

Abstract
Ischemic heart disease is the leading cause of death worldwide. An improved understanding of the mechanisms involved in myocardial injury would allow intervention downstream in the pathway where certain drugs including natural products could be efficiently applied to target the end effectors of the cell death pathway. Green tea polyphenols (GTPs) have potent anti-oxidative capabilities, which may account for their beneficial effects in preventing oxidative stress associated with ischemia injury. Although studies have provided convincing evidence to support the protective effects of GTPs in cardiovascular system, the potential end effectors that mediate cardiac protection are only beginning to be addressed. Proteomics analyses widely used to identify the protein targets for many cardiovascular diseases have advanced the discovery of the signaling mechanism for GTPs-mediated cardio-protection. This review focuses on putative triggers, mediators, and end effectors for the GTPs-mediated cardio-protection signaling pathways engaged in myocardial ischemia crisis, allowing a promising natural product to be used for ameliorating oxidative stress associated with ischemic heart diseases.
Keywords: Cardio-protection, Green tea polyphenols (GTPs), Ischemic heart disease, Oxidative stress, Proteomics
1. Introduction
Green tea polyphenols (GTPs) have attracted much interest in prevention of atherosclerosis and cardiovascular diseases [1-7]. Epidemiological studies have established a close correlation between the consumption of green tea and protection against cardiovascular diseases and risk factors [8-12]. Other experimental studies on myocardial ischemia injury have also suggested that the cardio-protective effect of GTPs is associated with the scavenging of active-oxygen radicals, the modulation of redox- sensitive transcription factors (e.g., NFκB, AP-1), the reduction of STAT-1 activation and Fas receptor expression, an increase in NO production, the exertion of positive inotropic effects, and the modulation of myofilament Ca2+ sensitivity [13-21]. However, limited information is known for the potential end effectors in the GTPs-conducted signaling pathways for cardiac protection. This review intends to increase our understanding on the GTPs-mediated cardio-protective mechanism by which molecular targeting for their anti-oxidative interventions on myocardial ischemic disorder is discussed.
2. Anti-oxidative capacities of GTPs
Oxidative stress describing an imbalance between the generation and clearance of reactive oxygen species (ROS) in cells has been associated with hypoxia or myocardial ischemia, and likely contributes to the progression of cardiovascular diseases [22]. Accumulating evidence also indicates that redox-sensitive signaling pathways via the effects of generation of ROS or reactive nitrogen species (RNS) or reactive lipid derived aldehydes (LDAs) are essentially involved in the pathological stress of heart cells [23]. Accordingly, molecular targeting for anti-oxidative interventions on redox signaling pathways may provide a therapeutic approach to ameliorate the risk and progression for heart diseases.
GTPs have potent antioxidant and radical-scavenging properties, which may partially account for their cardio-protective effects [24]. In vitro, they have been shown to scavenge ROS or RNS, chelate metal ions, prevent the activation of redox-sensitive transcription factors, inhibit ROS generating enzymes, and increase antioxidant enzymes [25, 26]. The major catechins in GTPs include epicatechin (EC), epigallocatechin (EGC), epicatechin- 3-gallate (ECG), and epigallocatechin-3-gallate (EGCG) [27, 28]. These compounds (i.e. biologically active polyphenolic flavonoids) contain two or more aromatic rings, each bearing at least one aromatic hydroxyl connected with a carbon bridge. EGCG is the most physiologically potent compound, and primarily accounts for the biological effects of green tea [2]. Studies with a cell line of H9c2 rat cardiomyoblasts associated with H2O2- induced oxidative stress also demonstrated the protective role of EGCG against oxidative injury and cell death caused by ROS and cytosolic Ca2+ overload in cardiac cells [29-31].
3. GTPs mediated cardiac protection against myocardial ischemic injury
3.1. Animal models for cardiac adaptation to oxidative stress and myocardial ischemia
Two different myocardial ischemia models (Figure 1) associated with chronic myocardial infarction (MI) and transient ischemiareperfusion (IR) were created in rats by ligating the left anterior descending coronary (LAD) for studying myocardial ischemic injury [13, 14]. In the MI model, severe myocardial infarction was found in post-MI rats [14], while the IR model involving brief regional ischemia for 20 min followed by subsequent reperfusion showed no severe infarcted injury [13]. These findings suggested that brief regional ischemia followed reperfusion may lead to activate pathways that either preserve cell viability (preconditioning) or lead to cell death (IR injury). In contrast, irreversible MI caused by death of myocytes, presumably as a result of both necrosis and apoptosis, mostly appears within the infarct and periinfarct regions [32-35].
Fig. 1.
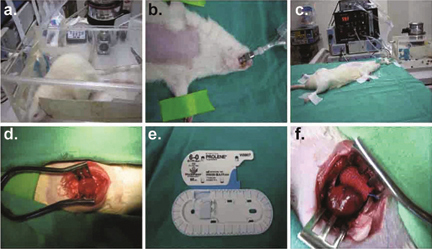
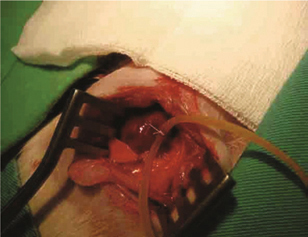
Myocardial ischemia models of chronic myocardial infarction (MI) and transient ischemia-reperfusion (IR) created in rats by ligating the left anterior descending coronary (LAD).
Reperfusion injury of ischemic tissue is known to be accompanied by the production of ROS and Ca2+ overload in injured cardiomyocytes [36-43]. The rise in cytosolic Ca2+ levels could induce mitochondrial Ca2+ ([Ca2+]m) accumulation via the mitochondrial Ca2+ uniporter and the increased ROS production. Both the [Ca2+]m overload and increased ROS generation would induce opening of the mitochondrial permeability transition pore (mPTP) and rupture of the plasma membrane, triggering cell death [44, 45].
3.2. Pretreatment of GTPs protects myocardial ischemia injury in post-IR rats
A previous study by Miwa et al. using isolated hearts perfused with a Langendorff’s apparatus showed that GTPs pre-treatment (1 mM, 35 ml/day for 14 days), administered orally prior to surgery, could protect hearts from oxidative stress after reperfusion and avoid cell edema [46]. This result suggested that GTPs might be used as a novel method for preparative cardiac surgery in the future [46]. In addition, other study using a post-IR model in rats also demonstrated that GTPs pretreatment for 4 hours prior to IR injury protects cardiomyocytes by preventing cytosolic Ca2+ overload, myofibril disruption, and alterations in adherens and gap junction protein expression and distribution [13].
3.3. GTPs attenuate myocardial remodeling injury in post-MI rats
Myocardial infarction (MI) largely resulting from cardiac ischemic injury often undergoes to cardiac remodeling process, which may cause secondary damage to the heart tissue by excessive ROS and free radicals [47]. A previous study with post MI rat model showed that GTPs reduced heart tissue remodeling injury, avoided ventricular hypertrophy, reduced infarct size, as well as significantly improved the left ventricular functions [14]. Using the same post MI model, the rate of intracellular free radicals produced in cardiomyocytes extracted from post MI rats with GTPs treatment for 3 days, 2 weeks, and 3 weeks all became slower in comparison with post MI cells without GTPs (Figure 2). With the increase of GTPs feeding period the rate of intracellular free radical production was also significantly reduced. In addition, GTPs treatment could help maintain the activity of SOD in cells located at the remote region of the heart in the post MI rats for the time periods from 3 days to 3 weeks, while in post MI group without GTPs treatment the SOD activity was found to be significantly decreased in cardiac tissues of rats suffered from post MI for 3 weeks (Figure 3). However, the measured SOD mRNA level in myocardial tissues was not significantly different in control rats, post MI rats with or without GTPs treatments. For measuring another anti-oxidant enzyme, heme oxygenase-1 (HO-1), the mRNA level was found significantly reduced in cardiac non-infarcted area (remote region) for post MI rats without GTPs treatment, while GTPs treatment prevented from the decrease of mRNA expression in myocardial tissues.
Fig. 2.
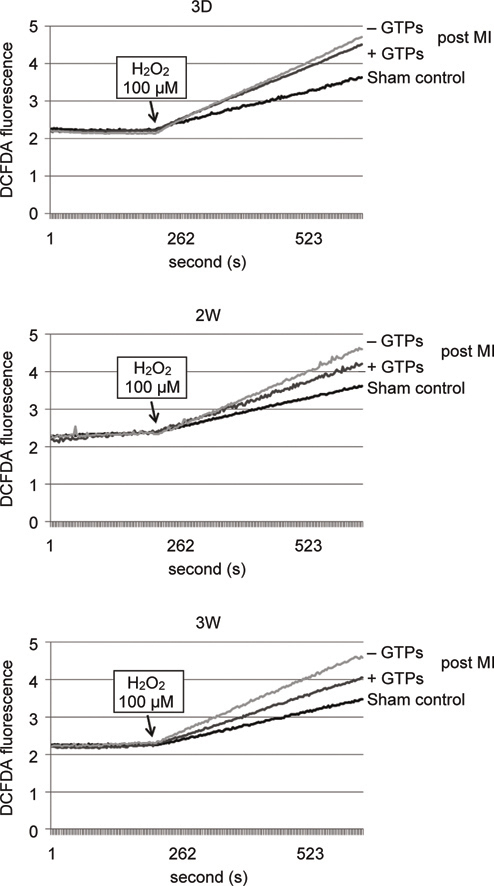
The rate of intracellular ROS produced in cardiomyocytes extracted from post MI rats with or without GTPs treatment for 3 days (3D), 2 weeks (2W), and 3 weeks (3W). Intracellular ROS formation was measured by the fluorescence changes of 2’,7’-dichlorofluorescin diacetate (DCF-DA) in cardiomyocytes with fluorescence spectrophotometry. The fluorescence excitation maximum for DCF-DA was 495 nm, and the corresponding emission maximum was 527 nm.
Fig. 3.
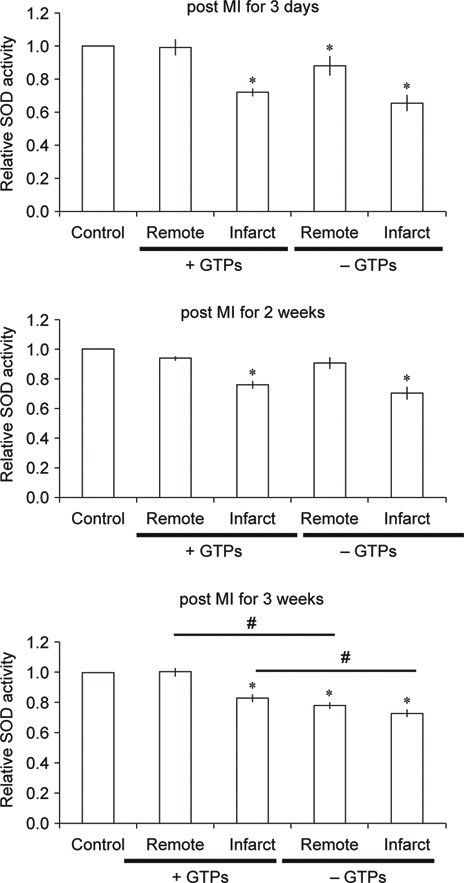
The relative activity of SOD in cardiac tissues of sham control, post MI rats with or without GTPs treatment for 3 days, 2 weeks, and 3 weeks.
To further examine the events for the oxidative stress in myocardial cells, 4-hydroxynonenal (4HNE) post-translational modification on myocardial proteins were determined in the hearts of control rats, post MI rats with or without GTPs treatments. Results showed that 4-HNE modified proteins were increased in myocardial tissues for the post MI rats without GTPs treatment, but no significant difference with GTPs treatments, as compared to sham controls.
3.4. Ischemic preconditioning cardiac protection signaling pathways
Ischemic preconditioning (IPC) is one of the most effective cardio-protection in which short periods of IR in the heart confer resistance to a subsequent prolonged ischemic stress [36, 40, 48- 51]. Many mediators and effectors have been shown to be essential for IPC and include the ATP-dependent mitochondrial K+ channels (KATP channel), PKC, tyrosine protein kinases (TPK), and adenosine, bradykinin, adrenergic and muscarinic receptor, NO donors, and phosphodiesterase inhibitors, and endotoxins, cytokines, and ROS. IPC initiates a number of cardio-protective events depending on the intervening time period between the protective stimulus (IPC) and the index IR injury [36, 40, 50]. Acute IPC (min to hrs) is mediated by the posttranslational modification of proteins, while “second window” IPC (days) induces protection by de novo protein synthesis [51].
Cardiac protection involving a memory (preconditioning) might be attributed to trigger mitochondrial swelling that causes enhanced substrate oxidation and ROS production, leading to redox activation of PKC, which inhibits GSK-3β [52]. Alternatively, TPK or certain G-protein coupled receptor (GPCR)-dependent activation elicits cell protection by inhibiting GSK-3β, via Akt and mTOR pathways, PKC pathways, or PKA pathways [44]. The convergence of these pathways via inhibition of GSK-3β on the end effector to limit mPTP induction is the general mechanism of cardiomyocyte protection [52]. Recent reports also provided evidence for that the cardio-protection of GTPs against oxidative stress associated with myocardial ischemic injury is caused by reducing cytosolic Ca2+ overload and generation of ROS via the Akt/GSK-3β/β-catenine and caveolae signaling both in vivo myocardial ischemia injury [13, 14] and In vitro H2O2-induced oxidative stress models [29-30].
3.5. The GPCR-dependent signaling pathways for cardiac protection
The GPCR-dependent mechanism initiates a downstream signaling cascade involving TPK, PI3K/Akt, NOS, activation of KATP channel, generation of ROS, activation of PKC isoforms, GSK-3β, and MAPK, and inhibition of the opening of the mPTP [49-52]. Although these components are considered to play a role in cardiac protection, it still remains to be resolved as to how signaling networks interact spatially and temporally in producing such protection. In particular, little is known about the regions to which proteins translocate and the molecules with which they interact. In many cases, the signals from GPCRs to target proteins are mediated via lipid signals [53].
3.6. Caveolae /lipid rafts involved in cardiac protection
Membrane lipids forms organized and dynamic structures based on interactions between membrane lipids and proteins including caveoli with clear morphology, dynamic rafts of different sizes and specific annular lipid layers surrounding proteins due to mutual affinity of lipids and proteins [54, 55]. It is proposed that these rafts function as platforms for the attachment of proteins when membranes are moved around inside the cell and during signal transduction [54, 55]. It is generally accepted that the structural and functional properties of rafts require an intact microtubule and actin filament; both are the primary interacting partners of caveolae/lipid rafts [56, 57].
Many of the properties of rafts have been inferred from detergent-resistant membranes (DRMs) that occur in nonionic detergent (e.g. Triton X-100) lysates of animal cells [54-55]. Lipid rafts, enriched in cholesterol and sphingolipids, form one such microdomain along with a subset of lipid rafts, caveolae, enriched in the protein caveolin (Cav) [54, 55]. In the membrane raft model, the DRMs represent poorly solubilized rafts [55], and the composition of the DRMs has served as a guide to the structural and functional properties of rafts. These domains selectively and dynamically gather or exclude signaling proteins, and the activity and specificity of membrane proteins is regulated by interaction partners [54, 55]. The “Cav/lipid raft signaling hypothesis” postulates that the regulation of signal transduction events occurs as a result of interaction of signaling proteins with a “Cav scaffolding domain”, an interaction that is hypothesized to inhibit such pathways by sequestering components away from signal transduction partners [53-58].
A growing body of data indicates that multiple signal transduction events in the heart occur via plasma membrane receptors located in signaling microdomains [59-61]. In the heart, a key Cav is Cav-3, whose scaffolding domain is thought to serve as an anchor for other proteins [62, 63]. Immunoprecipitation with anticaveolin antibodies indicated that several GPCRs, and their cognate heterotrimeric G proteins and effectors, localize to lipid rafts/ caveolae in neonatal cardiac myocytes [64]. Using In vitro and in vivo models of IR injury, it has been shown that that the volatile anesthetic, isoflurane, modifies cardiac myocyte sarcolemmal membrane structure and composition and that activation of Src and phosphorylation of Cav-1 contribute to cardiac protection [62, 63]. Thus, multiple signal transduction events in the heart occur via plasma membrane receptors located in signaling microdomains [65]. A recent study using In vitro oxidative stress model in H9c2 cells has demonstrated that Cav/lipid rafts involve in GTPs-mediated Akt/GSK-3β signaling for cardio-protection during oxidative stress [30]. This study also proposed a hypothetical model with interaction networks based on the identified proteins in EGFP (enhanced green fluorescence protein) expressed cells (Figure 4). It is very likely that GTPs may act to protect cardiac cells from oxidative stress and ischemic injury through lipid rafts.
Fig. 4.
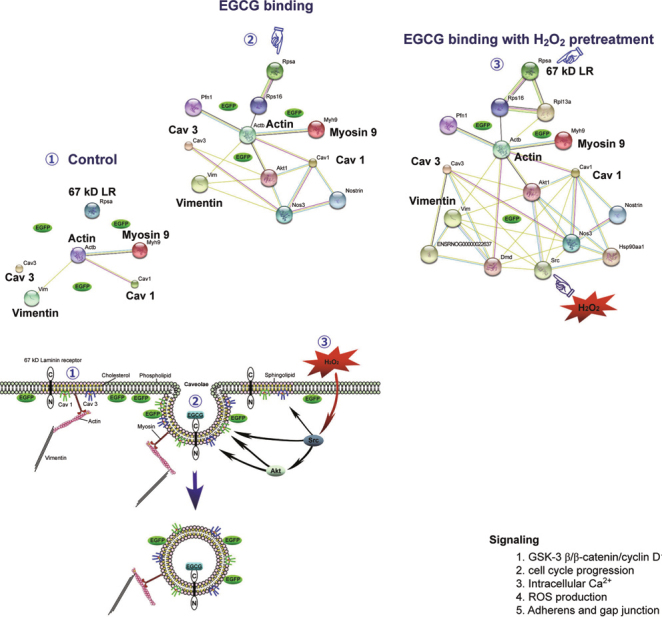
A hypothetical model for interaction networks obtained with identified proteins in EGFP-expressing H9c2 cells. The proteins identified were imported into the EMBL Search Tool for the Retrieval of Interacting Proteins (STRING) database, and an interaction map generated was used to construct the hypothetical mechanism for EGCG-induced fluorescence changes in EGFP-expressing H9c2 cells with or without H2O2 pretreatment [30].
3.7. Convergence of signaling pathways for cardio-protection on GSK-3β
Numerous cardio-protective drugs are shown to converge on GSK-3β [44, 52, 66-69]. The phosphorylation and inhibition of GSK-3β lead to inhibition or delayed activation of mPTP, a key regulator of apoptosis [52]. A recent study using isolated perfused working rat hearts subjected to global IR demonstrated that addition of a selective inhibitor of GSK-3β prior to ischemia or at the onset of reperfusion improves recovery of left ventricle work by reducing proton production and attenuating the intracellular Ca2+ overload [66]. In addition, previous studies have suggested that IPC results in phosphorylation and inhibition of GSK-3β [44], and that drugs that inhibit GSK-3β are cardio-protective [44, 52]. β-catenin is a transcriptional activator that activates target genes in the nucleus [70, 71]. Growth factors promote β-catenin signaling by inhibiting its phosphorylation by GSK-3β, resulting in a reduction of its degradation by the proteasome and its subsequent activation in the nucleus [72, 73]. Cyclin D1 is one of target genes that might be activated by β-catenin for cell proliferation [69]. Consistently, inhibition on the β-catenin signaling pathway would lead to a decrease in cyclin D1 expression in cells that prevent cell cycle progression into S phase [68]. Another route for inhibition of GSK-3β on the β-catenin signaling pathway is to modulate the cell-cell adhesion and communication via adherens and gap junction proteins (i.e. Cx43) [13]. Evidence also suggested that GTPs pretreatment acts to protect heart from IR injury through PI3K/Akt survival pathway to limit GSK-3β activity in cardiac cells [13, 29, 30].
4. Molecular targeting for GTPs-mediated cardiac protections
4.1. Cardioproteomics application to discover signaling mechanisms involved in cardiovascular diseases
Proteomics is a new technology that allows the detection and the identification of several proteins at a given time in a sample [74- 76]. It combines several techniques, including 2-D gel electrophoresis, image analysis, and mass spectrometry. This technique has been extensively employed to identify proteins involved in cardiac regeneration in the infarcted myocardium [77], to analyze modifications in the plasma protein map during an acute coronary syndrome [78], to analyze the role of complement in myocardial IR and its effect on myocardial protein expression [79]. In an in vivo dog model of myocardial IR [80], 2-D gel electrophoresis was used to identify changes in the level of four metabolic enzymes and a contractile protein. In an in vivo rabbit model of cardiac IR, Schwertz et al. [81] found 10 protein spots that were differentially expressed: two as the protective proteins SOD and αB-crystallin More recently [82], a proteomic approach was also used to study of the effects of ramipril on post-infarction left ventricular remodeling in the rabbit. In an In vitro rat model [83], 8 protein spots with altered expression after cardiac ischemia or IR were found: 5 protein spots as the endoplasmic reticulum enzyme, one as 60 kDa heat shock protein and two as mitochondrial elongation factor Tu. In the mouse, a model of permanent ischemia and a model of IR were used to identify changes in cardiac protein expression after in vivo MI by 2-D gel electrophoresis combined with mass spectrometry [84]. Using the H9c2 cell model of H2O2-induced oxidative stress for a proteomics study, Chou et al. [85] showed that oxidative stress triggers tyrosine phosphorylation on target proteins associated with cell-cell junctions, the actin cytoskeleton, and cell adhesion in cardiac cells.
4.2. Cardioproteomics exploring GTPs-mediated anti-oxidative intervention in H9c2 cardiomyoblasts
To identify the potential proteins for the GTPs-mediated cardioprotection, cardiac proteomics study was performed in an H2O2- induced oxidative stress model of myocardial ischemia injury [29]. In this model, 8 proteins associated with metabolism, electron transfer, redox regulation, signal transduction, RNA binding and transcription regulation were identified to take part in EGCGameliorating H2O2-induced injury to H9c2 cells. H2O2 exposure increased oxidative stress evidenced by increases in ROS and cytosolic Ca2+ overload, increases in glycolytic protein, α-enolase (Eno 1), decreases in antioxidant protein, peroxiredoxin-4 (Prdx4), as well as decreases in mitochondrial proteins, including aldehyde dehydrogenase-2 (Aldh2), ornithine aminotransferase (Oat), and succinate dehydrogenase ubiquinone flavoprotein subunit (Sdha). All of these effects were reversed by EGCG pre-treatment. In addition, EGCG attenuated the H2O2-induced increases of Type II inositol 3,4-bisphosphate 4-phosphatase (Inpp4b) and relieved its subsequent inhibition of downstream signaling for Akt and GSK-3β/cyclin D1 in H9c2 cells. Pre-treatment with EGCG or GSK-3β inhibitor (SB 216763) significantly improved the H2O2-induced suppression on cell viability, phosphorylation of pAkt (S473) and pGSK-3β (S9), and level of cyclin D1 in cells. Finally, EGCG counteracted the H2O2-induced decreases in heterogeneous nuclear ribonucleoprotein K (HnrnpK), playing a role in cell cycle progression, but increases in double-stranded RNA-binding protein Staufen homolog 2 (Stau2), involving RNA functions. These findings suggest that GTPs might act to protect cardiac cells from oxidative stress through Akt survival pathway to inhibit the GSK-3β effect on cardiac cell death pathway (Figure 5).
Fig. 5.
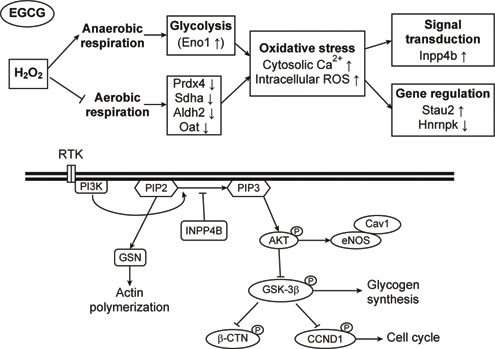
The putative mechanism for EGCG-conducted cardioprotection against H2O2-induced oxidative stress through the Akt/GSK-3β /caveolae pathways in cultured H9c2 cells. EGCG is hypothesized to protect cardiac cells from oxidative stress by PI3K/Akt survival pathway to attenuate the GSK-3β signaling on cardiac cell death.
4.3. Cardioproteomics identifying GTPs-mediated cardio-protection against myocardial ischemia stress in post LAD rats
It has been shown that LAD ligation for 3 days caused acute myocardial ischemia (AMI) and impairment of myocardial functions, while myocardial remodeling occurring in the rats after LAD ligation for 2 or 3 weeks [14]. Proteomic analysis has allowed to identify the molecular targets in the myocardium associated with disturbance by ischemia stress but protection by GTPs in post MI rats for 3 days (AMI) or 2-3 weeks (remodeling).
In AMI model associated with post LAD ligation for 3 days, 10 proteins involved in the functions of myocardial ischemia stress (i.e. Chloride intracellular channel protein 1 (CICP1); Endoplasmin), cytoskeletal organization (i.e. Tropomyosin alpha-3 chain, tpm3), mitochondria metabolism (i.e. 2-oxoglutarate dehydrogenase, Enoyl-CoA hydratase), redox signaling (i.e. Ribonuclease inhibitor (Rnh1), 14-3-3 protein θ), and acute inflammation (i.e. Serine protease inhibitors A3N, A3K) were identified for the GTPs-mediated cardio-protection against AMI injury (Figure 6). The data suggested that the activation of NF-κb transcription factor and inhibition on PI3K/Akt signaling might account for the AMI-induced stress, and such redox signaling events could be prevented by GTPs (Figure 6).
Fig. 6.
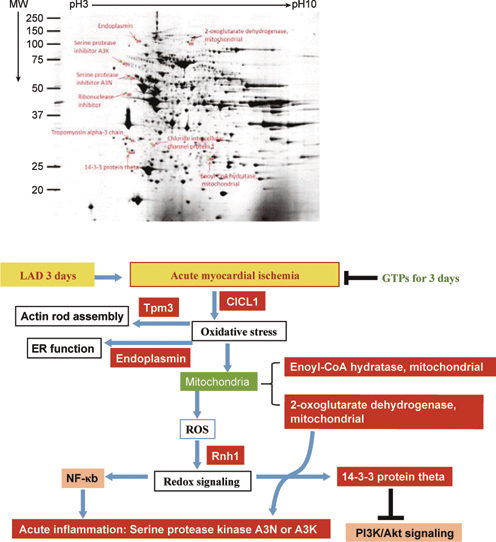
Proteomics analysis on molecular identification for targets involved in the GTPs-mediated cardio-protection against AMI in post LAD rats for 3 days.
In remodeling model with post LAD ligation for 2 weeks, 14 proteins associated with chaperone proteins (i.e. Heat shock protein 75 kDa, mitochondrial; 60 kDa heat shock protein, mitochondrial), muscle proteins (i.e. cardiac troponin T, desmin), lipid metabolism (i.e. Carnitine O-acetyltransferase), mitochondria functions (i.e. ES1 protein homolog, Electron transfer flavoprotein subunit β, Fumarate hydratase, ATP synthase subunit α, ATP synthase subunit β, inner membrane protein fragment), developmental protein (i.e. dihydropyrimidinase-related protein 2), and stress related adaptor protein (i.e. 14-3-3 protein ε) were identified for the GTPs-mediated cardio-protection against the myocardial remodeling after ischemia stress (Figure 7). These data suggest that during myocardial ischemia remodeling cardiac cells disturbed by mitochondria dysfunction associated with alterations of lipid metabolism trigger chaperone/stress response via the adaptor protein (14-3-3 protein ε) resulting in cytoskeleton reorganization and contractile apparatus disruption. Such stress-induced redox signaling for myocardial ischemia remodeling could be improved by GTPs. Consistently, myocardial remodeling with post LAD for 3 weeks also identified 10 proteins associated with cytoskeletal function, energy metabolism (i.e. electron transport chain, citric acid cycle, and fatty acid oxidation), and redox regulation (Figure 8).
Fig. 7.
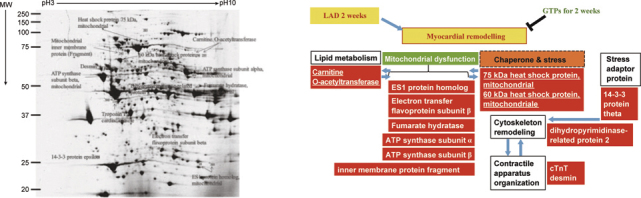
Proteomics analysis identifying molecular targets involved in the GTPs-mediated cardio-protection against myocardial remodeling in post LAD rats for 2 weeks.
Fig. 8.
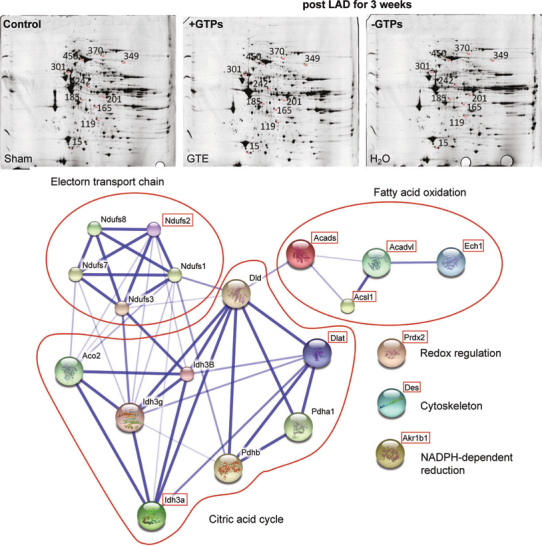
Molecular identification and hypothetical protein-protein interactions for proteins involved in myocardial remodeling of post MI for 3 weeks with or without GTPs in rats. Identified proteins are Peroxiredoxin-2 (Prdx2) (Spot 15), Delta(3,5)-Delta(2,4)- dienoyl-CoA isomerase, mitochondrial, (Ech1) (Spot 119), Aldose reductase (Akr1b1) (Spot 165), Isocitrate dehydrogenase [NAD] subunit alpha, mitochondrial (Idh3a) (Spot 185), Short-chain specific acyl-CoA dehydrogenase, mitochondrial (Acads) (Spot 201), NADH dehydrogenase [ubiquinone] iron-sulfur protein 2, mitochondrial (Ndufs2) (Spot 242), Desmin (Des)(Spot 301), Very longchain specific acyl-CoA dehydrogenase, mitochondrial (Acadvl) (Spot 349), Long-chain-fatty-acid--CoA ligase 1 (Acsl1) (Spot 370), Dihydrolipoyllysine-residue acetyltransferase component of pyruvate dehydrogenase complex, mitochondrial (Dlat0 (Spot 450).
5. Perspectives
Green tea, being rich in polyphenols (GTPs), is a natural choice for its myocardial protection against ischemia or oxidative stress. Cardiac proteomics have allowed to reveal the underlying mechanisms for the actions of GTPs exerting their favorable cardioprotective effects. Currently, the important findings illustrating the potential end effectors involved in cardiac protection by GTPs include: (1) EGCG exerting cardio-protection against H2O2- induced oxidative stress through the Akt/GSK-3β/caveolae pathways in cardiac cells, (2) GTPs preventing their activation of NF-κb and their inhibition on PI3K/Akt signaling for the AMI stress, (3) GTPs ameliorating mitochondria dysfunction associated with alterations of lipid metabolism, chaperone-induced stress response, and the adaptor 14-3-3 ε protein signaling for cytoskeleton remodeling /contractile apparatus disruption during post MI remodeling. It appears promising to apply this natural product as a therapeutic approach to treat ischemic heart diseases in the near future.
6. Acknowledgements
This work was supported by the National Science Council of Taiwan government (to Y-M L., Grants: NSC 100-2320-B-005- 001, NSC 101-2320-B-005-001), and also supported by cooperative projects between the Taichung Veterans General Hospital and the NCHU (TCVGH-NCHU 103S0514).
7. Conflicts of interest statement
The authors declare that they have no conflicting interests.
References
- [1].Mak JC. Potential role of green tea catechins in various disease therapies: Progress and promise. Clin Exp Pharmacol Physiol. 2012;39:265–73. doi: 10.1111/j.1440-1681.2012.05673.x. [DOI] [PubMed] [Google Scholar]
- [2].Liou YM, Hsieh SR, Wu TJ. Green tea and cardiac health. CAB Reviews: Perspectives in Agriculture, Veterinary Science, Nutrition and Natural Resources. 2009;4:020. doi: 10.1079/pavsnnr202116039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jochmann N, Baumann G, Stangl V. Green tea and cardiovascular disease: from molecular targets towards human health. Curr Opin Clin Nutr Metab Care. 2008;11:758–65. doi: 10.1097/MCO.0b013e328314b68b. [DOI] [PubMed] [Google Scholar]
- [4].Wolfram S. Effects of green tea and EGCG on cardiovascular and metabolic health. J Am Coll Nutr. 2007;26:373S–88S. doi: 10.1080/07315724.2007.10719626. [DOI] [PubMed] [Google Scholar]
- [5].Stangl V, Dreger H, Stangl K, Lorenz M. Molecular targets of tea polyphenols in the cardiovascular system. Cardiovasc Res. 2007;73:348–58. doi: 10.1016/j.cardiores.2006.08.022. [DOI] [PubMed] [Google Scholar]
- [6].Sumpio BE, Cordova AC, Berke-Schlessel DW, Qin F. the “Asian paradox,” and cardiovascular disease. J Am Coll Surg. 2006;202:813–25. doi: 10.1016/j.jamcollsurg.2006.01.018. [DOI] [PubMed] [Google Scholar]
- [7].Stangl V, Lorenz M, Stangl K. The role of tea and tea flavonoids in cardiovascular health. Mol Nutr Food Res. 2006;50:218–28. doi: 10.1002/mnfr.200500118. [DOI] [PubMed] [Google Scholar]
- [8].Kuriyama S. The relation between green tea consumption and cardiovascular disease as evidenced by epidemiological studies. J Nutr. 2008;138:1548S–53S. doi: 10.1093/jn/138.8.1548S. [DOI] [PubMed] [Google Scholar]
- [9].Kuriyama S, Shimazu T, Ohmori K, Kikuchi N, Nakaya N, Nishino Y. Green tea consumption and mortality due to cardiovascular disease, cancer, and all causes in Japan: the Ohsaki study. JAMA. 2006;296:1255–65. doi: 10.1001/jama.296.10.1255. [DOI] [PubMed] [Google Scholar]
- [10].Mukamal KJ, Maclure M, Muller JE, Sherwood JB, Mittleman MA. Tea consumption and mortality after acute myocardial infarction. Circulation. 2002;105:2476–81. doi: 10.1161/01.CIR.0000017201.88994.F7. [DOI] [PubMed] [Google Scholar]
- [11].Hirano R, Momiyama Y, Takahashi R, Taniguchi H, Kondo K, Nakamura H. Comparison of green tea intake in Japanese patients with and without angiographic coronary artery disease. Am J Cardio. 2002;90:1150–53. doi: 10.1016/S0002-9149(02)02787-X. [DOI] [PubMed] [Google Scholar]
- [12].Peters U, Poole C, Arab L. Does tea affect cardiovascular disease? A meta-analysis. Am J Epidemiol. 2001;154:495–503. doi: 10.1093/aje/154.6.495. [DOI] [PubMed] [Google Scholar]
- [13].Liou YM, Hsieh SR, Wu TJ, Chen JY. Green tea extract given before regional myocardial ischemia-reperfusion in rats improves myocardial contractility by attenuating calcium overload. Pflugers Arch. 2010;460:1003–14. doi: 10.1007/s00424-010-0881-6. [DOI] [PubMed] [Google Scholar]
- [14].Shieh SR, Tsai DC, Chen JY, Tsai SW, Liou YM. Green tea extract protects rats against myocardial infarction associated with left anterior descending coronary artery ligation. Pflugers Arch. 2009;458:631–642. doi: 10.1007/s00424-009-0655-1. [DOI] [PubMed] [Google Scholar]
- [15].Dreger H, Lorenz M, Kehrer A, Baumann G, Stangl K, Stangl V. Characteristics of catechin- and theaflavin-mediated cardioprotection. Exp Biol Med (Maywood) 2008;233:427–33. doi: 10.3181/0710-RM-292. [DOI] [PubMed] [Google Scholar]
- [16].Li D, Yang C, Chen Y. Identification of a PKCε-dependent regulation of myocardial contraction by epicatechin-3-gallate. Am J Physiol Heart Circ Physiol. 2008;294:345–53. doi: 10.1152/ajpheart.00785.2007. [DOI] [PubMed] [Google Scholar]
- [17].Liou YM, Kuo SC, Hsieh SR. Differential effects of a green teaderived polyphenol (-)-epigallocatechin-3-gallate on the acidosis-induced decrease in the Ca2+ sensitivity of cardiac and skeletal muscle. Pflugers Arch. 2008;456:787–800. doi: 10.1007/s00424-008-0456-y. [DOI] [PubMed] [Google Scholar]
- [18].Lorenz M, Hellige N, Rieder P. Positive inotropic effects of epigallocatechin- 3-gallate (EGCG) involve activation of Na+/H+ and Na+/ Ca2+ exchangers. Eur J Heart Fail. 2008;10:439–45. doi: 10.1016/j.ejheart.2008.03.004. [DOI] [PubMed] [Google Scholar]
- [19].Hirai M, Hotta Y, Ishikawa N, Wakida Y, Fukuzawa Y, Isobe F. Protective effects of EGCg or GCg, a green tea catechin epimer, against postischemic myocardial dysfunction in guinea-pig hearts. Life Sci. 2007;80:1020–32. doi: 10.1016/j.lfs.2006.11.032. [DOI] [PubMed] [Google Scholar]
- [20].Townsend PA, Scarabelli TM, Pasini E, Gitti G, Menegazzi M, Suzuki H. Epigallocatechin-3-gallate inhibits STAT-1 activation and protects cardiac myocytes from ischemia/reperfusion-induced apoptosis. FASEB J. 2004;18:1621–23. doi: 10.1096/fj.04-1716fje. [DOI] [PubMed] [Google Scholar]
- [21].Nakagawa T, Yokozawa T. Direct scavenging of nitric oxide and superoxide by green tea. Food Chem Toxicol. 2002;40:1745–50. doi: 10.1016/S0278-6915(02)00169-2. [DOI] [PubMed] [Google Scholar]
- [22].Santos CX, Anilkumar N, Zhang M, Brewer AC, Shah AM. Redox signaling in cardiac myocytes. Free Radic Biol Med. 2011;50:777–93. doi: 10.1016/j.freeradbiomed.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wall SB, Oh JY, Diers AR, Landar A. Oxidative modification of proteins: an emerging mechanism of cell signaling. Front Physiol. 2012;3:369. doi: 10.3389/fphys.2012.00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Frei B, Higdon JV. Antioxidant activity of tea polyphenols in vivo: evidence from animal studies. J Nutr. 2003;133:3275–84. doi: 10.1093/jn/133.10.3275S. [DOI] [PubMed] [Google Scholar]
- [25].Wu CC, Hsu MC, Hsieh CW, Lin JB, Lai PH, Wung BS. Upregulation of heme-oxygenase-1 by epigallocatechin-3-gallate via the phosphatidylinositol 3-kinase/Akt and ERK pathways. Life Sci. 2006;78:2889–97. doi: 10.1016/j.lfs.2005.11.013. [DOI] [PubMed] [Google Scholar]
- [26].Rice-Evans CA, Miller NJ, Paganga G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic Biol Med. 1996;20:933–56. doi: 10.1016/0891-5849(95)02227-9. [DOI] [PubMed] [Google Scholar]
- [27].Beecher GR. Overview of dietary flavonoids: nomenclature, occurrence and intake. J Nutr. 2003;133:3248S–54S. doi: 10.1093/jn/133.10.3248S. [DOI] [PubMed] [Google Scholar]
- [28].Graham HN. Green tea composition, consumption, and polyphenol chemistry. Prev Med. 1992;21:334–50. doi: 10.1016/0091-7435(92)90041-F. [DOI] [PubMed] [Google Scholar]
- [29].Chen WC, Hsieh SR, Chiu CH, Hsu BD, Liou YM. Molecular identification for epigallocatechin-3-gallate-mediated antioxidant intervention on the H2O2-induced oxidative stress in H9c2 rat cardiomyoblasts. J Biomed Sci. 2014;21:56. doi: 10.1186/1423-0127-21-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hsieh SR, Hsu CS, Lu CH, Chen WC, Chiu CH, Liou YM. Epigallocatechin- 3- gallate- mediated cardioprotection by Akt/GSK-3β/caveolin signalling in H9c2 rat cardiomyoblasts. J Biomed Sci. 2013;20:86. doi: 10.1186/1423-0127-20-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sheng R, Gu ZL, Xie ML, Zhou WX, Guo CY. Epigallocatechin gallate protects H9c2 cardiomyoblasts against hydrogen dioxidesinduced apoptosis and telomere attrition. Eur J Pharmacol. 2010;641:199–206. doi: 10.1016/j.ejphar.2010.05.054. [DOI] [PubMed] [Google Scholar]
- [32].Kumar D, Jugdutt BI. Apoptosis and oxidants in the heart. J Lab Clin Med. 2003;142:288–97. doi: 10.1016/S0022-2143(03)00148-3. [DOI] [PubMed] [Google Scholar]
- [33].Anversa P, Cheng W, Liu Y, Leri A, Redaelli G, Kajstura J. Apoptosis and myocardial infarction. Basic Res Cardiol. 1998;93(3):8–12. doi: 10.1007/s003950050195. [DOI] [PubMed] [Google Scholar]
- [34].Anversa P, Kajstura J. Myocyte cell death in the diseased heart. Circ Res. 1998;82:1231–33. doi: 10.1161/01.RES.82.11.1231. [DOI] [PubMed] [Google Scholar]
- [35].Anversa P, Olivetti G, Leri A, Liu Y, Kajstura J. Myocyte cell death and ventricular remodeling. Curr Opin Nephrol Hypertens. 1997;6:169–76. doi: 10.1097/00041552-199703000-00011. [DOI] [PubMed] [Google Scholar]
- [36].Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007;59:418–58. doi: 10.1124/pr.107.06002. [DOI] [PubMed] [Google Scholar]
- [38].Corbucci GG, Perrino C, Donato G, Ricchi A, Lettieri B, Troncone G. Transient and reversible deoxyribonucleic acid damage in human left ventricle under controlled ischemia and reperfusion. J Am Coll Cardiol. 2004;43:1992–99. doi: 10.1016/j.jacc.2004.01.040. [DOI] [PubMed] [Google Scholar]
- [39].Bolli R, Marbán E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev. 1999;79:609–34. doi: 10.1152/physrev.1999.79.2.609. [DOI] [PubMed] [Google Scholar]
- [40].Heyndrickx GR, Millard RW, McRitchie RJ, Maroko PR, Vatner SF. Regional myocardial functional and electrophysiological alterations after brief coronary artery occlusion in conscious dogs. J Clin Invest. 1975;56:978–85. doi: 10.1172/JCI108178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Depre C, Vatner SF. Cardioprotection in stunned and hibernating myocardium. Heart Fail Rev. 2007;12:307–317. doi: 10.1007/s10741-007-9040-3. [DOI] [PubMed] [Google Scholar]
- [42].McCully JD, Wakiyama H, Hsieh YJ, Jones M, Levitsky S. Differential contribution of necrosis and apoptosis in myocardial ischemia/ reperfusion injury. Am J Physiol Heart Circ Physiol. 2004;286:1923–35. doi: 10.1152/ajpheart.00935.2003. [DOI] [PubMed] [Google Scholar]
- [43].Koerner JE, Anderson BA, Dage RC. Protection against postischemic myocardial dysfunction in anesthetized rabbits with scavengers of oxygen-derived free radicals: superoxide dismutase plus catalase, N-2-mercaptopropionyl glycine and captopril. J Cardiovasc Pharmacol. 1991;17:185–91. doi: 10.1097/00005344-199102000-00002. [DOI] [PubMed] [Google Scholar]
- [44].Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res. 2006;70:240–53. doi: 10.1016/j.cardiores.2006.01.017. [DOI] [PubMed] [Google Scholar]
- [45].Pain T, Yang XM, Critz SD, Yue Y, Nakano A, Liu GS. Opening of mitochondrial K(ATP) channels triggers the preconditioned state by generating free radicals. Circ Res. 2000;87:460–66. doi: 10.1161/01.RES.87.6.460. [DOI] [PubMed] [Google Scholar]
- [46].Miwa S, Yamazaki K, Hyon SH, Komeda M. A novel method of ‘preparative’ myocardial protection using green tea polyphenol in oral uptake. Interact Cardiovasc Thorac Surg. 2004;3:612–5. doi: 10.1016/j.icvts.2004.06.013. [DOI] [PubMed] [Google Scholar]
- [47].Hori M, Nishida K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc Res. 2009;81:457–64. doi: 10.1093/cvr/cvn335. [DOI] [PubMed] [Google Scholar]
- [48].Baines CP, Goto M, Downey JM. Oxygen radicals released during ischemic preconditioning contribute to cardioprotection in the rabbit myocardium. J Mol Cell Cardiol. 1997;29:207–16. doi: 10.1006/jmcc.1996.0265. [DOI] [PubMed] [Google Scholar]
- [49].Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36. doi: 10.1161/01.CIR.74.5.1124. [DOI] [PubMed] [Google Scholar]
- [50].Fryer RM, Schultz JEJ, Hsu AK, Gross GJ. Importance of PKC and tyrosine kinase in single or multiple cycles of preconditioning in rat hearts. Am J Physiol Heart Circ Physiol. 1999;276:1229–35. doi: 10.1152/ajpheart.1999.276.4.H1229. [DOI] [PubMed] [Google Scholar]
- [51].Pagliaro P, Gattullo D, Rastaldo R, Losano G. Ischemic preconditioning: from the first to the second window of protection. Life Sci. 2001;69:1–15. doi: 10.1016/S0024-3205(01)01113-4. [DOI] [PubMed] [Google Scholar]
- [52].Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–49. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kukkonen JP. A ménage à trois made in heaven: G-protein-coupled receptors, lipids and TRP channels. Cell Calcium. 2011;50:9–26. doi: 10.1016/j.ceca.2011.04.005. [DOI] [PubMed] [Google Scholar]
- [54].Allen JA, Halverson-Tamboli RA, Rasenick MM. Lipid raft microdomains and neurotransmitter signaling. Nat Rev Neurosci. 2007;8:128–40. doi: 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- [55].Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–72. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- [56].Chichili G, Rodgers W. Cytoskeleton-membrane interactions in membrane raft structure. Cell Mol Life Sci. 2009;66:2319–28. doi: 10.1007/s00018-009-0022-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Head BP, Patel HH, Roth DM, Murray F, Swaney JS, Niesman IR. Microtubules and actin microfilaments regulate lipid raft/ caveolae localization of adenylyl cyclase signaling components. J Biol Chem. 2006;281:26391–9. doi: 10.1074/jbc.M602577200. [DOI] [PubMed] [Google Scholar]
- [58].Li PL, Zhang Y, Yi F. Lipid raft redox signaling platforms in endothelial dysfunction. Antioxid Redox Signal. 2007;9:1457–70. doi: 10.1089/ars.2007.1667. [DOI] [PubMed] [Google Scholar]
- [59].Das M, Das DK. Lipid Raft in Cardiac Health and Disease. Curr Cardiol Rev. 2009;5:105–11. doi: 10.2174/157340309788166660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Michel V, Bakovic M. Lipid rafts in health and disease. Biol Cell. 2007;99:129–40. doi: 10.1042/BC20060051. [DOI] [PubMed] [Google Scholar]
- [61].Jin S, Zhou F, Katirai F, Li PL. Lipid raft redox signaling: molecular mechanisms in health and disease. Antioxid Redox Signal. 2011;15:1043–83. doi: 10.1089/ars.2010.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Horikawa YT, Patel HH, Tsutsumi YM, Jennings MM, Kidd MW, Hagiwara Y. Caveolin-3 expression and caveolae are required for isoflurane induced cardiac protection from hypoxia and ischemia/ reperfusion injury. J Mol Cell Cardiol. 2008;44:123–30. doi: 10.1016/j.yjmcc.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Patel HH, Tsutsumi YM, Head BP, Niesman IR, Jennings M, Horikawa Y. Mechanisms of cardiac protection from ischemia/reperfusion injury: a role for caveolae and caveolin-1. FASEB J. 2007;21:1565–74. doi: 10.1096/fj.06-7719com. [DOI] [PubMed] [Google Scholar]
- [64].Rapacciuolo A, Suvarna S, Barki-Harrington L, Luttrell LM, Cong M, Lefkowitz RJ. Protein kinase A and G protein-coupled receptor kinase phosphorylation mediates beta-1 adrenergic receptor endocytosis through different pathways. J Biol Chem. 2003;278:35403–11. doi: 10.1074/jbc.M305675200. [DOI] [PubMed] [Google Scholar]
- [65].Steinberg SF. β2-Adrenergic receptor signaling complexes in cardiomyocyte caveolae/lipid rafts. J Mol Cell Cardiol. 2004;37:407–15. doi: 10.1016/j.yjmcc.2004.04.018. [DOI] [PubMed] [Google Scholar]
- [66].Juhaszova M, Zorov DB, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Role of glycogen synthase kinase-3beta in cardioprotection. Circ Res. 2009;104:1240–52. doi: 10.1161/CIRCRESAHA.109.197996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Omar MA, Wang L, Clanachan AS. Cardioprotection by GSK-3 inhibition: role of enhanced glycogen synthesis and attenuation of calcium overload. Cardiovasc Res. 2010;86:478–86. doi: 10.1093/cvr/cvp421. [DOI] [PubMed] [Google Scholar]
- [68].Vigneron F, Dos Santos P, Lemoine S, Bonnet M, Tariosse L, Couffinhal T, Duplaà C, Jaspard-Vinassa B. GSK-3β at the crossroads in the signalling of heart preconditioning: implication of mTOR and Wnt pathways. Cardiovasc Res. 2011;90:49–56. doi: 10.1093/cvr/cvr002. [DOI] [PubMed] [Google Scholar]
- [69].Takahashi-Yanaga F, Sasaguri T. GSK-3beta regulates cyclin D1 expression: a new target for chemotherapy. Cell Signal. 2008;20:581–9. doi: 10.1016/j.cellsig.2007.10.018. [DOI] [PubMed] [Google Scholar]
- [70].Dohn MR, Brown MV, Reynolds AB. An essential role for p120- catenin in Src- and Rac1-mediated anchorage-independent cell growth. J Cell Biol. 2009;184:437–50. doi: 10.1083/jcb.200807096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Huelsken J, Birchmeier W. New aspects of Wnt signaling pathways in higher vertebrates. Curr Opin Genet Dev. 2001;11:547–53. doi: 10.1016/S0959-437X(00)00231-8. [DOI] [PubMed] [Google Scholar]
- [72].Daugherty RL, Gottardi CJ. Phospho-regulation of Beta-catenin adhesion and signaling functions. Physiology. 2007;22:303–9. doi: 10.1152/physiol.00020.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Dashwood WM, Carter O, Al-Fageeh M, Li Q, Dashwood RH. Lysosomal trafficking of beta-catenin induced by the tea polyphenol epigallocatechin-3-gallate. Mutat Res. 2005;591:161–72. doi: 10.1016/j.mrfmmm.2005.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Guo Y, Fu Z, Van Eyk J. A Proteomic Primer for the Clinician. Proc Am Thorac Soc. 2007;4:9–17. doi: 10.1513/pats.200608-156JG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].McGregor E, Dunn MJ. Proteomics of the Heart: Unraveling Disease. Circ Res. 2006;98:309–21. doi: 10.1161/01.RES.0000201280.20709.26. [DOI] [PubMed] [Google Scholar]
- [76].Arab S, Gramolini AO, Ping P, Kislinger T, Stanley B, van Eyk J. Cardiovascular Proteomics: Tools to Develop Novel Biomarkers and Potential Applications. J Am Coll Cardiol. 2006;48:1733–41. doi: 10.1016/j.jacc.2006.06.063. [DOI] [PubMed] [Google Scholar]
- [77].Scobioala S, Klocke R, Kuhlmann M, Tian W, Hasib L, Milting H. Up-regulation of nestin in the infarcted myocardium potentially indicates differentiation of resident cardiac stem cells into various lineages including cardiomyocytes. FASEB J. 2009;22:1021–31. doi: 10.1096/fj.07-8252com. [DOI] [PubMed] [Google Scholar]
- [78].Mateos-Cáceres PJ, García-Méndez A, Farré AL, Macaya C, Núñez A, Gómez J. Proteomic analysis of plasma from patients during an acute coronary syndrome. J Am Coll Cardiol. 2004;44:1578–83. doi: 10.1016/j.jacc.2004.06.073. [DOI] [PubMed] [Google Scholar]
- [79].Buerke B, Schwertz H, Längin T, Buerke U, Prondzinsky R, Platsch H. Proteome analysis of myocardial tissue following ischemia and reperfusion-Effects of complement inhibition. Biochimica et Biophysica Acta. 2006;1764:1536–45. doi: 10.1016/j.bbapap.2006.03.008. [DOI] [PubMed] [Google Scholar]
- [80].Sawicki G, Jugdutt BI. Detection of regional changes in protein levels in the in vivo canine model of acute heart failure following ischemia-reperfusion injury: functional proteomics studies. Proteomics. 2004;4:2195–2202. doi: 10.1002/pmic.200300746. [DOI] [PubMed] [Google Scholar]
- [81].Schwertz H, Langin T, Platsch H, Richert J, Bomm S, Schmidt M. Two-dimensional analysis of myocardial protein expression following myocardial ischemia and reperfusion in rabbits. Proteomics. 2002;2:988–95. doi: 10.1002/1615-9861(200208)2:8<988::AID-PROT988>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- [82].Chen CY, Lee BC, Hsu HC, Lin HJ, Chao CL, Lin YH. A proteomic study of the effects of ramipril on post-infarction left ventricular remodelling in the rabbit. Euro J Heart Fail. 2008;10:740–8. doi: 10.1016/j.ejheart.2008.06.001. [DOI] [PubMed] [Google Scholar]
- [83].Sakai J, Ishikawa H, Kojima S, Satoh H, Yamamoto S, Kanaoka M. Proteomic analysis of rat heart in ischemia and ischemia-reperfusion using fluorescence two-dimensional difference gel electrophoresis. Proteomics. 2003;3:1318–24. doi: 10.1002/pmic.200300432. [DOI] [PubMed] [Google Scholar]
- [84].De Celle T, Vanrobaeys F, Lijnen P, Blankesteijn WM, Heeneman S, Van Beeumen J. Alterations in mouse cardiac proteome after in vivo myocardial infarction: permanent ischaemia versus ischaemia-reperfusion. Exp Physiol. 2005;90:593–606. doi: 10.1113/expphysiol.2005.030296. [DOI] [PubMed] [Google Scholar]
- [85].Chou HC, Chen YW, Lee TR, Wu FS, Chan HT, Lyu PC. Proteomics study of oxidative stress and Src kinase inhibition in H9c2 cardiomyocytes: a cell model of heart ischemia-reperfusion injury and treatment. Free Radic Biol Med. 2010;49:96–108. doi: 10.1016/j.freeradbiomed.2010.04.001. [DOI] [PubMed] [Google Scholar]