

Abstract
The interaction between the immune system and pathogens is often characterised as a predator–prey interaction. This characterisation ignores the fact that both require host resources to reproduce. Here, we propose novel theory that considers how these resource requirements can modify the interaction between the immune system and pathogens. We derive a series of models to describe the energetic interaction between the immune system and pathogens, from fully independent resources to direct competition for the same resource. We show that increasing within-host resource supply has qualitatively distinct effects under these different scenarios. In particular, we show the conditions for which pathogen load is expected to increase, decrease or even peak at intermediate resource supply. We survey the empirical literature and find evidence for all three patterns. These patterns are not explained by previous theory, suggesting that competition for host resources can have a strong influence on the outcome of disease.
Keywords: Consumer–resource theory, epidemiology, immunology, modelling
The interaction between the immune system and pathogens is often conceptualised as a predator–prey interaction (Fenton & Perkins 2010). While this analogy is useful, it overlooks the fact that both immune proliferation and pathogen replication depend on within-host resources. This joint dependence may be an additional source of antagonism between the immune system and pathogens. For example, if both the immune system and pathogens use the same resource, or if there is a metabolic link between their resources, then the two interact not only as predator and prey but also as competitors.
It is well known that both the immune system and pathogens depend on host resources. Detailed studies of the immune system have measured the metabolic cost of mounting an immune response. For example, vaccination with protein antigen has been shown to increase metabolic rate between 15 and 30% (Lochmiller & Deerenberg 2000). Similarly, many studies have documented changes in host resources caused by pathogen infection (Rivero et al. 2007; Frost et al. 2008). For example, Hall et al. (2009) showed that patterns of parasite virulence across a food gradient could be explained as a consequence of pathogen resource acquisition strategy.
Moreover, previous authors have noted that resource depletion by pathogens can reduce the ability of the host to mount an immune response (Koski & Scott 2001; Cornet & Sorci 2010). If the immune system competes for resources with pathogens, then the reciprocal interaction, whereby the immune system depletes resources and reduces the ability of the pathogen to replicate, is also possible.
Gaining insight into how resource dependence affects within-host dynamics is important because host internal resource abundance depends, in part, on the external environment (i.e. host food availability). Since the dynamics of food in the environment will depend on host population dynamics, internal resource dependence creates the potential for among-host dynamics to influence within-host dynamics. The reciprocal feedback, from within-host to among-host dynamics, is an emerging area of research and is known to affect, for example, the evolution of virulence (Mideo et al. 2008).
Here, we develop general theory to explore the consequences of resource dependence for within-host dynamics of the immune system and pathogens. We derive a series of models describing the resource interaction between our two protagonists, from each having separate resources (fully independent) to the two depending on the same resource (fully antagonistic). These models are qualitatively distinct both from existing models of within-host dynamics and from other consumer–resource models. Because increasing resources should have both costs and benefits for the pathogen (Beisel 1982), we focus our analysis on the response of pathogen load to increasing resource supply. We show that pathogen load will increase if the immune system and pathogens have independent resources, or if the pathogen is able to suppress the immune response by preempting the resource use of the immune system. If, on the other hand, the immune system can preempt the pathogen's resource use, we show that the pathogen load will decrease. Finally, if they compete for the same within-host resource, we show that pathogen load peaks at intermediate resource supply. To investigate whether similar resource-based patterns exist in the empirical literature, we also present the results of a literature survey documenting the response of pathogens to changes in host food across a wide range of host–pathogen systems.
Material and Methods
Theory often assumes that the immune–pathogen interaction can be modelled as a predator–prey interaction. For example, if I is the number of immune cells and N is the number of pathogens, then a simple model for this interaction is:
| 1 |
| 2 |
In this model, aB is the baseline proliferation rate of the immune system, fI is the contact rate between immune and pathogen cells (resulting in pathogen death), aI quantifies the additional induction of the immune system caused by immune–pathogen contacts, m is the per capita loss rate of immune cells, fN is the replication rate of the pathogen, and d is the background pathogen loss rate.
To incorporate resource dependence, we allow aB, aI and fN to be functions of some internal host resource R [i.e. aB = aB (R), aI = aI (R) and fN = fN (R)]. Here, we assume these internal resources take the form of within-host energy. Extending the predator–prey analogy, we consider four possible ‘food web topologies’ describing the interaction between energy, immunity and pathogens (Fig. 1).
Figure 1.
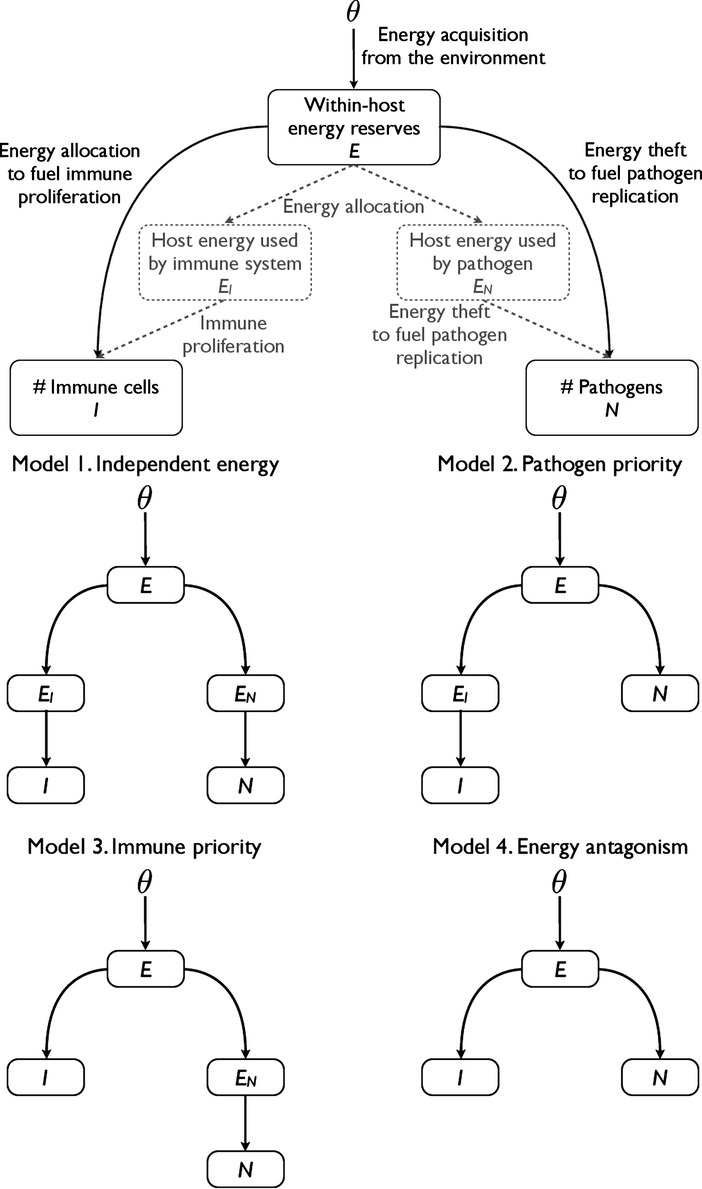
Depiction of the possible topologies of the interaction between energy E, the immune system I and pathogens N. Energy from the reserves can be used directly to fuel immune proliferation or pathogen replication, or it can first be allocated to intermediate energy bins EI and EN that fuel proliferation and replication.
All four models assume that the host acquires energy from the environment at the constant rate θ. This energy then enters a pool of metabolisable reserves E that is the proximate source of energy for all metabolic processes, including immune proliferation and pathogen replication. We consider two possible pathways from the energy reserves E to either immune cells I or pathogens N: energy in E can be used directly to fuel either immune proliferation or pathogen replication, or it can first be allocated to intermediate energy ‘bins’, EI or EN, that fuel immune proliferation and pathogen replication, respectively. These bins represent temporary storage pools of reserve energy that has been allocated by the host. Bear in mind that, whereas EI represents energy the host is allocating for immune proliferation (e.g. an ‘immune bin’), EN should not be thought of as a ‘pathogen bin’. Rather, the energy in EN is intended for a host metabolic process but is diverted by the pathogen. The four models analysed here represent all possible combinations of the different immune and pathogen pathways.
Independent resource model
We describe the independent resource model in full detail, as all other models are simplications of it. The biological interpretation of model parameters is given in Table1. The full model is:
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
The host acquires energy from the environment at a constant rate θ. This energy enters the energy reserves E, and is then non-reversibly allocated to fuel all metabolic processes (eqn 3). We assume that energy is used at a constant per capita rate r by all non-epidemiological processes. Energy flows to two other energy ‘bins’, EI and EN, at the per capita rates rI and rN. These serve as the proximate resources for the immune system and pathogen, respectively.
Table 1.
Model parameters, their biological interpretation and values for this study. ‘Flow rate’ in the interpretations implies an energy flow rate. Parameter values were chosen so that the pathogen-free equilibrium values 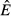 and
and 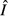 were identical across all models
were identical across all models
| Parameter | Interpretation | 1. Independent energy | 2. Pathogen priority | 3. Immune priority | 4. Energy antagonism |
|---|---|---|---|---|---|
| θ | Energy acquisition rate | 0.1–30.0 | 0.1–30.0 | 0.1–30.0 | 0.1–30.0 |
| r | Flow rate to non-epidemiological processes | 1.0 | 2.0 | 1.0 | 2.0 |
| rI | Flow rate to immune energy bin | 1.0 | 1.0 | ||
| rN | Flow rate to pathogen energy bin | 1.0 | 1.0 | ||
| a | Flow rate from pathogen bin to host processes | 1.0 | 1.0 | ||
| aB | Baseline flow rate to immune system | 1.0 | 1.0 | 1.0 | 1.0 |
| aI | Induced flow rate to immune system per pathogenkilled | 1.0 | 1.0 | 1.0 | 1.0 |
| εI | Cost of producing immune cell | 2.0 | 2.0 | 2.0 | 2.0 |
| εN | Cost of producing pathogen | 1.0 | 1.0 | 1.0 | 1.0 |
| fI | Killing rate per immune cell | 2.0 | 2.0 | 2.0 | 2.0 |
| fN | Energy consumption rate per pathogen | 20.0 | 20.0 | 20.0 | 20.0 |
| mI | Immune cell background mortality rate | 1.0 | 1.0 | 1.0 | 1.0 |
| mN | Pathogen background mortality rate | 0.1 | 0.1 | 0.1 | 0.1 |
Energy in the EI-bin is used to fuel immune proliferation at a baseline per capita rate aB (eqn 4). Immune–pathogen encounters occur at the rate fIIN, resulting in additional energy flow to immune proliferation. aI quantifies the amount of energy per pathogen killed. Additional immune cells I are produced at an energetic cost of εI (eqn 5). These cells die at a constant background rate mI.
Energy in the EN-bin is used by the host at a constant per capita rate a (eqn 6). The pathogen steals energy at the constant pathogen-specific rate fN. Conceptually, the pathogen has a Type I functional response. Additional pathogens N are produced at an energetic cost of εN (eqn 7). Pathogens die at a constant background rate of mN, and are killed by the immune system at the rate fIIN.
The key feature of this model is the separate resources for the immune system and pathogens, so that they do not interact through resources. Biologically, such a situation might arise if pathogenic infection is highly localised (e.g. tissue-specific). This model represents a simple extension of the standard predator–prey-type models used in epidemiology.
Pathogen priority model
| 8 |
| 9 |
| 10 |
| 11 |
In this model, the pathogen is assumed to steal energy directly from the reserves. This eliminates the equation for EN and replaces the allocation to EN in the dE/dt equation with the pathogen's functional response. The pathogen potentially weakens the immune response by reducing the flow of energy to EI, thereby indirectly reducing the immune proliferation rate.
Immune priority model
| 12 |
| 13 |
| 14 |
| 15 |
In this model, the immune system uses energy directly from the reserves. This eliminates the equation for EI and replaces the allocation to EI in the dE/dt equation with the baseline and induced allocations to immune proliferation. Here, the immune system reduces pathogen load both by direct killing and by indirectly reducing the flow of energy to EN.
Energy antagonism model
| 16 |
| 17 |
| 18 |
This model eliminates both the EI and EN bins. The immune system and pathogens are now directly competing for the same resource. This complexity makes it hard to predict how such a system would respond to changes in key energetic parameters.
Results
We focus on how changing the energy acquisition rate θ affects the equilibrium system state for each model. Expressions for the equilibrium pathogen load 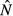 and immune abundance
and immune abundance 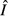 can be found in Table2.
can be found in Table2.
Table 2.
Expressions for the equilibrium pathogen load 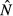 and immune abundance
and immune abundance 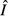 for each of the four models. Ecological interpretations of these expressions in terms of resource gain and loss rates (for
for each of the four models. Ecological interpretations of these expressions in terms of resource gain and loss rates (for 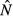 ) and pathogen replication and immune killing rates (for
) and pathogen replication and immune killing rates (for 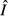 ) can be found in eqns (19) and (20) in the main text.
) can be found in eqns (19) and (20) in the main text. 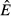 or
or 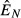 is the equilibrium pathogen resource abundance, as defined in Appendix S1
is the equilibrium pathogen resource abundance, as defined in Appendix S1
| Model name | Pathogen load, 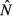
|
Immune cells, 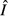
|
|---|---|---|
| 1. Independent energy model | 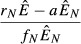 |
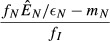 |
| 2. Pathogen priority model | 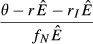 |
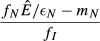 |
| 3. Immune priority model | 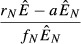 |
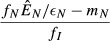 |
| 4. Energy antagonism model | 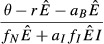 |
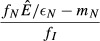 |
The expressions for both equilibria have intuitive ecological interpretations:
| 19 |
| 20 |
From these expressions, high pathogen load can result from high rates of resource gain [numerator of (19)], which fuels rapid replication rates, or slow rates of resource depletion [denominator of (19)], which allows the pathogen population to build up without suppressing resource abundance too much. High immune abundance can result from high rates of pathogen replication [numerator of (20)] or low rates of immune killing [denominator of (20)], both of which result in high pathogen loads and thus, increased immune induction.
The expressions are quite similar among the models, reflecting the broad similarities in model structure. The lone noticeable difference among the expressions in Table2 is in the denominator of the expression for equilibrium pathogen load for the energy antagonism model (model 4). For all models, the pathogen's resource is depleted by pathogen energy theft (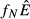 or
or 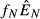 ); in the energy antagonism model, there is an additional depletion of energy caused by the proliferation of the immune system. The use of the pathogen's resource by the immune system acts to reduce the equilibrium pathogen load.
); in the energy antagonism model, there is an additional depletion of energy caused by the proliferation of the immune system. The use of the pathogen's resource by the immune system acts to reduce the equilibrium pathogen load.
How does increased energy acquisition by the host (θ) affect the pathogen and immune abundances? Our analysis shows that the four models make qualitatively different predictions (Fig. 2; see Appendix S1 for analytical details):
For the independent energy model, both equilibrium pathogen load
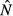 and immune abundance
and immune abundance 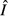 are increasing functions of acquisition rate θ.
are increasing functions of acquisition rate θ.For the pathogen priority model,
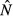 is an increasing function of θ, whereas
is an increasing function of θ, whereas 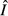 is constant.
is constant.For the immune priority model,
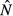 peaks at very low values of acquisition rate, so pathogen load tends to be a decreasing function of θ, whereas
peaks at very low values of acquisition rate, so pathogen load tends to be a decreasing function of θ, whereas 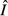 is an increasing function of θ.
is an increasing function of θ.For the energy antagonism model, equilibrium pathogen load
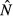 peaks at an intermediate energy acquisition rate θ, whereas equilibrium immune abundance
peaks at an intermediate energy acquisition rate θ, whereas equilibrium immune abundance 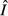 is an increasing function of θ.
is an increasing function of θ.
Figure 2.

Equilibrium model predictions as energy acquisition rate θ increases. Panels a–d show the equilibrium abundances 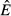 (solid),
(solid), 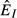 (dashed) and
(dashed) and 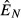 (dot-dash). The proximate resource for the immune system is highlighted in dark grey and the proximate resource for the pathogen is highlighted in light grey. Panels e–h show the immune abundances when the pathogen is present (solid, N+) and absent (dashed, N−). Note that increasing energy acquisition increases the number of immune cells even in the absence of infection because of the baseline immune allocation parameter aB. Panels i–l show the pathogen load. Panels m–p show the net gain rate (solid, point-up triangles) and per capita loss rate (dashed, point-down triangles) of the pathogen's resource (the numerator and denominator, respectively, of the equations in Table2). Note that these quantities have different units (1/time vs. 1/pathogens/time). Parameter values are given in Table1.
(dot-dash). The proximate resource for the immune system is highlighted in dark grey and the proximate resource for the pathogen is highlighted in light grey. Panels e–h show the immune abundances when the pathogen is present (solid, N+) and absent (dashed, N−). Note that increasing energy acquisition increases the number of immune cells even in the absence of infection because of the baseline immune allocation parameter aB. Panels i–l show the pathogen load. Panels m–p show the net gain rate (solid, point-up triangles) and per capita loss rate (dashed, point-down triangles) of the pathogen's resource (the numerator and denominator, respectively, of the equations in Table2). Note that these quantities have different units (1/time vs. 1/pathogens/time). Parameter values are given in Table1.
We show that these predictions can be understood by considering how increasing acquisition rate θ affects the energy reserve E, which serves as the proximate source of energy for both the immune system and pathogens. Figure 2 plots equilibrium values for each variable as acquisition rate θ increases.
For the independent energy model (model 1), increasing energy acquisition θ greatly increases reserves 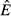 , which increases the abundances of both the immune (
, which increases the abundances of both the immune (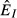 ) and pathogen (
) and pathogen (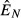 ) resources (Fig. 2a). This subsequently increases both immune abundance
) resources (Fig. 2a). This subsequently increases both immune abundance 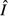 (Fig. 2e) and pathogen load
(Fig. 2e) and pathogen load 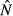 (Fig. 2i). These predictions are ecologically intuitive. In the absence of nonlinearity, increasing the availability of a resource will increase the abundance of its consumer. Note that the immune abundance increases with energy acquisition exactly as it does in the absence of pathogens because at equilibrium the immune abundance depends only on the amount of energy in E.
(Fig. 2i). These predictions are ecologically intuitive. In the absence of nonlinearity, increasing the availability of a resource will increase the abundance of its consumer. Note that the immune abundance increases with energy acquisition exactly as it does in the absence of pathogens because at equilibrium the immune abundance depends only on the amount of energy in E.
For the pathogen priority model (model 2), increasing energy acquisition θ has no effect on the abundance of the pathogen resource (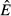 ; Fig. 2b). This indicates that any increase in energy acquisition leads directly to an increase in pathogen load (Fig. 2j) – the pathogen is preempting energy allocation to the immune system. The constant abundance of reserves
; Fig. 2b). This indicates that any increase in energy acquisition leads directly to an increase in pathogen load (Fig. 2j) – the pathogen is preempting energy allocation to the immune system. The constant abundance of reserves 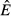 but increasing pathogen load
but increasing pathogen load 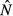 lead to declining abundance of the immune resource (
lead to declining abundance of the immune resource (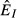 ) and a constant immune abundance
) and a constant immune abundance 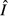 (Fig. 2b,f). Immune abundance is constant because, despite more frequent immune–pathogen encounters as θ increases, the declining immune resource means there is not enough energy to translate these encounters into increased immune proliferation. Thus, pathogen energy preemption suppresses the immune response.
(Fig. 2b,f). Immune abundance is constant because, despite more frequent immune–pathogen encounters as θ increases, the declining immune resource means there is not enough energy to translate these encounters into increased immune proliferation. Thus, pathogen energy preemption suppresses the immune response.
For the immune priority model (model 3), increasing energy acquisition θ increases both the immune (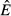 ) and pathogen (
) and pathogen (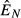 ) resource (Fig. 2c). The pathogen resource increases because the increase in immune abundance (Fig. 2g) suppresses the pathogen abundance (Fig. 2k). This implies that less energy is stolen by the pathogen, so the pathogen resource is able to increase. The immune resource preemption suppresses pathogen replication even as the resource increases. Note that pathogen load does increase over a very narrow range of acquisition rate θ values, which we discuss below.
) resource (Fig. 2c). The pathogen resource increases because the increase in immune abundance (Fig. 2g) suppresses the pathogen abundance (Fig. 2k). This implies that less energy is stolen by the pathogen, so the pathogen resource is able to increase. The immune resource preemption suppresses pathogen replication even as the resource increases. Note that pathogen load does increase over a very narrow range of acquisition rate θ values, which we discuss below.
The energy antagonism model (model 4) combines features of both the pathogen priority and immune priority results. When acquisition rate θ is low, pathogen energy consumption suppresses the immune response. The abundances of the resource (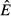 ) and immune cells remain constant while pathogen load increases, as in the pathogen priority model (compare panels d, h and l at low θ to panels b, f and j). As θ increases, the pathogen is less able to suppress the immune response, the abundances of the immune resource and immune cells increase and the pathogen declines, as in the immune priority model (compare panels d, h and l at high θ to panels c, g and k).
) and immune cells remain constant while pathogen load increases, as in the pathogen priority model (compare panels d, h and l at low θ to panels b, f and j). As θ increases, the pathogen is less able to suppress the immune response, the abundances of the immune resource and immune cells increase and the pathogen declines, as in the immune priority model (compare panels d, h and l at high θ to panels c, g and k).
Both the immune priority model and the energy antagonism model predict a peak in pathogen load. Mathematically, the peak occurs because the net gain rate of pathogen resource is a decelerating function of energy acquisition rate θ, whereas the per capita loss rate is an accelerating function of θ (the numerators and denominators of Table2; see Fig. 2o,p). Over low values of θ, the increase in the numerator (net gain rate) outweighs the increase in the denominator (per capita loss rate) and pathogen load increases; but as θ increases, the increase in the denominator outweighs the increase in the numerator and pathogen load decreases. Biologically, the peak occurs because of resource use by the immune system, with differences in topology affecting where pathogen load peaks. For the immune priority model, because the immune system has access to energy ‘upstream’ of the pathogen the peak occurs for very low values of θ. Therefore, increasing θ is more likely to decrease pathogen load. For the energy antagonism model, the pathogen's resource acquisition can suppress the immune response, increasing the range of θ values over which pathogen load increases.
The differences between the two models can be seen in Fig. 3. The solid line gives the acquisition rate θ where pathogen load peaks for the immune priority and energy antagonism models, for different values of pathogen energy consumption rate fN and immune attack rate fI. The hatched regions of the plots show parameter combinations where increasing acquisition rate will increase pathogen load. These plots show that increasing acquisition rate is more likely to increase pathogen load under the energy antagonism model. The predictions of the immune priority model are not very sensitive to variation in either pathogen consumption rate or immune killing rate. In contrast, for the energy antagonism model, increasing pathogen energy consumption rate increases the value of energy acquisition rate where pathogen load peaks, whereas increasing immune killing rate decreases it.
Figure 3.
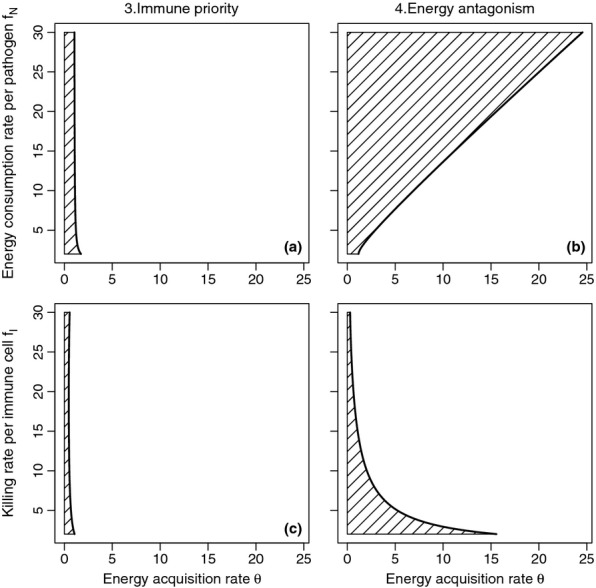
The regions of parameter space where increasing energy acquisition rate θ increases pathogen load for the immune priority and energy antagonism models. The solid line in all four panels shows the θ values where pathogen load peaks. Panels a and b show this peak for varying values of pathogen energy consumption rate fN, whereas panels c and d show the peak for varying values of immune killing rate fI. For parameter combinations in the hatched regions, increasing θ increases pathogen load.
Literature survey
To evaluate whether empirical studies support the general prediction that pathogen response to host resources can vary, we surveyed the literature for studies that varied the quantity of the host's diet (corresponding to variation in θ in our models) and measured the impact on pathogens. Figure 4 shows that all three outcomes predicted by our theory have been observed experimentally, including peak load at intermediate food (Lambrechts et al. 2006; Sadd 2011). A full list of empirical studies and search criteria can be found in Appendix S2. Included studies fit two criteria. First, we included only experimental studies that controlled both host food and pathogen exposure; however, correlational studies also indicate that pathogens can increase or decrease in response to changes in host diet (Scrimshaw et al. 1959; Bize et al. 2008). Second, we included only studies using parasites that can undergo population dynamics in a single host; this excludes most macroparasites, including ectoparasites and intestinal parasites, whose offspring typically disperse away from the focal host. Again, studies using such parasites indicate that parasite abundance can increase or decrease in response to changes in host diet (Murray et al. 1998; Tschirren et al. 2007). The fact that both correlational field studies and experiments with macroparasites show that parasite abundance varies with host diet suggests that energy may generally be an important driver of within-host dynamics.
Figure 4.
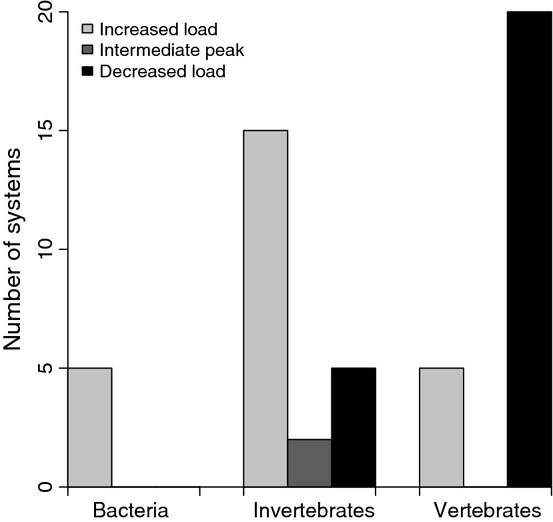
The number of host–pathogen systems showing an increase, decrease or intermediate peak in pathogen abundance as host food is increased, when the host is a bacteria, invertebrate or vertebrate. All three model-predicted responses can be found in the empirical literature.
Of the 52 studies that met our criteria, about half (25) documented a decrease in pathogen load, while the other half (25) documented an increase. Two showed an intermediate peak. Pathogens infecting invertebrate hosts were much more likely to increase in response to increased host food, whereas pathogens infecting vertebrates were much more likely to decrease.
Discussion
The goal of this study was threefold: (1) to motivate the need for an extended epidemiological theory that considers the role of within-host resources, (2) to provide such a theoretical framework by developing and analysing several alternative models of the interaction between resources, the immune system and pathogens and (3) to summarise resource-based empirical patterns in the existing literature. By demonstrating that the dynamics of resources, immune cells and pathogens are qualitatively different under the different model structures introduced here, this work provides guidance to future empirical studies that can more directly test the plausibility of these models.
Experiments showing that poor nutrition, whether through diet quantity or quality, leads to a weakened immune response go back to the early decades of the 20th century (Clark 1950; Scrimshaw et al. 1959; Good et al. 1976) and continue, with increasing sophistication, to the present (Moret & Schmid-Hempel 2000; Kristan 2008; Ayres & Schneider 2009). Studies explicitly testing the influence of host resources on pathogen abundance are less common, primarily because it is difficult to disentangle the direct effect of host resources on pathogen replication from the indirect effect of host resources on pathogen mortality, mediated by the immune system. However, recognition of the fact that host diet has both positive and negative effects on pathogens has been recognised for decades (Scrimshaw et al. 1959; Beisel 1982), and can be seen in the results of our empirical survey (Fig. 4).
However, existing models of within-host dynamics cannot predict these responses, as they typically assume that pathogen abundance is limited either by the immune system only (Antia et al. 1994; Alizon & van Baalen 2008; Fenton & Perkins 2010) or by resources only (Hoshen et al. 2000; Gilchrist et al. 2004). Even models incorporating both an immune system and pathogen resource do not consider that the immune system might also be resource limited (Wodarz et al. 2000; Mideo et al. 2011). It therefore seems reasonable that an extended theory is needed.
The models we propose above bracket a range of possible levels of resource interaction between hosts and pathogens, from fully independent (separate resources for pathogens and immune system) to fully antagonistic (competing for the same resource). These models are qualitatively distinct from previous attempts to model the immune–pathogen interaction as either a predator–prey system or as a consumer–resource system (e.g. Antia et al. 1994; Hoshen et al. 2000). In particular, because immune proliferation requires both pathogens and energy, the interaction is fundamentally different from a predator–prey interaction. In predator–prey systems, killed prey lead directly to increased predators. Here, killed pathogens (‘prey’) only indirectly lead to increased immune cells (‘predators’): even an immune system encountering and killing many pathogens cannot proliferate without energy. This suggests that our models are distinct from both standard predator–prey theory and from ecological theory that combines both predation and resource competition, such as intraguild predation theory (Holt & Polis 1997).
Our models are also distinct from models of resource competition. V. H. Smith and colleagues have shown how concepts like R* and resource ratio theory might apply to pathogens (Smith 1993,2007; Smith & Holt 1996; Smith et al. 2005). For example, applying the R* concept to our models, it is possible to calculate the equilibrium energy abundance (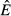 ) for the immune-only and pathogen-only cases. The value of
) for the immune-only and pathogen-only cases. The value of 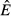 in the immune-only system determines whether the pathogen can persist (the pathogen must have a positive per capita growth rate when the system is at the immune-only equilibrium). However, the value of
in the immune-only system determines whether the pathogen can persist (the pathogen must have a positive per capita growth rate when the system is at the immune-only equilibrium). However, the value of 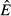 in the pathogen-only system is not meaningful because the immune system has no minimum resource requirement. As long as there is any energy in the system, baseline allocation ensures that immune cells will be produced. The pathogen cannot drive the immune system to extinction through competition for resources.
in the pathogen-only system is not meaningful because the immune system has no minimum resource requirement. As long as there is any energy in the system, baseline allocation ensures that immune cells will be produced. The pathogen cannot drive the immune system to extinction through competition for resources.
We suggest the immune–pathogen interaction is fundamentally distinct from any consumer–resource interaction and that our models provide a unique perspective on this interaction. Here, we show that the models make qualitatively different predictions regarding how increasing resource acquisition rate impacts immune abundance and pathogen load. Perhaps unsurprisingly, most of the models (the exception being the pathogen priority model) predict that immune abundance will increase with increasing resources. This prediction fits with intuition and with studies demonstrating the energetic cost of mounting an immune response (Lochmiller & Deerenberg 2000). More interesting are the predictions for pathogen load. Both the independent energy and pathogen priority models predict that pathogen load will increase in response to increased resource acquisition, whereas the energy antagonism model predicts a peak in pathogen load at intermediate resource acquisition and the immune priority model predicts that pathogen load will decrease over much of parameter space. Moreover, equilibrium pathogen loads vary considerably with model topology, suggesting that energetics may affect infection duration (discussed below).
These differences in predictions reflect the action of different ecological mechanisms. The independent energy model predicts that both immune abundance and pathogen load will increase because the resources are independent, so increasing energy acquisition increases the reproduction rate of both species. The pathogen priority model predicts that immune abundance will be constant and low because of pathogenic suppression of the immune response through resource preemption. The immune priority model predicts declining pathogen loads because the immune system can suppress pathogen replication through resource preemption, in addition to killing the pathogen directly. The energy antagonism model shows features of both the pathogen priority and immune priority models, causing pathogen load to peak at intermediate energy acquisition rates.
Pathogenic suppression of the immune response is fairly well-documented (Schmid-Hempel 2008). This most commonly takes the form of direct manipulation of the immune response, but resource consumption by the pathogen can also reduce the energy available for mounting a costly immune response (Koski & Scott 2001; Cornet & Sorci 2010). The converse possibility, that the immune response might deplete resources required by the pathogen, has not been considered experimentally.
However, several lines of evidence support the hypothesis that the immune response can deplete pathogen resources. Life history theory predicts that, when energy is limiting, the up-regulation of any physiological process that requires energy will force reallocation of resources from other processes (Sheldon & Verhulst 1996). Immune challenge experiments that expose hosts to a novel antigen have shown that there are significant energy costs of immune up-regulation (for reviews, see Klasing 1988; Lochmiller & Deerenberg 2000). Basal metabolic rate can increase by 15–30% upon exposure to non-fever inducing challenge; fever can increase this between 10 and 20% for each degree of temperature increase. Whole-body glucose consumption can increase by 68%. Nitrogen excretion increases as amino acids are depleted from muscle tissue for use in immune protein synthesis. These changes to energetic processes can have substantial impacts on other aspects of life history, including loss of body mass (Ots et al. 2001), reduction of lipid stores (DiAngelo et al. 2009), and reduced reproductive investment (Cai et al. 2009). All of this points to a significant reallocation cost of immune up-regulation, one that could easily impinge on pathogen resource availability as hypothesised by the immune priority and energy antagonism models.
For some systems, we have sufficient knowledge of the biology to hypothesise a most likely model structure among those studied here. For example, malaria parasites (Plasmodium) use red blood cells (RBCs) as a resource; while the relevant immune resource is unclear, it is not RBCs (hosts typically increase RBC production during infection-induced anaemia), suggesting that this system best fits the independent energy model. Moreover, Mideo et al. (2011) have shown that Plasmodium dynamics cannot be explained without considering both immune killing and resource density.
Castrating parasites, those that cause chronic infection with no host reproduction, appear to fit many of the assumptions of the pathogen priority model. In particular, parasite abundance increases with increased host food in these systems (Sandland & Minchella 2003; Ebert et al. 2004; Seppälä et al. 2008; Hall et al. 2009). For Daphnia, at least, the immune response appears to play very little role in limiting parasite reproduction (Mucklow et al. 2004; Auld et al. 2010). Moreover, existing models for parasitic castration (e.g. Hall et al. 2007) share the essential features of the pathogen priority model: in these models, the parasite's resource is ‘upstream’ of all other metabolic processes.
For many systems, however, the data are insufficient to determine which of the proposed model structures is most likely. The empirical data (Fig. 4) document a previously unrecognised pattern that clearly relates infection outcomes to within-host resource dynamics. These data suggest that further empirical investigation is warranted, and our models provide a conceptual framework to guide such investigation. In particular, the approach taken by Mideo et al. (2011) of confronting competing models that include resource, immune and pathogen dynamics with empirical data using model fitting will be a powerful tool for understanding the role of within-host resources in particular systems.
The empirical literature survey reveals that the response of pathogens to changes in host food varies across systems. The most obvious difference is between invertebrates and vertebrates, with pathogens infecting invertebrates much more likely to increase in response to increases in host food than pathogens infecting vertebrates. The reasons for this difference are interesting as they may reflect fundamental differences between vertebrate and invertebrate immune systems, the most obvious being the presence of adaptive immunity in vertebrates. In particular, leucocytic cytokines of the vertebrate immune system are known to play an important role in regulating many of the metabolic consequences of immune up-regulation (Klasing 1988). No known molecules play a similar role in invertebrates. Alternatively, pathogens in invertebrates may be more likely to be resource-limited. Vertebrates tend to be larger than invertebrates and ingestion increases with body size (Kooijman 2010), so it may be that pathogens in vertebrate hosts are energy saturated, so that their abundances are regulated only by the immune response, whereas pathogen abundance in invertebrates is regulated by both resource availability and the immune response. Similar arguments might also explain why pathogens in bacteria always increased in abundance with host food, as bacteria have only very rudimentary immune systems and their small size may predispose their pathogens to resource limitation. The existence of resource-based patterns in infection outcome suggests that further work is warranted, and the models developed here can serve as a conceptual framework for such experiments.
The models developed here can provide a novel perspective on other open questions in epidemiology. For example, decreased food consumption is a common response to infection, and many studies have suggested that anorexia might be an adaptive host response (Murray & Murray 1979; Exton 1997; Ayres & Schneider 2009). The models proposed here provide a theoretical framework for exploring the adaptive value of anorexia. Our results already suggest that anorexia will be beneficial whenever the pathogen can preempt resource use by the immune system.
Models that incorporate within-host energy can provide new insight into the question of whether an infection is acute or chronic. In most epidemiological theory, the duration of infection is a modelling assumption (Alizon & van Baalen 2008). The models presented here are models of chronic infection, in part because it is known empirically that chronic infections can have dramatic effects on host nutritional status (Beisel 1982). However, the differences in equilibrium pathogen load predicted by each model suggest that differences in energetic structure may influence infection duration. In particular, if there are independent resources or the immune system can preempt the pathogen, acute infections are more likely as pathogen load is very low, whereas if the pathogen can suppress the immune response through resource consumption, chronic infections are more likely. Moreover, theory suggests several mechanisms that can produce acute infection, including immortal immune cells (Alizon & van Baalen 2008), time delays in the immune response (Fenton et al. 2006) or immune ‘memory’ (Wodarz et al. 2000). Each of these could be affected by within-host resources.
This model could also be used to understand the ecological and evolutionary feedbacks between within-host and among-host processes. In recent years, there has been considerable interest in using ‘nested’ models to explicitly link within- and among-host processes. Such models can provide insight on the emergence of trade-offs between epidemiological parameters and the consequences of conflicting selection (Gilchrist & Sasaki 2002). However, in most cases the nesting of the within-host model into the among-host model is ‘inessential’: within-host processes may affect among-host processes, but there is no reciprocal feedback (Mideo et al. 2008). The explicit inclusion of within-host resources in our models allows for essential nesting: within-host resource dynamics are affected by host ingestion from the environment, but the dynamics of environmental resources are affected by among-host processes. There is a feedback from among-host processes to within-host processes mediated by host resources. Such feedbacks will certainly affect the evolution of both host and pathogen traits. We expect that understanding the ecological and evolutionary implications of such feedbacks will be a rich vein for future study. In general, we suggest that broadening the ecological view of the interaction between the immune system and pathogens to include resources will help to understand and manage disease systems (Smith & Holt 1996; Smith et al. 2005).
Acknowledgments
Our most sincere thanks go to Dr P. Thrall and three anonymous referees for detailed and very helpful comments. This material is based upon work supported by a Queen's University Senate Advisory Research Committee Postdoctoral Fellowship (to CEC) and by the National Science Foundation under Award No. DBI-1103593 (to CEC). WAN acknowledges support from the NSERC Discovery programme.
Authorship
All authors contributed to study design, CEC performed the research and wrote the first draft of the manuscript, and all authors contributed substantially to revisions.
Supporting Information
Additional Supporting Information may be downloaded via the online version of this article at Wiley Online Library (www.ecologyletters.com).
Supplementary
References
- Alizon S. van Baalen M. Acute or chronic? Within-host models with immune dynamics, infection outcome, and parasite evolution. Am. Nat. 2008;172:E244–E256. doi: 10.1086/592404. [DOI] [PubMed] [Google Scholar]
- Antia R, Levin BR. May RM. Within-host population dynamics and the evolution and maintenance of microparasite virulence. Am. Nat. 1994;144:457–472. [Google Scholar]
- Auld SKJR, Scholefield JA. Little TJ. Genetic variation in the cellular response of Daphnia magna (Crustacea: Cladocera) to its bacterial parasite. Proc. R. Soc. Lond. B. 2010;277:3291–3297. doi: 10.1098/rspb.2010.0772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres JS. Schneider DS. The role of anorexia in resistance and tolerance to infections in Drosophila. PLoS Biol. 2009;7:e1000150. doi: 10.1371/journal.pbio.1000150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisel WR. Synergism and antagonism of parasitic diseases and malnutrition. Rev. Infect. Dis. 1982;4:746–750. doi: 10.1093/4.4.746. [DOI] [PubMed] [Google Scholar]
- Bize P, Jeanneret C, Klopfenstein A. Roulin A. What makes a host profitable? Parasites balance host nutritive resources against immunity. Am. Nat. 2008;171:107–118. doi: 10.1086/523943. [DOI] [PubMed] [Google Scholar]
- Cai X-Q, Yang M, Zhong W-Q. Wang D-H. Humoral immune response suppresses reproductive physiology in male Brandt's voles (Lasiopodomys brandtii. Zoology. 2009;112:69–75. doi: 10.1016/j.zool.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Clark PF. Influence of nutrition in experimental infection. Ann. Rev. Microbiol. 1950;4:343–358. doi: 10.1146/annurev.mi.04.100150.002015. [DOI] [PubMed] [Google Scholar]
- Cornet S. Sorci G. Parasite virulence when the infection reduces the host immune response. Proc. R. Soc. Lond. B. 2010;277:1929–1935. doi: 10.1098/rspb.2010.0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiAngelo JR, Bland ML, Bambina S, Cherry S. Birnbaum MJ. The immune response attenuates growth and nutrient storage in Drosophila by reducing insulin signaling. PNAS. 2009;106:20853–20858. doi: 10.1073/pnas.0906749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert D, Carius HJ, Little T. Decaestecker E. The evolution of virulence when parasites cause host castration and gigantism. Am. Nat. 2004;164:S19–S32. doi: 10.1086/424606. [DOI] [PubMed] [Google Scholar]
- Exton MS. Infection-induced anorexia: active host defence strategy. Appetite. 1997;29:369–383. doi: 10.1006/appe.1997.0116. [DOI] [PubMed] [Google Scholar]
- Fenton A, Lello J. Bonsall MB. Pathogen responses to host immunity: the impact of time delays and memory on the evolution of virulence. Proc. R. Soc. Lond. B. 2006;273:2083–2090. doi: 10.1098/rspb.2006.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton A. Perkins SE. Applying predator-prey theory to modelling immune-mediated, within-host interspecific parasite interactions. Parasitology. 2010;137:1027–1038. doi: 10.1017/S0031182009991788. [DOI] [PubMed] [Google Scholar]
- Frost PC, Ebert D. Smith VH. Bacterial infection changes the elemental composition of Daphnia magna. J. Anim. Ecol. 2008;77:1265–1272. doi: 10.1111/j.1365-2656.2008.01438.x. [DOI] [PubMed] [Google Scholar]
- Gilchrist MA, Coombs D. Perelson AS. Optimizing within-host viral fitness: infected cell lifespan and virion production rate. J. Theor. Biol. 2004;229:281–288. doi: 10.1016/j.jtbi.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Gilchrist MA. Sasaki A. Modeling host parasite coevolution: a nested approach based on mechanistic models. J. Theor. Biol. 2002;218:289–308. doi: 10.1006/jtbi.2002.3076. [DOI] [PubMed] [Google Scholar]
- Good RA, Fernandes G, Yunis EJ, Cooper WC, Jose DC, Kramer TR. Nutritional deficiency, immunologic function, and disease. Am. J. Pathol. 1976;84:599–614. [PMC free article] [PubMed] [Google Scholar]
- Hall SR, Simonis JL, Nisbet RM, Tessier AJ. Cáceres CE. Resource ecology of virulence in a planktonic host-parasite system: an explanation using dynamic energy budgets. Am. Nat. 2009;174:149–162. doi: 10.1086/600086. [DOI] [PubMed] [Google Scholar]
- Hall SR, Sivars-Becker L, Becker C, Duffy MA, Tessier AJ. Cáceres CE. Eating yourself sick: transmission of disease as a function of foraging ecology. Ecology. 2007;10:207–218. doi: 10.1111/j.1461-0248.2007.01011.x. [DOI] [PubMed] [Google Scholar]
- Holt RD. Polis GA. A theoretical framework for intraguild predation. Am. Nat. 1997;149:745–764. [Google Scholar]
- Hoshen MB, Heinrich R, Stein WD. Ginsburg H. Mathematical modelling of the within-host dynamics of Plasmodium falciparum. Parasitology. 2000;121:227–235. doi: 10.1017/s0031182099006368. [DOI] [PubMed] [Google Scholar]
- Klasing KC. Nutritional aspects of leukocytic cytokines. J. Nutr. 1988;118:1434–1446. doi: 10.1093/jn/118.12.1436. [DOI] [PubMed] [Google Scholar]
- Kooijman SALM. Dynamic Energy Budget Theory for Metabolic Organization. New York: Cambridge University Press; 2010. , 3. [Google Scholar]
- Koski KG. Scott ME. Gastrointestinal nematodes, nutrition and immunity: breaking the negative spiral. Ann. Rev. Nutr. 2001;21:297–321. doi: 10.1146/annurev.nutr.21.1.297. [DOI] [PubMed] [Google Scholar]
- Kristan DM. Calorie restriction and susceptibility to intact pathogens. Age. 2008;30:147–156. doi: 10.1007/s11357-008-9056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts L, Chavatte JM, Snounou G. Koella JC. Environmental influence on the genetic basis of mosquito resistance to malaria parasites. Proc. R. Soc. Lond. B. 2006;273:1501–1506. doi: 10.1098/rspb.2006.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochmiller RL. Deerenberg C. Trade-offs in evolutionary immunology: just what is the cost of immunity? Oikos. 2000;88,:87–98. [Google Scholar]
- Mideo N, Alizon S. Day T. Linking within- and between-host dynamics in the evolutionary epidemiology of infectious diseases. Trends Ecol. Evol. 2008;23:511–517. doi: 10.1016/j.tree.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Mideo N, Savill NJ, Chadwick W, Schneider P, Read AF. Day T. Causes of variation in malaria infection dynamics: insights from theory and data. Am. Nat. 2011;178:174–188. doi: 10.1086/662670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moret Y. Schmid-Hempel P. Survival for immunity: the price of immune activation for bumblebee workers. Science. 2000;290:1166–1168. doi: 10.1126/science.290.5494.1166. [DOI] [PubMed] [Google Scholar]
- Mucklow PT, Vizoso DB, Jensen KH, Refardt D. Ebert D. Variation in phenoloxidase activity and its relation to parasite resistance within and between populations of Daphnia magna. Proc. R. Soc. Lond. B. 2004;271:1175–1183. doi: 10.1098/rspb.2004.2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray DL, Keith LB. Cary JR. Do parasitism and nutritional status interact to affect production in snowshoe hares. Ecology, 1998;79,:1209–1222. [Google Scholar]
- Murray MJ. Murray AB. Anorexia of infection as a mechanism of host defense. Am. J. Clin. Nutr. 1979;32:593–596. doi: 10.1093/ajcn/32.3.593. [DOI] [PubMed] [Google Scholar]
- Ots I, Kerimov AB, Ivankina EV, Ilyina TA. Hõrak P. Immune challenge affects basal metabolic activity in wintering great tits. Proc. R. Soc. Lond. B. 2001;268:1175–1181. doi: 10.1098/rspb.2001.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero A, Agnew P, Bedhomme S, Sidobre C. Michalakis Y. Resource depletion in Aedes aegypti mosquitoes infected by the microsporidia Vavraia culicis. Parasitology. 2007;134:1355–1362. doi: 10.1017/S0031182007002703. [DOI] [PubMed] [Google Scholar]
- Sadd BM. Food-environment mediates the outcome of specific interaction between a bumblebee and its trypanosome parasite. Evolution. 2011;65:2995–3001. doi: 10.1111/j.1558-5646.2011.01345.x. [DOI] [PubMed] [Google Scholar]
- Sandland GJ. Minchella DJ. Effects of diet tits Echinostoma revolutum infection on energy allocation patterns in juvenile Lymnaea elodes snails. Oecologia. 2003;134:479–486. doi: 10.1007/s00442-002-1127-x. [DOI] [PubMed] [Google Scholar]
- Schmid-Hempel P. Parasite immune evasion: a momentous molecular war. Trends Ecol. Evol. 2008;23:318–326. doi: 10.1016/j.tree.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Scrimshaw NS, Taylor CE. Gordon JE. Interactions of nutrition and infection. Am. J. Med. Sci. 1959;237:367–403. [PubMed] [Google Scholar]
- Seppälä O, Liljeroos K, Karvonen A. Jokela J. Host condition as a constraint for parasite reproduction. Oikos. 2008;117:749–753. [Google Scholar]
- Sheldon BC. Verhulst S. Ecological immunology: costly parasite defences and trade-offs in evolutionary ecology. Trends Ecol. Evol. 1996;11:317–321. doi: 10.1016/0169-5347(96)10039-2. [DOI] [PubMed] [Google Scholar]
- Smith VH. Resource competition between host and pathogen. Bioscience. 1993;43:21–30. [Google Scholar]
- Smith VH. Host resource supplies influence the dynamics and outcome of infectious disease. Integr. Comp. Biol. 2007;47:310–316. doi: 10.1093/icb/icm006. [DOI] [PubMed] [Google Scholar]
- Smith VH. Holt RD. Resource competition and within-host disease dynamics. Trends Ecol. Evol. 1996;11:386–389. doi: 10.1016/0169-5347(96)20067-9. [DOI] [PubMed] [Google Scholar]
- Smith VH, Jones TP. Smith MS. Host nutrition and infectious disease: an ecological view. Front. Ecol. Environ. 2005;3:268–274. [Google Scholar]
- Tschirren B, Bischoff LL, Saladin V. Richner H. Host condition and host immunity affect parasite fitness in a bird-ectoparasite system. Func. Ecol. 2007;21:372–378. [Google Scholar]
- Wodarz D, Page KM, Arnaout RA, Thomsen AR, Lifson JD. Nowak MA. A new theory of cytotoxic T-lymphocyte memory: implications for HIV treatment. Philos. Trans. R. Soc. Lond. B. 2000;355:329–343. doi: 10.1098/rstb.2000.0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary