

Abstract
Recombinant WT human cardiac actin (WT actin) was expressed using the baculovirus/insect cell expression system, purified, and used to reconstitute the thin-filament of bovine cardiac muscle fibers, together with bovine cardiac tropomyosin (Tm) and troponin (Tn). Effects of [Ca2+], [ATP], [phosphate] and [ADP] on tension and tension transients were studied at 25°C by using sinusoidal analysis, and the results were compared with those of native fibers and fibers reconstituted with purified bovine cardiac actin (BVC actin). In actin filament reconstituted fibers (without Tm/Tn), those reconstituted with WT actin showed exactly the same active tension as those reconstituted with purified BVC actin (WT: 0.75±0.06 T0, N=11; BVC: 0.73±0.07 T0, N=12, where T0 is tension of original fibers before extraction). After Tm/Tn reconstitution, fibers reconstituted with WT actin generated 0.85±0.06 T0 (N=11) compared to 0.98±0.04 T0 (N=12) recovered by those reconstituted with BVC actin. In the presence of Tm/Tn, WT actin reconstituted fibers showed exactly the same Ca2+ sensitivity as those of the native fibers and BVC actin reconstituted fibers (pCa50: native fibers: 5.69±0.01, N=10; WT: 5.69±0.02, N=11; BVC: 5.68±0.02, N=12). Sinusoidal analysis showed that the cross-bridge kinetics were the same among native fibers, BVC actin reconstituted fibers, and WT actin reconstituted fibers, followed by reconstitution of Tm/Tn. These results demonstrate that baculovirus/insect cell expressed actin has no significant differences from tissue purified actin and can be used for thin-filament reconstitution assays. One hypertrophic cardiomyopathy (HCM) causing actin mutant A331P actin was also expressed and studied similarly, and the results were compared to those of the WT actin. In the reconstituted fibers, A331P significantly decreased the tension both in the absence of Tm/Tn (0.55±0.03 T0, N=13) and in their presence (0.65±0.02 T0, N=13) compared to those of the WT (0.75±0.06 T0 and 0.85±0.06 T0, respectively, N=11). A331P also showed decreased pCa50 (5.57±0.03, N=13) compared to that of WT (5.69±0.02, N=11). The cross-bridge kinetics and its distribution were similar between WT and A331P actin reconstituted fibers, indicating that force/cross-bridge was decreased by A331P. In conclusion, A331P causes a weakened cross-bridge force, which leads to a decreased active tension, reduces left-ventricular ejection fraction, and eventually results in the HCM phenotype.
Keywords: Actin, Hypertrophic Cardiomyopathy, A331P, Ca2+ sensitivity, cross-bridge kinetics, actin-myosin interaction
1. Introduction
In adult human heart, α-cardiac actin (hereafter, abbreviated as actin) accounts for more than 80% of the total actin and is critical for maintaining normal structure and function of the sarcomere [1]. Mutations of actin have been found in patients suffering from hypertrophic cardiomyopathy (HCM), a serious heart disease characterized by abnormal thickening of the left ventricle and/or inter ventricular septum, affects 1:500 individuals, and results in sudden cardiac death (1–2%) in young adults [2]. So far, 11 missense mutations in actin (H88Y, R95C, E99K, P164A, Y166C, A230V, S271F, A295S, M305L, R312C, and A331P) have been indentified to cause HCM [3–10], and two (R312H and A361G) to cause dilated cardiomyopathy (DCM) [11] in humans. Fig. 1 shows an actin dimer structure in F-form along one strand of the two stranded F-actin helix. The position of the HCM-causing mutation A331P, which is addressed in this report is also denoted in the figure. Despite the vital importance of actin in the heart muscle, the relationship between actin mutation and HCM disease phenotype remains to be investigated.
Figure 1. Actin dimer structure in F-form and the position of A331P mutation.

The N and C terminus of each actin monomer are labeled. S1–S4 represent each subdomain in the actin monomer. A331P mutation is marked in red in both actin monomers. Modified from [40](PDB ID:4a7f)
The functional consequences of human actin mutations were seldom studied because of the difficulties involved in expressing recombinant actin. Actin expressed in E. coli could not fold properly and only formed inclusion bodies [12]. Yeast actin has 86% amino acid sequence identity with human cardiac actin, hence is not an excellent model, because actin is an extremely conserved protein. An in vitro transcription/translation system was also used for actin expression, but the yield was too low (only at the picogram level) to carry out functional analysis [13]. During the past few years, the baculovirus infected insect cell expression system has been used to generate recombinant human actin protein for research. With this system G-actin (all α, β, and γ isoforms) folds correctly, and readily polymerizes to form F-actin [14–16]. In this study, a new improved method was used to ensure the robust expression and specific purification of un-tagged recombinant actin [17]. The most notable improvements included the using of P10 promoter and DNase-I column plus polymerization/de-polymerization purification [17]. Previous studies using polyhedrin promoter generally had lower yield than those using P10 promoter [14, 18]. Compared to previously used Q-sepharose columns [19], DNase-I columns specifically bind with actin monomer and further improve the yield and purity of the recombinant actin. Using this improved method, the yield of recombinant actin would be satisfactory for functional analysis in the reconstituted fibers.
Several actin mutants expressed using baculovirus/insect system have been characterized [15, 19–21]. Transgenic (Tg) mouse models for actin mutations E99K and E361G have been generated and myocardium and single myofibrils from the transgenic models were used to test the functional consequences of these mutations [22–25]. Actin was also purified from the transgenic models and applied to in vitro motility analysis [22, 23]. However, for the HCM-causing actin mutants, a comprehensive functional analysis including cross-bridge kinetics in a muscle fiber environment has not been performed. As the backbone of both contraction and its regulatory processes, as well as a key structural element in the sarcomere, actin interacts with many other sarcomeric proteins. These interactions are essential for sarcomere functions. A single molecule approach (in vitro motility assay) cannot characterize all these interactions. It also has a problem of requiring low ionic strength (~50 mM), which is not physiological (~200 mM). Therefore, it is of vital importance that the effect of disease causing actin mutations be evaluated in the muscle fiber system under the physiological conditions.
Skinned (permeabilized) papillary muscle fibers from transgenic (Tg) mouse models with HCM mutations in sarcomeric proteins have been used to study the pCa-tension relationship [23, 24]. However, myocardium from Tg models are affected by adaptive responses that include interstitial fibrosis and sarcomere disarray [22, 23], as well as post-translational modifications, notably phosphorylation. These secondary effects also affect contractile and regulatory properties, making it difficult to isolate the immediate effect of the mutation that triggers the downstream events and finally lead to a cardiomyopathy phenotype. To address this problem in a real muscle fiber system and avoid the difficulties with transgenic models, we applied the thin-filament extraction/reconstitution technique to create cardiac muscle fibers containing HCM-causing actin mutants without changing other factors which may interfere with muscle function [26, 27]. Thus, any measured abnormality in the reconstituted cardiac fibers is the immediate effect of the mutation. In the past, such a thin-filament extraction/reconstitution technique has been successfully employed to study the functional consequences of Tm and TnT mutations that cause HCM or DCM [28–30]. The reconstituted fibers is also very stable, making it possible for us to apply prolonged activations to enable sinusoidal analysis and to characterize the elementary steps of the cross-bridge cycle [26].
In this study, we demonstrate that 1) reconstituted fibers using recombinant WT actin possess the same contractile and regulatory functions as those of native fibers and fibers reconstituted with BVC actin; 2) an actin mutant A331P decreases tension of reconstituted fibers in the presence/absence of Tm/Tn; 3) A331P shows a decreased Ca2+ sensitivity compared to that of WT. These results are discussed in the context of the actin-Tm-myosin interaction and the early molecular pathogenesis of HCM.
2. Material and Methods
2.1. Experimental Material and the Thin-Filament Extraction and Reconstitution Technique
Bundles of trabecular muscles were collected from fresh cow hearts, skinned (permeabilized) and further dissected to thin fibers (~2 mm long and 90–110 μm thick; sometimes called ‘strips’) as described previously [26]. The thin-filament extraction/reconstitution technique was performed as described previously [26, 28, 31].
2.2. WT and mutant actin expression and purification from sf-21 cells
Recombinant baculovirus containing human WT and mutant actin cDNA sequence were constructed as described previously [17]. In short, human cardiac actin cDNA sequence was cloned into pAcUW2Bmod and cotransfected into sf-21 cells with Bsu36I-digested BacPAK6 viral DNA (Clontech) to generate recombinant baculovirus. The insertion of WT and mutant actin sequence into pAcUW2Bmod was confirmed by direct sequencing.
Low passage Sf-21 cells (Invitrogen) cultured in SF900 III medium (Invitrogen) with 1% penicillin/streptomycin were infected with the recombinant virus (MOI=2). 2 liters of Sf-21 cells (~ 4×109 cells) were harvested 72 h after infection by centrifuging the medium at 3000 rpm for 15 min. Purification of actin using DNase I and DE-52 columns was performed as described [14, 16, 17]. In short, the cells were lysed by sonication in cell-lysis buffer, consisting of 1 M Tris-HCl, pH 7.5, 0.5 mM MgCl2, 0.5 mM ATP, 4% Triton X-100, 1 mg/ml Tween 20, 1 mm DTT, and a protease inhibitor mixture (lima bean protease inhibitor, leupeptin, aprotinin, antipain, TLCK, TPCK, E-64, each at 1.25 μg/ml). The cell lysate was cleared by centrifugation at 50,000 rpm for 80 min at 2°C. The cell lysate was then applied to a DNase-I column. After loading the lysate, G-buffer (10 mM Tris-HCl, pH 7.5, 0.2 mM CaCl2, and 0.2 mM ATP and a protease inhibitor mixture of pepstatin, chymostatin, BAEE, leupeptin, aprotinin, antipain, TLCK, TPCK, each at 0.5 μg/ml and PMSF at 0.625 mM) was used to thoroughly wash the column. 1.0 M of NaCl in G-buffer was then used to wash out cofilin from the DNase-I column. Actin was eluted from the DNase-I column to a connected DE-52 column using 50% formamide in G buffer. After thoroughly washing the DE-52 column with G-buffer, actin was finally eluted using 0.3 M KCl in G-buffer. Eluted actin was then dialyzed against G-buffer (4 liter) overnight to remove excess KCl.
The following morning, actin was concentrated to less than 1 ml in volume by using Dextran T150 (average MW=150,000). An additional polymerization/de-polymerization cycle was applied to the concentrated actin as the last step of purification. The quality of purified actin was examined by SDS-PAGE (Fig. 2A). Purified G-actin (in solution form) was stored on ice in a refrigerator (4°C). To ensure the freshness of actin, the purified recombinant actin was used within 4 days.
Figure 2. Purification and polymerization of recombinant actin.
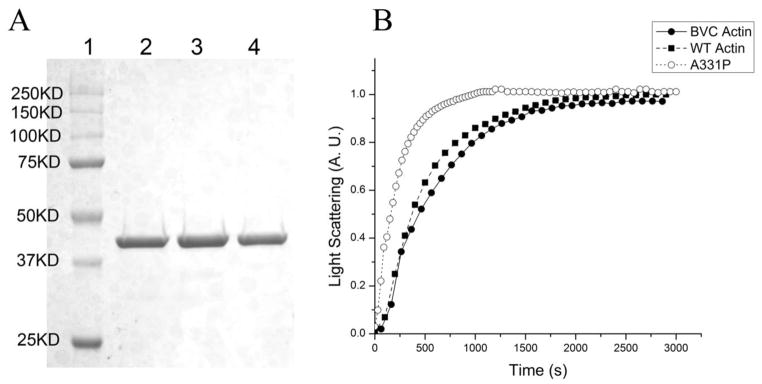
(A) SDS-PAGE of purified actins. Lane 1: Precision plus protein dual color standards (Bio-rad); Lane 2: BVC actin; Lane 3: WT actin; Lane 4: A331P actin. (B) Polymerization of 0.2mg/ml G-actin, monitored as the increase of light scattering signal. All light scattering signals were normalized to the saturation level (at ~2800 s) of WT actin.
2.3. Other proteins and solutions
Bovine cardiac actin, Tm and Tn were purified from bovine hearts as described [32, 33]. For muscle fiber studies, the standard activating solution (5S8P) contained (mM:) 6 K2CaEGTA, 5.8 Na2MgATP, 1.36 Na2K2ATP, 15 Na2CP (creatine phosphate), 4 KH2PO4, 4 K2HPO4, 0.7 NaAc (Ac=acetate), 73 KAc, 10 NaN3, 10 MOPS, 160 U/ml creatine kinase, and pH adjusted to 7.00. The ionic strength of this solution was 200 mM, pCa 4.66, [Mg2+] 1 mM, [MgATP2−] 5 mM, and [Pi] 8 mM, where Pi=phosphate. All the other experimental solutions were prepared as described in Table S2 in Ref. [28].
2.4. Actin polymerization assay
Polymerization of G-actin was induced by adding 6 μl of 20x F-salt (10 mM Tris, 40 mM MgCl2, 1 M KCl, 0.1 mM DTT and 0.1 mM CaCl2) into 114 μl 0.2 mg/ml G-actin under 25°C. Polymerization was measured by following the increase in light scattering of the sample in a FluoroMax-3 fluorescence spectrometer (HORIBA Jobin Yvon Inc). Both the excitation and emission wavelengths were set to 360 nm with the slit widths set at 1 nm.
2.5. pCa-Tension study
After Tm/Tn reconstitution, pCa-tension studies were performed as described [30] with pCa ranging from 8.0 to 4.66. The tension at pCa 8.0 was called low Ca2+ tension (TLC), and the tension at pCa 4.66 was called high Ca2+ tension (THC). Tact is the Ca2+ activatable tension (Tact=THC−TLC), and Ca50 represents the Ca2+ concentration at half-maximum tension (0.5 Tact). pCa50 (= −log10Ca50) represents Ca2+-sensitivity, and nH (Hill factor) represents the cooperativity. These are defined in Eq. S1 (Supplementary Materials). Tension baseline was defined as that measured in the relaxing solution, which contains 6 mM EGTA, no added Ca, 40 mM BDM, and at 0°C. There are no detectable actively cycling cross-bridges under these conditions. All tension measurement was performed at 25°C as an increment from this baseline tension.
2.6. Sinusoidal analysis
The elementary steps of the cross-bridge cycle based on six states (Scheme S1 in Supporting Materials) were characterized by sinusoidal analysis performed as described [28, 34, 35]. This method yielded the complex modulus data Y(f), which is a frequency response function. Y(f) was fitted to Eq. S2 (Supporting Materials) which contains two exponential processes, and their apparent rate constants 2πb (medium speed) and 2πc (fast speed) were derived [36]. The effects of [ATP], [ADP], and phosphate (Pi) on 2πb and 2πc were studied, the results were fitted to Eqs. S3 and S4, and the kinetic constants that characterize elementary steps of the cross-bridge cycle consisting of six states were derived [34].
3. Results
3.1. Purification and polymerization of the recombinant actin
The purity of recombinant actin was examined by SDS-PAGE and is shown in Fig. 2A. Purified BVC actin (Lane 2) was used as the control. WT actin (Lane 3) and A331P actin (Lane 4) are also shown. The typical yield of recombinant actin reached 0.6–0.7 mg per liter of cells. The N-terminus of the recombinant actin in this study was not tagged to ensure the proper folding of actin.
The ability for actin to polymerize is critical for cardiac actin function and the reconstitution of the thin-filament. To determine if the recombinant actin polymerized normally, light scattering was performed and the results are shown in Fig. 2B. Purified BVC actin was used as the control (––●––). Fig. 2B shows that WT actin (--■--) has slightly faster polymerization kinetics than that of BVC actin. The HCM causing mutant A331P actin (--○--) polymerized even faster than BVC actin or WT actin. The maximal light scattering signal was almost identical between WT actin and A331P actin, but slightly lower in BVC actin (Fig. 2B). These results demonstrate that the recombinant actin proteins retained the ability to polymerize; hence they can be used for the thin-filament reconstitution assays.
3.2. Tension generation and kinetic analysis of purified insect actin reconstituted fibers
A previous study has demonstrated that endogenous insect actin accounted for a small fraction of purified actin [19]. To examine the effect of insect actin, purified insect actin was used to reconstitute the actin filament of the cardiac fibers, followed by reconstitution with BVC Tm/Tn. The results are summarized in Table 1. In the absence of Tm/Tn, insect actin generated 0.52±0.05 T0 (N=6), which compares to 0.73±0.07 T0 (N=12) of BVC actin reconstituted fibers. In the presence of Tm/Tn at pCa 4.66, tension decreased to 0.39±0.05 T0 (N=6) in insect actin, which compares to 0.98±0.04 T0 (N=12) of BVC actin. Thus, tension generated by actomyosin interaction is significantly diminished in insect actin. The allosteric tension caused by Tm/Tn and Ca2+ is negative, showing the negative allosteric effect, implying a mismatch between actin and Tm. Many kinetic constants (K2, k−4, K4, K5, and some more) are significantly and distinctively different from three other actin reconstituted fibers. In particular, the Pi association constant (K5) was by far the largest of all the fibers used for experiments. Consequently, it can be said that the insect actin behaved very differently from other actins (BVC, WT, A331P) in the reconstituted fibers, and that contaminating insect actin (if any) does not alter the results obtained from the recombinant actin reconstituted fibers.
Table 1.
Comparison among native fibers, and four forms of actin reconstituted fibers
| Parameter (unit) | Native fiber | BVC actin | WT actin | A331P actin | Insect actin |
|---|---|---|---|---|---|
| Ta (T0) | N/A | 0.73±0.07 (12) | 0.75±0.06 (11) | 0.55±0.03*§ (13) | 0.52±0.05*§ (6) |
| THC (T0) | N/A | 0.98±0.04 (12) | 0.85±0.06 (11) | 0.65±0.02*§ (13) | 0.39±0.05*§ (6) |
| TLC (T0) | 0.06±0.01 (10) | 0.08±0.02 (12) | 0.08±0.01 (11) | 0.05±0.01*§ (13) | 0.07±0.02 (6) |
| Tact (T0) | 0.94±0.01 (10) | 0.91±0.02 (12) | 0.76±0.06 (11) | 0.61±0.03*#§ (13) | 0.31±0.06 *#§ (6) |
| Rigor Stiffness (MPa) | 2.12±0.15 (10) | 2.23±0.21 (12) | 2.24±0.17 (11) | 0.92±0.09*#§ (13) | 1.62±0.46 (5) |
| YHC (MPa) | 0.94±0.08 (10) | 0.92±0.10 (12) | 0.82±0.12 (11) | 0.44±0.04*#§ (13) | 0.62±0.12 (5) |
| YLC (MPa) | 0.19±0.03 (10) | 0.30±0.05 (12) | 0.24±0.05 (11) | 0.14±0.01*§ (13) | 0.20±0.04 (5) |
| Yact (MPa) | 0.73±0.06 (10) | 0.62±0.08 (12) | 0.59±0.08 (11) | 0.30±0.03*#§ (13) | 0.42±0.15 (5) |
| pCa50 | 5.69±0.01(10) | 5.68±0.02 (12) | 5.69±0.02 (11) | 5.57±0.03*#§ (13) | 5.53±0.02*#§ (4) |
| Hill Factor (Cooperativity) | 3.22±0.18 (10) | 2.71±0.23 (12) | 2.6±0.3 (11) | 2.33±0.34# (13) | 2.62±0.19# (4) |
| K0 (mM−1) | 17.7±1.3 (9) | 69.6±8.3# (6) | 35.04±6.3#§ (6) | 31.0±5.44#§ (7) | 84.4 (1) |
| K1 (mM−1) | 1.26±0.29 (8) | 2.10±0.44# (8) | 2.07±0.28# (10) | 1.64±0.18 (7) | 2.77±0.96 (4) |
| K2 | 0.88±0.13 (8) | 1.00±0.15 (8) | 0.78±0.13 (10) | 0.69±0.10 (7) | 0.40±0.06*#§ (4) |
| K4 | 0.70±0.08 (7) | 0.68±0.16 (7) | 0.76±0.14 (8) | 0.92±0.16 (7) | 1.92±0.15*#§ (4) |
| K5 (mM−1) | 0.26±0.04 (7) | 0.19±0.05 (7) | 0.22±0.04 (8) | 0.17±0.04 (7) | 1.06±0.34*#§ (4) |
| k2 (s−1) | 14.46±2.16 (8) | 21.3±3.0*# (8) | 11.99±1.42§ (10) | 8.44±1.65§ (7) | 4.99±0.59*#§ (4) |
| k−2 (s−1) | 16.83±2.28 (8) | 19.8±2.1 (8) | 16.83±1.53 (10) | 12.24±1.34 (7) | 13.0±1.7 (4) |
| k4 (s−1) | 6.75±0.79 (7) | 7.45±0.97 (7) | 7.75±0.75 (8) | 13.19±1.73*#§ (7) | 12.1±1.9*#§ (4) |
| k−4 (s−1) | 9.29±0.70 (7) | 12.29±1.89 (7) | 11.94±1.94 (8) | 15.84±2.44# (7) | 6.21±0.7*#§ (4) |
Notes. WT actin and A331P actin are recombinant proteins produced by baculovirus infected insect cells. T0: Tension of original bovine cardiac muscle fibers at the standard activating conditions (5S8P) at pCa 4.66. T0=26.85±1.13 KPa (n=41). Only the first row (Ta) was measured in the absence of Tm/Tn, whereas others were measured after reconstitution with Tm/Tn. Ta: Tension of the actin filament reconstituted fibers at the standard activating conditions (5S8P). THC: Tension at pCa 4.66. TLC: Tension at pCa 8. All experiments were performed at 25°C. All the data are shown as mean ± S. E. M.
Significantly different from WT actin (p<0.05);
Significantly different from native fibers (p<0.05);
Significantly different from BVC actin reconstituted fibers (p<0.05).
3.3. Tension generation of BVC actin, WT actin, and A331P actin reconstituted fibers
The recombinant actin proteins were used to reconstitute the thin filament as described in the Methods. Isometric tension was measured with the standard activating solution. The pCa of this solution was 4.66, and all experiments were performed at 25°C. T0 (active tension of native fibers before extraction) averaged 26.9±1.1 kPa (N=41). The results of the tension study are summarized in Fig. 3A and B. In the absence of Tm/Tn, tension of actin-filament reconstituted fibers (Ta) was similar between BVC actin (0.73±0.07 T0, N=12) and WT actin (0.75±0.06 T0, N=11) (Fig. 3A) [26]. After Tm and Tn reconstitution (Fig. 3B), the tension of BVC actin reconstituted fibers (THC) reached 0.98±0.04 T0 (N=12), and that of WT actin reconstituted fibers reached 0.85±0.06 T0 (N=11). The increase of active tension due to the reconstitution of Tm/Tn is called positive allostery. The results from BVC actin and WT actin reconstituted fibers are not significantly different. A331P actin reconstituted fibers showed significantly decreased tension both in the absence of Tm/Tn (Ta=0.55±0.03 T0, N=13) and in their presence (THC=0.65±0.03 T0, N=13). In the presence of Tm/Tn, the stiffness (Y∞ defined after Eq. S2 in Supporting Materials) of the reconstituted fibers during activation (YHC) was also measured and shown in Fig. 3C, which exhibited a similar trend with THC. Rigor was induced by washing out ATP from the Ca2+ activated preparations using the rigor solution (Table S2, Ref [28]), stiffness was measured at 100 Hz, and plotted in Fig. 3C. There was no significant difference in rigor stiffness among native fibers, BVC actin, and WT actin reconstituted fibers, but A331P actin reconstituted fibers showed significantly decreased rigor stiffness compared with that of WT actin reconstituted fibers (Fig. 3C).
Figure 3. Summary of isometric tension and stiffness.
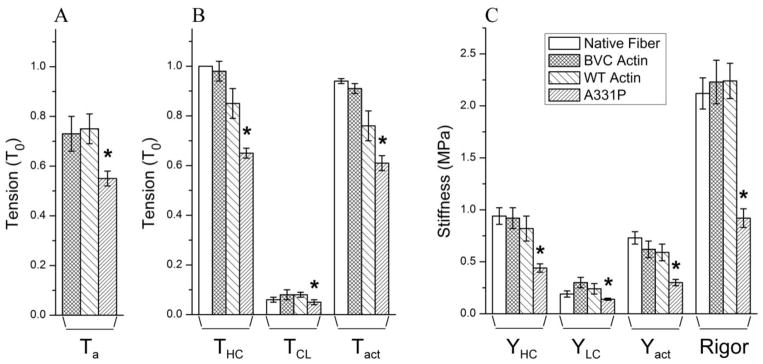
(A) Active isometric tension of the actin-filament reconstituted cardiac muscle fibers in the absence of Tm/Tn (Ta). (B) Isometric tension of the reconstituted fibers in the presence of Tm/Tn. In (A) and (B), tension was measured and normalized to T0 (initial tension of native fibers before extraction, T0=26.9±1.1 KPa, N=41). THC (high Ca2+ tension at pCa 4.66) and TLC (low Ca2+ tension at pCa 8) were measured from the absolute baseline. Tact=THC−TLC. (C) Stiffness (Y) of the reconstituted fibers in the presence of Tm/Tn. Subscripts HC, LC, and act corresponds to those of tension. Rigor was induced from fully reconstituted and Ca2+ activated fibers by washing out ATP, but YLC was not subtracted. Other than the absolute baseline, all experiments were performed at 25°C. *Significantly different from WT actin (p<0.05).
Our previous investigations have demonstrated that there are some actively cycling cross-bridges during relaxation (at pCa 8.0) in cardiac muscle fibers; their number increases with HCM mutants of Tm [28], and decreases with DCM mutants of Tm [29]. Therefore, tension (TLC) and stiffness (YLC) at pCa 8.0 were measured to evaluate the effect of recombinant actins. WT actin reconstituted fibers showed the same TLC (0.08±0.01 T0, N=11) as that of the BVC actin reconstituted fibers (0.08±0.02 T0, N=12) and native fibers (0.06±0.01 T0, N=10). A331P actin reconstituted fibers showed significantly reduced TLC (0.05±0.01 T0, N=13) compared to that of WT (Fig. 3B). The same trend was observed with YLC (Fig. 3C). Tact was defined as THC−TLC, which is Ca2+ activatable tension. A331P caused a significant decrease in Tact compared to that of WT. The relaxed stiffness (YLC) remained unchanged among native fibers, BVC actin, and WT actin reconstituted fibers (Fig. 3C). A331P actin reconstituted fibers showed significantly decreased YLC compared to that of WT (Fig. 3C).
3.4. pCa-tension study
To determine the effect of recombinant actins (WT and A331P) on Ca2+-sensitivity (pCa50) and cooperativity (nH), tension of the thin filament-reconstituted fibers was studied as a function of [Ca2+]. pCa-tension plots comparing native fibers, those reconstituted with BVC actin, WT actin, and A331P actin are shown in Fig. 4. The curves in this Fig. 4 are the results of fitting the data to Eq. S1 in Supporting Materials. Native fibers, BVC actin reconstituted fibers, and WT actin reconstituted fibers exhibited almost the same pCa-tension relationship as shown in Fig. 4A. The Ca2+ sensitivity of fibers reconstituted with WT actin (5.69±0.02, N=11) was exactly the same as that of the native fibers (5.69±0.01, N=10) and BVC actin reconstituted fibers (5.68±0.02, N=12) (Fig. 4C). Cooperativity (nH) was also calculated by averaging the nH of individual curves (Fig. 4D). Cooperativity of the WT actin (2.6±0.3, N=11) and BVC actin (2.7±0.2, N=12) were slightly less than that of the native fibers (3.2±0.2, N=10), but without significance. The HCM causing mutant A331P shifted the pCa-tension curve towards the right and decreased pCa50 (5.57±0.03, N=13) compared to that of WT actin (Fig. 4B and C). Cooperativity of A331P (2.3±0.3, N=13) remained unchanged compared to that of WT actin (2.6±0.3, N=11) (Fig. 4D).
Figure 4. pCa-tension study comparing native fibers, fibers reconstituted with three forms of actin, followed by reconstitution of Tm/Tn.
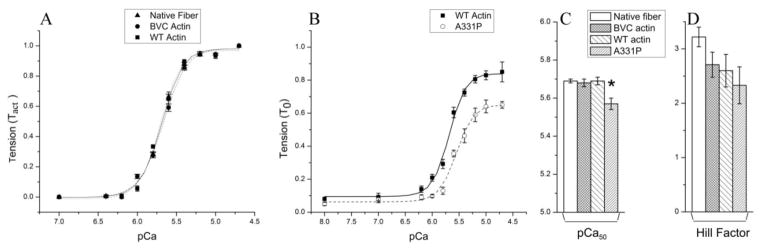
(A) pCa-tension plots comparing native fibers (▲, N=10), and fibers reconstituted with BVC actin (●, N=12) and WT actin (■, N=11). TLC was subtracted from tension and normalized against Tact; (B) pCa-tension plots comparing fibers reconstituted with A331P actin (○, N=13) and WT actin (■, N=11). In (A) and (B), symbols represent the mean ± SE, and continuous curves are the results of fitting the averaged data to Eq. 1 of Ref. [28]. Tension was normalized against T0 (initial tension of native fibers before thin-filament extraction). (C) Results of Ca2+ sensitivity (pCa50), and (D) cooperativity (nH) are compared for native fibers, BVC actin reconstituted fibers, WT actin reconstituted fibers, and A331P actin reconstituted fibers. The value of the pCa50 and nH were calculated by averaging the fitted parameters of each curve. *Significantly different from WT actin reconstituted fibers (p<0.05).
3.5. Sinusoidal analysis and cross-bridge kinetics in the presence of Tm/Tn
Sinusoidal analysis was performed to characterize the cross-bridge kinetics and the elementary steps of the cross-bridge cycle [28, 34, 35]. Fig. 5 shows the complex modulus data of native fibers, and reconstituted fibers with BVC actin, WT actin, and A331P actin, followed by the reconstitution with Tm/Tn, and the data were collected in the standard activating solution at 25°C. Also plotted are the baseline records collected at 0°C in the relaxing solution that contained 40 mM BDM. Two apparent rate constants (2πb and 2πc) and their magnitudes (B and C) were deduced from the complex modulus data and shown in Fig. 6. There were no significant differences in either 2πb or 2πc among native fibers, BVC actin reconstituted fibers, and WT actin reconstituted fibers (Fig. 6A). A331P caused a significantly slower 2πc compared to that of the WT (Fig. 6A). The magnitude B was similar among native fibers, and BVC, WT, and A331P actin reconstituted fibers (Fig. 6B). The magnitude C decreased in A331P, but without significance (Fig. 6B). 2πb and 2πc were studied as functions of [MgATP] and [Pi], and the results are plotted in Fig. 7. It can be seen that these rate constants from native fibers, BVC actin reconstituted fibers, and WT actin reconstituted fibers showed almost the same dependence on [ATP] and [Pi], and the fitted curves overlapped with each other (Fig. 7 A and C). A331P showed small changes from WT, but these changes were not significant (Fig. 7B and D).
Figure 5. The complex modulus data of native fibers, BVC actin reconstituted fibers, WT actin reconstituted fibers, and A331P actin reconstituted fibers, followed by reconstitution of Tm/Tn.

The complex modulus data were measured in the standard activation solution (5S8P) at pCa 4.66 and 25°C. (A)–(B): Dynamic modulus; (C)–(D): Phase shift; (E)–(F): Nyquist plots. In (A)–(F), continuous curves are the results of fitting the data to Eq. 2 of Ref. [28] based on two exponential processes. Baseline records were taken in the relaxing solution containing 40 mM BDM without added Ca2+ at 0°C. ▲: Native fibers; ●: BVC actin reconstituted fibers; ■: WT actin reconstituted fibers; ○: A331P actin reconstituted fibers. Continuous curves represent theoretical projections.
Figure 6. (A) The apparent rate constants 2πa and 2πb, and (B) the magnitude parameters of exponential processes B and C under the standard activating conditions.

These data were deduced by fitting the complex modulus data to Eq. 2 in Ref [28]. Error bars represent standard errors (SE). *Significantly different from WT actin reconstituted fibers (p<0.05).
Figure 7. Apparent rate constants of native fibers, BVC actin reconstituted fibers, WT actin reconstituted fibers, and A331P actin reconstituted fibers followed by reconstitution with Tm/Tn.

Two apparent rate constants, 2πb and 2πc, are plotted against [Pi] and [MgATP]. Symbols represent the mean ± SE. Continuous curves were generated by fitting the data to Eq. 3 (A–B) or Eq. 4 (C–D) of ref [28]. A–B: 2πb is plotted against [Pi]. C–D: 2πc is plotted against [MgATP]. (▲: Native fibers; ●: BVC actin reconstituted fibers; ■: WT actin reconstituted fibers; ○: A331P actin reconstituted fibers; N=7–10).
The rate and association constants (collectively referred to as “kinetic constants”) of the elementary steps as defined in Scheme S1 in Supporting Materials were deduced by fitting the ligand concentration dependences ([Pi], [MgATP], and [MgADP]) on the apparent rate constants [34] and plotted in Fig. 8. Compared to those of the native fibers, both BVC and WT actin reconstituted fibers exhibited significant increases (p<0.05) in K0 and K1 (See Table 1 for details). Compared with those of the BVC actin reconstituted fibers, WT actin showed no significant difference in the kinetic constants, except for ~50% decrease in K0 and a ~55% decrease in k2 (p<0.05). Compared to those of the WT actin, A331P showed no significant difference in the equilibrium constants, except a ~70% increase in k4 (p<0.05).
Figure 8. Kinetic constants of the native fibers, and reconstituted fibers with BVC actin, WT actin, and A331P actin followed by reconstitution with Tm/Tn.
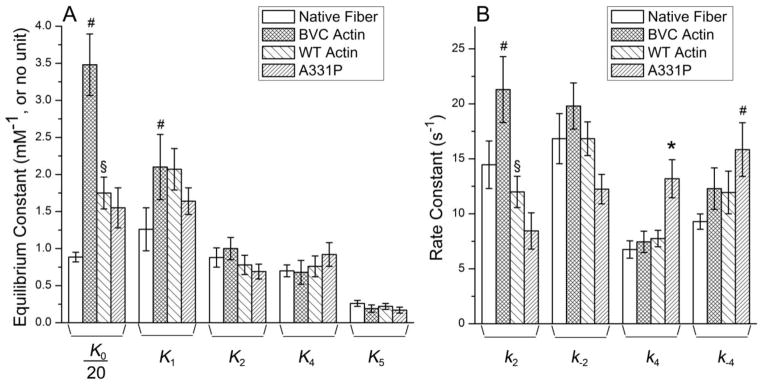
The kinetic constants of the cross-bridge cycle (Scheme 1) are compared among native fibers, WT and recombinant actin reconstituted fibers. (A) Equilibrium constants. Note that K0 was divided by 20. (B) Rate constants. *Significantly different from WT actin reconstituted fibers (p<0.05); #Significantly different from native fibers (p<0.05); §Significantly different from BVC actin reconstituted fibers (p<0.05).
3.6. Cross-bridge distribution and force per cross-bridge in the presence of Tm/Tn
Actin directly interacts with myosin and is one of the key molecules for force generation. Mutations in actin, especially those at residues directly involved in the AM interface, are likely to affect AM binding. Altered AM binding may either change the number of force generating cross-bridges, or the force generated by each cross-bridge. To discriminate between these possibilities, we calculated the distribution of cross-bridges in each state (Fig. 9) under the standard activating conditions (5 mM MgATP, 8 mM Pi, 0.02 mM MgADP) and as described [37]. This requires mainly equilibrium constants shown in Fig. 7A [38]. The distributions of cross-bridges were just about the same among all preparations tested, except that the AM state (Scheme S1 in Supplementary Material) of native fibers was larger than those of exogenous actin reconstituted fibers. Att indicates the sum of all strongly attached (force generating) cross-bridges: Att=AMD+AM+AM*S+AM*DP+AM*D. The percentage of the Att state was similar among all preparations (native fibers: 73±1.9%; BVC actin reconstituted fibers: 76±1.7%; WT actin reconstituted fibers: 75±1.6%; A331P actin reconstituted fibers: 77±1.5%), indicating the number of force generating cross-bridges was not changed significantly by the reconstitution process (BVC actin reconstituted fibers vs. native fibers), recombinant actin (WT vs. BCV), or the mutation (A331P vs. WT). Consequently, it can be concluded that in the presence of Tm and Tn, A331P decreased tension per cross-bridge (=THC/Xatt) by 25±7% (± with error propagation) compared to that of WT actin. The simultaneous decrease in stiffness during standard activation and during rigor induction (Fig. 3C) also indicates that the cross-bridge force was weakened with the mutant actin A331P.
Figure 9. Calculated cross-bridge distributions based on equilibrium constants in Fig. 8A at the standard activating condition.

([Pi]=8 mM, [MgATP]=5 mM, [MgADP]=0.02 mM). All the cross-bridge states are defined in Scheme S1 in Supporting Materials, where A=actin, M=myosin, S=ATP, D=ADP, and P=phosphate. Det indicates the sum of weakly attached states (AMS, AMDP) and detached states (MS, MDP). Att indicates the sum of all strongly attached (force generating) cross-bridges: Att=AMD+AM+AM*S+AM*DP+AM*D. #Significantly different from native fibers (p<0.05). The distribution was calculated on each fiber, and averaged among the fibers.
4. Discussion
4.1. Using baculovirus/insect cell expressed actin for thin-filament reconstitution
G-actin generated by the baculovirus/Sf-21 cell expression system polymerizes to F-actin [14] (see also Fig. 2B) and interacts normally with myosin in vitro [19]. Before using the recombinant actin for thin-filament reconstitution and study the effect of actin mutation on real muscle fibers, we have to demonstrate that recombinant WT actin works in the same way as does purified BVC actin, and that the reconstituted fibers (together with Tm/Tn) possess the same contractile and regulatory properties as those of native fibers. As we have shown, recombinant WT actin was fully functional in a real muscle fiber system, and that the WT actin reconstituted fibers showed no major differences in both contraction and regulation when compared with both native fibers and BVC actin reconstituted fibers (Table 1, Figs. 3–9).
One HCM causing actin mutant, A331P, was also synthesized to study the mutation’s effect on contraction and regulation. Because A331P lies in a polymerization interface that leads in one direction to the nucleotide binding cleft, and in the other direction to the barbed end of the monomer (Fig. 1), it is necessary to assess its polymerization kinetics to examine any defects. In the light scattering assay, the increase of signal strength indicates the formation of F-actin. As shown in Fig. 2B, the signal strength of A331P and WT was almost the same when the polymerization curve reached the plateau in 2800 sec, indicating that A331P polymerized to the same extent as WT actin did. This result demonstrates that A331P actin polymerized to form F-actin, as WT and BVC actins did when human cardiac actin sequence was used. This result is at variance from that using yeast actin containing A331P mutation, in which a serious polymerization defect was reported [39]. Apparently, the difference must reside in the difference in the sequence between yeast actin and cardiac actin.
Reconstitution of the thin filament by using insect actin demonstrates that insect actin behaved very differently from purified BVC actin and recombinant WT actin, as described in section 3.2 and Table 1. The results of BVC actin and WT actin as shown in Figs. 3–7 are not significantly different, whereas active tension was significantly decreased with insect actin, indicating that the effect of contaminating insect actin (if any) in the WT actin preparation is negligible in our thin filament reconstituted fibers. Similarly, the kinetic analysis shows that many of the kinetic constants (K2, k−4, K4, and K5 in particular) are very different from WT actin reconstituted fibers, whereas they are similar among BVC actin, WT actin, and A331P actin reconstituted fibers. Furthermore, K5 (Pi association constant) is largest in insect actin (1.06±0.34), smallest in A331P actin (0.17±0.04), and in between in BVC actin (0.19±0.05) and WT actin (0.22±0.04), indicating that the data on A331P actin is not a combined effect of insect actin and BVC (or WT) actin. These results demonstrate that the contaminating insect actin in the recombinant actin preparations is negligible; Trybus and her colleagues used a specific monoclonal antibody for β-cytoplasmic actin to detect the insect actin and showed there was only a small carryover of the insect actin [19]. Consequently, we conclude that the effects observed with A331P actin reconstituted fibers (lower Ta, Tact, THC, Yact, and YHC) are the true effects of A331P actin, and not those of insect actin. All of these results demonstrate that contaminating insect actin (if any) does not interfere with our functional studies.
4.2. A331P leads to HCM phenotype through decreasing the contractility of the affected myocardium
A331P is located within a highly conserved loop region of actin, which contains both hydrophobic and ionic residues (326–336: KIKIIAPPERK) [40]. Within this region, a yeast actin mutant P332A (next to A331P, but carrying the reverse mutation seen with A331P) was shown to increase filament stability without changing the polymerization kinetics [41, 42]. P332A in γ-non-muscle actin was shown to cause early onset of deafness in humans [43]. In the absence of Ca2+ in the blocked-state, this region was shown to be critical for actin-Tm binding [44]. In F-actin, two clusters of charged residues (K326, K328 and R147 as well as D25, R28 and E334) located in subdomain 1 were shown to directly interact with Tm [45]. Point mutations, K326A, K328A, and E334A were individually shown to eliminate actin’s ability to bind to Tm in vitro [44], which further provided evidence that the binding force between actin and Tm is primarily electrostatic [46]. Actin residues D25, R28, and P333 also form a bulge which defines the azimuthal edge of blocked-state Tm on actin, and P333 was shown to be at the high radius of the bulge [47]. During Ca2+ activation process, Tm has to slide over P333 in order to reach the closed-state position [47]. It is very likely that the A331P mutation may change the surface topology of this region by introducing an extra kink. P333 may be pushed even further away from the actin surface in order to accommodate the extra kink introduced by the A331P mutation, which may lead to a higher bulge on the actin surface and make it more difficult for Tm to move from the blocked-state position to the closed-state position. The topology change caused by the A331P mutation may also shorten the distance between actin and Tm. The strength of the electrostatic interaction between two charged residues is determined by their distance. If A331P mutation shortens the distance between actin and Tm, it could cause an increased actin-Tm interaction, and lead to an over-inhibition of Tm on the AM interaction. The over inhibition will result in decreased Ca2+ sensitivity and decreased relaxed tension (TLC) as we have observed (Figs. 3B and 4, Table 1). In the strongly bound state, this region of actin is known to be a part of the actin-Tm-myosin interface [40], which directly interacts with myosin segment R405-K415 through hydrophobic interaction, and at the same time, maintains electrostatic interaction with both Tm and myosin [40, 48]. During the strongly bound state, actin residues D311, K315, K328 and E334 (all of which surround A331) are known to be in close proximity to E286 and R288 of myosin, and R178, E181 and R182 of Tm [40]. If the A331P mutation favors the actin-Tm binding, it may well shift the loop region further away from myosin and lead to a decreased AM interaction, which could result in a decreased force/cross-bridge in A331P actin reconstituted fibers, as we have observed.
Based on a limited patient study, A331P was reported to be an HCM-causing mutation [3]. However, the functional consequences A331P has on reconstituted fibers appeared to be very similar with those generally found in DCM causing mutations. There is a generally held view that the Ca2+ sensitivity change determines the disease phenotype: increased Ca2+ sensitivity and/or contractility in the myocardium results in HCM, and decreased Ca2+ sensitivity and/or contractility results in DCM [49–51]. Our finding on A331P is at variance to this view. There may be three possibilities to account for this difference:
A331P may not be an HCM causing mutation. There has been no large cohort of patients or transgenic models to firmly prove that A331P leads to HCM. Therefore, it is possible that A331P was actually a DCM-causing mutation, or not a disease causing mutation at all. The HCM phenotype of the A331P mutation-bearing patient may have been caused by other reasons, such as a mutation in another sarcomeric gene(s). However, this possibility is unlikely for the following reasons. First, the symptom of the proband was typical of HCM with a solid diagnosis [3], and there has been no report of a DCM patient manifesting an HCM symptom. Second, A331P caused a substantial decrease in Ca2+ sensitivity and contractility (~25%) in the reconstituted fibers (Figs. 3, 4B and 4C). It would be very unlikely that there exist some other causes that could reverse these effects to result in an increase in Ca2+ sensitivity and hypercontractility. It has been reported that 3–5% of HCM patients may carry double or compound mutations, either in the same gene or different genes [52]. But these compound mutations appear to cause more severe phenotypes, and do not correct to each other [52].
A331P is an HCM-causing exception to the above-mentioned hypothesis on Ca2+ sensitivity, joining a group of exceptions. Our previous studies showed that an HCM causing Tm mutation D175N, DCM causing Tm mutations E40K and E54K, and DCM causing TnT mutation ΔK210 did not change the Ca2+ sensitivity of the reconstituted fibers [27–30]. Similarly, two HCM causing Tm mutations, V95A and D175N, have been shown to cause a decreased contractility (Tact) in the reconstituted fibers [28]. Independent studies from other labs using different approaches also reported exceptions to the above-mentioned view [53–55]. Our current study demonstrates that in the thin-filament reconstituted fibers, A331P caused significant decreases in Ca2+ sensitivity and contractility (both THC and Tact, ~25% decrease) compared to those of WT actin (Fig. 3 and 4), which further adds to the existing list of exceptions. We believe this second possibility is very likely to be the case, because theoretically it is possible that decreased Ca2+ sensitivity and contractility could lead to HCM. It has long been proposed that HCM phenotype could be triggered by the decreased contractility as a compensatory mechanism [56]. The most convincing evidence for compensatory hypertrophy comes from the fact that the cardiac hypertrophy in HCM patients is restricted to the left ventricle and interventricular septum, despite the ubiquitous expression of the mutant protein in both ventricles. Studies of human HCM patients also showed that decreased contractility may cause pressure overload on the myocardium, thereby stimulating the expression of stress responsive trophic factors in cardiomyocytes to lead into the compensatory hypertrophy [57–59]. The increase of the myocardium mass in the left ventricle may in turn help compensate for the loss of cardiac contractility in HCM patients. Decreased force generation (either THC or Tact) has been observed in many DCM causing mutations. The reason why decreased contractility doesn’t stimulate hypertrophy in DCM patients remains unknown. It has been shown that insulin-like growth factor-1 (IGF-1), which is associated with cardiac hypertrophy in HCM patients, relieves DCM symptoms in animal models [60, 61].
The Ca2+ sensitivity change may not be the absolute measure to distinguish HCM from DCM, as shown in our recent works [28, 29] as well as others [53–55]. Previous experiments that showed the characteristic Ca2+ sensitivity shift [62, 63] was generally carried out in 0 mM Pi and low ionic strength solutions (~150 mM), but the shift disappeared when the experiments were carried out in physiological ionic strength solutions (~200 mM) and Pi concentrations (8 mM) [29]. Thus, the Ca2+ sensitivity change observed under non-physiological conditions (0 mM Pi and 150 mM IS) may not occur under the physiological conditions (~ 6 mM Pi and ~ 215 mM IS) [64–66]. The levels of phosphorylation change of TnI and cMyBP-C are also known to cause the shift [67, 68], but the phosphorylation level change in transgenic mice may be secondary to the HCM or DCM development.
4.3. A331P decreases the cross-bridge force, but doesn’t change the cross-bridge kinetics
A six-state cross-bridge model (Scheme S1 in Supporting Materials) [38] was used to characterize the elementary steps of the cross-bridge cycle [69]. Unlike previously characterized Tm and TnT mutants, A331P doesn’t cause any significant changes in most of the kinetic constants compared to WT (Fig. 8). There were no significant changes in ADP dissociation, ATP binding, force generation, and Pi release steps. One exception was an increase in k4 (Fig. 8B), but this was compensated by an increase in k-4, giving rise to an unchanged equilibrium constant K4 (Fig. 8A). The equilibrium constants approximately determine the population of cross-bridges in each state, and thereby determine isometric tension [38]. Unaltered equilibrium constants give rise to unaltered number of cross-bridges in the strongly attached states (Fig. 9). Because Tact of the mutant A331P was about 2/3 of that of WT, it can be concluded that the cross-bridge force is also reduced to about 2/3 with the actin mutant A331P. This is most likely caused by the weakened AM interaction by the mutation, which is also consistent with the reduced active and rigor stiffness (Fig. 3C) in the presence of regulatory proteins, and reduced active tension (Ta) in the absence of regulatory proteins (Fig. 3A). Another possibility is the weakened actin-actin interaction results in the increased series compliance, hence less tension and stiffness. Because the step size is fixed by the geometry of the cross-bridges, the unitary force becomes less with increased series compliance. An argument against this possibility is that the light scattering kinetics (Fig. 2B) was faster, but normal otherwise with A331P actin compared to WT actin. However, the light scattering study was performed under the unloaded condition, and may not represent the isometric condition.
The effect of the A331P mutation appears small and localized, and it does not induce any major structural changes in either actin or myosin. Tm-actin binding in the absence of Ca2+ appears to be affected by A331P mutation and leads to a decreased TLC (Fig. 3B) and Ca2+ sensitivity (Fig. 4C). In the presence of Ca2+, Tm may uncover a major AM binding region that includes A331P, and shift actin to a position to interact with myosin. Therefore, the channel for ATP entry, ADP dissociation, or Pi release in the actin-myosin complex is not blocked by the mutated actin molecule, and every step of the cross-bridge cycle appears to happen normally.
It may be argued that the above conclusions are model dependent, because we used the six state cross-bridge model shown in Scheme S1 (Supporting Materials). However, this is not the case in the current analysis, because there are no big changes in equilibrium constants, and major changes with A331P are the active tension (Tact) and stiffness (Yact). These results can be used to arrive at the same conclusion that force/cross-bridge is reduced with the actin mutation. The cross-bridge model similar to ours has been used by many investigators for more than 30 years [70, 71], and the results based on our model [34] generally agree with those obtained using caged ATP [72], caged Pi [73], pressure-release experiments [74], and with the step that generate force [34, 73, 74].
5. Conclusion
The baculovirus/insect cell expressed actin was fully functional and can be used for thin-filament reconstitution studies. Reconstituted fibers using WT actin possessed the same contractile and regulatory properties as those of the native fibers and BVC actin reconstituted fibers. HCM causing actin mutation A331P significantly decreased the contractility and the Ca2+ sensitivity of the reconstituted fibers. The HCM phenotype in ACTC A331P bearing patients may be a compensatory effect for the decreased contractility.
Supplementary Material
Sf-21 cell expressed actins were polymerized in myocardium to form the thin filament
WT actin reconstituted myocardium behaved exactly the same as the native myocardium
A331P decreased the tension and pCa50 of reconstituted fibers compared to WT actin
Cross-bridge kinetics of reconstituted fibers were similar between A331P and WT actin
Diminished contractility with A331P actin must cause the characteristic HCM phenotype
Acknowledgments
We thank Drs. Kuo-Kuang Wen and Sarah E. Bergeron in the University of Iowa, Dept. of Biochemistry, for providing technique help on recombinant actin synthesis and characterization. This work was supported by grants from NIH HL70041 (MK), AHA 13GRNT16810043 (MK) and HSFC NA 6469 (JFD). No additional external funding was received for this study.
Footnotes
Disclosures
None
The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of funding organizations, which had no role in design, data collection and analyses, preparation of the manuscript, or decision to publish.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Fan Bai, Email: Fan-Bai@uiowa.edu.
Hannah M. Caster, Email: casterha@gmail.com, Hannah-Caster@uiowa.edu.
Peter A. Rubenstein, Email: Peter-Rubenstein@uiowa.edu.
John F. Dawson, Email: jdawso01@uoguelph.ca.
Masataka Kawai, Email: Masataka-Kawai@uiowa.edu.
References
- 1.Vandekerckhove J, Bugaisky G, Buckingham M. Simultaneous expression of skeletal muscle and heart actin proteins in various striated muscle tissues and cells. A quantitative determination of the two actin isoforms. J Biol Chem. 1986;261:1838–43. [PubMed] [Google Scholar]
- 2.Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet. 2004;363:1881–91. doi: 10.1016/S0140-6736(04)16358-7. [DOI] [PubMed] [Google Scholar]
- 3.Olson TM, Doan TP, Kishimoto NY, Whitby FG, Ackerman MJ, Fananapazir L. Inherited and de novo mutations in the cardiac actin gene cause hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2000;32:1687–94. doi: 10.1006/jmcc.2000.1204. [DOI] [PubMed] [Google Scholar]
- 4.Mogensen J, Perrot A, Andersen PS, Havndrup O, Klausen IC, Christiansen M, et al. Clinical and genetic characteristics of alpha cardiac actin gene mutations in hypertrophic cardiomyopathy. J Med Genet. 2004;41:e10. doi: 10.1136/jmg.2003.010447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Driest SL, Ellsworth EG, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation. 2003;108:445–51. doi: 10.1161/01.CIR.0000080896.52003.DF. [DOI] [PubMed] [Google Scholar]
- 6.Mogensen J, Klausen IC, Pedersen AK, Egeblad H, Bross P, Kruse TA, et al. Alpha-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J Clin Invest. 1999;103:R39–43. doi: 10.1172/JCI6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, et al. Shared genetic causes of cardiac hypertrophy in children and adults. The New England journal of medicine. 2008;358:1899–908. doi: 10.1056/NEJMoa075463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monserrat L, Hermida-Prieto M, Fernandez X, Rodriguez I, Dumont C, Cazon L, et al. Mutation in the alpha-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non-compaction, and septal defects. European heart journal. 2007;28:1953–61. doi: 10.1093/eurheartj/ehm239. [DOI] [PubMed] [Google Scholar]
- 9.Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, et al. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clinic proceedings Mayo Clinic. 2008;83:630–8. doi: 10.4065/83.6.630. [DOI] [PubMed] [Google Scholar]
- 10.Kaski JP, Syrris P, Esteban MT, Jenkins S, Pantazis A, Deanfield JE, et al. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circulation Cardiovascular genetics. 2009;2:436–41. doi: 10.1161/CIRCGENETICS.108.821314. [DOI] [PubMed] [Google Scholar]
- 11.Olson TM, Michels VV, Thibodeau SN, Tai YS, Keating MT. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science. 1998;280:750–2. doi: 10.1126/science.280.5364.750. [DOI] [PubMed] [Google Scholar]
- 12.Frankel S, Condeelis J, Leinwand L. Expression of Actin in Escherichia-Coli - Aggregation, Solubilization, and Functional-Analysis. Journal of Biological Chemistry. 1990;265:17980–7. [PubMed] [Google Scholar]
- 13.Vang S, Corydon TJ, Borglum AD, Scott MD, Frydman J, Mogensen J, et al. Actin mutations in hypertrophic and dilated cardiomyopathy cause inefficient protein folding and perturbed filament formation. Febs Journal. 2005;272:2037–49. doi: 10.1111/j.1742-4658.2005.04630.x. [DOI] [PubMed] [Google Scholar]
- 14.Rutkevich LA, Teal DJ, Dawson JF. Expression of actin mutants to study their roles in cardiomyopathy. Canadian Journal of Physiology and Pharmacology. 2006;84:111–9. doi: 10.1139/Y05-140. [DOI] [PubMed] [Google Scholar]
- 15.Muller M, Mazur AJ, Behrmann E, Diensthuber RP, Radke MB, Qu Z, et al. Functional characterization of the human alpha-cardiac actin mutations Y166C and M305L involved in hypertrophic cardiomyopathy. Cellular and molecular life sciences : CMLS. 2012;69:3457–79. doi: 10.1007/s00018-012-1030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergeron SE, Zhu M, Thiem SM, Friderici KH, Rubenstein PA. Ion-dependent Polymerization Differences between Mammalian beta- and gamma-Nonmuscle Actin Isoforms. Journal of Biological Chemistry. 2010;285:16087–95. doi: 10.1074/jbc.M110.110130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yates SP, Otley MD, Dawson JF. Overexpression of cardiac actin with baculovirus is promoter dependent. Arch Biochem Biophys. 2007;466:58–65. doi: 10.1016/j.abb.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 18.Anthony Akkari P, Nowak KJ, Beckman K, Walker KR, Schachat F, Laing NG. Production of human skeletal alpha-actin proteins by the baculovirus expression system. Biochem Biophys Res Commun. 2003;307:74–9. doi: 10.1016/s0006-291x(03)01133-1. [DOI] [PubMed] [Google Scholar]
- 19.Bookwalter CS, Trybus KM. Functional consequences of a mutation in an expressed human alpha-cardiac actin at a site implicated in familial hypertrophic cardiomyopathy. J Biol Chem. 2006;281:16777–84. doi: 10.1074/jbc.M512935200. [DOI] [PubMed] [Google Scholar]
- 20.Mundia MM, Demers RW, Chow ML, Perieteanu AA, Dawson JF. Subdomain location of mutations in cardiac actin correlate with type of functional change. PLoS One. 2012;7:e36821. doi: 10.1371/journal.pone.0036821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Debold EP, Saber W, Cheema Y, Bookwalter CS, Trybus KM, Warshaw DM, et al. Human actin mutations associated with hypertrophic and dilated cardiomyopathies demonstrate distinct thin filament regulatory properties in vitro. J Mol Cell Cardiol. 2010;48:286–92. doi: 10.1016/j.yjmcc.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song W, Dyer E, Stuckey D, Leung MC, Memo M, Mansfield C, et al. Investigation of a transgenic mouse model of familial dilated cardiomyopathy. J Mol Cell Cardiol. 2010;49:380–9. doi: 10.1016/j.yjmcc.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 23.Song W, Dyer E, Stuckey DJ, Copeland O, Leung MC, Bayliss C, et al. Molecular mechanism of the E99K mutation in cardiac actin (ACTC gene) that causes apical hypertrophy in man and mouse. J Biol Chem. 2011 doi: 10.1074/jbc.M111.252320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song W, Vikhorev PG, Kashyap MN, Rowlands C, Ferenczi MA, Woledge RC, et al. Mechanical and energetic properties of papillary muscle from ACTC E99K transgenic mouse models of hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2013;304:H1513–24. doi: 10.1152/ajpheart.00951.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vikhorev PG, Wilkinson R, Song WH, Copeland O, Marston SB, Ferenczi MA. DCM-Causing Mutation E361G in Actin Slows Myofibril Relaxation Kinetics and Uncouples Myofibril Ca2+ Sensitivity from Protein Phosphorylation. Biophysical Journal. 2013;104:312a-a. [Google Scholar]
- 26.Kawai M, Ishiwata S. Use of thin filament reconstituted muscle fibres to probe the mechanism of force generation. J Muscle Res Cell Motil. 2006;27:455–68. doi: 10.1007/s10974-006-9075-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bai F, Wang L, Kawai M. A study of tropomyosin’s role in cardiac function and disease using thin-filament reconstituted myocardium. J Muscle Res Cell Motil. 2013;34:295–310. doi: 10.1007/s10974-013-9343-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bai F, Weis A, Takeda AK, Chase PB, Kawai M. Enhanced active cross-bridges during diastole: molecular pathogenesis of tropomyosin’s HCM mutations. Biophys J. 2011;100:1014–23. doi: 10.1016/j.bpj.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bai F, Groth HL, Kawai M. DCM-Related Tropomyosin Mutants E40K/E54K Over-Inhibit the Actomyosin Interaction and Lead to a Decrease in the Number of Cycling Cross-Bridges. PloS one. 2012;7:e47471. doi: 10.1371/journal.pone.0047471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bai F, Caster HM, Pinto JR, Kawai M. Analysis of the Molecular Pathogenesis of Cardiomyopathy-Causing cTnT Mutants I79N, ΔE96, and ΔK210. Biophys J. 2013;104:1979–88. doi: 10.1016/j.bpj.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujita H, Yasuda K, Niitsu S, Funatsu T, Ishiwata S. Structural and functional reconstitution of thin filaments in the contractile apparatus of cardiac muscle. Biophys J. 1996;71:2307–18. doi: 10.1016/S0006-3495(96)79465-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holroyde MJ, Robertson SP, Johnson JD, Solaro RJ, Potter JD. The calcium and magnesium binding sites on cardiac troponin and their role in the regulation of myofibrillar adenosine triphosphatase. J Biol Chem. 1980;255:11688–93. [PubMed] [Google Scholar]
- 33.Fujita H, Lu X, Suzuki M, Ishiwata S, Kawai M. The effect of tropomyosin on force and elementary steps of the cross-bridge cycle in reconstituted bovine myocardium. J Physiol. 2004;556:637–49. doi: 10.1113/jphysiol.2003.059956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawai M, Halvorson HR. Two step mechanism of phosphate release and the mechanism of force generation in chemically skinned fibers of rabbit psoas. Biophys J. 1991;59:329–42. doi: 10.1016/S0006-3495(91)82227-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujita H, Sasaki D, Ishiwata S, Kawai M. Elementary steps of the cross-bridge cycle in bovine myocardium with and without regulatory proteins. Biophy J. 2002;82:915–28. doi: 10.1016/S0006-3495(02)75453-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawai M, Brandt PW. Sinusoidal analysis: a high resolution method for correlating biochemical reactions with physiological processes in activated skeletal muscles of rabbit, frog and crayfish. J Muscle Res Cell Motil. 1980;1:279–303. doi: 10.1007/BF00711932. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Y, Swamy PM, Humphries KA, Kawai M. The effect of partial extraction of troponin C on the elementary steps of the cross-bridge cycle in rabbit psoas muscle fibers. Biophys J. 1996;71:2759–73. doi: 10.1016/S0006-3495(96)79469-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawai M, Saeki Y, Zhao Y. Crossbridge scheme and the kinetic constants of elementary steps deduced from chemically skinned papillary and trabecular muscles of the ferret. Circ Res. 1993;73:35–50. doi: 10.1161/01.res.73.1.35. [DOI] [PubMed] [Google Scholar]
- 39.Wong WW, Doyle TC, Cheung P, Olson TM, Reisler E. Functional studies of yeast actin mutants corresponding to human cardiomyopathy mutations. J Muscle Res Cell Motil. 2001;22:665–74. doi: 10.1023/a:1016354308436. [DOI] [PubMed] [Google Scholar]
- 40.Behrmann E, Muller M, Penczek PA, Mannherz HG, Manstein DJ, Raunser S. Structure of the rigor actin-tropomyosin-myosin complex. Cell. 2012;150:327–38. doi: 10.1016/j.cell.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bryan KE, Wen KK, Zhu M, Rendtorff ND, Feldkamp M, Tranebjaerg L, et al. Effects of human deafness gamma-actin mutations (DFNA20/26) on actin function. J Biol Chem. 2006;281:20129–39. doi: 10.1074/jbc.M601514200. [DOI] [PubMed] [Google Scholar]
- 42.Bryan KE, Rubenstein PA. Allele-specific effects of human deafness gamma-actin mutations (DFNA20/26) on the actin/cofilin interaction. J Biol Chem. 2009;284:18260–9. doi: 10.1074/jbc.M109.015818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu M, Yang T, Wei S, DeWan AT, Morell RJ, Elfenbein JL, et al. Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26) American journal of human genetics. 2003;73:1082–91. doi: 10.1086/379286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barua B, Fagnant PM, Winkelmann DA, Trybus KM, Hitchcock-DeGregori SE. A periodic pattern of evolutionarily conserved basic and acidic residues constitutes the binding interface of actin-tropomyosin. J Biol Chem. 2013;288:9602–9. doi: 10.1074/jbc.M113.451161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li XE, Tobacman LS, Mun JY, Craig R, Fischer S, Lehman W. Tropomyosin position on F-actin revealed by EM reconstruction and computational chemistry. Biophys J. 2011;100:1005–13. doi: 10.1016/j.bpj.2010.12.3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown JH, Zhou ZC, Reshetnikova L, Robinson H, Yammani RD, Tobacman LS, et al. Structure of the mid-region of tropomyosin: Bending and binding sites for actin. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:18878–83. doi: 10.1073/pnas.0509269102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lehman W, Orzechowski M, Li XE, Fischer S, Raunser S. Gestalt-binding of tropomyosin on actin during thin filament activation. J Muscle Res Cell Motil. 2013;34:155–63. doi: 10.1007/s10974-013-9342-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, Holmes KC, et al. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 1993;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- 49.Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, Potter JD. Mutations in Troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function? J Mol Cell Cardiol. 2010;48:882–92. doi: 10.1016/j.yjmcc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 50.Dweck D, Hus N, Potter JD. Challenging current paradigms related to cardiomyopathies. Are changes in the Ca2+ sensitivity of myofilaments containing cardiac troponin C mutations (G159D and L29Q) good predictors of the phenotypic outcomes? J Biol Chem. 2008;283:33119–28. doi: 10.1074/jbc.M804070200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marston SB. How Do Mutations in Contractile Proteins Cause the Primary Familial Cardiomyopathies? J Cardiovasc Transl. 2011;4:245–55. doi: 10.1007/s12265-011-9266-2. [DOI] [PubMed] [Google Scholar]
- 52.Keren A, Syrris P, McKenna WJ. Hypertrophic cardiomyopathy: the genetic determinants of clinical disease expression. Nat Clin Pract Cardiovasc Med. 2008;5:158–68. doi: 10.1038/ncpcardio1110. [DOI] [PubMed] [Google Scholar]
- 53.Mettikolla P, Calander N, Luchowski R, Gryczynski I, Gryczynski Z, Zhao J, et al. Cross-bridge kinetics in myofibrils containing familial hypertrophic cardiomyopathy R58Q mutation in the regulatory light chain of myosin. Journal of theoretical biology. 2011;284:71–81. doi: 10.1016/j.jtbi.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee S, Lu R, Muller-Ehmsen J, Schwinger RH, Brixius K. Increased Ca2+ sensitivity of myofibrillar tension in ischaemic vs dilated cardiomyopathy. Clinical and experimental pharmacology & physiology. 2010;37:1134–8. doi: 10.1111/j.1440-1681.2010.05443.x. [DOI] [PubMed] [Google Scholar]
- 55.Wolff MR, Buck SH, Stoker SW, Greaser ML, Mentzer RM. Myofibrillar calcium sensitivity of isometric tension is increased in human dilated cardiomyopathies: role of altered beta-adrenergically mediated protein phosphorylation. J Clin Invest. 1996;98:167–76. doi: 10.1172/JCI118762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marian AJ, Roberts R. The Molecular Genetic Basis for Hypertrophic Cardiomyopathy. J Mol Cell Cardiol. 2001;33:655–70. doi: 10.1006/jmcc.2001.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marian AJ. Pathogenesis of diverse clinical and pathological phenotypes in hypertrophic cardiomyopathy. The Lancet. 2000;355:58–60. doi: 10.1016/s0140-6736(99)06187-5. [DOI] [PubMed] [Google Scholar]
- 58.Hasegawa K, Fujiwara H, Koshiji M, Inada T, Ohtani S, Doyama K, et al. Endothelin-1 and its receptor in hypertrophic cardiomyopathy. Hypertension. 1996;27:259–64. doi: 10.1161/01.hyp.27.2.259. [DOI] [PubMed] [Google Scholar]
- 59.Li RK, Li G, Mickle DA, Weisel RD, Merante F, Luss H, et al. Overexpression of transforming growth factor-beta1 and insulin-like growth factor-I in patients with idiopathic hypertrophic cardiomyopathy. Circulation. 1997;96:874–81. doi: 10.1161/01.cir.96.3.874. [DOI] [PubMed] [Google Scholar]
- 60.Welch S, Plank D, Witt S, Glascock B, Schaefer E, Chimenti S, et al. Cardiac-specific IGF-1 expression attenuates dilated cardiomyopathy in tropomodulin-overexpressing transgenic mice. Circ Res. 2002;90:641–8. doi: 10.1161/01.res.0000013780.77774.75. [DOI] [PubMed] [Google Scholar]
- 61.Nagaya N, Kangawa K, Itoh T, Iwase T, Murakami S, Miyahara Y, et al. Transplantation of mesenchymal stem cells improves cardiac function in a rat model of dilated cardiomyopathy. Circulation. 2005;112:1128–35. doi: 10.1161/CIRCULATIONAHA.104.500447. [DOI] [PubMed] [Google Scholar]
- 62.Chang AN, Harada K, Ackerman MJ, Potter JD. Functional Consequences of Hypertrophic and Dilated Cardiomyopathy-causing Mutations in α-Tropomyosin. J Biol Chem. 2005;280:34343–9. doi: 10.1074/jbc.M505014200. [DOI] [PubMed] [Google Scholar]
- 63.Mirza M, Robinson P, Kremneva E, Copeland On, Nikolaeva O, Watkins H, et al. The Effect of Mutations in α-Tropomyosin (E40K and E54K) That Cause Familial Dilated Cardiomyopathy on the Regulatory Mechanism of Cardiac Muscle Thin Filaments. J Biol Chem. 2007;282:13487–97. doi: 10.1074/jbc.M701071200. [DOI] [PubMed] [Google Scholar]
- 64.Roth K, Hubesch B, Meyerhoff DJ, Naruse S, Gober JR, Lawry TJ, et al. Noninvasive Quantitation of Phosphorus Metabolites in Human-Tissue by Nmr-Spectroscopy. Journal of Magnetic Resonance. 1989;81:299–311. [Google Scholar]
- 65.Opie LH, Mansford KR, Owen P. Effects of increased heart work on glycolysis and adenine nucleotides in the perfused heart of normal and diabetic rats. Biochem J. 1971;124:475–90. doi: 10.1042/bj1240475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Godt RE, Maughan DW. On the composition of the cytosol of relaxed skeletal muscle of the frog. Am J Physiol. 1988;254:C591–604. doi: 10.1152/ajpcell.1988.254.5.C591. [DOI] [PubMed] [Google Scholar]
- 67.Buscemi N, Foster DB, Neverova I, Van Eyk JE. p21-activated kinase increases the calcium sensitivity of rat triton-skinned cardiac muscle fiber bundles via a mechanism potentially involving novel phosphorylation of troponin I. Circ Res. 2002;91:509–16. doi: 10.1161/01.res.0000035246.27856.53. [DOI] [PubMed] [Google Scholar]
- 68.Kooij V, Boontje N, Zaremba R, Jaquet K, dos Remedios C, Stienen GJ, et al. Protein kinase C alpha and epsilon phosphorylation of troponin and myosin binding protein C reduce Ca2+ sensitivity in human myocardium. Basic research in cardiology. 2010;105:289–300. doi: 10.1007/s00395-009-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kawai M, Zhao Y. Cross-bridge scheme and force per cross-bridge state in skinned rabbit psoas muscle fibers. Biophys J. 1993;65:638–51. doi: 10.1016/S0006-3495(93)81109-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taylor EW. Mechanism of actomyosin ATPase and the problem of muscle contraction. CRC Crit Rev Biochem. 1979;6:103–64. doi: 10.3109/10409237909102562. [DOI] [PubMed] [Google Scholar]
- 71.Geeves MA, Goody RS, Gutfreund H. Kinetics of acto-S1 interaction as a guide to a model for the cross-bridge cycle. J Muscle Res Cell Motil. 1984;5:351–61. doi: 10.1007/BF00818255. [DOI] [PubMed] [Google Scholar]
- 72.Goldman YE, Hibberd MG, Trentham DR. Relaxation of rabbit psoas muscle fibres from rigor by photochemical generation of adenosine-5′-triphosphate. J Physiol. 1984;354:577–604. doi: 10.1113/jphysiol.1984.sp015394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dantzig J, Goldman Y, Millar NC, Lacktis J, Homsher E. Reversal of the cross-bridge force-generating transition by the photogeneration of phosphate in rabbit psoas muscle fibers. J Physiol. 1992;451:247–78. doi: 10.1113/jphysiol.1992.sp019163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fortune NS, Geeves MA, Ranatunga KW. Tension responses to rapid pressure release in glycerinated rabbit muscle fibers. Proc Natl Acad Sci (USA) 1991;88:7323–7. doi: 10.1073/pnas.88.16.7323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.