

Abstract
Paroxysmal kinesigenic dyskinesia (PKD), a rare paroxysmal movement disorder often misdiagnosed as epilepsy, is characterized by recurrent, brief dyskinesia attacks triggered by sudden voluntary movement. Pathophysiological mechanism of PKD remains not well understood. Ion channelopathy has been suggested, since the disease responds well to ion channel blockers. Mutations in proline-rich transmembrane protein 2 (PRRT2) were recently identified in patients with familial PKD. To extend these genetic reports, we studied a family with clinical manifestations of familial PKD responding well to low dose carbamazepine. Therapeutic dose ranged from 1.5 to 2.0 mg/ kg/day, below that in seizure control. One insertion mutation c.649_650insC (p.P217fsX7) was identified in three patients of the family. This study avers PRRT2’s high sensitivity for PKD phenotype. Identification of genes underlying pathogenesis will enhance diagnosis and treatment. Function of PRRT2 and its role in PKD warrant further investigation.
Keywords: Carbamazepin, Paroxysmal dyskinesia, PRRT2, Mutation
1. Introduction
Paroxysmal kinesigenic dyskinesia (PKD, OMIM 128000) is a rare paroxysmal movement disorder, often misdiagnosed as epilepsy and characterized by recurrent, brief attacks of dyskinesia triggered by sudden voluntary movement. Onset is around early teenage [1]. Sudden movement after a prolonged rest period is the most common precipitating factor, duration of attack usually brief, lasting seconds up to five minutes [2-5]. They can occur many times daily, but frequency and severity of attack seem to decrease with age. Brain imaging and electroencephalography (EEG) of such patients are usually normal [1, 6] Pathophysiological mechanism of PKD remains not well understood, but studies suggest it as a type of reflex epilepsy [7-9]. Ion channelopathy has been cited; this disease responds well to ion channel blockers [10]. Mutations of proline-rich transmembrane protein 2 (PRRT2) have recently been identified in cases of familial PKD [11-15]. PRRT2 protein, expressed strongly in a developing nervous system and localized to axons, interacts with synaptic protein SNAP 25, and may play a role in synaptic regulation [11, 14]. We report a family with clinical manifestations of familial PKD responding to low-dose CBZ. One insertion mutation c.649_650insC (p.P217fsX7) was identified in three patients of the family.
2. Patients and Methods
2.1. Clinical Report
A twelve-year-old male had two-year history of paroxysmal movement disorder induced by sudden movement or brief exercise; birth, delivery, and development were unremarkable, without significant medical history. Attacks were first evident at age 10, after sudden arm movements; these entailed dystonic flexion and greater internal rotation of left arm and left leg, as well as finger posturing. He was fully aware of his surroundings during the attacks (4-5 times/day), lasting 10-20 (rarely 30) seconds. General physical and neurologic examinations proved normal. Magnetic resonance imaging of the brain, interictal electroencephalogram, and laboratory workup yielded unremarkable results; carbamazepine was prescribed upon diagnosis of paroxysmal kinesigenic dyskinesia. At 200 mg daily (10 mg/kg/day), the patient attained complete resolution of signs that has persisted for the past seven years, no side effects from medication. His father and brother, both diagnosed as PKD, had the same symptoms (Fig. A) ; Table 1 summarizes clinical manifestations. Both were prescribed CBZ as monotherapy (dosage 200 mg/day) to attain total resolution for the past seven years.
Table 1.
Clinical Summary of Three Patients with Paroxysmal Kinesigenic Dyskinesia.
| Members of family | Patient 1 | Patient 2 | Patient 3 |
|---|---|---|---|
| Age (years) | 46 | 14 | 12 |
| Gender | male | male | male |
| Age of onset (years) | 10 | 10 | 10 |
| Clinical feature Dystonia | + | + | + |
| Chreoathetosis | - | + | + |
| Distribution Bilateral or alternating sides | + | Left side only | Left side only |
| Frequency | variable | 4-5times/day | 4-5times/day |
| Precipitating factor Sudden movement | + | + | + |
| Anxiety | + | - | + |
| EEG/MRI | -- | AbN/N | AbN/N |
| Epilepsy | - | - | - |
| AED | CBZ | CBZ | CBZ |
| Response to AED | -- | Effective | Effective |
EEG: eletroencephalogram
MRI: magnetic resonance image
AED: Antiepileptic drug CBZ: carbamazepine
AbN: abnormal
N: normal
2.2. Mutation Analysis
After written consent, genomic DNA was extracted from peripheral leukocytes by AxyPrep Blood Genomic DNA kit (Axygen Biosciences, Union City, CA). Our mutation analysis of PRRT2 used direct sequencing after PCR amplification (primer sequences available on request) in three patients, with amplified fragments directly sequenced by BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA) and run on ABI PRISM 3130_l Genetic Analyzer (Applied Biosystems). Results show p.R217Pfs*8 (c.649_650insC) in three patients (Fig. B).
Fig. 1.
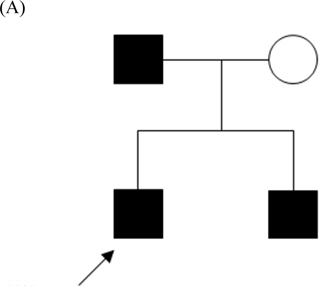
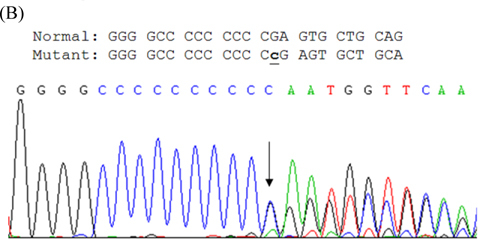
(A) Pedigree of PKC/PKD Taiwanese family: persons designated by sex, disease status (filled symbols represent patients, open symbols normal persons). Index case indicated by arrow. (B) Sequencing results of mutation in PRRT2 gene of index case. Arrow indicates one-base C inserted at nucleotide 650 (c.650insC), causing protein translation shift and stopping after seventh residue.
3. Discussion
PKD is the most common form of paroxysmal dyskinesia. Clinical features of our patients resembled cases reported previously: e.g., precipitating factors, patterns of attack, age of onset [1, 2, 4, 5, 6, 16]. Overall, family members had clinical features similar to those of individuals in literature. Controversial surrounds pathophysiology of involuntary movements. It is still uncertain whether symptoms relate to epileptic seizure or are dysfunction of basal ganglia [17, 18, 19]. Some propose this disorder as epileptic syndrome, based on prodromata preceding attacks, as well as its response to anticonvulsants. Interictal EEG abnormalities of reported PKD cases include sporadic epileptic discharge or slow rhythm [7, 8, 9]. None of our recordings showed any such abnormalities. Also, consciousness during attacks was always preserved.
Patients with PKD attacks seem to respond well to anticonvulsants: lamotrigine, phenytoin, valproate, oxacarbazepine, and especially CBZ [5, 6, 20, 21, 22], whose mechanism is blockade of ion conduction through voltagedependent ion channels of the neuron. Physiology of PKD is still uncertain, but ion channelopathy is considered; prior studies reported PKD patients sensitive to ion channel blockers like CBZ [6, 21]. CBZ is widely used, being inexpensive and broad-spectrum in seizure controls; we found 100mg/day effectively controlling PKD. Therapeutic dose ranged from 1.5 to 2.0 mg/kg/day, lower than that in seizure control. No patients treated with CBZ had intellectual impairment or decline of school performance during follow-up.
Seventeen PRRT2 mutations were recently identified in patients with familial or apparently sporadic PKD/IC from several ethnic groups [11-15, 23], the vast majority (11/17) premature termination or frameshift mutation resulting in truncation of PRRT protein, the rest missense mutations. Interestingly, p.R217P*8 arose in about 80% of PKD patients with PRRT2 mutations [11-15, 23]. All identified families displayed autosomally dominant inheritance. Function of PRRT2 protein is poorly characterized. Recent in vivo and in vitro studies demonstrated that PRRT2 is highly expressed in the developing nervous system and localized on cell membrane, predominantly in axons [11, 14]. It is not surprising that truncating mutations can significantly reduce protein expression and/or cause loss of transmembrane property, which conceivably impairs PRRT2 protein function [11, 14]. This study highlights frequency of PRRT2 mutations in a Taiwanese cohort with idiopathic PKD. For this benign neurological condition, low dose of CBZ is adequate to provide good control.
Acknowledgments
This study was supported in part by the China Medical University Hospital (grant number DMR-103-034).
References
- [1].Houser MK, Soland VL, Bhatia KP, Quinn NP, Marsden CD. Paroxysmal inesigenic choreoathetosis: a report of 26 patients. J Neurol. 1999;246:120–26. doi: 10.1007/s004150050318. [DOI] [PubMed] [Google Scholar]
- [2].Demirkiran M, Jankovic J. Paroxysmal dyskinesias: clinical features and classification. Ann Neurol. 1995;38:571–79. doi: 10.1002/ana.410380405. [DOI] [PubMed] [Google Scholar]
- [3].Bhatia KP. The paroxysmal dyskinesias. J Neurol. 1999;246:149–55. doi: 10.1007/s004150050325. [DOI] [PubMed] [Google Scholar]
- [4].Nagamitsu S, Matsuishi T, Hashimoto K. Multicenter study of paroxysmal dyskinesias in Japan: clinical and pedigree analysis. Mov Disord. 1999;4:658–63. doi: 10.1002/1531-8257(199907)14:4<658::AID-MDS1016>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- [5].Zorzi G, Conti C, Erba A, Granata T, Angelini L, Nardocci N. Paroxysmal yskinesias in childhood. Pediatr Neurol. 2003;28:168–72. doi: 10.1016/S0887-8994(02)00512-X. [DOI] [PubMed] [Google Scholar]
- [6].Hwang WJ, Lu CS, Tsai JJ. Clinical manifestations of 20 Taiwanese patients with paroxysmal kinesigenic dyskinesia. Acta Neurol Scand. 1998;98:340–45. doi: 10.1111/j.1600-0404.1998.tb01745.x. [DOI] [PubMed] [Google Scholar]
- [7].Ohmori I, Ohtsuka Y, Ogino T, Yoshinaga H, Oka E. Relationship between paroxysmal kinesigenic choreoathetosis and epilepsy. Neuropediatrics. 2002;33:15–20. doi: 10.1055/s-2002-23594. [DOI] [PubMed] [Google Scholar]
- [8].Tan LC, Tan AK, Tjia H. Paroxysmal kinesigenic choreoathetosis in Singapore and its relationship to epilepsy. Clin Neurol Neurosurg. 1998;100:187–92. doi: 10.1016/S0303-8467(98)00038-9. [DOI] [PubMed] [Google Scholar]
- [9].Hirata K, Katayama S, Saito T. Paroxysmal kinesigenic choreoathetosis with abnormal electroencephalogram during attacks. Epilepsia. 1991;32:492–94. doi: 10.1111/j.1528-1157.1991.tb04682.x. [DOI] [PubMed] [Google Scholar]
- [10].Ptacek LJ. Channelopathies: ion channel disorders of muscle as a paradigm for paroxysmal disorders of the nervous system. Neuromuscul Disord. 1997;7:250–55. doi: 10.1016/S0960-8966(97)00046-1. [DOI] [PubMed] [Google Scholar]
- [11].Chen WJ, Lin Y, Xiong ZQ, Wei W, Ni W. Exome sequencing dentifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic yskinesia. Nat Genet. 2011;43:1252–55. doi: 10.1038/ng.1008. [DOI] [PubMed] [Google Scholar]
- [12].Wang JL, Cao L, Li XH, Hu ZM, Li JD. Identification of PRRT2 asthe causative gene of paroxysmal kinesigenic dyskinesias. Brain. 2012;134:490–98. doi: 10.1093/brain/awr289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li J, Zhu X, Wang X, Sun W, Feng B. Targeted genomic sequencing identifies PRRT2 mutations as a cause of paroxysmal kinesigenic choreoathetosis. J Med Genet. 2012;49:76–78. doi: 10.1136/jmedgenet-2011-100635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lee HY, Huang Y, Bruneau N, Roll P, Roberson EDO. Mutations in the novel protein PRRT2 cause paroxysmal kinesigenic dyskinesia with infantile convulsion. Cell Reports. 2012;1:1–11. doi: 10.1016/j.celrep.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Heron SE, Grinton BE, Kivity S, Afawi Z, Zuberi SM. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am J Hum Genet. 2012;90:152–60. doi: 10.1016/j.ajhg.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Choi IS, Kim JH, Jung WY. Paroxysmal kinesigenic choreoathetosis. Yonsei Med J. 1996;37:68–71. doi: 10.3349/ymj.1996.37.1.68. [DOI] [PubMed] [Google Scholar]
- [17].Ko CH, Kong CK, Ngai WT, Ma KM. Ictal (99m)Tc ECD SPECT in paroxysmal kinesigenic choreoathetosis. Pediatr Neurol. 2001;24:225–27. doi: 10.1016/S0887-8994(00)00257-5. [DOI] [PubMed] [Google Scholar]
- [18].Perlmutter JS, Raichle ME. Pure hemidystonia with basal ganglion abnormalities on positron emission tomography. Ann Neurol. 1984;15:228–33. doi: 10.1002/ana.410150303. [DOI] [PubMed] [Google Scholar]
- [19].Hayashi R, Hanyu N, Yahikozawa H, Yanagisawa N. Ictal muscle discharge pattern and SPECT in paroxysmal kinesigenic choreoathetosis. Electromyogr Clin Neurophysiol. 1997;37:89–94. [PubMed] [Google Scholar]
- [20].Wein T, Andermann F, Silver K, Dubeau F, Andermann E, Rourke-Frew F, Keene D. Exquisite sensitivity of paroxysmal kinesigenic choreoathetosis to carbamazepine. Neurology. 1996;47:1104–06. doi: 10.1212/WNL.47.4.1104-a. [DOI] [PubMed] [Google Scholar]
- [21].Gokcay A, Gokcay F. Oxcarbazepine therapy in paroxysmal kinesigenic choreoathetosis. Acta Neurol Scand. 2000;101:344–45. doi: 10.1034/j.1600-0404.2000.9l3331.x. [DOI] [PubMed] [Google Scholar]
- [22].Uberall MA, Wenzel D. Effectiveness of lamotrigine in children with paroxysmal kinesigenic choreoathetosis. Dev Med Child Neurol. 2000;42:699–700. doi: 10.1017/S0012162200001286. [DOI] [PubMed] [Google Scholar]
- [23].Liu Q, Qi Z, Wan XH, Li JY, Shi L. Mutations in PRRT2 result in paroxysmal dyskinesias with marked variability in clinical expression. J Med Genet. 2012;49:79–82. doi: 10.1136/jmedgenet-2011-100653. [DOI] [PubMed] [Google Scholar]