ, Wells RG, Iredale JP. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology. 2011;53:1192–1205. doi: 10.1002/hep.24108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, Le QT, Giaccia AJ. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouw JK, Yui Y, Damiano L, Bainer RO, Lakin JN, Acerbi I, Ou G, Wijekoon AC, Levental KR, Gilbert PM, et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat Med. 2014;20:360–367. doi: 10.1038/nm.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Wei X, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Condenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005;5:231–237. doi: 10.1038/nrc1560. [DOI] [PubMed] [Google Scholar]
- Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J, Muthuswamy SK, Brugge JS. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat Cell Biol. 2003;5:733–740. doi: 10.1038/ncb1026. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E, Reed JC. Anchorage dependence, integrins, and apoptosis. Cell. 1994;77:477–478. doi: 10.1016/0092-8674(94)90209-7. [DOI] [PubMed] [Google Scholar]
- Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore AP, Metcalfe AD, Romer LH, Streuli CH. Integrin-mediated survival signals regulate the apoptotic function of Bax through its conformation and subcellular localization. J Cell Biol. 2000;149:431–446. doi: 10.1083/jcb.149.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahir N, Lakins JN, Russell A, Ming W, Chatterjee C, Rozenberg GI, Marinkovich MP, Weaver VM. Autocrine laminin-5 ligates alpha6beta4 integrin and activates RAC and NFkappaB to mediate anchorage-independent survival of mammary tumors. J Cell Biol. 2003;163:1397–1407. doi: 10.1083/jcb.200302023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Yamamoto Y, Ibusuki M, Fujiwara S, Yamamoto S, Tomita S, Nakano M, Murakami K, Iyaka K, Iwase H. Evaluation of tumor stiffness by elastography is predictive for pathologic complete response to neoadjuvant chemotherapy in patients with breast cancer. Ann Surg Oncol. 2012;19:3042–3049. doi: 10.1245/s10434-012-2343-1. [DOI] [PubMed] [Google Scholar]
- Lewis JM, Truong TN, Schwartz MA. Integrins regulate the apoptotic response to DNA damage through modulation of p53. Proc Natl Acad Sci USA. 2002;99:3627–3632. doi: 10.1073/pnas.062698499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubovskaya VM, Cance WG. FAK and p53 protein interactions. Anticancer Agents Med Chem. 2011;11:617–619. doi: 10.2174/187152011796817619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ST, Chen XL, Lim Y, Hanson DA, Vo TT, Howerton K, Larocque N, Fisher SJ, Schlaepfer DD, Ilic D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008;29:9–22. doi: 10.1016/j.molcel.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver VM, Lelievre S, Lakins JN, Chrenek MA, Jones JC, Giancotti F, Werb Z, Bissell MJ. beta4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell. 2002;2:205–216. doi: 10.1016/s1535-6108(02)00125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ST, Chen L, Wang HJ, Tang XD, Fang DC, Yang SM. hTERT promotes the invasion of telomerase-negative tumor cells in vitro. Int J Oncol. 2009;35:329–336. [PubMed] [Google Scholar]
- Liu T, Chung MJ, Ullenbruch M, Yu H, Jin H, Hu B, Choi YY, Ishikawa F, Phan SH. Telomerase activity is required for bleomycin-induced pulmonary fibrosis in mice. J Clin Invest. 2007;117:3800–3809. doi: 10.1172/JCI32369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunimura C, Kikuchi K, Ahmed N, Shimizu A, Yasumoto S. Telomerase activity in a specific cell subset co-expressing integrinbeta1/EGFR but not p75NGFR/bcl2/integrin beta4 in normal human epithelial cells. Oncogene. 1998;17:187–197. doi: 10.1038/sj.onc.1201916. [DOI] [PubMed] [Google Scholar]
- Zhou J, Schmid T, Schnitzer S, Brune B. Tumor hypoxia and cancer progression. Cancer Lett. 2006;237:10–21. doi: 10.1016/j.canlet.2005.05.028. [DOI] [PubMed] [Google Scholar]
- Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- Avraamides CJ, Garmy-Susini B, Varner JA. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008;8:604–617. doi: 10.1038/nrc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama SK, Yamada SS, Chen WT, Yamada KM. Analysis of fibronectin receptor function with monoclonal antibodies: roles in cell adhesion, migration, matrix assembly, and cytoskeletal organization. J Cell Biol. 1989;109:863–875. doi: 10.1083/jcb.109.2.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Cell-matrix adhesion in vascular development. J Thromb Haemost. 2007;5(Suppl. 1):32–40. doi: 10.1111/j.1538-7836.2007.02569.x. [DOI] [PubMed] [Google Scholar]
- Bonanno E, Iurlaro M, Madri JA, Nicosia RF. Type IV collagen modulates angiogenesis and neovessel survival in the rat aorta model. In Vitro Cell Dev Biol Anim. 2000;36:336–340. doi: 10.1290/1071-2690(2000)036<0336:TICMAA>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Stratman AN, Malotte KM, Mahan RD, Davis MJ, Davis GE. Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood. 2009;114:5091–5101. doi: 10.1182/blood-2009-05-222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schor AM, Schor SL, Allen TD. Effects of culture conditions on the proliferation, morphology and migration of bovine aortic endothelial cells. J Cell Sci. 1983;62:267–285. doi: 10.1242/jcs.62.1.267. [DOI] [PubMed] [Google Scholar]
- Montesano R, Orci L, Vassalli P. In vitro rapid organization of endothelial cells into capillary-like networks is promoted by collagen matrices. J Cell Biol. 1983;97:1648–1652. doi: 10.1083/jcb.97.5.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Agarwal S. Mechanical signals activate vascular endothelial growth factor receptor-2 to upregulate endothelial cell proliferation during inflammation. J Immunol. 2010;185:1215–1221. doi: 10.4049/jimmunol.0903660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammoto A, Connor KM, Mammoto T, Yung CW, Huh D, Aderman CM, Mostoslavsky G, Smith LE, Ingber DE. A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature. 2009;457:1103–1108. doi: 10.1038/nature07765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingber D, Folkman J. Inhibition of angiogenesis through modulation of collagen metabolism. Lab Invest. 1988;59:44–51. [PubMed] [Google Scholar]
- Jones RA, Kotsakis P, Johnson TS, Chau DY, Ali S, Melino G, Griffin M. Matrix changes induced by transglutaminase 2 lead to inhibition of angiogenesis and tumor growth. Cell Death Differ. 2006;13:1442–1453. doi: 10.1038/sj.cdd.4401816. [DOI] [PubMed] [Google Scholar]
- Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- Wang Y, McNiven MA. Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK-p130Cas complex. J Cell Biol. 2012;196:375–385. doi: 10.1083/jcb.201105153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaty BT, Condeelis J. Digging a little deeper: the stages of invadopodium formation and maturation. Eur J Cell Biol. 2014 doi: 10.1016/j.ejcb.2014.07.003. doi: 10.1016/j.ejcb.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh A, Ruppender NS, Branch KM, Sewell-Loftin MK, Lin J, Boyer PD, Candiello JE, Merryman WD, Guelcher SA, Weaver AM. Sensing and modulation of invadopodia across a wide range of rigidities. Biophys J. 2011;100:573–582. doi: 10.1016/j.bpj.2010.12.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parri M, Chiarugi P. Rac and Rho GTPases in cancer cell motility control. Cell Commun Signal. 2010;8:23. doi: 10.1186/1478-811X-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135:510–523. doi: 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- Chen JC, Zhuang S, Nguyen TH, Boss GR, Pilz RB. Oncogenic Ras leads to Rho activation by activating the mitogen-activated protein kinase pathway and decreasing Rho-GTPase-activating protein activity. J Biol Chem. 2003;278:2807–2818. doi: 10.1074/jbc.M207943200. [DOI] [PubMed] [Google Scholar]
- Xia M, Land H. Tumor suppressor p53 restricts Ras stimulation of RhoA and cancer cell motility. Nat Struct Mol Biol. 2007;14:215–223. doi: 10.1038/nsmb1208. [DOI] [PubMed] [Google Scholar]
- Celià-Terrassa T, Meca-Cortés O, Mateo F, de Paz AM, Rubio N, Arnal-Estapé A, Ell BJ, Bermudo R, Díaz A, Guerra-Rebollo M, et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest. 2012;122:1849–1868. doi: 10.1172/JCI59218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803–36810. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- Menon S, Beningo KA. Cancer cell invasion is enhanced by applied mechanical stimulation. PLoS One. 2011;6:e17277. doi: 10.1371/journal.pone.0017277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak A, Kumar S. Independent regulation of tumor cell migration by matrix stiffness and confinement. Proc Natl Acad Sci USA. 2012;109:10334–10339. doi: 10.1073/pnas.1118073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup MW, Laklai H, Acerbi I, Owens P, Gorska AE, Chytil A, Aakre M, Weaver VM, Moses HL. Stromally derived lysyl oxidase promotes metastasis of transforming Growth factor-beta deficient mouse mammary carcinoma. Cancer Res. 2013;73:5336–5346. doi: 10.1158/0008-5472.CAN-13-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leight JL, Wozniak MA, Chen S, Lynch ML, Chen CS. Matrix rigidity regulates a switch between TGF-beta1-induced apoptosis and epithelial-mesenchymal transition. Mol Biol Cell. 2012;23:781–791. doi: 10.1091/mbc.E11-06-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondareva A, Downey CM, Ayres F, Liu W, Boyd SK, Hallgrimsson B, Jirik FR. The lysyl oxidase inhibitor, beta-aminopropionitrile, diminishes the metastatic colonization potential of circulating breast cancer cells. PLoS One. 2009;4:e5620. doi: 10.1371/journal.pone.0005620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol. 2010;10:712–723. doi: 10.1038/nri2852. [DOI] [PubMed] [Google Scholar]
- Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, Mecham RP, Senior RM, Shapiro SD. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest. 2006;116:753–759. doi: 10.1172/JCI25617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyaard L. The inhibitory collagen receptor LAIR-1 (CD305) J Leukoc Biol. 2008;83:799–803. doi: 10.1189/jlb.0907609. [DOI] [PubMed] [Google Scholar]
- Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- O'Connor RS, Hao X, Shen K, Bashour K, Akimova T, Hancock WW, Kam LC, Milone MC. Substrate rigidity regulates human T cell activation and proliferation. J Immunol. 2012;189:1330–1339. doi: 10.4049/jimmunol.1102757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel M, Schwartz L. The metabolic advantage of tumor cells. Mol Cancer. 2011;10:70. doi: 10.1186/1476-4598-10-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillies RJ, Robey I, Gatenby RA. Causes and consequences of increased glucose metabolism of cancers. J Nucl Med. 2008;49(Suppl. 2):24S–42S. doi: 10.2967/jnumed.107.047258. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassian AR, Coloff JL, Brugge JS. Extracellular matrix regulation of metabolism and implications for tumorigenesis. Cold Spring Harb Symp Quant Biol. 2011;76:313–324. doi: 10.1101/sqb.2011.76.010967. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hochwald SN. The role of FAK in tumor metabolism and therapy. Pharmacol Ther. 2014;142:154–163. doi: 10.1016/j.pharmthera.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- Zhou J, Mochizuki T, Smeets H, Antignac C, Laurila P, de Paepe A, Tryggvason K, Reeders ST. Deletion of the paired alpha 5(IV) and alpha 6(IV) collagen genes in inherited smooth muscle tumors. Science. 1993;261:1167–1169. doi: 10.1126/science.8356449. [DOI] [PubMed] [Google Scholar]
- Slater SD, McGrath JA, Hobbs C, Eady RA, McKee PH. Expression of mutant p53 gene in squamous carcinoma arising in patients with recessive dystrophic epidermolysis bullosa. Histopathology. 1992;20:237–241. doi: 10.1111/j.1365-2559.1992.tb00962.x. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, Pinkel D, Bissell MJ, Werb Z. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 1999;98:137–146. doi: 10.1016/s0092-8674(00)81009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izbicki G, Segel MJ, Christensen TG, Conner MW, Breuer R. Time course of bleomycin-induced lung fibrosis. Int J Exp Pathol. 2002;83:111–119. doi: 10.1046/j.1365-2613.2002.00220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stramer BM, Mori R, Martin P. The inflammation-fibrosis link? A Jekyll and Hyde role for blood cells during wound repair. J Invest Dermatol. 2007;127:1009–1017. doi: 10.1038/sj.jid.5700811. [DOI] [PubMed] [Google Scholar]
- Dangerfield J, Larbi KY, Huang MT, Dewar A, Nourshargh S. PECAM-1 (CD31) homophilic interaction up-regulates alpha6beta1 on transmigrated neutrophils in vivo and plays a functional role in the ability of alpha6 integrins to mediate leukocyte migration through the perivascular basement membrane. J Exp Med. 2002;196:1201–1211. doi: 10.1084/jem.20020324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco C, Britto K, Wong E, Hou G, Zhu SN, Chen M, Cybulsky MI, Bendeck MP. Discoidin domain receptor 1 on bone marrow-derived cells promotes macrophage accumulation during atherogenesis. Circ Res. 2009;105:1141–1148. doi: 10.1161/CIRCRESAHA.109.207357. [DOI] [PubMed] [Google Scholar]
- Wesley RB, 2nd, Meng X, Godin D, Galis ZS. Extracellular matrix modulates macrophage functions characteristic to atheroma: collagen type I enhances acquisition of resident macrophage traits by human peripheral blood monocytes in vitro. Arterioscler Thromb Vasc Biol. 1998;18:432–440. doi: 10.1161/01.atv.18.3.432. [DOI] [PubMed] [Google Scholar]
- Perri RT, Kay NE, McCarthy J, Vessella RL, Jacob HS, Furcht LT. Fibronectin enhances in vitro monocyte-macrophage-mediated tumoricidal activity. Blood. 1982;60:430–435. [PubMed] [Google Scholar]
- Stahl M, Schupp J, Jager B, Schmid M, Zissel G, Muller-Quernheim J, Prasse A. Lung collagens perpetuate pulmonary fibrosis via CD204 and M2 macrophage activation. PLoS One. 2013;8:e81382. doi: 10.1371/journal.pone.0081382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel NR, Bole M, Chen C, Hardin CC, Kho AT, Mih J, Deng L, Butler J, Tschumperlin D, Fredberg JJ, et al. Cell elasticity determines macrophage function. PLoS One. 2012;7:e41024. doi: 10.1371/journal.pone.0041024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung A, Cui X, Audeh W, Giuliano A. Current status of anti-human epidermal growth factor receptor 2 therapies: predicting and overcoming herceptin resistance. Clin Breast Cancer. 2013;13:223–232. doi: 10.1016/j.clbc.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwick E, Bange J, Ullrich A. Receptor tyrosine kinase signalling as a target for cancer intervention strategies. Endocr Relat Cancer. 2001;8:161–173. doi: 10.1677/erc.0.0080161. [DOI] [PubMed] [Google Scholar]
- Waghray M, Yalamanchili M, di Magliano MP, Simeone DM. Deciphering the role of stroma in pancreatic cancer. Curr Opin Gastroenterol. 2013;29:537–543. doi: 10.1097/MOG.0b013e328363affe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Liao S, Diop-Frimpong B, Chen W, Goel S, Naxerova K, Ancukiewicz M, Boucher Y, Jain RK, Xu L. TGF-beta blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc Natl Acad Sci USA. 2012;109:16618–16623. doi: 10.1073/pnas.1117610109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–515. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- Thompson CB, Shepard HM, O'Connor PM, Kadhim S, Jiang P, Osgood RJ, Bookbinder LH, Li X, Sugarman BJ, Connor RJ, et al. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol Cancer Ther. 2010;9:3052–3064. doi: 10.1158/1535-7163.MCT-10-0470. [DOI] [PubMed] [Google Scholar]
- Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M, Mikels A, Vaysberg M, Ghermazien H, Wai C, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. 2010;16:1009–1017. doi: 10.1038/nm.2208. [DOI] [PubMed] [Google Scholar]
- Golubovskaya VM. Targeting FAK in human cancer: from finding to first clinical trials. Front Biosci (Landmark Ed) 2014;19:687–706. doi: 10.2741/4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rombouts K, Carloni V. The fibrotic microenvironment as a heterogeneity facet of hepatocellular carcinoma. Fibrogenesis Tissue Repair. 2013;6:17. doi: 10.1186/1755-1536-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup MW, Laklai H, Acerbi I, Owens P, Gorska AE, Chytil A, Aakre M, Weaver VM, Moses HL. Stromally derived lysyl oxidase promotes metastasis of transforming growth factor-beta-deficient mouse mammary carcinomas. Cancer Res. 2013;73:5336–5346. doi: 10.1158/0008-5472.CAN-13-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]