

Abstract
Objective
Electromyography (EMG) is used routinely to diagnose neuromuscular dysfunction in a wide range of peripheral neuropathies, myopathies, and neuromuscular degenerative diseases including motor neuron diseases such as amyotrophic lateral sclerosis (ALS). Definitive neurological diagnosis may also be indicated by the analysis of pathological neuromuscular innervation in motor-point biopsies. Our objective in this study was to preempt motor-point biopsy by combining live imaging with electrophysiological analysis of slow degeneration of neuromuscular junctions (NMJs) in vivo.
Methods
We combined conventional needle electromyography with fiber-optic confocal endomicroscopy (CEM), using an integrated hand-held, 1.5-mm-diameter probe. We utilized as a test bed, various axotomized muscles in the hind limbs of anaesthetized, double-homozygous thy1.2YFP16: WldS mice, which coexpress the Wallerian-degeneration Slow (WldS) protein and yellow fluorescent protein (YFP) in motor neurons. We also tested exogenous vital stains, including Alexa488-α-bungarotoxin; the styryl pyridinium dye 4-Di-2-Asp; and a GFP conjugate of botulinum toxin Type A heavy chain (GFP-HcBoNT/A).
Results
We show that an integrated EMG/CEM probe is effective in longitudinal evaluation of functional and morphological changes that take place over a 7-day period during axotomy-induced, slow neuromuscular synaptic degeneration. EMG amplitude declined in parallel with overt degeneration of motor nerve terminals. EMG/CEM was safe and effective when nerve terminals and motor endplates were selectively stained with vital dyes.
Interpretation
Our findings constitute proof-of-concept, based on live imaging in an animal model, that combining EMG/CEM may be useful as a minimally invasive precursor or alternative to motor-point biopsy in neurological diagnosis and for monitoring local administration of potential therapeutics.
Introduction
Diagnosis of neuromuscular or neurodegenerative disease is frequently based on clinical examination supplemented by data obtained on motor unit or muscle function via surface or needle electromyography (EMG).1–3 For instance, diagnosis of amyotrophic lateral sclerosis (ALS) and other motor neuron diseases is currently informed by a combination of clinical evaluation and interpretation of functional deficits, low-resolution imaging (X-rays and MRI), analysis of cerebrospinal fluid, genotyping for familial forms of the disease, and EMG.4–10 Diagnosis of ALS and other neuromuscular diseases may also be corroborated by histopathological and electrophysiological analysis of motor-point biopsies: that is, explants of whole muscles or muscle tissue containing intramuscular nerve branches and neuromuscular junctions (NMJs).11–20 Subsequent treatments may be systemic but topical administration of drugs or other treatments, including therapeutic neurotoxins such as botulinum toxin (BoNTx), is also indicated in some cases.21–24 Methods that enable localization and direct visualization of NMJs in situ may therefore facilitate analysis and interpretation of EMG, without recourse to an invasive motor-point biopsy, and may also improve the targeting and efficacy of potential treatments.25
We show here, using mice, that live imaging of intramuscular axons and NMJs, with a hand-held confocal endomicroscope (confocal endomicroscopy, CEM) can readily be combined with EMG recording, enabling simultaneous morphological and physiological appraisal of neuromuscular synaptic pathology in situ. We took advantage of two useful phenotypes in this study: transgenic expression of yellow fluorescent protein (YFP) in motor neurons, driven by the thy1.2 promoter26–28; and slow synaptic degeneration induced by axotomy in the WldS (Wallerian-degeneration Slow) natural mouse mutant.27,29–32 We have shown previously that synaptic degeneration can be observed and monitored effectively in vivo using CEM27,33 in double homozygotes of these two lines (thy1.2YFP16: WldS); also in the thy1.2YFP16: hSOD1G93A transgenic mouse model of ALS; and in an ENU mutant with defective axon regeneration.27,34 Our extended goals in this study were therefore first, to combine CEM with EMG using an integrated probe; second, to use the integrated probe to extend the time course and detail of longitudinal description of neuromuscular synaptic degeneration in vivo; and third to make progressive steps toward identifying potential fluorochromes that could label NMJs with high contrast, by localized infusion rather than transgenic expression of fluorescent proteins. We also present data supporting the safety of the method from the standpoints of mechanical damage, phototoxicity, or neuromuscular paralysis.
Together, our findings constitute proof-of-concept that combining simultaneous EMG recording and CEM imaging may be a safe approach for observing the form and function of NMJs in situ. The data suggest that this approach could be further developed and translated to the routine study of neuromuscular synaptic structure and function, either in healthy human subjects; or as a precursor or alternative, in some conditions, to motor-point biopsy for evaluation of neuromuscular synaptic pathology; and/or as a method for accurate delivery and monitoring of effective treatments, in cases of neurodegenerative or neuromuscular disease.35
Methods
Animals
All animals (mice and rats) were housed in cages of six or fewer, and maintained with Local Ethical Committee approval and with the approval under UK Home Office Project and Personal Licences, in accordance with the United Kingdom Animals (Scientific procedures) Act, 1986. Mice of the thy1.2YFP16:C57Bl6 and thy1.2YFP16:WldS strains were obtained from in-house breeding colonies (University of Edinburgh) generated initially using the thy1.2YFP16 line obtained originally from Jackson Laboratories (Bar Harbor, ME) and C57Bl6 and the innocuous mutant WldS line obtained originally from Harlan-Olac (Bicester, UK). Due to the age dependency of the WldS phenotype27,32,36 only animals aged 5–10 weeks were included in this study. At the end of the experiments, animals were sacrificed by cervical dislocation. In some experiments, Sprague–Dawley rats weighing 100–300 g were used instead of mice.
Sciatic nerve section
Animals were prepared for surgery with a subcutaneous injection of Vetergesic (0.05 mg kg−1 Alstoe-Sogeval UK Ltd.). They were then anaesthetized by inhalation of isoflurane and oxygen (2–5%, 0.4–1 L/min; Merial Animal Health, Harlow, UK). The area surrounding the sciatic notch was shaved and wiped with a sterilizing solution. A small incision was made, and then blunt dissection was used to expose the sciatic nerve. A small section (~2 mm) of the nerve was removed. The wound was then closed with 7/0 silk suture (Ethicon, Livingston, UK).
Repeated visualization of nerve terminals and axons27
A Cellvizio confocal endomicroscope (Mauna Kea Technologies, Paris, France) fitted with an S-1500 1.5-mm-diameter Proflex optical fiber probe was used to monitor axonal and synaptic degeneration in anaesthetized mice. Utilization of thy1.2YFP16 transgenic mice26,27 permitted us to observe axonal and neuromuscular synaptic degeneration within the same, reanaesthetized animals at several time points. A small incision was made in the hind limb, exposing the tibial nerve, and NMJs of muscles in the vicinity, including flexor digitorum longus and gastrocnemius. The probe was micromanipulated by hand, placing its tip in direct contact with the exposed muscle surface. Fluorescence excitation was provided by the 488 nm wavelength from the inbuilt diode laser. Fluorescence emission (500–650 nm) was collected through the same optical fibers at 12 video frames per second. Images were acquired and processed using ImageCell (Mauna Kea Technologies), Adobe Photoshop (USA), or ImageJ software (downloadable from http://imagej.nih.gov/ij/), running on Apple Mac G5 or Powerbook computers. To study the degeneration of nerve terminals following sciatic nerve lesion, thy1.2YFP16: WldS mice (N = 32) and thy1.2YFP16: C57Bl6 (N = 9 mice) were reanaesthetized and NMJs visualized up to four times over a 7-day period post axotomy. The amount of innervation in these images was estimated from the proportion of axon terminal branches that supplied a discernible motor axon terminal. To test whether CEM imaging might itself produce phototoxic damage, we imaged NMJ clusters in three mice with intact sciatic nerves, repeatedly over a 7-day period. In addition, to monitor the acute effects of intense illumination, we monitored recovery from photobleaching of YFP. Four thy1.2YFP16:WldS mice were anaesthetized and tibial nerve exposed. Maximal illumination was applied for 3–5 min. The CEM probe was held in place with a micromanipulator, and the nerve then briefly imaged at reduced laser power, at 10-min intervals over the following 60 min.
EMG recording
Compound muscle action potentials (CMAP) were recorded using a monopolar EMG needle (50 × 0.45 mm; Ambu, St. Ives, UK) attached to the CEM probe via a small length of silicone rubber tubing (3.0 mm external diameter, 1.5 mm internal diameter; Portex-Smiths Medical, Ashford, UK Ltd.) and inserted into muscles alongside the CEM probe. A reference electrode was positioned in the foot and a ground electrode was inserted rectally. The sciatic nerve was exposed via a small incision at the thigh level, and placed on a bipolar hook electrode. Stimulation pulses of 0.2-msec duration and up to 10 V in amplitude were delivered by a Digitimer DS2 stimulator (Digitimer, Welwyn Garden City, UK) via a Powerlab (ADInstruments Oxford, UK), driven by Scope software running on Apple Mac Powerbook. CMAP amplitude and latency were also measured using this software. Animals were culled either by overdose of anesthetic or by cervical dislocation after the final recording. In some cases, the tibial nerve and/or NMJs in the contralateral hind limb were imaged and EMG recorded at this stage.
Vital staining
Acetylcholine receptors (AChR) in postsynaptic membranes were stained using a fluorescent Alexa488 conjugate of α-bungarotoxin (Alexa488-α-BTX).37,38 Lumbrical muscles were dissected from the hind feet of C57Bl6 mice (N = 3) and incubated with Alexa488-α-BTX (Invitrogen-Life Technologies, Paisley, UK, UK; 5 μg/mL) in mammalian physiological saline (MPS), containing 120 mmol/L NaCl, 5 mmol/L KCl, 2 mmol/L CaCl2, 1 mmol/L MgCl2, 0.4 mmol/L NaH2PO4, 23.8 mmol/L NaHCO3, 5.6 mmol/L d-glucose. The muscles were then washed twice with MPS for 10 min and then fixed in 4% paraformaldehyde (PFA) for 20 min. After washing twice again with MPS for 10 min, the muscle was mounted in Mowiol (Calbiochem, Merck Millipore Watford, UK) and imaged using a Nikon Eclipse/Bio-Rad Nikon: Kingston-upon-Thames, UK; BioRad: Hemel Hempstead, UK. Radiance confocal microscope using a 40× oil immersion lens. For live imaging in vivo, Alexa488-α-BTX (5 μg/mL) was injected into the interstitial spaces of the inner leg of six mice, using an Inoject EMG needle (30 × 0.36 mm; Ambu) attached to the Cellvizio probe (see Fig. 4). The solution was left on the muscle for 20 min before washing away the excess with saline. CEM and/or EMG were performed before and after application of the solution.
Figure 4.

Motor nerve terminals can be identified using CEM following infusion of 4-Di-2-Asp. (A and B) Images of fluorescent motor nerve terminals following local infusion of 4-Di-2-Asp in mouse (A) and rat (B) muscle. The contrast is less than with transgenic expression of YFP and axons are unlabeled but motor nerve terminals are discernible (arrows). (C and D) EMG recordings of averaged CMAP (14 and 20 sweeps, respectively) obtained before local administration of 4-Di-2-Asp and 60 min later. There was no discernible effect on functional response. (E and F) Intracellular recordings of evoked EPPs; and (G and H) spontaneous MEPPs before and 60 min after adding 4-Di-2-Asp (20 μmol/L) to isolated FDB muscles. (I) Amplitude of EPPs decreased significantly over time (r2 = 0.43, F = 15.98, P < 0.0001). (J) Spontaneous MEPPs also decreased in amplitude (r2 = 0.29, F = 7.57, P < 0.02). (K) Quantal content of EPPs also declined, and (L) spontaneous MEPP frequency increased progressively over the period of incubation in the dye (summary data in I–L are based on recordings from three to five muscles at each time point. *P < 0.05, ANOVA, post hoc Bonferroni test). None of these effects was evident when the 4-Di-2-Asp concentration was reduced to 10 μmol/L (see Fig. S2). CMAPs, compound muscle action potentials; EMG, electromyography; NMJ, neuromuscular junctions; CEM, confocal endomicroscopy; FDB, flexor digitorum brevis.
In two muscles, motor nerve terminals were labeled using the styryl pyridinium dye 4-Di-2-Asp (4-(4-diethylaminostyryl)-1-methylpyridinium iodide, Sigma Aldrich, 20 μmol/L in MPS Gillingham, UK). This cationic mitochondrial dye was previously used extensively to reimage mouse motor nerve terminals in vivo, until it was superseded by transgenic expression of YFP.26,39–41 The muscles were pinned in Sylgard-lined Petri dishes and bathed in dye solution for 15 min, followed by a 5-min wash in MPS. The muscles were then mounted in Mowiol and images were captured into OpenLab software (Perkin Elmer, Coventry, UK) on an Apple Mac G5 computer via a Hamamatsu Orca-ER camera. Hamamatsu Photonics UK Ltd., Welwyn Garden City, UK Further image processing was performed in Adobe Photoshop. For live imaging by CEM, a higher concentration of 4-Di-2-Asp (40 μmol/L) was injected into intramuscular spaces of eight mice, as described above for Alexa488-α-BTX. Lower concentrations produced weaker fluorescence that was difficult to detect with CEM. Motor nerve terminals were imaged in all mice and EMG recorded in three of the mice for up to 60 min. Nerve terminal and postsynaptic endplate imaging using 4-Di-2-asp and Alexa488-α-BTX, respectively, were repeated in three rats.
Botulinum toxin labeling
GFP-labeled botulinum toxin Type A heavy chain (HcBoNTx/A) has been previously shown to label nerve terminals both after direct muscle application and repeated in vivo injections.42 We explored a similar approach, with the aim of developing a nontoxic stain that could be used to image nerve terminals in vivo using CEM. The C-terminal binding domain (HC) of type A botulinum neurotoxin was expressed as a GFP fusion protein from the plasmid pET29-GFP-HC/A (a kind gift of Professor J. Oliver Dolly, Dublin), in Rossetta™ 2(DE3)pLysS (NEB). Bacterial cultures were grown in LB media at 37°C, 220 RMP to an OD580 0.7, at which point isopropyl b-d-1-thiogalactopyranoside (IPTG) was added to 0.5 mmol/L, the temperature reduced to 18–20°C, and the incubation continued for a further 16–18 h. Bacteria were harvested by centrifugation at 7500g for 10 min at 4°C. The bacterial pellet was washed once and then resuspended in a solution of 10 mmol/L TrisHCl pH 8.5, 10 mmol/L imidazol, 300 mmol/L NaCl, and Complete™ – EDTA protease inhibitors (Roche Burgess Hill, UK)(binding buffer), at 1 mL per 10 mL starting culture. This cell suspension was subject to two freeze/thaw cycles and then sonicated on ice to reduce viscosity (Soniprep 150; MSE/Sanyo London, UK). The cell lysate was then centrifuged at 7500g for 20 min at 4°C, and the supernatant loaded on to a 1 mL His-TALON™ gravity flow column (Clontech Saint-Germain-en-Lay, France). The column was washed with 10× column volumes of binding buffer, and eluted in 10 mmol/L TrisHCl pH 8.5, 150 mmol/L imidazol, and Complete™–EDTA protease inhibitors. GFP fluorescence was used to identify fractions containing recombinant protein, which were pooled and extensively dialyzed against phosphate-buffered saline (pH 7.5).
In an initial test of the GFP-tagged HcBoNTx/A product, we dissected lumbrical muscle fans (deep lumbrical muscles 1–4 attached to their common calcaneus tendon of origin but detached from their insertions) and triangularis sterni muscles from five C57 Bl6 mice, pinned them in a Sylgard-lined Petri dish and bathed them in GFP-HcBoNTx (400 μg/mL) in MPS for 90 min. Preparations were then washed twice for 10 min in normal MPS. AChR at motor endplates were then counterstained with rhodamine–bungarotoxin (TRITC-α-BTX; 5 μg/mL in MPS for 15 min), and then washed twice for 10 min in MPS. The muscles were then mounted in Mowiol and imaged with conventional confocal microscopy (as above). For live imaging, 10 mice were injected subcutaneously in the inner leg with 3 μL of 0.5 or 1 mg/mL GFP-HcBoNTx/A in MPS using a Hamilton syringe Bonaduz, Switzerland. After an hour, the mice were anesthetized and the muscles imaged by CEM as described above.
Measurement of synaptic transmission
Wild-type mice (C57/Bl6, N = 9 mice) were used to test whether 4-Di-2-Asp (10–20 μmol/L, nine muscles) or GFP-HcBoNTx/A (400 μg/mL, two muscles from one mouse) impair neuromuscular transmission. The hind limbs of animals sacrificed by cervical dislocation were removed and the skin stripped. The limbs were pinned on to a Sylgard-lined Petri dish, and immersed in fresh oxygenated MPS equilibrated with 95% oxygen 5% CO2. Flexor digitorum brevis (FDB) nerve-muscle preparations were dissected and used for electrophysiological analysis.32,43 The muscles were pinned in a Sylgard-lined recording chamber. The tibial nerve supply was stimulated using a glass suction electrode connected to a Digitimer DS2 stimulator, controlled by a Digitimer D4030 programmer. Electrode glass (1.5 mm o.d. × 0.86 mm i.d., Harvard Apparatus, Edenbridge, UK) was pulled on a Sutter Flaming/Brown micropipette puller (P87;) and filled with 4mol/L potassium acetate. Microelectrode tip resistances were around 30–70 MΩ. Muscle fibers were impaled and EPPs recorded using an Axoclamp 2B amplifier (Axon Instruments Molecular Devices, Wokingham, UK). The percentage of innervated fibers was estimated from impalements of 30 randomly selected fibers, by scoring the number of fibers giving an evoked response upon supramaximal stimulation of the tibial nerve. A CED micro1401 interface transferred data to a computer (Dell Bracknell, UK), and EPPs were analyzed using WinWCP software (Strathclyde Electrophysiological Software, University of Strathclyde, UK). For quantal analysis, trains of 30 EPPs were recorded from each muscle fiber at 0.5–1 Hz. WinWCP software was used to analyze quantal content using the variance method.43,44 The Mclachlan–Martin correction factor ‘f’ for non-linear summation45 was set to 0.3 and the reversal potential was assumed to be −10 mV. Spontaneous MEPP activity was recorded for approximately 100 s for each fiber using Spike-2 software (Cambridge Electronic Design, Cambridge UK). Recordings were analyzed using Minianalysis (Synaptosoft, Atlanta GA), with auto detection of MEPPs. As resting membrane potential (RMP) affects MEPP amplitude, recordings were only taken from fibers with RMP more negative than −60 mV.
Statistical analysis
Data presented in the text and figures are mean ± SEM unless stated otherwise. GraphPad prism (GraphPad Software Inc. San Diego, CA) was used for statistical analysis. Normality was assumed and significance (P < 0.05) was evaluated using either unpaired t-tests or one-way ANOVA with Bonferroni post hoc tests, where appropriate.
Results
Longitudinal study of neuromuscular synaptic degeneration
We coupled a monopolar EMG needle to a CEM S-1500 probe and used this to monitor NMJ form, function, and degeneration in vivo (Fig. 1A; Video S1). High-contrast, selective fluorescence of intramuscular axons and motor terminals in thy1.2YFP16 transgenic mice was readily detected using this probe.
Figure 1.

Simultaneous EMG recording and live CEM imaging does not damage axons or NMJ. (A) standard monopolar EMG needle electrode secured to a 1.5-mm-diameter Proflex fiber-optic probe. (B–E) CEM images of motor axons in the tibial nerve in a thy1.2YFP16:WldS mouse before (0 min) and 5 min after full-power laser illumination, bleaching YFP fluorescence in the vicinity of the CEM probe tip, with subsequent recovery over the following 20–60 min. The moving front of recovery suggests that this was at least partly by axonal transport of YFP into the bleached region. (F and G) CEM images of left-side tibial nerve axons in the same reanaesthetized mouse, imaged 7 days apart. (H and I) CEM images of the same cluster of NMJs imaged 7 days apart; insets show EMG responses recorded in the same vicinity. (J) Peak amplitudes of EMG responses measured in the vicinity where images of motor nerve terminals were obtained 7 days apart. The differences were not significant (n = 3 mice; P > 0.05; paired t-test). Overall, these data suggest that CEM caused no axonal or presynaptic terminal damage and the peak amplitude of the evoked EMG response remained within the normal range. EMG, Electromyography; NMJ, neuromuscular junctions; CEM, confocal endomicroscopy; YFP, yellow fluorescent protein.
Intense, acute illumination of YFP-labeled axons via the 488-nm laser in the CEM control unit did not appear to impair axonal structure or function. In four mice, we used maximum laser power to illuminate the exposed tibial nerve for 5 min, which produced photobleaching of the illuminated region (Fig. 1B). Reexamination over the following 60 min with reduced laser power showed that fluorescence recovered along a moving front, presumably by a combination of spontaneous recovery of fluorescence46 and by effective transport of fluorescent protein from adjacent, unilluminated regions through the illuminated region47 (Fig. 1C–E).
We also examined the effect of illumination on the morphology of tibial nerve axons and NMJs, and on EMG responses to tibial nerve stimulation at multiple time points in the same animals (Fig. 1F–I). In three mice, there was no discernible fragmentation or any other evidence of degeneration of tibial nerve axons (Fig. 1F and G) or NMJs (Fig. 1H and I). EMG responses were also unimpaired (Fig. 1J): that is, the mean difference in the peak amplitude of the compound muscle action potential (CMAP) between measurements on the same region of the same mice, 7 days apart was 0.19 ± 0.02 mV (n = 3; P > 0.05, paired t-test). Thus, neither axonal nor NMJ structure, nor nerve conduction and neuromuscular transmission were significantly impaired by repeated visualization of the same neuromuscular synapses, 7 days apart.
Next, we obtained CMAPs and CEM images from thy1.2YFP16: WldS mice immediately then up to 7 days after axotomy by section of the sciatic nerve (Fig. 2). As expected,27,32 at 2 days post axotomy, there were no overt signs of deterioration of motor nerve terminal structure and muscles contracted vigorously in response to stimulation (Fig. 2A and B; Video S2). However, CMAP were discernibly reduced in amplitude (Fig. 2B and E), consistent with our previous observations of demise in physiological response before frank denervation of muscle fibers.27,31,32 EMG recording and revisualization of NMJs 5–7 days post axotomy (Fig. 2C and D) showed progressive deterioration of CMAP amplitude (Fig. 2E) and an increase in nerve-excitation threshold, measured by the stimulus intensity required to evoke a CMAP response (Fig. 2F). CMAP amplitude decreased from its initial value, 18.53 ± 0.67 mV (n = 32 muscles), to 11.61 ± 3.48 mV at 48 h (n = 4), 9.37 ± 2.66 mV at 3 days (n = 5), 4.39 ± 1.21 mV at 5 days (n = 7), and 5.65 ± 2.00 mV at 7 days (n = 7; F = 24.76; df = 54; P < 0.0001, ANOVA, Bonferroni post hoc t = 7.01, P < 0.001). The threshold for excitation was unchanged up to 5 days post axotomy (control: 0.52 ± 0.09 V, n = 13 muscles; 2 days axotomy: 0.37 ± 0.09 V, n = 3; 3 days axotomy: 0.45 ± 0.12 V, n = 4; 5 days axotomy: 0.85 ± 0.20 V, n = 7; P > 0.05 one-way ANOVA). At 7 days post axotomy, the threshold was significantly increased compared to controls (2.44 ± 1.09 V n = 5, F = 3.89; df = 31; P < 0.02, one-way ANOVA; Bonferroni post hoc t = 3.68, P < 0.05; Fig. 2F).
Figure 2.

Combining EMG and CEM gives complementary data on slow neuromuscular synaptic degeneration. (A–D) CEM Images (above) and EMG recordings of CMAPs (below) from thy1.2YFP16: WldS mice at sequential times after sciatic nerve section, indicated in days after sciatic nerve section. The WldS gene protects axons and nerve terminals from rapid Wallerian degeneration, so that most nerve terminals are still intact and the EMG appears normal or only slightly reduced for at least 2 days post axotomy (Panel B is a still frame from Video S2). By 5 days, several motor nerve terminals had degenerated (arrows) and there was a significant reduction in amplitude of the evoked EMG. By 6–7 days, most NMJ had degenerated and the EMG response was substantively reduced. (E) decline in EMG response; and (F) increase in stimulus intensity threshold required to evoke an EMG response with time after axotomy in thy1.2YFP16: WldS mice. (G) Quantification of loss of motor axon terminals from NMJ, over the same 2- to 7-day period. Numbers embedded in each column are the numbers of muscles studied in that group. Asterisks indicate significance at P < 0.05 (ANOVA and post hoc Bonferroni tests). Scale bars in A–D, 50 μm. EMG, Electromyography; NMJ, neuromuscular junctions; CEM, confocal endomicroscopy; YFP, yellow fluorescent protein; CMAPs, compound muscle action potentials.
Morphological indicators of progressive muscle fiber denervation matched the deterioration of CMAPs measured physiologically. Still frames from CEM video recordings obtained at each time point were selected and the percentage of endplates (n = 20–30 sampled in each muscle) showing clearly visible nerve terminals or intramuscular axon stumps were scored in order to provide a morphological estimate of nerve terminal degeneration (Fig. 2G). In control muscles, 100 ± 0% intramuscular axons had a visible nerve terminal (n = 3 muscles). At 2 days, 98.21 ± 1.04% of axon branches were tipped by an arborized axon terminal (n = 4), compared with 86.67 ± 6.67% at 3 days (n = 3) and 66.91 ± 8.04% at day 5 post axotomy (n = 7). By 7 days, the reduction in the number of visible nerve terminals compared to controls was substantial (22.46 ± 6.55%, n = 6, P < 0.001, t = 7.11, one-way ANOVA with Bonferroni post hoc test, F = 21.25, total df, 22). Thus, the amount of presynaptic degeneration estimated morphologically using CEM was similar to estimates made more directly by measuring motor endplate occupancy using conventional fluorescence or confocal microscopy.27,32
We also recorded EMG responses from axotomized YFP16 Bl6 animals (wild type for WldS) at 0, 24, and 48 h (Fig. S1). As expected,27,32,48,49 the normal and rapid rate of Wallerian degeneration in these mice caused the amplitudes of recorded CMAPs to decrease precipitously: from 17.58 ± 0.80 mV (n = 14 muscles) at the time of nerve section; to 0.86 ± 0.53 mV, (n = 5) at 24 h and 0.59 ± 0.59 mV at 48 h (n = 4; F = 124.0, df = 22; P < 0.001, one-way ANOVA).
Together, the data suggest that CEM is not inherently toxic or disruptive to the structural and functional integrity of axons or motor nerve terminals; and, when combined with EMG, this live imaging method can provide complementary morphological and physiological data in longitudinal studies of slow, progressive synaptic degeneration.
Exogenous vital staining of NMJs
Translating CEM/EMG recording into a clinical context will require labeling of NMJs with high-contrast fluorochromes, without recourse to transgenic expression of fluorescent proteins. We first tested the feasibility of this in six mice by local infusion of Alexa488-α-BTX, to label postsynaptic acetylcholine receptors (Fig. 3). As expected and indicated by simultaneous EMG recording, labeling AChR with this irreversible toxic ligand blocked synaptic transmission within 20 min of application, both in vivo and in vitro (Fig. 3B; Video S3). Bright, high-contrast fluorescent endplates were subsequently discernible in both mice (Fig. 3C and D) and rats (data not shown). Alexa488-α-BTX was also applied to muscles in thy1.2YFP16: WldS mice, 5 days after sciatic nerve section. The fluorescence intensity of Alexa488-α-BTX-labeled endplates was similar to that of YFP fluorescence of the axons. Alexa488-α-BTX staining also confirmed that several axons had lost their motor nerve terminals (Fig. 3E and F; compare with Fig. 2).
Figure 3.
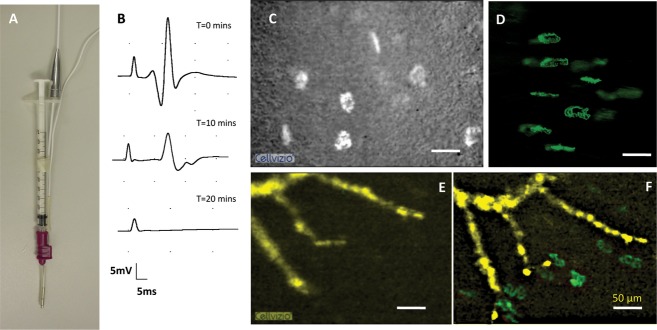
NMJ can be visualized using CEM after local injection of fluorochromes. (A) Combined CEM/EMG and hypodermic needle probe (1 mL syringe) used to exogenously administer fluorochromes in vivo. (B) EMG recordings following instillation of Alexa488-α-BTX. As expected, the labeled toxin produced progressive and eventually complete neuromuscular block. (C) CEM image of fluorescent motor endplates after local infusion of Alexa488-α-BTX. (D) Standard confocal microscopic image of NMJ labeled by infusion with Alexa488-α-BTX for comparison. (E) Intramuscular axons in a thy1.2YFP16: WldS mouse 5 days after axotomy. Axons were pseudocolored yellow. (F) The same group of intramuscular axons reimaged after infusion of Alexa488-α-BTX. Endplates were masked and then pseudocolored green, to differentiate their denervated state, denuded of overlying motor nerve terminals. Compare, for example, with Figures 1H and 2A. EMG, electromyography; NMJ, neuromuscular junctions; CEM, confocal endomicroscopy.
Having established that a fluorescent ligand could be locally administered to NMJs and provide sufficient brightness and contrast to allow motor endplates to be visualized readily by CEM, we next asked whether high-contrast labeling of motor nerve terminals could be obtained using other exogenous fluorescent dyes. We explored this possibility initially using the cationic styryl pyridinium dye 4-Di-2-Asp, which has previously been used to label NMJs in vivo.39,50 This dye labeled nerve terminals when injected in vivo at a concentration of 40 μmol/L both in mice (Fig. 4A; Video S4) and rats (Fig. 4B, Video S5).
4-Di-2-Asp has been reported to have no overtly toxic effects on NMJs and only weak anticholinergic activity at acute concentrations of 15–50 μmol/L, based on its effect on spontaneous miniature endplate potential amplitudes and efferent activity in cell types other than muscle.51–53 There was no overt effect of 4-Di-2-Asp on CMAPs recorded with the adjoined EMG electrode (17.32 ± 0.69 mV at 0 min (n = 3 muscles), 17.68 ± 0.17 mV at 60 min (n = 3), t-test, P = 0.32, t = 0.506, df = 4; Fig. 4C and D). To reevaluate any potential toxicity, however, we measured both spontaneous and evoked neuromuscular transmission in isolated FDB nerve-muscle preparations bathed in 4-Di-2-Asp (10–20 μmol/L) in normal MPS. At a concentration of 10 μmol/L, we observed no significant effect on MEPP amplitude or frequency, nor were the amplitudes of EPPs significantly attenuated (Fig. S2). However, when the concentration of 4-Di-2-Asp was increased to 20 μmol/L, endplate potential peak amplitude slowly and progressively declined (initially: 25.77 ± 1.21 mV, n = 4 muscles; at 10 min: 24.43 ± 1.24 mV, n = 5; at 20 min: 22.20 ± 2.28 mV, n = 5; at 30 min: 21.91 ± 2.10 mV, n = 5; and at 60 min: 15.6 ± 2.48 mV, n = 4; linear regression r2 = 0.43, F = 15.98, P < 0.0001; Fig. 4E, F, and I). MEPP amplitude was also reduced (0 min: 1.01 ± 0.17 mV, n = 3 muscles; 10 min: 0.84 ± 0.10 mV, n = 5; 20 min: 0.67 ± 0.11 mV, n = 5; 30 min: 0.86 ± 0.22 mV, n = 4; 60 min: 0.42 ± 0.08 mV, n = 4; linear regression: r2 = 0.29, F = 7.57, P < 0.02; Fig. 4G–H and J). Quantal analysis of the EPPs showed no compelling evidence for any substantive decrease in transmitter release and the slight, nonsignificant downward trend could have been due to the loss of signal:noise ratio of uniquantal responses, as MEPP amplitude declined and the smallest responses became buried in the recording noise (0 min: m = 48.09 ± 5.80 quanta, n = 4 muscles; 10 min: m = 48.66 ± 8.52, n = 5; 20 min: 38.42 ± 4.82, n = 5; 30 min: 33.88 ± 4.91, n = 5; 60 min: 32.17 ± 4.62, n = 4; P > 0.05 one-way ANOVA, Fig. 4K). However, there was evidence of a presynaptic effect based on a progressive increase in spontaneous transmitter release (Fig. 4H and L). MEPP frequency at 0 mins was 0.33 ± 0.02 s−1 (n = 3 muscles), and was not significantly different to MEPP frequency at 10 min (0.34 ± 0.06 s−1, n = 5) or 20 min (0.47 ± 0.06 s−1, n = 5). The MEPP frequency became more variable between fibers at 30 mins (1.80 ± 1.08 MEPPs s−1, n = 4). By 60 min, mean MEPP frequency was significantly increased (3.96 ± 0.97 MEPPs s−1, n = 4; P < 0.05 ANOVA).
Taken together, this analysis is consistent with a weak anticholinergic effect of 4-Di-2-Asp, reported previously, at concentrations above 20 μmol/L.52 Thus, although not overtly toxic, low concentrations of 4-Di-2-Asp may cause mild impairment of neuromuscular transmission in vivo, due to reduced MEPP amplitude and an increase in spontaneous MEPP frequency; but, based on the persistence of CMAPs in EMG recordings, this appears not to have been sufficient to impair overall neuromuscular function. Further evaluation of these anticholinergic effects may be worthwhile, however, in order to establish whether 4-Di-2-Asp might be a suitable fluorescent marker for vital staining of NMJs in humans.
Finally, we evaluated the potential utility of fluorescent conjugates of inactivated botulinum toxin type A. The native toxin blocks synaptic transmission via enzymic cleavage of the synaptic protein SNAP-25, which is essential for vesicular exocytosis and transmitter release.53–55 BoNTx/A binds to receptors on motor nerve terminal membranes via its heavy chain and removing the light chain renders the protein nontoxic.56,57 We therefore tested a GFP-tagged heavy chain construct of BoNTx/A (GFP-HcBoNTx/A)42 for toxicity and efficacy of staining and visualization of motor nerve terminals (see Methods). Conventional confocal microscopy and microelectrode recordings of EPPs revealed high-contrast staining of motor nerve terminals without blocking neuromuscular transmission (Fig. 5). Bath application of GFP-HcBoNTx/A (1 mg/mL) to two FDB nerve-muscle preparations (n = 1 mouse) for up to 1 h had no effect on EPP amplitude (20.40 ± 7.78 mV [SD] compared with 18.51 ± 4.22 mV; n = 30 muscle fibers in each case; P > 0.05 t-test) or quantal content (m = 49.16 ± 26.05 compared with 63.68 ± 23.07; Fig. 6B and C; P > 0.05 t-test).
Figure 5.
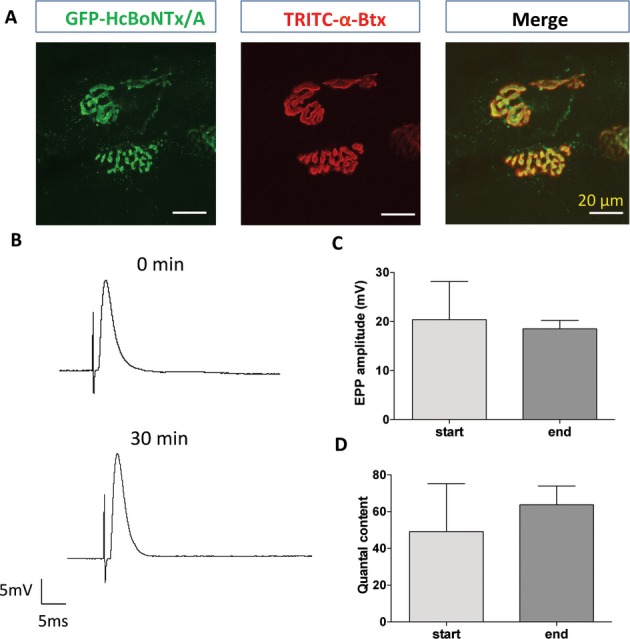
Motor nerve terminals imaged after labeling with GFP conjugates of HcBoNTx/A. (A) Conventional confocal microscopic images of motor nerve terminals (green, left) labeled with GFP-HcBoNTx/A, and motor endplates labeled with TRITC-α-bungarotoxin (red); together with a merged image showing complete coherence of labeled pre- and postsynaptic structures. (B) Intracellular recordings of evoked EPPs showed no evidence of impairment in synaptic physiology after labeling with GFP-HcBoNTx/A; and (C and D) there was no significant change in either peak amplitude or quantal content of EPPs 30–60 min after administration of the fluorochrome (mean ± SD, n = 2 muscles from one mouse, 30 fibers sampled in each).
Figure 6.

Fluorescence of motor nerve terminals labeled with GFP-tagged HcBoNTx/A was weak. (A) Identical setting of laser power and photomultiplier gain and black level in a conventional confocal microscope were used to image motor terminals in thy1.2YFP16 mouse muscle (left column) and wild-type muscles labeled by infusion of GFP-HcBoNTx/A (middle column). The fluorescence intensity of simultaneous TRITC-α-BTX labeling is shown as a positive control for microscope setting (middle row) and merged images (bottom row). Increasing the power of the 488-nm argon laser rendered terminals more discernible (right column) but the brightness was still less than that of transgenic expression of YFP. (B) Low power image of motor nerve terminals (arrows) in a muscle labeled by infusion of GFP-tagged HcBoNTx/A. The background intensity is high due to the maximum setting of the laser power. (C) Digital zoom on of one motor nerve terminal visualized using CEM in a muscle labeled by GFP-tagged HcBoNTx/A. Background subtraction and contrast stretching were used to enhance the differentiation of the labeled terminal against background.
Unfortunately, fluorescence intensity of motor terminals exogenously labeled with GFP-HcBoNTx/A was much weaker than the endogenous fluorescence of motor terminals in thy1.2YFP16 mice. To compare the fluorescence with that of YFP, we imaged nerve terminals in triangularis sterni (TS) muscles dissected from thy1.2YFP16 mice and non-YFP mice, labeled with GFP-HcBoNTx/A. We used identical settings of laser power, photomultiplier gain, and offset and scanning rate. The images in Figure 6 show that fluorescence was about 10 times weaker in terminals labeled with GFP-HcBoNTx/A compared with that of the YFP transgenics (Fig. 6). GFP-HcBoNTx/A (1 mg/mL) in vivo produced barely discernible nerve terminal staining (Fig. 6B and C; Video S6). The fluorescence was therefore too weak and its detection too unreliable for us to undertake a longitudinal study using this form of labeling of motor nerve terminals using GFP-HcBoNTx/A.
Discussion
We have demonstrated here that combining live imaging and electromyographic recording provides complementary information about the structure and function of motor units and the integrity of their neuromuscular connections, under conditions where both may be compromised: specifically, during progressive neuromuscular synaptic degeneration. This was technically achievable for three main reasons: first, the size and mechanical flexibility of the endomicroscopy probe; second, utilization of transgenic mice that strongly express YFP in motor neurons, yielding high-contrast, selective vital staining of axons and motor nerve terminals; and third, utilization of a natural mutation that delays synaptic degeneration. These advantages also define the principle limitations that must be confronted before extending and translating the method to clinical applications.
Probe limitations
Although fitted with a flexible, slender (1.5 mm diameter) probe and with an acquisition video frame rate (12 fps) that permits smooth real-time visualization, our CEM/EMG recording approach is limited, optically, by light sensitivity and a nominal working distance of zero: that is, maximum acuity is obtained at the probe’s interface. The principle advantage of this is that high-contrast images of axons and motor nerve terminals are obtainable by gliding the probe face over a muscle surface in the region of the NMJs. One disadvantage is that, in a clinical context, neuromuscular pathology may lie much deeper and, therefore, more invasive penetration of the probe tip may be necessary in order to visualize neuromuscular defects. A further risk is that locating NMJs, especially deep within muscle, might require multiple penetrations with the probe. The number and accuracy of penetrations required would most likely be improved upon with experience, perhaps supplemented by preliminary surface and/or single-fiber needle EMG recording (SFEMG),1,4,58,59 thus identifying the most likely locations of NMJs in the regions of interest.
Fluorochromes
As in our previous study, we obtained very good contrast and morphological delineation, within the specification of a 5-μm spatial resolution, using transgenic expression of YFP in motor neurons.26,27 Finding safe fluorochromes that readily stain motor axons and their endings with high-contrast and high quantum yield, thus visualizing a similar degree of structural detail, remains perhaps the most significant challenge facing extension of CEM to diagnostic neurology. However, we have made progress toward this goal in this study. Postsynaptic staining of acetylcholine receptors by injection and local infusion of a fluorescent conjugate of α-BTX yielded images of comparable intensity and contrast to presynaptic labeling with YFP. But the concentrations used to ensure such visualization blocked neuromuscular transmission, and the affinity of α-BTX for its receptor, were such that dissociation would take several days after its administration in vivo60–62 and meanwhile, such binding may also destabilize AChR and provoke internalization of the BTX-bound complexes.63–65 Moreover, although endplate morphology is in register with, and thus reflects, presynaptic terminal structure in healthy muscle, postsynaptic staining gives no direct clue regarding the integrity of presynaptic terminals in disease or following nerve damage (with or without reinnervation).
We also obtained discernible staining of motor nerve terminals using the vital dye 4-Di-2-Asp, which – prior to the advent of transgenic expression of fluorescent proteins in motor neurons using the thy1.2 promoter26–28 – was a fluorochrome of choice for vital staining of motor nerve terminals in longitudinal studies of NMJ morphology in vivo.39,40,52 We also obtained images using a GFP conjugate of the botulinum toxin heavy chain. However, the intensity of fluorescence of motor nerve terminals labeled by injection and infusion in vivo with this fluorochrome was substantively less than with 4-Di-2-Asp and considerably less than the fluorescence of transgenically expressed YFP (see Fig. 6). Fluorescent conjugates of botulinum toxin heavy chain remain attractive as candidate neuromuscular fluorochromes, since the active form (containing the enzymically active light chain) is already licensed for clinical and cosmetic applications. However, the data from this study suggest that GFP-HcBoNTx/A does not have a sufficient photon yield for detection by CEM, at least in our hands. Perhaps by increasing the photon yield of fluorescent ligands bound to the BoNTx/A heavy chain, or to its receptor in nerve terminal membranes, will enable this attractive molecular target to be exploited more effectively and to achieve its full potential for applications to CEM imaging. Quantum yield of fluorescence could be increased by uniting HcBoNTx/A with other conjugates.66 Alternatively, targeting fluorescent ligands to other receptors in presynaptic terminals could improve the contrast and intensity of the motor nerve terminal fluorescence. Seeking highly fluorescent molecular probes that compete for the botulinum toxin heavy chain receptor may be a fruitful strategy.67 A promising alternative approach may be to utilize aptamer screening for empirical selection of suitable fluorochromes. This approach has proved successful in identifying fluorescent compounds that effectively stain axons in peripheral nerve but unfortunately not, thus far, at NMJs.68.
Monitoring axonal and synaptic degeneration in vivo
Homozygous WldS mutant mice display a remarkable, covert phenotype: protection from Wallerian degeneration that delays axonal and synaptic degeneration by an order of magnitude compared with wild-type mice.27,36,69 Thus, while Wallerian degeneration is complete within 1–3 days of axotomy in wild-type mice, almost all of the intramuscular axons and motor nerve terminals remain wholly intact for about 3 days after section of the sciatic nerve and only then degenerate, in a progressive fashion, via a process of asynchronous retraction of synaptic boutons from motor endplates.31,32,69 This phenotype, which evidently arises via substitution of the stable enzymic activity of the chimeric WldS protein for unstable axonal Nmnat2,70–72 is of considerable practical utility for development of methods such as those described in this study.27 We also showed in a previous report that CEM enables longitudinal study and visualization of progressive synaptic degeneration in the SOD1 mouse model of ALS and in a mouse model of disease with slow axon regeneration.27 Thus, utilization of WldS mice and other mouse models has enabled us to identify and evaluate asynchronous synaptic degeneration in both investigator-triggered and spontaneous synaptic degeneration. Additional experimental validation is necessary to establish further the optical limitations of the method: for instance, whether collateral and motor nerve terminal sprouting can be visualized, following either chronic paralysis or partial denervation.38,53,55
Translational potential of EMG/CEM
Electromyography (EMG) is widely used as a diagnostic tool for neuromuscular disorders, including motor neuron diseases such as ALS.2,73–75 Clinical applications of CEM have, thus far, included investigation of the GI tract,76 urinary tract,77 respiratory tract78, and cardiovascular system.79 The present findings lay a foundation for extending CEM to neurological investigation and using it, in association with conventional EMG recording, for diagnosis and monitoring the efficacy of potential treatments of neuromuscular disease.
What are the likely benefits of combining EMG and CEM in a clinical context? First, direct visualization of neuromuscular synaptic morphology in situ could preempt or complement histopathological investigation by motor-point biopsy. Motor-point biopsies have been most frequently used in diagnosis and characterization of several forms of myasthenia.15–17,19,20,80,81 Thus, CEM may be a viable alternative to motor-point biopsy in these conditions. Second, a combination of CEM and EMG may facilitate earlier diagnosis of neurodegenerative axonopathies such as ALS.35,82–84 EMG analysis allows earlier diagnosis and shows abnormalities in nonsymptomatic regions in ALS patients.2,85,86 However, most of our knowledge regarding NMJ pathology in ALS derives from limited data obtained via motor-point biopsy12–14 or postmortem tissue.82,83 Although it has been argued that neuromuscular synaptic degeneration is an early sign of disease in animal models,41,87–89 the evidence for this in human ALS is less compelling and indirect.41,82,83,86 An efficient imaging method for defining, detecting, and monitoring disease onset and progression at NMJs at different stages may facilitate diagnosis of different forms of the disease and establish the role of the NMJ in disease onset and progression.
Finally, there is intense contemporary interest in possibilities for transplanting differentiated cells derived from human embryonic stem (ES) cells or inducible pluripotent stem (iPS) cells, either as vehicles for delivery of therapeutic cargoes or for functional regeneration of axons and NMJs.90–94 Since it is possible to express fluorescent protein in these cells,90,92,93,95–97 CEM/EMG may facilitate the localized delivery of cell-based therapeutics, contributing to development of effective, novel treatments for diseases like ALS.
Conclusion
In summary, CEM is minimally invasive, and it can be applied without causing overt mechanical or photochemical damage to motor axons and their terminals. It provides novel morphological data at 5-μm resolution, via a probe that is smaller in diameter than most needle-biopsy probes, and can be utilized at the same time as EMG recording, via integration of standard needle electrodes. CEM, in combination with EMG recording, may preempt, or constitute an attractive alternative to, motor-point biopsy for visualization of neuromuscular synaptic integrity. CEM may also be used to monitor the localized administration of potential therapeutics. Thus, adjoining EMG recording with CEM-based live, real-time imaging of NMJs in humans would constitute a powerful and refined approach to research, clinical diagnosis, follow-up investigations, and monitoring the efficacy of treatment of neuromuscular disease.
Acknowledgments
This study was supported by grants from the MRC (G0401091), the Motor Neurone Disease Association (GR6065) and the RS MacDonald Trust. We thank Professor J. Oliver Dolly for the gift of GFP-HcBoNTx/A construct; Mr Derek Thomson for expert technical assistance and genotyping; and Mr Sultan Abdulkarim Almdallaleh and Mr Nour Eddin Farouq Alshaaer for assistance with expression and purification of GFP-tagged constructs. We are grateful to Professor Michael Eddleston MD PhD for helpful comments on the manuscript.
Author Contributions
R. B. assisted design and management of the project; acquired and analyzed data; and co-wrote the manuscript. K. N. D. acquired and analyzed data. P. A. S. generated key reagents; assisted the design of the study; and co-wrote the manuscript. R. R. R. designed and directed the study; managed the project, reviewed and co-analyzed the data; and co-wrote the manuscript. All coauthors have seen and agree with the contents of the manuscript. ICMJE requirements for authorship have been met. Each author believes it represents honest work.
Conflict of Interest
None of the authors has any financial or nonfinancial associations that may be perceived as relevant to the submitted manuscript, including personal, professional, political, institutional, religious, or other associations that may be perceived as relevant.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Axotomized wild-type motor terminals degenerate rapidly. (A and B) CEM Images of intramuscular axons in wild-type (thy1.2YFP16: C57Bl6) mice 24 h and 48 h, respectively, after sciatic nerve section. (C) EMG recording 24 h after nerve section in the vicinity of intramuscular axons like those shown in A, showing records in response to progressively increasing stimulus intensity, from 1 to 30 V. The large spike is a stimulus artifact due to stimulation at over 30 V intensity applied to the tibial nerve: there was no evoked CMAP. (D) Peak EMG amplitude in control (n = 14 muscles), and 1 day (n = 5) and 2 days (n = 4) after axotomy. Responses were lost within 24–48 h. Almost all motor nerve terminals degenerated within 24 h in these mice, with concomitant loss of evoked neuromuscular responses.
Low concentrations of 4-Di-2-Asp (10 μmol/L) have no effect on neuromuscular transmission. (A) EPPs and (B) MEPPs recorded after 60-min incubation. There was no significant linear regression over 1 h of incubation in: (C) EPP amplitude, (D) MEPP amplitude, (E) quantal content, or (F) MEPP frequency. Each data point in C–F shows the mean ± SEM and the calculated best fit linear regression. n = 4 muscles, three muscle fibers sampled in each muscle at each time point. The slopes were not significantly different (P > 0.05, linear regression in all four cases.)
CEM of intramuscular axons and motor nerve terminals in a control thy1.2YFP16: WldS mouse. The Proflex probe was glided over the surface of the muscle while capturing images in real time at 12 fps, while the sciatic nerve was supramaximally stimulated. Twitching of the muscle is discernible.
CEM of motor nerve terminals imaged in real time at 12 fps during sciatic nerve stimulation; thy1.2YFP16: WldS mouse, 2 days after sciatic nerve section. Note the intermittent twitching of the muscle produced by stimulation of the distal, tibial nerve stump.
CEM of motor endplates imaged in real time at 12 fps, following instillation of Alexa488-α-bungarotoxin. The fluorescence is due to postsynaptic labeling of AChR.
CEM of motor nerve terminals imaged in real time at 12 fps following instillation of 4-Di-2-Asp to label motor nerve terminals in a nontransgenic (C57Bl6) mouse.
CEM of motor nerve terminals imaged in real time at 12 fps following instillation of 4-Di-2-Asp to label motor nerve terminals in a Sprague–Dawley rat.
CEM of motor nerve terminals imaged in real time at 12 fps following instillation GFP-HcBoNTx/A, to label motor nerve terminals in a nontransgenic (C57Bl6) mouse.
References
- Saitou K, Masuda T, Michikami D, et al. Innervation zones of the upper and lower limb muscles estimated by using multichannel surface EMG. J Hum Ergol (Tokyo) 2000;29:35–52. [PubMed] [Google Scholar]
- Krarup C. Lower motor neuron involvement examined by quantitative electromyography in amyotrophic lateral sclerosis. Clin Neurophysiol. 2011;122:414–422. doi: 10.1016/j.clinph.2010.06.027. [DOI] [PubMed] [Google Scholar]
- Narayanaswami P, Spieker AJ, Mongiovi P, et al. Utilizing a handheld electrode array for localized muscle impedance measurements. Muscle Nerve. 2012;46:257–263. doi: 10.1002/mus.23307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falla D, Dall’Alba P, Rainoldi A, et al. Location of innervation zones of sternocleidomastoid and scalene muscles–a basis for clinical and research electromyography applications. Clin Neurophysiol. 2002;113:57–63. doi: 10.1016/s1388-2457(01)00708-8. [DOI] [PubMed] [Google Scholar]
- Rutkove SB, Caress JB, Cartwright MS, et al. Electrical impedance myography correlates with standard measures of Als severity. Muscle Nerve. 2014;49:441–443. doi: 10.1002/mus.24128. [DOI] [PubMed] [Google Scholar]
- Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7:639–649. doi: 10.1038/nrneurol.2011.153. [DOI] [PubMed] [Google Scholar]
- Bromberg MB. An electrodiagnostic approach to the evaluation of peripheral neuropathies. Phys Med Rehabil Clin N Am. 2013;24:153–168. doi: 10.1016/j.pmr.2012.08.020. [DOI] [PubMed] [Google Scholar]
- Bromberg MB. Electrodiagnostic studies in clinical trials for motor neuron disease. J Clin Neurophysiol. 1998;15:117–128. doi: 10.1097/00004691-199803000-00004. [DOI] [PubMed] [Google Scholar]
- Rajagopalan V, Yue GH, Pioro EP. Brain white matter diffusion tensor metrics from clinical 1.5T MRI distinguish between ALS phenotypes. J Neurol. 2013;260:2532–2540. doi: 10.1007/s00415-013-7012-1. [DOI] [PubMed] [Google Scholar]
- Pioro EP, Majors AW, Mitsumoto H, et al. 1H-MRS evidence of neurodegeneration and excess glutamate + glutamine in ALS medulla. Neurology. 1999;53:71–79. doi: 10.1212/wnl.53.1.71. [DOI] [PubMed] [Google Scholar]
- Ben AmmarA, Soltanzadeh P, Bauche S, et al. A mutation causes MuSK reduced sensitivity to agrin and congenital myasthenia. PLoS One. 2013;8:e53826. doi: 10.1371/journal.pone.0053826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornskov EK, Norris FH, Jr, Mower-Kuby J. Quantitative axon terminal and end-plate morphology in amyotrophic lateral sclerosis. Arch Neurol. 1984;41:527–530. doi: 10.1001/archneur.1984.04050170073021. [DOI] [PubMed] [Google Scholar]
- Bjornskov EK, Dekker NP, Norris FH, Jr, Stuart ME. End-plate morphology in amyotrophic lateral sclerosis. Arch Neurol. 1975;32:711–712. doi: 10.1001/archneur.1975.00490520081016. [DOI] [PubMed] [Google Scholar]
- Maselli RA, Wollman RL, Leung C, et al. Neuromuscular transmission in amyotrophic lateral sclerosis. Muscle Nerve. 1993;16:1193–1203. doi: 10.1002/mus.880161109. [DOI] [PubMed] [Google Scholar]
- Slater CR, Fawcett PR, Walls TJ, et al. Pre- and post-synaptic abnormalities associated with impaired neuromuscular transmission in a group of patients with ‘limb-girdle myasthenia’. Brain. 2006;129(Pt 8):2061–2076. doi: 10.1093/brain/awl200. [DOI] [PubMed] [Google Scholar]
- Slater CR, Lyons PR, Walls TJ, et al. Structure and function of neuromuscular junctions in the vastus lateralis of man. A motor point biopsy study of two groups of patients. Brain. 1992;115(Pt 2):451–478. [PubMed] [Google Scholar]
- Selcen D, Shen XM, Milone M, et al. GFPT1-myasthenia: clinical, structural, and electrophysiologic heterogeneity. Neurology. 2013;81:370–378. doi: 10.1212/WNL.0b013e31829c5e9c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes J. Miniature end-plate potentials in neuromuscular disease: an electrophysiological investigation of motor-point muscle biopsies. J Neurol Neurosurg Psychiatry. 1971;34:521–526. doi: 10.1136/jnnp.34.5.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmqvist D, Hofmann WW, Kugelberg J, Quastel DM. An electrophysiological investigation of neuromuscular transmission in myasthenia gravis. J Physiol. 1964;174:417–434. doi: 10.1113/jphysiol.1964.sp007495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niks EH, Kuks JB, Wokke JH, et al. Pre- and postsynaptic neuromuscular junction abnormalities in musk myasthenia. Muscle Nerve. 2010;42:283–288. doi: 10.1002/mus.21642. [DOI] [PubMed] [Google Scholar]
- Evidente VG, Adler CH. An update on the neurologic applications of botulinum toxins. Curr Neurol Neurosci Rep. 2010;10:338–344. doi: 10.1007/s11910-010-0129-z. [DOI] [PubMed] [Google Scholar]
- Molenaers G, Fagard K, Van Campenhout A, Desloovere K. Botulinum toxin A treatment of the lower extremities in children with cerebral palsy. J Child Orthop. 2013;7:383–387. doi: 10.1007/s11832-013-0511-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. Clinical uses of botulinum neurotoxins: current indications, limitations and future developments. Toxins (Basel) 2012;4:913–939. doi: 10.3390/toxins4100913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Maio M. Therapeutic uses of botulinum toxin: from facial palsy to autonomic disorders. Expert Opin Biol Ther. 2008;8:791–798. doi: 10.1517/14712598.8.6.791. [DOI] [PubMed] [Google Scholar]
- Van Campenhout A, Molenaers G. Localization of the motor endplate zone in human skeletal muscles of the lower limb: anatomical guidelines for injection with botulinum toxin. Dev Med Child Neurol. 2011;53:108–119. doi: 10.1111/j.1469-8749.2010.03816.x. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Wong F, Fan L, Wells S, et al. Axonal and neuromuscular synaptic phenotypes in Wld(S), SOD1(G93A) and ostes mutant mice identified by fiber-optic confocal microendoscopy. Mol Cell Neurosci. 2009;42:296–307. doi: 10.1016/j.mcn.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Caroni P. Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J Neurosci Methods. 1997;71:3–9. doi: 10.1016/s0165-0270(96)00121-5. [DOI] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, et al. Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- Conforti L, Wilbrey A, Morreale G, et al. Wld S protein requires Nmnat activity and a short N-terminal sequence to protect axons in mice. J Cell Biol. 2009;184:491–500. doi: 10.1083/jcb.200807175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry JA, Ribchester RR. Persistent polyneuronal innervation in partially denervated rat muscle after reinnervation and recovery from prolonged nerve conduction block. J Neurosci. 1995;15:6327–6339. doi: 10.1523/JNEUROSCI.15-10-06327.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingwater TH, Thomson D, Mack TG, et al. Age-dependent synapse withdrawal at axotomised neuromuscular junctions in Wld(s) mutant and Ube4b/Nmnat transgenic mice. J Physiol. 2002;543(Pt 3):739–755. doi: 10.1113/jphysiol.2002.022343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco G, Ribchester RR. Confocal microendoscopy of neuromuscular synapses in living mice. Curr Protocol Mouse Biol. 2012;2:1–8. doi: 10.1002/9780470942390.mo110144. [DOI] [PubMed] [Google Scholar]
- Mackenzie FE, Romero R, Williams D, et al. Upregulation of PKD1L2 provokes a complex neuromuscular disease in the mouse. Hum Mol Genet. 2009;18:3553–3566. doi: 10.1093/hmg/ddp304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadon-Nachum M, Melamed E, Offen D. The, “dying-back” phenomenon of motor neurons in ALS. J Mol Neurosci. 2011;43:470–477. doi: 10.1007/s12031-010-9467-1. [DOI] [PubMed] [Google Scholar]
- Perry VH, Brown MC, Tsao JW. The effectiveness of the gene which slows the rate of Wallerian degeneration in C57BL/Ola mice declines with age. Eur J Neurosci. 1992;4:1000–1002. doi: 10.1111/j.1460-9568.1992.tb00126.x. [DOI] [PubMed] [Google Scholar]
- Costanzo EM, Barry JA, Ribchester RR. Co-regulation of synaptic efficacy at stable polyneuronally innervated neuromuscular junctions in reinnervated rat muscle. J Physiol. 1999;521(Pt 2):365–374. doi: 10.1111/j.1469-7793.1999.00365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo EM, Barry JA, Ribchester RR. Competition at silent synapses in reinnervated skeletal muscle. Nat Neurosci. 2000;3:694–700. doi: 10.1038/76649. [DOI] [PubMed] [Google Scholar]
- Lichtman JW, Magrassi L, Purves D. Visualization of neuromuscular junctions over periods of several months in living mice. J Neurosci. 1987;7:1215–1222. doi: 10.1523/JNEUROSCI.07-04-01215.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Lichtman JW. In vivo visualization of the growth of pre- and postsynaptic elements of neuromuscular junctions in the mouse. J Neurosci. 1990;10:894–908. doi: 10.1523/JNEUROSCI.10-03-00894.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer AM, Sanes JR, Lichtman JW. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J Comp Neurol. 2005;490:209–219. doi: 10.1002/cne.20620. [DOI] [PubMed] [Google Scholar]
- Ho M, Chang LH, Pires-Alves M, et al. Recombinant botulinum neurotoxin A heavy chain-based delivery vehicles for neuronal cell targeting. Protein Eng Des Sel. 2011;24:247–253. doi: 10.1093/protein/gzq093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribchester RR, Thomson D, Wood NI, et al. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington’s disease mutation. Eur J Neurosci. 2004;20:3092–3114. doi: 10.1111/j.1460-9568.2004.03783.x. [DOI] [PubMed] [Google Scholar]
- Harris JB, Ribchester RR. The relationship between end-plate size and transmitter release in normal and dystrophic muscles of the mouse. J Physiol. 1979;296:245–265. doi: 10.1113/jphysiol.1979.sp013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan EM, Martin AR. Non-linear summation of end-plate potentials in the frog and mouse. J Physiol. 1981;311:307–324. doi: 10.1113/jphysiol.1981.sp013586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnecker D, Voigt P, Hellwig N, Schaefer M. Reversible photobleaching of enhanced green fluorescent proteins. Biochemistry. 2005;44:7085–7094. doi: 10.1021/bi047881x. [DOI] [PubMed] [Google Scholar]
- Bilsland LG, Sahai E, Kelly G, et al. Deficits in axonal transport precede ALS symptoms in vivo. Proc Natl Acad Sci USA. 2010;107:20523–20528. doi: 10.1073/pnas.1006869107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater CR. Time course of failure of neuromuscular transmission after motor nerve section. Nature. 1966;209:305–306. doi: 10.1038/209305b0. [DOI] [PubMed] [Google Scholar]
- Winlow W, Usherwood PN. Ultrastructural studies of normal and degenerating mouse neuromuscular junctions. J Neurocytol. 1975;4:377–394. doi: 10.1007/BF01261371. [DOI] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Lichtman JW. Long-term synapse loss induced by focal blockade of postsynaptic receptors. Nature. 1994;372:519–524. doi: 10.1038/372519a0. [DOI] [PubMed] [Google Scholar]
- Dawkins R, Keller SL, Sewell WF. Pharmacology of acetylcholine-mediated cell signaling in the lateral line organ following efferent stimulation. J Neurophysiol. 2005;93:2541–2551. doi: 10.1152/jn.01283.2004.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrassi L, Purves D, Lichtman JW. Fluorescent probes that stain living nerve terminals. J Neurosci. 1987;7:1207–1214. doi: 10.1523/JNEUROSCI.07-04-01207.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogozhin AA, Pang KK, Bukharaeva E, et al. Recovery of mouse neuromuscular junctions from single and repeated injections of botulinum neurotoxin A. J Physiol. 2008;586:3163–3182. doi: 10.1113/jphysiol.2008.153569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo G, Rossetto O, Catsicas S, et al. Identification of the nerve terminal targets of botulinum neurotoxin serotypes A, D, and E. J Biol Chem. 1993;268:23784–23787. [PubMed] [Google Scholar]
- de Paiva A, Meunier FA, Molgo J, et al. Functional repair of motor endplates after botulinum neurotoxin type A poisoning: biphasic switch of synaptic activity between nerve sprouts and their parent terminals. Proc Natl Acad Sci USA. 1999;96:3200–3205. doi: 10.1073/pnas.96.6.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zurawski TH, Meng J, et al. Novel chimeras of botulinum and tetanus neurotoxins yield insights into their distinct sites of neuroparalysis. FASEB J. 2012;26:5035–5048. doi: 10.1096/fj.12-210112. [DOI] [PubMed] [Google Scholar]
- Daniels-Holgate PU, Dolly JO. Productive and non-productive binding of botulinum neurotoxin A to motor nerve endings are distinguished by its heavy chain. J Neurosci Res. 1996;44:263–271. doi: 10.1002/(SICI)1097-4547(19960501)44:3<263::AID-JNR7>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Sanders DB, Stalberg EV. AAEM minimonograph #25: single-fiber electromyography. Muscle Nerve. 1996;19:1069–1083. doi: 10.1002/(SICI)1097-4598(199609)19:9<1069::AID-MUS1>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Stalberg E, Schwartz MS, Trontelj JV. Single fibre electromyography in various processes affecting the anterior horn cell. J Neurol Sci. 1975;24:403–415. doi: 10.1016/0022-510x(75)90166-5. [DOI] [PubMed] [Google Scholar]
- Jansen JK, Van Essen DC. Re-innervation of rat skeletal muscle in the presence of alpha-bungarotoxin. J Physiol. 1975;250:651–667. doi: 10.1113/jphysiol.1975.sp011075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomp JJ, van Kempen GT, Molenaar PC. The upregulation of acetylcholine release at endplates of alpha-bungarotoxin-treated rats: its dependency on calcium. J Physiol. 1994;478(Pt 1):125–136. doi: 10.1113/jphysiol.1994.sp020236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomp JJ, van Kempen GT, Molenaar PC. Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin-treated rats. J Physiol. 1992;458:487–499. doi: 10.1113/jphysiol.1992.sp019429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau EG, Akaaboune M. The dynamics of recycled acetylcholine receptors at the neuromuscular junction in vivo. Development. 2006;133:4485–4493. doi: 10.1242/dev.02619. [DOI] [PubMed] [Google Scholar]
- Xu R, Salpeter MM. Rate constants of acetylcholine receptor internalization and degradation in mouse muscles. J Cell Physiol. 1999;181:107–112. doi: 10.1002/(SICI)1097-4652(199910)181:1<107::AID-JCP11>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Akaaboune M, Culican SM, Turney SG, Lichtman JW. Rapid and reversible effects of activity on acetylcholine receptor density at the neuromuscular junction in vivo. Science. 1999;286:503–507. doi: 10.1126/science.286.5439.503. [DOI] [PubMed] [Google Scholar]
- Couesnon A, Molgo J, Connan C, Popoff MR. Preferential entry of botulinum neurotoxin A Hc domain through intestinal crypt cells and targeting to cholinergic neurons of the mouse intestine. PLoS Pathog. 2012;8:e1002583. doi: 10.1371/journal.ppat.1002583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Peet NP, Butler MM, et al. Small molecule inhibitors as countermeasures for botulinum neurotoxin intoxication. Molecules. 2011;16:202–220. doi: 10.3390/molecules16010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney MA, Crisp JL, Nguyen LT, et al. Fluorescent peptides highlight peripheral nerves during surgery in mice. Nat Biotechnol. 2011;29:352–356. doi: 10.1038/nbt.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingwater TH, Ribchester RR. The relationship of neuromuscular synapse elimination to synaptic degeneration and pathology: insights from WldS and other mutant mice. J Neurocytol. 2003;32:863–81. doi: 10.1023/B:NEUR.0000020629.51673.f5. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Freeman MR. Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci. 2010;33:245–267. doi: 10.1146/annurev-neuro-060909-153248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inghilleri M, Iacovelli E. Clinical neurophysiology in ALS. Arch Ital Biol. 2011;149:57–63. doi: 10.4449/aib.v149i1.1264. [DOI] [PubMed] [Google Scholar]
- Joyce NC, Carter GT. Electrodiagnosis in persons with amyotrophic lateral sclerosis. PM R. 2013;5(5 suppl):S89–S95. doi: 10.1016/j.pmrj.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daube JR, Rubin DI. Needle electromyography. Muscle Nerve. 2009;39:244–270. doi: 10.1002/mus.21180. [DOI] [PubMed] [Google Scholar]
- Berzosa M, Wallace MB. Surveillance of Barrett’s esophagus: why biopsy if you can endomicroscopy. Gastrointest Endosc. 2014;79:222–223. doi: 10.1016/j.gie.2013.11.019. [DOI] [PubMed] [Google Scholar]
- Wu K, Liu JJ, Adams W, et al. Dynamic real-time microscopy of the urinary tract using confocal laser endomicroscopy. Urology. 2011;78:225–231. doi: 10.1016/j.urology.2011.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs FS, Zirlik S, Hildner K, et al. Confocal laser endomicroscopy for diagnosing lung cancer in vivo. Eur Respir J. 2013;41:1401–1408. doi: 10.1183/09031936.00062512. [DOI] [PubMed] [Google Scholar]
- Huang C, Kaza AK, Hitchcock RW, Sachse FB. Identification of nodal tissue in the living heart using rapid scanning fiber-optics confocal microscopy and extracellular fluorophores. Circ Cardiovasc Imaging. 2013;6:739–746. doi: 10.1161/CIRCIMAGING.112.000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujino A, Maertens C, Ohno K, et al. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc Natl Acad Sci USA. 2003;100:7377–7382. doi: 10.1073/pnas.1230273100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selcen D, Milone M, Shen XM, et al. Dok-7 myasthenia: phenotypic and molecular genetic studies in 16 patients. Ann Neurol. 2008;64:71–87. doi: 10.1002/ana.21408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley WG, Good P, Rasool CG, Adelman LS. Morphometric and biochemical studies of peripheral nerves in amyotrophic lateral sclerosis. Ann Neurol. 1983;14:267–277. doi: 10.1002/ana.410140304. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Glass JD. Axonal degeneration in motor neuron disease. Neurodegener Dis. 2007;4:431–442. doi: 10.1159/000107704. [DOI] [PubMed] [Google Scholar]
- Costa J, Swash M, de Carvalho M. Awaji criteria for the diagnosis of amyotrophic lateral sclerosis:a systematic review. Arch Neurol. 2012;69:1410–1416. doi: 10.1001/archneurol.2012.254. [DOI] [PubMed] [Google Scholar]
- Aggarwal A, Nicholson G. Detection of preclinical motor neurone loss in SOD1 mutation carriers using motor unit number estimation. J Neurol Neurosurg Psychiatry. 2002;73:199–201. doi: 10.1136/jnnp.73.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, et al. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006;9:408–419. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, et al. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson JB, Machado CB, Crossley M, et al. Optical control of muscle function by transplantation of stem cell-derived motor neurons in mice. Science. 2014;344:94–97. doi: 10.1126/science.1248523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohn DC, Miles GB, Rafuse VF, Brownstone RM. Transplanted mouse embryonic stem-cell-derived motoneurons form functional motor units and reduce muscle atrophy. J Neurosci. 2008;28:12409–12418. doi: 10.1523/JNEUROSCI.1761-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JA, Adams KL, Gundersen CB, Novitch BG. Functional neuromuscular junctions formed by embryonic stem cell-derived motor neurons. PLoS One. 2012;7:e36049. doi: 10.1371/journal.pone.0036049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- Feldman EL, Boulis NM, Hur J, et al. Intraspinal Neural Stem Cell Injections in ALS Subjects: phase I Trial Outcomes. Ann Neurol. 2014;75:363–73. doi: 10.1002/ana.24113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulis NM, Federici T, Glass JD, et al. Translational stem cell therapy for amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;8:172–176. doi: 10.1038/nrneurol.2011.191. [DOI] [PubMed] [Google Scholar]
- Gowing G, Svendsen CN. Stem cell transplantation for motor neuron disease: current approaches and future perspectives. Neurotherapeutics. 2011;8:591–606. doi: 10.1007/s13311-011-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrovo C, Bergamin N, Bates D, et al. In vivo tracking of murine adipose tissue-derived multipotent adult stem cells and ex vivo cross-validation. Int J Mol Imaging. 2013;2013:426961. doi: 10.1155/2013/426961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Axotomized wild-type motor terminals degenerate rapidly. (A and B) CEM Images of intramuscular axons in wild-type (thy1.2YFP16: C57Bl6) mice 24 h and 48 h, respectively, after sciatic nerve section. (C) EMG recording 24 h after nerve section in the vicinity of intramuscular axons like those shown in A, showing records in response to progressively increasing stimulus intensity, from 1 to 30 V. The large spike is a stimulus artifact due to stimulation at over 30 V intensity applied to the tibial nerve: there was no evoked CMAP. (D) Peak EMG amplitude in control (n = 14 muscles), and 1 day (n = 5) and 2 days (n = 4) after axotomy. Responses were lost within 24–48 h. Almost all motor nerve terminals degenerated within 24 h in these mice, with concomitant loss of evoked neuromuscular responses.
Low concentrations of 4-Di-2-Asp (10 μmol/L) have no effect on neuromuscular transmission. (A) EPPs and (B) MEPPs recorded after 60-min incubation. There was no significant linear regression over 1 h of incubation in: (C) EPP amplitude, (D) MEPP amplitude, (E) quantal content, or (F) MEPP frequency. Each data point in C–F shows the mean ± SEM and the calculated best fit linear regression. n = 4 muscles, three muscle fibers sampled in each muscle at each time point. The slopes were not significantly different (P > 0.05, linear regression in all four cases.)
CEM of intramuscular axons and motor nerve terminals in a control thy1.2YFP16: WldS mouse. The Proflex probe was glided over the surface of the muscle while capturing images in real time at 12 fps, while the sciatic nerve was supramaximally stimulated. Twitching of the muscle is discernible.
CEM of motor nerve terminals imaged in real time at 12 fps during sciatic nerve stimulation; thy1.2YFP16: WldS mouse, 2 days after sciatic nerve section. Note the intermittent twitching of the muscle produced by stimulation of the distal, tibial nerve stump.
CEM of motor endplates imaged in real time at 12 fps, following instillation of Alexa488-α-bungarotoxin. The fluorescence is due to postsynaptic labeling of AChR.
CEM of motor nerve terminals imaged in real time at 12 fps following instillation of 4-Di-2-Asp to label motor nerve terminals in a nontransgenic (C57Bl6) mouse.
CEM of motor nerve terminals imaged in real time at 12 fps following instillation of 4-Di-2-Asp to label motor nerve terminals in a Sprague–Dawley rat.
CEM of motor nerve terminals imaged in real time at 12 fps following instillation GFP-HcBoNTx/A, to label motor nerve terminals in a nontransgenic (C57Bl6) mouse.