

Abstract
Object
Ischemic brain injury is the leading cause for death and long-term disability in patients who suffer cardiac arrest and embolic stroke. Excitotoxicity and subsequent Ca2+-overload lead to ischemic neuron death. We explore a novel mechanism concerning the role of the excitatory extracellular calcium-sensing receptor (CaSR) in the induction of ischemic brain injury.
Method
Mice were exposed to forebrain ischemia and the actions of CaSR were determined after its genes were ablated specifically in hippocampal neurons or its activities were inhibited pharmacologically. Since the CaSR forms a heteromeric complex with the inhibitory type B γ-aminobutyric acid receptor 1 (GABABR1), we compared neuronal responses to ischemia in mice deficient in CaSR, GABABR1, or both, and in mice injected locally or systemically with a specific CaSR antagonist (or calcilytic) in the presence or absence of a GABABR1 agonist (baclofen).
Results
Both global and focal brain ischemia led to CaSR overexpression and GABABR1 downregulation in injured neurons. Genetic ablation of Casr genes or blocking CaSR activities by calcilytics rendered robust neuroprotection and preserved learning and memory functions in ischemic mice, partly by restoring GABABR1 expression. Concurrent ablation of Gabbr1 gene blocked the neuroprotection caused by the Casr gene knockout. Coinjection of calcilytics with baclofen synergistically enhanced neuroprotection. This combined therapy remained robust when given 6 h after ischemia.
Interpretation
Our study demonstrates a novel receptor interaction, which contributes to ischemic neuron death through CaSR upregulation and GABABR1 downregulation, and feasibility of neuroprotection by concurrently targeting these two receptors.
Introduction
Excitotoxicity and subsequent calcium overload is well known to contribute to ischemic neuron death.1–3 Ischemia leads to the loss of transmembrane ATPases and ion transporters/exchangers, leading to disruption of ionic gradients, and cell depolarization.1–4 This leads to excessive releases of the excitatory neurotransmitter glutamate, which, when bound to its receptors, leads to increased Ca2+ permeability.1,4–6 Accumulation of intracellular [Ca2+] ([Ca2+]i) to pathologically high levels promotes neuronal death.1,7,8
Several pharmaceutical approaches have been studied in the past, to target neuronal hyperactivity at the early phase of the disease.3,5,9,10 Antagonists against the NMDA (N-methyl-d-aspartate) receptors were neuroprotective in preclinical studies with a narrow temporal therapeutic window.6,11,12 These drugs, however, were ineffective or even produced negative outcomes when given at late stages of the disease.6,11,12 At the clinical level, trials of this class of compounds were disappointing.13
Another approach to reduce excitotoxicity is to increase inhibitory neurotransmission.14–18 γ-Aminobutyric acid (GABA) inhibits neuronal excitability in the brain by signaling through ionotropic (type A and C) GABA receptors and metabotropic (type B) GABA receptors (GABABRs).19,20 Reducing GABABR signaling in the brain induces neuronal hyperactivity and produces seizures and correlates with increased hippocampal neuron death, while GABABR agonists such as baclofen and CGP-35348 suppress seizures.19,20 Studies of animal models support the role of GABABRs in the injury response to ischemia. Transient global ischemia (TGI) in gerbils markedly reduced both GABABR1 and R2 transcripts in pyramidal cell layers of the hippocampus in conjunction with neuronal death.21,22 Brain biopsies from patients with temporal lobe epilepsy, trauma, anoxic injury, and other conditions also showed reduced GABABR1 expression.23 Baclofen, a specific GABABR agonist, has been shown to be neuroprotective,14,16,19,20,24–26 but only at a higher dose that also caused side effects.16
GABABRs (R1 and R2) are members of the family C of the G-protein coupled receptor (GPCR) superfamily, which also includes the extracellular Ca2+-sensing receptor (CaSR) and metabotropic glutamate receptors (mGluRs).27,28 The CaSR was originally identified in parathyroid cells where its activation suppresses the synthesis and secretion of parathyroid hormone and thereby controls systemic mineral homeostasis.29,30 The CaSR was latter found in many other tissues, including cartilage, bone, kidney, breast, intestinal epithelium, lung, and brains.31,32 The CaSR RNA transcript has been detected in many subdivisions of the brain, including hippocampus, neocortex, and hypothalamus.33 In cultured neurons, activation of CaSR stimulates different types of ion channels, activates phospholipase C (PLC), and increases [Ca2+]i in soma.34–37 At nerve terminals, the CaSR regulates membrane excitability38,39 and modulates neurotransmitter release.40,41 However, in vivo biological functions of these stimulatory pathways remain largely unexplored in the central nervous system (CNS). Although defects in neurite growth were observed in the global CaSR knockout (KO) mice42 and implicate a role for the CaSR in brain development, the severe metabolic phenotypes and early death of these KO mice29 preclude definitive assessment of the function of the CaSR in the brain, particularly during postnatal growth or in response to pathological conditions, like ischemia and traumatic brain injuries.
Interestingly, the CaSR and GABABR1 are coexpressed in many regions of the brain,33 including cortical and hippocampal neurons, in the form of CaSR/GABABR1 heteromeric complexes.27,43 Inhibition of GABABR1 expression robustly increased CaSR expression in hippocampal neurons.27 Coexpression of GABABR1 suppressed both total and cell-surface expression of CaSR protein and its signaling (PLC activation) in transfected HEK-293 cells, via direct posttranslational protein–protein interactions.27 These observations suggest that stoichiometric interactions of CaSR and GABABR1 may impact the excitatory states of neurons in physiological and/or pathological conditions. The current study took genetic and pharmacological approaches to define the interplay between CaSR and GABABR1 in neurons subjected to ischemic insults and developed preclinical regimens for neuroprotection by targeting these two receptors.
Materials and Methods
Animals and ischemic injury models
All experiments were performed on 4- or 6-month-old male mice in C57/B6 background or 9-month-old Sprague–Dawley rats (Charles River, Hollister, CA, USA) weighing between 270–300 g. HippCaSR−/− mice were generated by breeding the floxed-CaSR30 mice with Ca2+/calmodulin-dependent protein kinases II alpha subunit (CAMKIIa)-Cre mice,44 while HippCaSR−/−//GABABR1+/− and HippCaSR−/−//GABABR1−/− mice were produced by breeding HippCaSR−/− mice with floxed-GABABR1 mice.45 Mice were anesthetized with isoflurane (5% for induction, 2% for maintenance via a facemask) in a mixture of air/oxygen (3:1). Transient global cerebral ischemia (TGI) and middle cerebral artery occlusion (MCAO) were performed as described previously.27,46,47 For TGI, both common carotid arteries (CCA) were occluded for 10–15 min, followed by reperfusion. For MCAO in mice, a small craniotomy was made to open the dura and the MCA was occluded by short coagulation with a bipolar electrode at a segment proximal to the olfactory branch. In male rats, MCAO was induced by inserting an uncoated 30-mm segment of 3-0 nylon monofilament suture (Harvard Apparatus, Holliston, MA) with an enlarged tip into the stump of the CCA, and advanced into the internal carotid artery to 18–20 mm from the bifurcation of the internal and external carotid arteries to occlude the ostium of the MCA. The suture was left in the place for 2 h.47 Sham-operated mice or rats were subjected to the same surgical procedures and anesthetic exposure, with the exception that the CCA or MCA were not occluded. Core temperatures of the mice and rats were monitored and maintained by a heating blanket and rectal thermistor servo-loop. Mice and rats were recovered for 1–21 days.
Administration of calcilytics
CaSR antagonists (or calcilytics), NPS89636 and NPS2143, were provided by Dr. Edward Nemeth (MetisMedica, Toronto, Canada) or purchased from Tocris Bioscience (Bristol, UK). Intracerebroventricular (ICV) injections took place 30 min after TGI or Sham surgeries or at other times specified. Briefly, anesthetized mice were placed in a stereotaxic frame and calcilytics (1 ng in 1 μL phosphate-buffered saline (PBS)) or vehicle (0.1% Dimethyl sulfoxide (DMSO) in 1 μL PBS) was injected into the right lateral cerebral ventricle (stereotaxic coordinates: 1 mm caudal to bregma, 1.3 mm lateral to sagittal suture, and 2 mm in depth) at a speed of 2 μL/min via a burr hole. The needle was left in place for 5 min to allow drug diffusion into tissues before it was retrieved and then the hole in the skull was filled with bone wax. Intraperitoneal (IP) injections of calcilytics and/or baclofen (1 mg/kg body wt) were given at different time points after TGI or Sham surgeries and continued daily for a specified duration of time.
Hematoxylin and eosin and TUNEL staining
Brains were prepared and analyzed by terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) staining after 24 and 72 h of reperfusion as described previously.46 Brains were dissected, postfixed in 4% paraformaldehyde (PFA), perfused with 20% sucrose at 4°C for 24 h, frozen, and cryosectioned (10 μm). Hematoxylin and eosin (H&E)-stained sections were used to determine the extent of ischemic injury in hippocampal cornu ammonis (CA)1, CA3, and Dentate gyrus (DG). In order to determine the number of apoptotic neurons, adjacent brain sections were subjected to TUNEL using ApopTag Plus Peroxidase In Situ Apoptosis Detection Kit (EMD Millipore, Billerica, MA) following the manufacturer’s instructions. Fixed cryosections were incubated with terminal deoxynucleotidyl transferase (TdT) and digoxigenin-labeled dUTP, followed by a peroxidase-conjugated antidigoxigenin antibody and 3,3′-diaminobenzidine (DAB) substrate. For negative controls, sections were stained with the same procedures in the absence of TdT. TUNEL-(+) cells were counted in three separate fields of each region (CA1, CA3, and DG) of hippocampus from 4 to 6 mice in each group by an investigator blinded to the treatment groups. The number of TUNEL-(+) cells was normalized to total neuronal numbers (~1672, 1227, and 2558 in CA1, CA3, and DG, respectively, in the Sham control mice) acquired from adjacent sections stained with H&E.
Immunohistochemistry
To detect CaSR and GABABR1 expression, cryosections of brain were incubated sequentially with 0.1% H2O2 for 3 min, a blocking buffer (0.5% Triton X-100, 0.1% bovine serum albumin (BSA), 1.5% normal horse serum in PBS) for 30 min, and polyclonal rabbit anti-CaSR (10 μg/mL) or guinea pig anti-GABABR1 (5 μg/mL) antibodies overnight at 4°C.27,43 Biotinylated anti-rabbit IgG (5 μg/mL; Vector Laboratories, Burlingame, CA) or anti-guinea pig IgG (2 μg/mL; Sigma-Aldrich, St. Louis, MO), peroxidase-conjugated avidin (ABC Elite; Vector Laboratories), and DAB-substrate were used to amplify and detect immunoreactivity. For negative controls, anti-CaSR and anti-GABABR1 were preabsorbed with corresponding peptides (100-fold excess per molarity), against which the antibodies were raised, for 30 min at 37°C, or omitted in the assays. Sections were counterstained with hematoxylin or Cresyl violet. Specificity of the antibody was confirmed by the absence of signals in sections treated with peptide-preabsorbed antisera or with secondary antibodies alone (data not shown).
Visualization of cerebral vasculature
Mice were anesthetized with a cocktail of ketamine/acepromazine, heparinized, and sequentially perfused via the left ventricle with 25 mL of PBS, 4% PFA, and a colored (red) contrasting agent MICROFIL® (Flow Tech Inc., Carver, MA, USA). Following perfusion, mice were maintained at 4°C allowing Microfil to polymerize. Next day the brain was excised and fixed in cold PFA for 24 h, and imaged with a M205C stereo-microscope (Leica Microsystems Inc, Buffalo Grove, IL).
Neuronal culture and adenoviral infection
Mouse hippocampal neurons were isolated and cultured from floxed-GABABR1 mice as described previously.27,46 Cells were cultured in poly-d-lysine-coated (0.1 mg/mL) plastic dishes and/or glass coverslips in a neuron maintenance medium27,46 for 3 days and supplemented with cytosine β-d-arabinofuranoside (3 μmol/L; Sigma, St. Louis, MO, USA) for an additional 3–4 days. These neurons were then infected with adenoviruses (8 pfu/cell) carrying a cDNA encoding Cre recombinase (Ad-Cre; Microbix Inc., Toronto, Canada) or empty viral vector (Ad-Cont Microbix Inc., Toronto, Canada) as described previously.27,46
Immunoblotting
Preparation of crude membranes from brains and cultured neurons and immunoblotting were performed as described previously.27,46 Proteins were extracted from these membranes with 1% Nonidet P-40 in PBS in the presence of a protease inhibitor mixture (Complete; Roche Applied Science, Indianapolis, IN, USA). Fifty microgram hippocampus membrane proteins were electrophoresed on a sodiumdodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel, and transferred to polyvinylidine fluoride (PVDF) membranes. PVDF membranes were incubated with anti-CaSR (5 μg/mL), anti-GABABR1 (1 μg/mL), or anti-β-actin (0.5 μg/mL), followed by corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies. Immunoreactivities were detected by a SuperSignal West Dura chemiluminescent substrate (Thermo Scientific, Waltham, MA) and X-ray films (Eastman Kodak Company, Rochester, NY).
Behavior test
Spatial learning and memory were evaluated by the Morris Water Maze (MWM) test starting 14 days after TGI and specified treatments as described previously with modifications.48,49 Briefly, mice were trained to locate a platform in opaque water during two successive daily sessions with a 1-h intersession interval. During the cued test on day 1–2, a submerged but flagged platform was placed in a different quadrant in every session. On day 3–5 or 3–6, mice were trained to locate a hidden platform (no flag) at a fixed location. Latency, pathlength, swim speeds and percent time spent swimming in the zone containing the platform were recorded using a video tracking system (Noldus Information Technology, Tacoma, WA). On day 6 or 7, a 1-min probe trial, in which the platform was removed, was performed to test memory retention.
Statistical analysis
Statistics were performed by Student’s t-test for measurements of TUNEL-(+) neurons and by two-way repeated measure analysis of variance (RANOVA) for behavior tests. P-values <0.05 were considered significant. All data were expressed as mean ± SE.
Results
Ischemia induces CaSR overexpression and GABABR1 downregulation
We observed concurrent CaSR overexpression and GABABR1 downregulation that were closely associated with the death of hippocampal neurons in mice subjected to TGI procedures mimicking episodes of cardiac arrest by standard transient (15 min) occlusion of both CCA.46 The TGI-induced changes in CaSR and GABABR1 expression took place within 24 h of brain reperfusion (Fig. 1A and B) and sustained until neuronal death that usually occurred about 48–72 h after reperfusion in our mouse model (Fig. 1C).46 In the latter figure, blue/purple eosin nuclear staining demonstrates a significant reduction in the number of nuclei, which also show severe shrinkage, while the hematoxylin cytoplasm staining shows significantly reduced cellularity in all regions of hippocampus in mice 3 days after TGI. These morphological changes coincided with increases in the number of TUNEL-(+) neurons in the same regions of hippocampus in the injured mice. There is no remarkable change in brain morphology (H&E or TUNEL) in mice 1 day after TGI (Fig. 1C), suggesting that CaSR overexpression and GABA-B-R1 downregulation preceded neuronal injury after TGI. Similar overexpression of CaSR was also observed in cortical neurons at infarct sites in rats or mice subjected to focal brain ischemia caused by transient MCAO50 (Fig. 1D), representing a common ischemic injury response regardless of animal origin or ischemic methods. Interestingly, these changes in receptor expression were mitigated by a hypothermia regimen that also protected neurons from ischemic injury.46 The above data suggest that interplay between CaSR and GABABR1 critically regulates neuronal survival.
Figure 1.
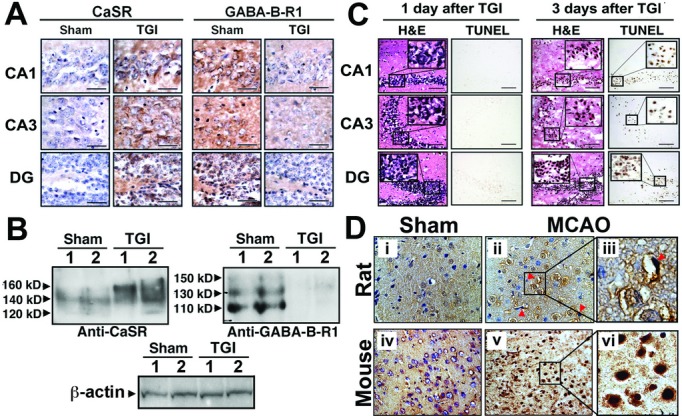
Ischemia induced CaSR overexpression, GABABR1 downregulation, and neuronal death in TGI and MCAO models. (A) Immunohistological detection and (B) immunoblotting of CaSR and GABABR1 in the hippocampi from 6 months old Sham-treated mice and those subjected to TGI by transient occlusion of both common carotid arteries for 15 min, followed by 1 day reperfusion. (A) CaSR and GABABR1 signals are depicted by brown DAB staining. Brain sections were counterstained with hematoxylin. Bar: 20 μm. (B) In the left panel, 120, 140, and 160 kDa protein bands depicted by CaSR-specific antisera represent the protein core and two highly glycosylated receptors, respectively. Similarly, in the right panel, 110, 130, and 150 kDa protein bands represent the protein core and glycosylated forms of the GABABR1, respectively. β-actin was used as loading control. Lanes 1 and 2 are samples from two separate mice. (C) H&E and TUNEL staining in the hippocampi of 6 months old TGI-treated mice, followed by 1 or 3 days reperfusion, indicates that neuronal apoptosis occurs after CaSR overexpression and GABABR1 downregulation. N = 6–8 mice/group. (D) Immunohistological detection of CaSR in cerebral cortex of 8-week-old rats (ii, iii) and 3-month-old mice (v, vi), which had been subjected to MCAO, shows profound increases in CaSR expression in the affected neurons when compared to sham-treated rats (i) and mice (iv). (iii) and (vi) are digitally enlarged views of the regions of interest boxed in panels (ii) and (v), respectively. CaSR signal is depicted by brown DAB staining. Sections are counterstained with hematoxylin. CaSR, Ca2+-sensing receptor; GABABR1, γ-aminobutyric acid receptor 1; TGI, transient global ischemia; MCAO, middle cerebral artery occlusion; DAB, 3,3′-diaminobenzidine; TUNEL, transferase dUTP nick end-labeling.
Blocking CaSR expression and activity renders robust neuroprotection
To determine whether the CaSR overexpression causally induces neuronal death following ischemia, we generated HippCaSR−/− mice (in C57/B6 background), whose Casr genes were deleted postnatally (3 weeks after birth) in most hippocampal neurons and some cortical neurons, but not hypothalamic neurons. These mice were made by breeding the floxed-CaSR mice, which carry loxP sequences flanking the exon 7 of the gene30 (Fig. 2A), with mice expressing Cre recombinase under the control of a CAMKIIa gene promoter that only becomes active 3 weeks after birth.44 This strategy is aimed to prevent confounding effects of CaSR KO on the brain during embryonic and early postnatal development, particularly in the hypothalamus where the CaSR mediates various neuroendocrine functions (Park-Sigal J, et al, unpubl. ms.). Genomic cDNA analyses confirmed the excision of CaSR gene alleles in the hippocampi of HippCaSR−/− mice (Fig. 2B). Immunohistochemistry with antisera against the exon 7–encoded C-terminal tail of the CaSR further confirmed the ablation of CaSR protein in all regions of the hippocampi in HippCaSR−/− mice (Fig. 2C). There was, however, no remarkable change in the number of neurons by H&E (Fig. 3A and B, Cont-Sham vs. HippCaSR−/−-Sham) and silver (data not shown) staining or in their apoptosis (Fig. 3C, Cont-Sham vs. HippCaSR−/−-Sham) by TUNEL staining in the HippCaSR−/− versus control (Cont) hippocampi. Nor did we observe apparent changes in their vasculature as indicated by the comparable vasculature patterns and sizes of their posterior communicating arteries (PCOM) (Fig. 2D) or in their neurobehaviors at a physiological state as assessed by MWM test (Fig. 2E). These data indicate unremarkable effects of postnatal CaSR KO (3 weeks after birth) on regular hippocampal functions in adult mice. The pathways compensating for the loss of CaSR functions in the HippCaSR−/− mice are unclear. Ablating CaSR expression, however, strongly protected the hippocampal neurons from TGI-induced injury, as indicated by significant preservation of neurons as visualized by H&E staining and reduced numbers of apoptotic cells as determined by TUNEL staining in all regions of hippocampi in TGI-treated HippCaSR−/− versus control mice (Fig. 3). In control (Cont) mice, TGI significantly reduced the numbers of neurons in the CA1, CA3, and DG regions of hippocampi by 55 ± 4%, 52 ± 3%, and 42 ± 3%, respectively (Fig. 3A, Cont-TGI vs. Cont-Sham; 3B, ▪), with 90 ± 4%, 78 ± 5%, and 69 ± 4% of the remaining neurons being TUNEL-positive [TUNEL-(+)] (Fig. 3C, Cont-TGI vs. Cont-Sham; 3D, ▪), indicating a severe injury response to TGI in Cont mice. In the hippocampi of the TGI-treated HippCaSR−/− mice, 70 ± 2%, 72 ± 4%, and 75 ± 2% of neurons were preserved (Fig. 3A, HippCaSR−/−-TGI vs. HippCaSR−/−-Sham; 3B, □) in the CA1, CA3, and DG, respectively, with only 50 ± 5%, 30 ± 4%, 22 ± 2% of them being TUNEL-(+) (Fig. 3C, HippCaSR−/−-TGI vs. HippCaSR−/−-Sham; 3D, □), indicating reduced injury responses in the absence of CaSR. These data support a causal role for the CaSR overexpression in inducing neuronal injury at least in a short-term response to ischemia.
Figure 2.
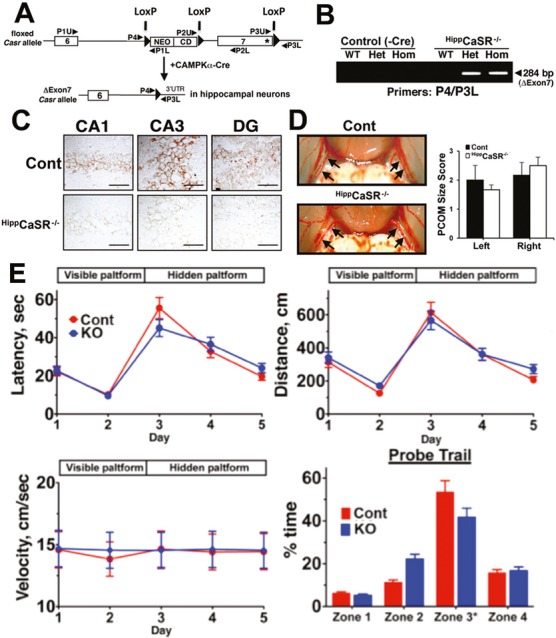
Generation and basal phenotype of HippCaSR−/− mice with CaSR KO targeted to hippocampus. (A) The HippCaSR−/− mice were made by breeding floxed-CaSR mice, which carried loxP sequences flanking the exon 7 of Casr genes, with CAMPKIIa-Cre mice expressing bacteriophage P1 Cre recombinase under the control of the CAMPKIIa gene promoter. (B) PCR analyses of genomic DNAs from hippocampi of control (Casrwt/wt, Casrwt/flox, and Casrflox/flox mice without Cre expression), heterozygous HippCaSR+/− (Het), and homozygous HippCaSR−/− (Hom) mice were performed with specific primers to confirm the excision of exon 7 as indicated by the presence of a 284-bp cDNA band (ΔExon7) in the Het and Hom KO mice. (C) Immunohistological detection of CaSR in the CA1, CA3, and DG of the hippocampi from 6 months old HippCaSR−/− and control (Cont) mice shows that CaSR expression, depicted by brown DAB staining, in different regions of hippocampus in Cont mice is abrogated in the same regions of the KO mice. Bar: 20 μm. (D) Vasculature patterns and sizes in Cont and HippCaSR−/− mice were examined after infusion with MICROFIL™ contrasting reagent (in red) (Flow Tech Inc., MA). Quantification scores for the sizes of left and right posterior communicating (PCOM) arteries shown in the right panel indicate comparable vascular sizes in the Cont and HippCaSR−/− mice. N = 4–6 mice. (E) Neurobehaviors were assessed by MWM tests on 4 months old male mice as described in Materials and Methods. Latency, distance traveled, and swimming speed during visible-platform (day 1–2) and hidden-platform (day 3–5) sections are not different between HippCaSR−/− (KO) and Cont littermates, indicating comparable ability of spatial learning. Nor did the % time spent in target zone (Zone 3), where the platform was set previously, differ in the final probe trial between the Cont and KO mice, suggesting comparable memory retention. Mean ± SE; n = 7 mice per group. CaSR, Ca2+-sensing receptor; KO, knockout; CA, cornu ammonis; DG, dentate gyrus; DAB, 3,3′-diaminobenzidine; MWM, Morris water maze.
Figure 3.
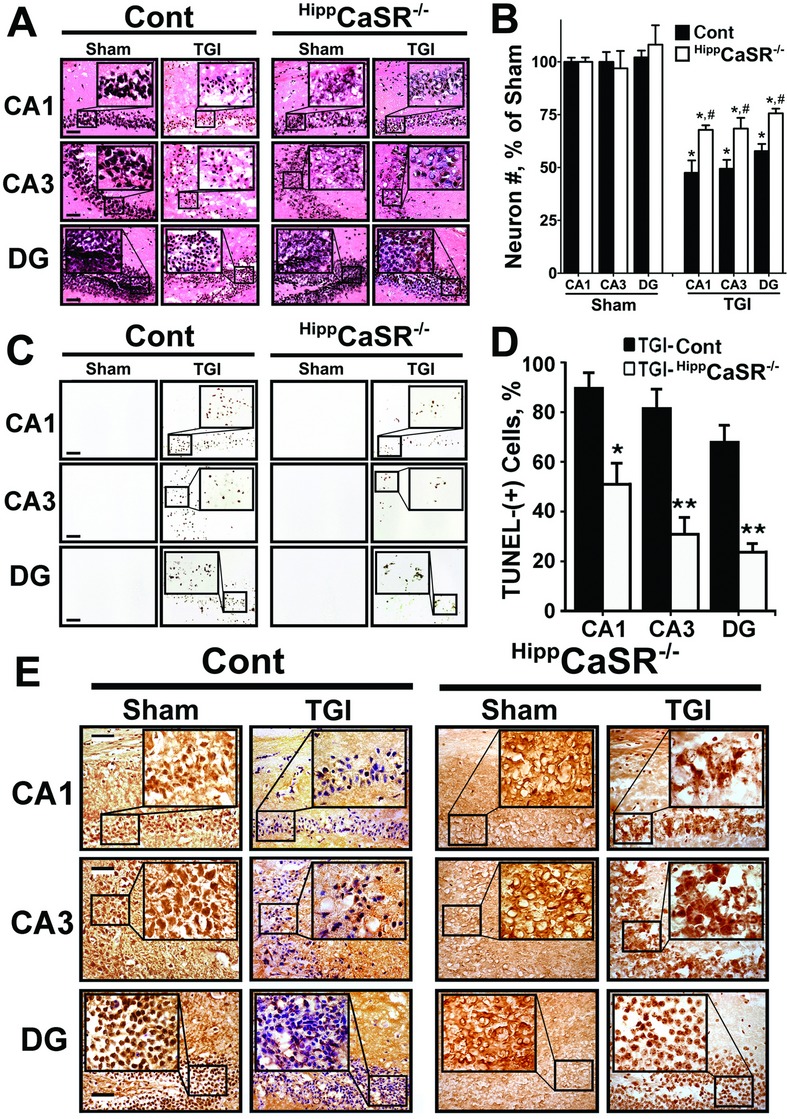
CaSR KO protected neurons from ischemic injury and restored GABABR1 expression in hippocampi of HippCaSR−/− mice. (A) Representative H&E-stained sections of hippocampi; (B) the % of neurons (normalized to the numbers of neurons in Sham-treated Cont mice) which were preserved; (C) representative TUNEL staining of hippocampi; (D) the percentage of TUNEL-(+) neurons normalized to neuronal numbers acquired from adjacent sections stained with H&E as represented in (A) which were shown; and (E) expression of GABABR1 protein, as assessed by immunohistochemistry, in 6 months old control (Cont) and HippCaSR-KO mice subjected to TGI (15 min) or Sham procedures. *P < 0.01 versus Sham in each animal group; #P < 0.01 versus TGI-Cont mice in (B); *P < 0.05, **P < 0.01 versus TGI-Cont in (D). Mean ± SE, N = 6–12 mice/group. Scale bar: 20 μm. CaSR, Ca2+-sensing receptor; KO, knockout; GABABR1, γ-aminobutyric acid receptor 1; TUNEL, transferase dUTP nick end-labeling; TGI, transient global ischemia.
We hypothesize that CaSR hyperactivity, as a result of receptor overexpression, stimulates intrinsic excitatory responses that contribute to neuronal death. We, therefore, tested whether two CaSR antagonists, namely calcilytics NPS89636 and NPS2413, which specifically inhibit CaSR-mediated signaling responses and intracellular Ca2+ mobilization in various cell types,51–53 protect against neuronal injury in wild-type (WT) C57/B6 mice. In WT mice, which were subjected to TGI, daily IP injections of vehicle (0.1% DMSO), and 72 h of reperfusion, the numbers of hippocampal neurons in the CA1, CA3, and DG were reduced by 53 ± 2%, 50 ± 2%, and 45 ± 3% (Fig. 4B, ▪) with 88 ± 3%, 67 ± 6%, and 43 ± 4% of them being TUNEL-(+) (Fig. 4A, DMSO; 4C, ▪), in the CA1, CA3, and DG, respectively. In contrast, daily IP injections of either NPS89636 or NPS2413 (1 mg/kg body wt), which began 1 h after the TGI procedure, significantly protected neurons from ischemic injury. In TGI mice injected with NPS89636, 59 ± 3%, 58 ± 2%, 64 ± 2% of hippocampal neurons was preserved in the CA1, CA3, and DG, respectively (Fig. 4B, ▪), with 57 ± 4%, 38 ± 2%, and 32 ± 1% of them being TUNEL-(+) (Fig. 4A, NPS89636; 4C, ▪). In TGI-treated NPS2143-injected mice, 68 ± 2%, 67 ± 2%, and 72 ± 1% of hippocampal neurons were preserved in the CA1, CA3, and DG, respectively (Fig. 4B, □), with only 39 ± 4%, 26 ± 2%, and 21 ± 1% of them being TUNEL-(+) (Fig. 4A, NPS-2143; 4C, □), indicating a greater potency of NPS2143 versus NPS89636 (P < 0.05) in neuroprotection. This is consistent with a lower half-maximal inhibitory concentration (IC50) for NPS2143 (43 nmol/L or 17 ng/mL) versus NPS89636 (271 nmol/L or 143 ng/mL) in blocking CaSR-mediated signaling responses.54–57 We did not observe significant changes in body temperatures of the mice with either calcilytics, excluding potential thermo effects on the neuronal responses.
Figure 4.
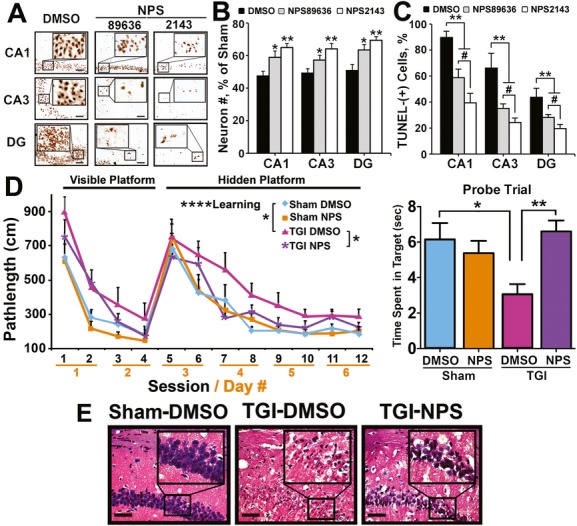
Intraperitoneal (IP) injections of calcilytics protected neurons against ischemic injury and preserved hippocampal functions. (A) Representative TUNEL staining of hippocampi; (B) the % of neurons (normalized to the numbers of neurons in Sham-treated DMSO-injected mice) which were preserved; and (C) the percentage of TUNEL-(+) neurons which were shown in the hippocampi of 6-month-old wild-type C57/B6 mice subjected to a TGI (15 min) procedure, daily IP injections of NPS89636, NPS2143 (1 mg/kg), or vehicle (DMSO), and 3 days of reperfusion. *P < 0.05; **P < 0.01 versus DMSO group in (B); **P < 0.01 versus TGI-DMSO, #P < 0.05 NPS89636 versus NPS2143 in (C). N = 6–12 mice/group. Scale bar: 20 μm. (D) MWM tests were performed on 4-month-old wild-type C57/B6 mice subjected to Sham or TGI (10 min) procedure with or without daily injections of NPS2143 (1 mg/kg) for 2 weeks, as described in Materials and Methods. Left panel shows the effects of TGI and NPS2143 on learning efficiency as indicated by the pathlength in finding visible and hidden platforms during the 6-day 12-session training. All animal groups showed significant learning (two-way ANOVA: main effect of learning: ****P < 0.0001) as indicated by progressively shortened path lengths in seeking a hidden platform in sessions 6–12. However, the learning ability was significantly (*P < 0.05) impaired in TGI-DMSO mice when compared to Sham-DMSO mice. Interestingly, IP injections of NPS2143 significantly (*P < 0.05) restored the TGI-induced loss of learning ability (TGI-DMSO vs. TGI-NPS). Right panel shows effects of TGI and NPS on memory retention or recall as indicated by the length of time spent swimming in the previous platform in the final probe trial. *P < 0.05, **P < 0.01. N = 9–17 per group. (E) Micrographs show representative H&E staining of hippocampi (CA1) from mice subjected to Sham or TGI (10 min) procedures, followed by daily IP injection of vehicle (DMSO) or NPS2143 (NPS) for 14 days and 1-week MWM test. Brain samples were collected 1 day after the completion of MWM test. TUNEL, transferase dUTP nick end-labeling; TGI, transient global ischemia; MWM, Morris Water Maze; ANOVA, analysis of variance; CA, cornu ammonis.
We next tested whether neuronal injury caused by TGI leads to functional deficits in the mice and whether calcilytics that inhibit CaSR activation and protect neurons against ischemia-induced apoptosis also preserve neurological function. In order to retain sufficient numbers of viable animals for the MWM test, the duration of TGI was shortened from 15 to 10 min to produce milder injury in those experiments. During MWM tests (Fig. 4D, left panel), both Sham and TGI mice, independent of treatment, learned to locate a visible (F3,132 = 51.9, P < 0.0001; sessions 1–4) or a hidden (F7,308 = 37.6, P < 0.0001; sessions 5–12) platform, as evidenced by traveling a progressively shortened pathlength to find the platform. However, TGI significantly worsened the performance in the tests as demonstrated by a prolonged pathlength to find the visible (F1,44 = 7.6, P < 0.01) or hidden (F1,44 = 5.7, P < 0.05) platform compared to the sham-operated mice (TGI-DMSO vs. Sham-DMSO). Daily IP injections of NPS2143 for 2 weeks, beginning 1 hr after TGI, significantly (post hoc analyses, P < 0.05) improved learning and memory in TGI mice (TGI-NPS vs. TGI-DMSO). During the probe trial (Fig. 4D, right panel), TGI reduced memory retention or recall in mice injected with DMSO as indicated by the significantly (P < 0.05) reduced time spent swimming in the previous platform location (Sham-DMSO vs. TGI-DMSO). This reduced memory retention was, however, significantly (P < 0.01) restored in TGI mice treated with NPS compound for 2 weeks (Fig. 4D, right panel: TGI-DMSO vs. TGI-NPS). Analyses of brain samples from mice subjected to MWM tests confirmed robust preservation of hippocampal neurons in mice, which have much improved learning and memory retention after NPS2143 treatment (Fig. 4E, TGI-NPS vs. TGI-DMSO). These data confirm the neuroprotective property of calcilytics to preserve brain functions by suppressing the CaSR activity in ischemic neurons.
Because systemic administration of calcilytics through an IP route could also inhibit CaSR functions in peripheral tissues, including parathyroid cells, this could produce a transient increase in serum PTH level and cause transient hypercalcemia.55 To ascertain that the observed neuroprotection was due to direct actions of calcilytics on the injured neurons but not due to secondary effects of other metabolic changes, we administered the compound directly into the right lateral ventricle of the brain (coordinates: 1 mm caudal to bregma, 1.3 mm lateral to sagittal suture, and 2 mm in depth) through an ICV route. The injections were performed ~30 min after the 15-min TGI or sham procedures. In mice, which were subjected to TGI, a single ICV injection of vehicle, and 3 days of reperfusion, ~45–55% of hippocampal neurons remained in the CA1, CA3, and DG (Fig. 5B, ▪) with ~60–85% of them being TUNEL-(+) (Fig. 5A, DMSO; 5C, ▪). In TGI mice injected with NPS89636, ~65–70% of hippocampal neurons were preserved (Fig. 5B, □) with only ~20–30% of them being TUNEL-(+) (Fig. 5A, NPS; 5C, □), supporting a direct action of calcilytics on the injured neurons. Apparently, the ICV regimen, albeit only a single dose, was more effective than multiple IP injections of the same compound in the above studies (Fig. 4B and C, ▪), suggesting that more focal and/or earlier delivery of the compound to the affected neurons may produce better outcomes. Nevertheless, our observations confirm that CaSR overexpression and overactivity critically mediate injury responses of neurons and that blocking CaSR expression or activity can effectively protect neurons from cell death due to ischemia.
Figure 5.
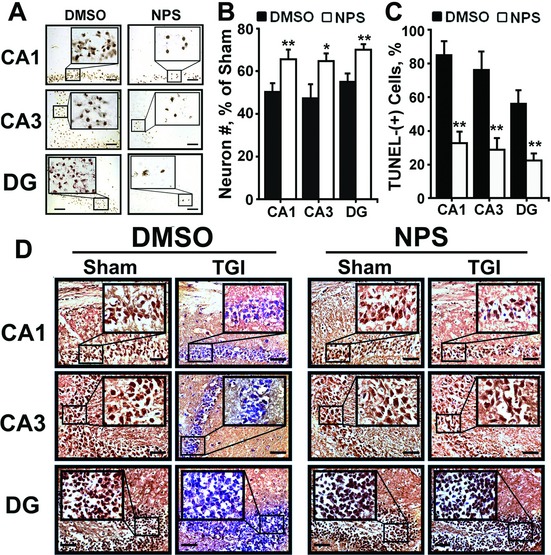
Intracerebroventricular (ICV) injections of calcilytic rendered more robust neuroprotection against ischemic injury and restored GABABR1 expression. (A) Representative TUNEL staining of hippocampi; (B) the % of neurons preserved; (C) the % of TUNEL-(+) neurons; and (D) expression of GABABR1 protein, as assessed by immunohistochemistry, in WT mice subjected to TGI (15 min), one single ICV injection of NPS89636 (1 ng in 1 μL PBS) or vehicle (DMSO), and 3 days reperfusion. Injections were performed 30 min after TGI. *P < 0.05; **P < 0.01 versus DMSO group; N = 6–12 mice/group. Scale bar: 20 μm. GABABR1, γ-aminobutyric acid receptor 1; TUNEL, transferase dUTP nick end-labeling; WT, wild-type; TGI, transient global ischemia.
CaSR overexpression and overactivity downregulate GABABR1 expression
To test whether CaSR overexpression/overactivity exacerbates neuronal injury, at least in part, by downregulating GABABR1 expression, we examined the effects of blocking CaSR expression and/or activity on GABABR1 expression in the hippocampi of HippCaSR−/− and Cont mice. As shown in Figure 3E, ischemia decreased GABABR1 expression in the hippocampal neurons (particularly in their cell bodies stained in blue) in Cont mice (Cont-Sham vs. Cont-TGI) and this effect was averted, although not completely, in the hippocampi of HippCaSR−/− mice (Cont-TGI vs. HippCaSR−/−-TGI). Similar reversal of the ischemic effect on GABABR1 expression in hippocampal neurons was also observed when CaSR activity was blocked with a single ICV dose of NPS89636 (Fig. 5D, DMSO-TGI vs. NPS-TGI) in WT mice. These data support the notion that CaSR overexpression and overactivity suppress GABABR1 expression in the injured neurons.
To determine whether restoring GABABR1 expression/signaling contributes to neuroprotection by the CaSR KO, we studied ischemic effects on hippocampi of HippCaSR−/−//GABABR1+/− and HippCaSR−/−//GABABR1−/− mice, which have one or two alleles of GABABR1 gene ablated, respectively, in the background of homozygous CaSR KO by breeding HippCaSR−/− mice with floxed-GABABR1 mice.45 All HippCaSR−/−//GABABR1−/− mice died before 3 months of age, preventing the study of their responses to TGI. Acute ablation of GABABR1 genes, by infecting hippocampal neurons cultured from homozygous floxed-GABABR1 mice58 with replication-incompetent adenoviruses expressing Cre recombinase cDNA (Ad-Cre), significantly knocked down the GABABR1 expression (Fig. 6A) and substantially increased the number of TUNEL-(+) neurons (Fig. 6C) along with robust CaSR expression (Fig. 6B) within 3 days of viral induction. These observations confirm a nonredundant role for the GABABR1 in supporting the survival of hippocampal neurons potentially by preventing aberrant overexpression of the excitatory CaSR.
Figure 6.
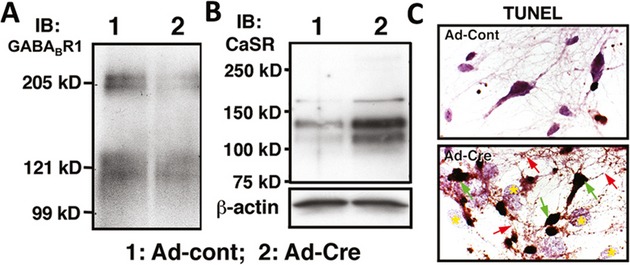
Effects of acute GABABR1 KO on the expression CaSR and GABABR1 and apoptosis in cultured hippocampal neurons. (A and B) Immunoblots of lysates from cultured floxed-GABABR1 hippocampal neurons, which had been infected with Ad-Cont (lane 1) or Ad-Cre (lane 2) adenoviruses for 3 days, were performed for the expression of (A) GABABR1 and (B) CaSR. (C) TUNEL staining indicates that the Ad-Cre-infected hippocampal neuron cultures show a profound increase in the number of apoptotic cells depicted by brown DAB staining (green arrows). Cells are counterstained with hematoxylin. GABABR1, γ-aminobutyric acid receptor 1; KO, knockout; CaSR, Ca2+-sensing receptor; TUNEL, transferase dUTP nick end-labeling; DAB, 3,3′-diaminobenzidine.
In contrast to HippCaSR−/−//GABABR1−/− mice, HippCaSR−/−//GABABR1+/− mice grew normally in body size and weight (data not shown), suggesting that expression of one single allele of Gabbr1 gene is sufficient to maintain cell survival in the absence of both alleles of Casr genes. This was indicated by the lack of TUNEL-(+) neurons in all regions of the hippocampi of the uninjured HippCaSR−/−//GABABR1+/− mice (Fig. 7A, Sham). We next used these mice to test whether one allele of Gabbr1 gene produces adequate amount of GABA-B-R1 and its downstream signaling responses to render the neuroprotection that we observed in the TGI-treated HippCaSR−/− mice (Fig. 7A, TGI: HippCaSR−/−). Interestingly, TGI profoundly reduced neuronal numbers (Fig. 7B, ▪ vs. □) and increased TUNEL-(+) cell numbers (Fig. 7A, TGI: HippCaSR−/− vs. HippCaSR−/−//GABABR1+/−; 7C, ▪ vs. □) in the hippocampi of HippCaSR−/−//GABABR1+/− versus HippCaSR−/− mice. Immunohistochemical analyses confirmed the profoundly reduced GABABR1 expression in the hippocampi of TGI-treated HippCaSR−/−//GABABR1+/− mice versus TGI-treated HippCaSR−/− mice (Fig. 7D). These studies suggest that expression of both Gabbr1 gene alleles is required for neuronal protection in the HippCaSR−/− mice in response to ischemia.
Figure 7.
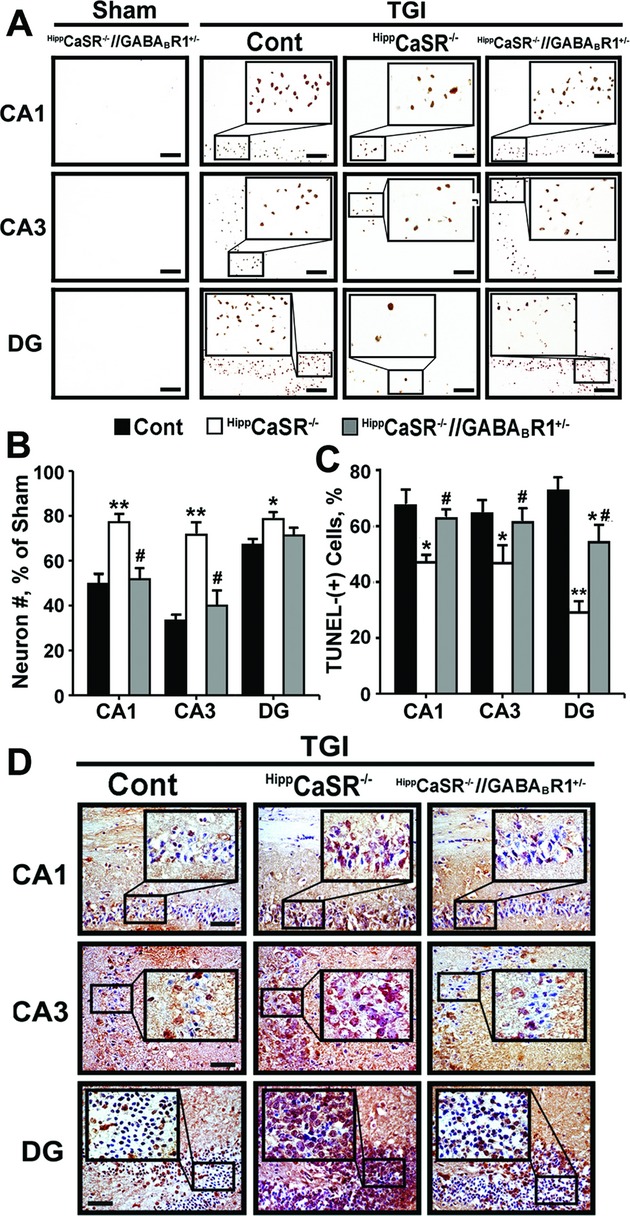
Concurrent heterozygous GABABR1 KO blunted the ability of CaSR KO to protect neurons against ischemic injury. (A) TUNEL staining of hippocampi; (B) the % of neurons preserved; and (C) the % of TUNEL-(+) neurons shown in hippocampi in control (Cont), HippCaSR−/−, and HippCaSR−/−//GABABR1+/− mice subjected to TGI (15 min) and in HippCaSR−/−//GABABR1+/− mice subjected to Sham procedure, followed by 3 days of reperfusion. *P < 0.05; **P < 0.01 versus TGI-treated Cont mice; #P < 0.01 versus HippCaSR−/− mice, N = 6–8 mice/group. Scale bar: 20 μm. (D) Representative immunohistochemical staining for the expression of GABABR1 in hippocampi of control (Cont), HippCaSR−/−, and HippCaSR−/−//GABABR1+/− mice subjected to TGI procedure, followed by 3 days of reperfusion. N = 6–8 mice/group. Scale bar: 20 μm. GABABR1, γ-aminobutyric acid receptor 1; KO, knockout; CaSR, Ca2+-sensing receptor; TUNEL, transferase dUTP nick end-labeling; TGI, transient global ischemia.
Synergistic neuroprotection by calcilytics and baclofen
The ability of calcilytics to restore GABABR1 expression in hippocampal neurons after ischemia is presumed to enhance the responsiveness of injured neurons to GABABR1 agonists, raising a possibility of using a combined calcilytics/baclofen treatment to further enhance neuroprotection. To test such a regimen, we compared TGI effects on hippocampi of WT mice injected with NPS2143, baclofen, or both. As shown in Figure 8A and B (Baclo vs. DMSO), daily IP injections of baclofen failed to preserve neurons in all region of hippocampi (Fig. 8C, Baclo vs. DMSO) and to reduce the number of TUNEL-(+) neurons (Fig. 8D, Baclo vs. DMSO) in the CA3 and DG of hippocampi in mice subjected to TGI and 3 days reperfusion. The baclofen had a modest but significant (P < 0.05) effect in reducing the number of TUNEL-(+) neurons in the CA1 (Fig. 8D, Baclo vs. DMSO). As anticipated, injections of NPS2143 profoundly preserved neurons (Fig. 8A and C, NPS vs. DMSO) and prevented their death (Fig. 8B and D, NPS vs. DMSO) after TGI. Coinjection of NPS2143 and baclofen preserved more neurons (Fig. 8A and C, NPS vs. NPS+Baclo) and further suppressed apoptosis (Fig. 8B and D, NPS vs. NPS+Baclo) in the CA1 (P < 0.05), when compared to the treatment of NPS alone, indicating synergism between these two compounds.
Figure 8.
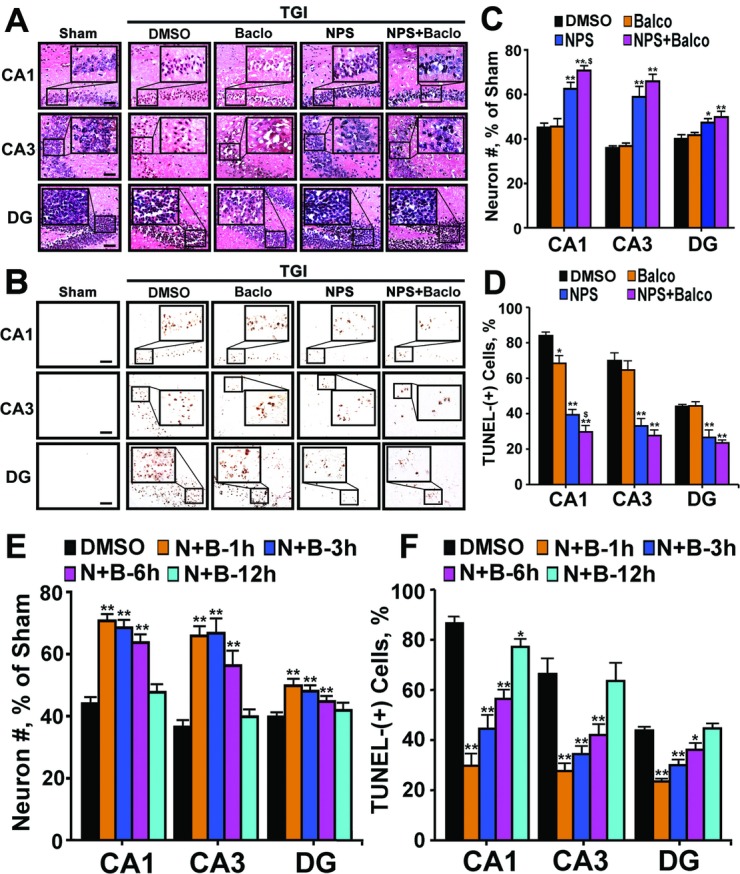
Synergy of calcilytic and baclofen in neuroprotection against ischemic injury. (A) H&E staining of hippocampi; (B) TUNEL staining of hippocampi; (C) the % of neurons preserved; and (D) the % of TUNEL-(+) neurons shown in the hippocampi of TGI-treated (15 min) mice injected with vehicle (DMSO), NPS2143 (NPS, 1 mg/kg), baclofen (Baclo, 1 mg/kg), or combination (NPS+Baclo), followed by 3 days of reperfusion. Injections began 1 h after TGI. *P < 0.05, **P < 0.01 versus TGI-DMSO; $P < 0.05 TGI-NPS+Baclo versus TGI-NPS in CA1; N = 6–12 mice/group. Scale bar: 20 μm. (E) The % of neuronal number (#) preserved and (F) the % of TUNEL-(+) neurons shown in the hippocampi of TGI-treated (15 min) mice injected with DMSO, or a combination of NPS2143 and baclofen (N+B) beginning 1, 3, 6, or 12 h after TGI procedure. **P < 0.01; *P < 0.05 versus DMSO group; N = 6–12 mice/group. TUNEL, transferase dUTP nick end-labeling; TGI, transient global ischemia; CA, cornu ammonis.
To explore the effective time window for the combined NPS2143/baclofen treatment, daily injections of the compounds were started at different times (1, 3, 6, and 12 h) after brain reperfusion. The neuroprotective effects of the combined calcilytics/baclofen treatment were comparable in preserving neurons (Fig. 8E, N+B-1 h vs. N+B-3 h) and in reducing apoptosis (Fig. 8F, N+B-1 h vs. N+B-3 h) when the injections were commenced at 1 and 3 h after TGI. When they were given to the mice 6 h after TGI, the effects of the compounds in preserving neurons (Fig. 8E, N+B-1 h vs. N+B-6 h) and blocking apoptosis (Fig. 8F, N+B-1 h vs. N+B-6 h) remained but with modest (~20–50%) reductions depending on the regions of hippocampus. When they were given to the mice 12 h after TGI, the effects of compounds on reducing apoptosis was only significant (P < 0.05) in CA1, but unremarkable in other regions of hippocampus (Fig. 8E and F, N+B-1 h vs. N+B-12 h). It appears that the combination of calcilytics and baclofen could produce an effective treatment window of >6 h. Future studies are needed to determine whether this time window can be further extended to improve the treatment by adjusting the doses of the compounds as well as the methods of their administration.
On the basis of the data presented herein and in previous studies, we propose a model for the actions of CaSR and GABABR1 in inducing neuronal hyperactivity and injury following ischemia (Fig. 9). Through unknown mechanisms, ischemia increases CaSR expression and signaling (1), which downregulates the GABABR1 expression, therefore reducing GABA signaling responses (2). Reduced GABABR1 expression further increases the CaSR expression and activity (3), initiating a feed-forward response that sustains neuronal hyperactivity (4). These sustained cellular responses eventually lead to permanent neuronal damage and cell death (5) and cause neurodegeneration and disabilities in learning, memory, cognition, or locomotors depending on the sites of injury (6).
Figure 9.
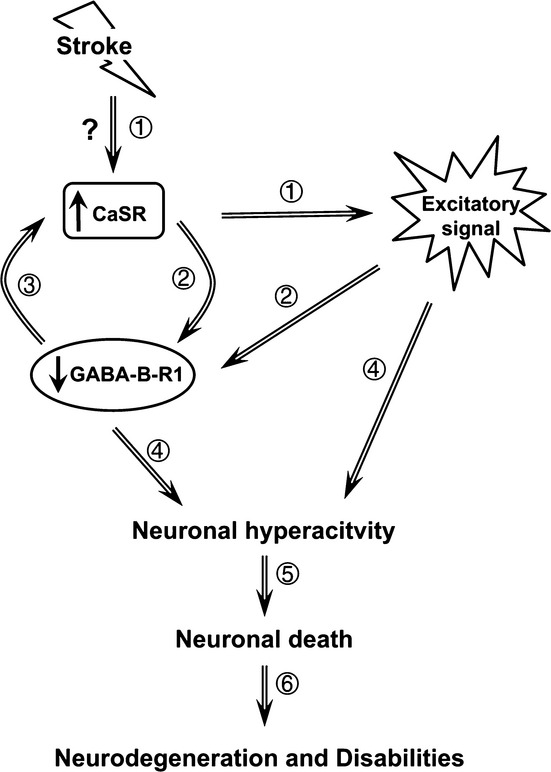
Working model for the induction of neuronal injury by overexpression of CaSR and downregulation of GABABR1 under ischemic conditions. CaSR, Ca2+-sensing receptor; GABABR1, γ-aminobutyric acid receptor 1.
Discussion
So far, there is no neuroprotective pharmaceutical available for treatment of global cerebral ischemia, which is a frequent complication of cardiac arrest. Pharmaceutical approaches with antagonists of NMDA receptors had been devised to prevent the development of neuronal injury and cell death by controlling neuronal hyperactivity at the early phase of the disease,9,11,12,59 but clinical trials of this class of compounds were disappointing.13 It was later proposed that NMDA receptors have biphasic actions – producing uncontrolled cell membrane depolarization in the early response to ischemia and functioning as a critical component of later neuronal repair pathway.11,12 These biphasic actions give a very narrow time window (first few minutes during the ischemia) for the treatment using this class of antagonist and make the treatment impractical in a clinical setting. These observations also suggest that other mechanisms mediating prolonged excitatory responses are likely involved in the progression of the disease. Our studies uncover a novel ischemia-induced injury cascade involving sustained upregulation of the excitatory CaSR and downregulation of the inhibitory GABABR1. On the basis of these findings, we further developed a novel combined therapy to promote neuroprotection against ischemia by concurrently targeting the CaSR and GABABR1 with calcilytics and baclofen, respectively. The temporal therapeutic window for intervention as demonstrated here is effective even when the compounds are administered after 6 h of ischemia onset. This time window is likely sufficient for treating subgroups of anoxic encephalopathy in patients suffering cardiac arrest and related conditions. Baclofen has been approved clinically for neurological disorders while calcilytics are in clinical trials for skeletal anabolism.54,60 Their pharmacokinetics and safety have been well characterized, so these compounds are readily available for trials to validate their neuroprotective effects in other animal models and eventually in human to determine its clinical relevance and feasibility.
In addition to treating ischemic injury, we have also shown calcilytics to be an effective neuroprotectant for traumatic brain injury in a controlled cortical impact mouse model,61 suggesting that CaSR overexpression and GABABR1 downregulation may be common injury responses leading to neuronal hyperactivity. It will be interesting to see if those molecular changes also occur in other conditions of neuronal hyperactivity, like Alzheimer’s and Parkinson’s diseases. The amyloid-β peptide, whose excessive production is closely associated with occurrence of Alzheimer’s disease, produces a CaSR-mediated activation of a Ca2+-permeable, nonselective cation channel and elevation in cytosolic Ca2+ in cultured hippocampal pyramidal neurons,62 supporting a link of the CaSR signaling to the development of the disease.
In addition to its clinical implication, our study reveals a new paradigm for interplay among members of the family C GPCR that have various functions in neurons as well as in peripheral tissues. The CaSR also coexpresses and physically interacts with GABABR2 and mGluRs (R1 and R5) in neurons.28 Changes in stoichiometric interactions among these receptors at subcellular domains in neurons could potentially alter excitatory states of neurons and vary their sensitivity and responsiveness to the corresponding ligands at different anatomical sites in the brain. This may help explain the heterogeneity of GABA(B) responses that contrasts with a very limited diversity of cloned GABABR subunits.63 Likewise, varied interactions of CaSR with GABA-B-R1 and mGluRs in subcellular domains of neurons (e.g., soma vs. neurite) may also explain the diverse signaling responses of the CaSR (e.g., activating different classes of ion channels or enzymes) recorded at different sites of the neuron.34–41
The ability of the CaSR to interact with unique members of family C GPCRs could further render its ability to mediate diverse neuronal functions in a cell context-dependent manner. For example, in taste and olfactory neurons, the CaSR is highly expressed along with various taste and vomeronasal receptors,64–69 which are also members of family C GPCR. If the CaSR is proven to physically interact with those specialized receptors, as it does with the GABABR1, it will suggest that the CaSR may mediate taste and olfactory functions in those cells.
In addition to the CNS, CaSR and GABABR1 are coexpressed in many peripheral tissues, including parathyroid gland, renal tubular cells, chondrocytes, and bone cells27,43 that control mineral and skeletal homeostasis. Interplay between those receptors is assumed to alter the responsiveness of those tissues to Ca2+ and perhaps GABA that is produced locally. Future studies are required to detail the interactions among those receptors in regulating diverse physiological functions and pathological responses. This information may provide new bases for the biased signaling of the CaSR and is required for designing more specific pharmaceutics targeting the CaSR in different tissues.
Acknowledgments
We thank Dolores Shoback at the Endocrine Unit and Lilly Jan and Shi-Bing Yang in the Department of Physiology, UCSF for their critical suggestions during the study; Zhiqiang Cheng for his assistance in assessing vasculature of the mice; Edward Nemeth at MetisMedica for providing the invaluable NPS89636 compound; and Bernhard Bettler at University of Basel for providing the critical floxed-GABABR1 mice for this study.
Authors Contributions
W. C., M. A. Y., and J. Y. K. designed the study; J. Y. K. and N. K. performed animal surgeries, histology, immunohistochemistry, and vasculature analyses; H. H., bred KO mice and performed neurobehavioral tests and analyses; J. L. designed and analyzed neurobehavioral experiments; C. L. T. generated floxed-CaSR and HippCaSR−/− mice and performed cell-culture and Western blotting; W. C. and J. Y. K. wrote the manuscript with critical input from all coauthors. All authors claim no conflict of interest. All animal models and cDNA constructs must be obtained through an MTA.
Conflict of Interest
None declared.
References
- Delorenzo RJ, Sun DA, Deshpande LS. Cellular mechanisms underlying acquired epilepsy: the calcium hypothesis of the induction and maintainance of epilepsy. Pharmacol Ther. 2005;105:229–266. doi: 10.1016/j.pharmthera.2004.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon B, Shorvon SD. Ischaemic stroke in adults and epilepsy. Epilepsy Res. 2009;87:1–11. doi: 10.1016/j.eplepsyres.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Stankowski JN, Gupta R. Therapeutic targets for neuroprotection in acute ischemic stroke: lost in translation? Antioxid Redox Signal. 2011;14:1841–1851. doi: 10.1089/ars.2010.3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Pathologically activated therapeutics for neuroprotection. Nat Rev Neurosci. 2007;8:803–808. doi: 10.1038/nrn2229. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Pathologically-activated therapeutics for neuroprotection: mechanism of NMDA receptor block by memantine and S-nitrosylation. Curr Drug Targets. 2007;8:621–632. doi: 10.2174/138945007780618472. [DOI] [PubMed] [Google Scholar]
- Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:e331–e339. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- Krnjevic K. Electrophysiology of cerebral ischemia. Neuropharmacology. 2008;55:319–333. doi: 10.1016/j.neuropharm.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Savitz SI. Introduction to cellular therapy: the next frontier for stroke therapeutics. Stroke. 2009;40(suppl 3):S141–S142. doi: 10.1161/STROKEAHA.108.535864. [DOI] [PubMed] [Google Scholar]
- Zaleska MM, Mercado ML, Chavez J, et al. The development of stroke therapeutics: promising mechanisms and translational challenges. Neuropharmacology. 2009;56:329–341. doi: 10.1016/j.neuropharm.2008.10.006. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Stefovska V, Turski L. Neuronal death enhanced by N-methyl-D-aspartate antagonists. Proc Natl Acad Sci USA. 2000;97:12885–12890. doi: 10.1073/pnas.220412197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1:383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- Muir KW, Lees KR. Excitatory amino acid antagonists for acute stroke. Cochrane Database Syst Rev. 2003;3:CD001244. doi: 10.1002/14651858.CD001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa C, Leone G, Saulle E, et al. Coactivation of GABA(A) and GABA(B) receptor results in neuroprotection during in vitro ischemia. Stroke. 2004;35:596–600. doi: 10.1161/01.STR.0000113691.32026.06. [DOI] [PubMed] [Google Scholar]
- Xu J, Li C, Yin XH, Zhang GY. Additive neuroprotection of GABA A and GABA B receptor agonists in cerebral ischemic injury via PI-3K/Akt pathway inhibiting the ASK1-JNK cascade. Neuropharmacology. 2008;54:1029–1040. doi: 10.1016/j.neuropharm.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Jackson-Friedman C, Lyden PD, Nunez S, et al. High dose baclofen is neuroprotective but also causes intracerebral hemorrhage: a quantal bioassay study using the intraluminal suture occlusion method. Exp Neurol. 1997;147:346–352. doi: 10.1006/exnr.1997.6637. [DOI] [PubMed] [Google Scholar]
- Babcock AM, Everingham A, Paden CM, Kimura M. Baclofen is neuroprotective and prevents loss of calcium/calmodulin-dependent protein kinase II immunoreactivity in the ischemic gerbil hippocampus. J Neurosci Res. 2002;67:804–811. doi: 10.1002/jnr.10169. [DOI] [PubMed] [Google Scholar]
- Kulinskii VI, Mikhel’son GV. Additivity and independence of neuroprotective effects of GABAA and GABAB receptor agonists in complete global cerebral ischemia. Bull Exp Biol Med. 2000;130:772–774. doi: 10.1007/BF02766091. [DOI] [PubMed] [Google Scholar]
- Bettler B, Kaupmann K, Mosbacher J, Gassmann M. Molecular structure and physiological functions of GABA(B) receptors. Physiol Rev. 2004;84:835–867. doi: 10.1152/physrev.00036.2003. [DOI] [PubMed] [Google Scholar]
- Ulrich D, Bettler B. GABA(B) receptors: synaptic functions and mechanisms of diversity. Curr Opin Neurobiol. 2007;17:298–303. doi: 10.1016/j.conb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Vollenweider F, Bendfeldt K, Maetzler W, et al. GABA(B) receptor expression and cellular localization in gerbil hippocampus after transient global ischemia. Neurosci Lett. 2006;395:118–123. doi: 10.1016/j.neulet.2005.10.079. [DOI] [PubMed] [Google Scholar]
- Cimarosti H, Kantamneni S, Henley JM. Ischaemia differentially regulates GABA(B) receptor subunits in organotypic hippocampal slice cultures. Neuropharmacology. 2009;56:1088–1096. doi: 10.1016/j.neuropharm.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz A, Arellano JI, DeFelipe J. GABABR1 receptor protein expression in human mesial temporal cortex: changes in temporal lobe epilepsy. J Comp Neurol. 2002;449:166–179. doi: 10.1002/cne.10287. [DOI] [PubMed] [Google Scholar]
- Zhou C, Li C, Yu HM, et al. Neuroprotection of gamma-aminobutyric acid receptor agonists via enhancing neuronal nitric oxide synthase (Ser847) phosphorylation through increased neuronal nitric oxide synthase and PSD95 interaction and inhibited protein phosphatase activity in cerebral ischemia. J Neurosci Res. 2008;86:2973–2983. doi: 10.1002/jnr.21728. [DOI] [PubMed] [Google Scholar]
- Epsztein J, Ben-Ari Y, Represa A, Crepel V. Late-onset epileptogenesis and seizure genesis: lessons from models of cerebral ischemia. Neuroscientist. 2008;14:78–90. doi: 10.1177/1073858407301681. [DOI] [PubMed] [Google Scholar]
- Kobuchi S, Shintani T, Sugiura T, et al. Renoprotective effects of gamma-aminobutyric acid on ischemia/reperfusion-induced renal injury in rats. Eur J Pharmacol. 2009;623:113–118. doi: 10.1016/j.ejphar.2009.09.023. [DOI] [PubMed] [Google Scholar]
- Chang W, Tu C, Cheng Z, et al. Complex formation with the Type B gamma-aminobutyric acid receptor affects the expression and signal transduction of the extracellular calcium-sensing receptor. Studies with HEK-293 cells and neurons. J Biol Chem. 2007;282:25030–25040. doi: 10.1074/jbc.M700924200. [DOI] [PubMed] [Google Scholar]
- Gama L, Wilt SG, Breitwieser GE. Heterodimerization of calcium sensing receptors with metabotropic glutamate receptors in neurons. J Biol Chem. 2001;276:39053–39059. doi: 10.1074/jbc.M105662200. [DOI] [PubMed] [Google Scholar]
- Brown EM, Gamba G, Riccardi D, et al. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature. 1993;366(December):575–580. doi: 10.1038/366575a0. [DOI] [PubMed] [Google Scholar]
- Chang W, Tu C, Chen TH, et al. The extracellular calcium-sensing receptor (CaSR) is a critical modulator of skeletal development. Sci Signal. 2008;1:ra1. doi: 10.1126/scisignal.1159945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi D, Brennan SC, Chang W. The extracellular calcium-sensing receptor, CaSR, in fetal development. Best Pract Res Clin Endocrinol Metab. 2013;27:443–453. doi: 10.1016/j.beem.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi D, Kemp PJ. The calcium-sensing receptor beyond extracellular calcium homeostasis: conception, development, adult physiology, and disease. Annu Rev Physiol. 2012;74:271–297. doi: 10.1146/annurev-physiol-020911-153318. [DOI] [PubMed] [Google Scholar]
- Ruat M, Molliver ME, Snowman AM, Snyder SH. Calcium sensing receptor: molecular cloning in rat and localization to nerve terminals. Proc Natl Acad Sci USA. 1995;92:3161–3165. doi: 10.1073/pnas.92.8.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano S, Brown EM, Chattopadhyay N. Calcium-sensing receptor in the brain. Cell Calcium. 2004;35:257–264. doi: 10.1016/j.ceca.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Ward DT. Calcium receptor-mediated intracellular signalling. Cell Calcium. 2004;35:217–228. doi: 10.1016/j.ceca.2003.10.017. [DOI] [PubMed] [Google Scholar]
- Awumey EM, Howlett AC, Putney JW, Jr, et al. Ca(2+) mobilization through dorsal root ganglion Ca(2+)-sensing receptor stably expressed in HEK293 cells. Am J Physiol Cell Physiol. 2007;292:C1895–C1905. doi: 10.1152/ajpcell.00404.2006. [DOI] [PubMed] [Google Scholar]
- Lu B, Zhang Q, Wang H, et al. Extracellular calcium controls background current and neuronal excitability via an UNC79-UNC80-NALCN cation channel complex. Neuron. 2010;68:488–499. doi: 10.1016/j.neuron.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Bergsman JB, Wang X, et al. Presynaptic external calcium signaling involves the calcium-sensing receptor in neocortical nerve terminals. PLoS One. 2010;5:e8563. doi: 10.1371/journal.pone.0008563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Bergsman JB, Harata NC, et al. Recordings from single neocortical nerve terminals reveal a nonselective cation channel activated by decreases in extracellular calcium. Neuron. 2004;41:243–256. doi: 10.1016/s0896-6273(03)00837-7. [DOI] [PubMed] [Google Scholar]
- Smith SM, Chen W, Vyleta NP, et al. Calcium regulation of spontaneous and asynchronous neurotransmitter release. Cell Calcium. 2012;52:226–233. doi: 10.1016/j.ceca.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyleta NP, Smith SM. Spontaneous glutamate release is independent of calcium influx and tonically activated by the calcium-sensing receptor. J Neurosci. 2011;31:4593–4606. doi: 10.1523/JNEUROSCI.6398-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizard TN, O’Keeffe GW, Gutierrez H, et al. Regulation of axonal and dendritic growth by the extracellular calcium-sensing receptor. Nat Neurosci. 2008;11:285–291. doi: 10.1038/nn2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Tu C, Rodriguez L, et al. Type B gamma-aminobutyric acid receptors modulate the function of the extracellular Ca2+-sensing receptor and cell differentiation in murine growth plate chondrocytes. Endocrinology. 2007;148:4984–4992. doi: 10.1210/en.2007-0653. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, et al. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Haller C, Casanova E, Muller M, et al. Floxed allele for conditional inactivation of the GABAB(1) gene. Genesis. 2004;40:125–130. doi: 10.1002/gene.20073. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kim N, Yenari MA, Chang W. Mild hypothermia suppresses calcium-sensing receptor (CaSR) induction following forebrain ischemia while increasing GABA-B receptor 1 (GABA-B-R1) expression. Transl Stroke Res. 2011;2:195–201. doi: 10.1007/s12975-011-0082-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, van Hoecke M, Tang XN, et al. Pyruvate protects against experimental stroke via an anti-inflammatory mechanism. Neurobiol Dis. 2009;36:223–231. doi: 10.1016/j.nbd.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama K, Suh SW, Hamby AM, et al. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci. 2006;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- Hong SM, Liu Z, Fan Y, et al. Reduced hippocampal neurogenesis and skill reaching performance in adult Emx1 mutant mice. Exp Neurol. 2007;206:24–32. doi: 10.1016/j.expneurol.2007.03.028. [DOI] [PubMed] [Google Scholar]
- Zhao H, Wang JQ, Shimohata T, et al. Conditions of protection by hypothermia and effects on apoptotic pathways in a rat model of permanent middle cerebral artery occlusion. J Neurosurg. 2007;107:636–641. doi: 10.3171/JNS-07/09/0636. [DOI] [PubMed] [Google Scholar]
- Davies SL, Gibbons CE, Vizard T, Ward DT. Ca2+-sensing receptor induces Rho kinase-mediated actin stress fiber assembly and altered cell morphology, but not in response to aromatic amino acids. Am J Physiol Cell Physiol. 2006;290:C1543–C1551. doi: 10.1152/ajpcell.00482.2005. [DOI] [PubMed] [Google Scholar]
- Dal Pra I, Chiarini A, Nemeth EF, et al. Roles of Ca2+ and the Ca2+-sensing receptor (CASR) in the expression of inducible NOS (nitric oxide synthase)-2 and its BH4 (tetrahydrobiopterin)-dependent activation in cytokine-stimulated adult human astrocytes. J Cell Biochem. 2005;96:428–438. doi: 10.1002/jcb.20511. [DOI] [PubMed] [Google Scholar]
- Dvorak MM, Siddiqua A, Ward DT, et al. Physiological changes in extracellular calcium concentration directly control osteoblast function in the absence of calciotropic hormones. Proc Natl Acad Sci USA. 2004;101:5140–5145. doi: 10.1073/pnas.0306141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Matheny CJ, Hoffman SJ, et al. An orally active calcium-sensing receptor antagonist that transiently increases plasma concentrations of PTH and stimulates bone formation. Bone. 2009;46:534–542. doi: 10.1016/j.bone.2009.09.028. [DOI] [PubMed] [Google Scholar]
- Nemeth EF, Delmar EG, Heaton WL, et al. Calcilytic compounds: potent and selective Ca2+ receptor antagonists that stimulate secretion of parathyroid hormone. J Pharmacol Exp Ther. 2001;299:323–331. [PubMed] [Google Scholar]
- Gowen M, Stroup GB, Dodds RA, et al. Antagonizing the parathyroid calcium receptor stimulates parathyroid hormone secretion and bone formation in osteopenic rats. J Clin Invest. 2000;105:1595–1604. doi: 10.1172/JCI9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharmalingam S, Daulat AM, Antflick JE, et al. Calcium-sensing receptor modulates cell adhesion and migration via integrins. J Biol Chem. 2011;286:40922–40933. doi: 10.1074/jbc.M111.265454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupmann K, Huggel K, Heid J, et al. Expression cloning of GABA(B) receptors uncovers similarity to metabotropic glutamate receptors [see comments] Nature. 1997;386:239–246. doi: 10.1038/386239a0. [DOI] [PubMed] [Google Scholar]
- Arvidsson A, Kokaia Z, Lindvall O. N-methyl-D-aspartate receptor-mediated increase of neurogenesis in adult rat dentate gyrus following stroke. Eur J Neurosci. 2001;14:10–18. doi: 10.1046/j.0953-816x.2001.01611.x. [DOI] [PubMed] [Google Scholar]
- Caltabiano S, Dollery CT, Hossain M, et al. Characterization of the effect of chronic administration of a calcium-sensing receptor antagonist, ronacaleret, on renal calcium excretion and serum calcium in postmenopausal women. Bone. 2013;56:154–162. doi: 10.1016/j.bone.2013.05.021. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kim N, Yenari MA, Chang W. Hypothermia and pharmacological regimens that prevent overexpression and overactivity of the extracellular calcium-sensing receptor protect neurons against traumatic brain injury. J Neurotrauma. 2013;30:1170–1176. doi: 10.1089/neu.2012.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye C, Ho-Pao CL, Kanazirska M, et al. Amyloid-beta proteins activate Ca(2+)-permeable channels through calcium-sensing receptors. J Neurosci Res. 1997;47:547–554. doi: 10.1002/(sici)1097-4547(19970301)47:5<547::aid-jnr10>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Pinard A, Seddik R, Bettler B. GABAB receptors: physiological functions and mechanisms of diversity. Adv Pharmacol. 2010;58:231–255. doi: 10.1016/S1054-3589(10)58010-4. [DOI] [PubMed] [Google Scholar]
- Bystrova MF, Romanov RA, Rogachevskaja OA, et al. Functional expression of the extracellular-Ca2+-sensing receptor in mouse taste cells. J Cell Sci. 2010;123(Pt 6):972–982. doi: 10.1242/jcs.061879. [DOI] [PubMed] [Google Scholar]
- Maruyama Y, Yasuda R, Kuroda M, Eto Y. Kokumi substances, enhancers of basic tastes, induce responses in calcium-sensing receptor expressing taste cells. PLoS One. 2012;7:e34489. doi: 10.1371/journal.pone.0034489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SanGabriel A, Uneyama H, Maekawa T, Torii K. The calcium-sensing receptor in taste tissue. Biochem Biophys Res Commun. 2009;378:414–418. doi: 10.1016/j.bbrc.2008.11.060. [DOI] [PubMed] [Google Scholar]
- Loretz CA, Pollina C, Hyodo S, Takei Y. Extracellular calcium-sensing receptor distribution in osmoregulatory and endocrine tissues of the tilapia. Gen Comp Endocrinol. 2009;161:216–228. doi: 10.1016/j.ygcen.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Flanagan JA, Bendell LA, Guerreiro PM, et al. Cloning of the cDNA for the putative calcium-sensing receptor and its tissue distribution in sea bream (Sparus aurata. Gen Comp Endocrinol. 2002;127:117–127. doi: 10.1016/s0016-6480(02)00035-7. [DOI] [PubMed] [Google Scholar]
- Ferry S, Traiffort E, Stinnakre J, Ruat M. Developmental and adult expression of rat calcium-sensing receptor transcripts in neurons and oligodendrocytes. Eur J Neurosci. 2000;12:872–884. doi: 10.1046/j.1460-9568.2000.00980.x. [DOI] [PubMed] [Google Scholar]