

Abstract
Objective
Mutations in nuclear-encoded mitochondrial DNA (mtDNA) polymerase (POLG) are known to cause autosomal dominant chronic progressive external ophthalmoplegia (adCPEO) with accumulation of multiple mtDNA deletions in muscles. However, no animal model with a heterozygous Polg mutation representing mtDNA impairment and symptoms of CPEO has been established. To understand the pathogenic mechanism of CPEO, it is important to determine the age dependency and tissue specificity of mtDNA impairment resulting from a heterozygous mutation in the Polg gene in an animal model.
Methods
We assessed behavioral phenotypes, tissue-specific accumulation of mtDNA deletions, and its age dependency in heterozygous PolgD257A knock-in mice carrying a proofreading-deficient mutation in the Polg.
Results
Heterozygous PolgD257A knock-in mice exhibited motor dysfunction in a rotarod test. Polg+/D257A mice had significant accumulation of multiple mtDNA deletions, but did not show significant accumulation of point mutations or mtDNA depletion in the brain. While mtDNA deletions increased in an age-dependent manner regardless of the tissue even in Polg+/+ mice, the age-dependent accumulation of mtDNA deletions was enhanced in muscles and in the brain of Polg+/D257A mice.
Interpretation
Heterozygous PolgD257A knock-in mice showed tissue-specific, age-dependent accumulation of multiple mtDNA deletions in muscles and the brain which was likely to result in neuromuscular symptoms. Polg+/D257A mice may be used as an animal model of adCPEO associated with impaired mtDNA maintenance.
Introduction
Chronic progressive external ophthalmoplegia (CPEO) is a mitochondrial neuromuscular disease presenting progressive external ophthalmoplegia and other neuromuscular symptoms. On the molecular level, the accumulation of multiple mtDNA deletions in muscles is characteristic of CPEO, which is distinct from other inherited mitochondrial diseases because of point mutations or copy number reduction of mtDNA in affected tissues.1–3 CPEO is a relatively late-onset disease accompanied by neurological symptoms such as ataxia, exercise intolerance, fatigue, optic atrophy, cognitive impairment, and mood disorders.4–18 Postmortem brain analyses showed increased accumulation of multiple mtDNA deletions in the brain.4,7 Thus, neurological symptoms in CPEO likely result from the accumulation of multiple mtDNA deletions in the brain.
Mutations in nuclear-encoded proteins involved in mtDNA maintenance cause inherited CPEO.3,8–11,18–21 The nuclear gene encoding the catalytic subunit of the mtDNA polymerase, known as DNA polymerase γ (Polg), plays a prominent role in replication and repair, which are essential for mtDNA synthesis.14 Heterozygous mutations in POLG cause autosomal dominant CPEO. Thus, a heterozygous mutant Polg knock-in mouse may be an appropriate animal model for CPEO. Homozygous knock-in mice that carry proofreading-deficient Polg (PolgD257A) exhibited marked accumulation of mtDNA point mutations and showed systemic physical abnormalities and reduced life span; these mice have been proposed to be an animal model of premature aging.22–27 However, heterozygous PolgD257A mice were reportedly normal.22–30
In this study, we examined heterozygous PolgD257A mice in detail. These mice exhibited motor dysfunction and accumulation of mtDNA deletions in the brain and muscles in an age-dependent and tissue-specific manner.
Materials and Methods
Animals and behavioral analysis
The PolgD257A mouse was described previously.23 In this study, Polg+/D257A mice were backcrossed for more than six generations into C57BL/6JJcl; next, selective breeding using 104 microsatellite markers covering the entire genome enabled the production of mutant mice with a genetic background similar to C57BL/6J for >99% of offspring by mating male Polg+/D257A mice with female C57BL/6JJcl mice purchased from CLEA Japan (Tokyo, Japan). We examined 34 weeks old male mice kept in cages with a running wheel for 7 weeks from 19 to 25 weeks. The details of a conventional behavioral test battery are described in Data S1. All animal experiments were performed in accordance with the protocols that had been approved by the Animal Experiment Committee of RIKEN (Wako, Saitama, Japan) and Shiga University of Medical Science (Otsu, Shiga, Japan). The number of animals used their suffering was minimized as much as possible.
Analysis of mtDNA point mutation
mtDNAs were purified from mice frontal lobe using the mtDNA Extractor CT kit (Wako Pure Chemical Industries, Osaka, Japan), which is based on the alkaline-SDS method for the plasmid DNA purification followed by removal of sodium iodide and RNA by RNaseA treatment. The 1782-bp fragment (#13557–15340) of mtDNA (NC_005089) from each genotype (n = 3) was digested using the restriction endonucleases XhoI and BglII and directly cloned into pBluescript SK+ plasmid DNA without polymerase chain reaction (PCR) amplification. From each mouse tissue, more than 12 independent clones were sequenced and the average mutation frequency was examined (Polg+/+ mice: 48 total clones, Polg+/D257A mice: 47 total clones, PolgD257A/D257A mice: 53 total clones). More than 21,000 nucleotides were analyzed from each mouse.
Southern blot analysis
Purified total mtDNA was either left undigested or digested by incubation with restriction endonuclease BglII (Takara Bio, Shiga, Japan). After electrophoresis and depurination, alkaline blotting was performed by capillary transfer.
mtDNAs were detected using the Digoxigein (DIG)-labeled DNA probe for mouse mtDNA, which was described previously.31 The DIG-labeled DNA probes for the control region (D-loop), ND1, and COX1 were synthesized using the PCR DIG Probe Synthesis Kit (Roche Diagnostics, Mannheim, Germany), and whole mtDNA probe was synthesized from Taq I-digested mouse whole mtDNA using random primers and the BcaBEST DIG labeling kit (Takara).
PCR analysis
All primer sequences are described in Table S1. For PCR, mouse mtDNA was amplified from 10 ng of purified mtDNA with the 0.2 μmol/L primers D3 and D4,31 which face outward from the D-loop of mtDNA, using SYBR Premix Ex Taq reagent (Takara Bio) with the following protocol: 95°C for 20 sec, 55°C for 20 sec, and 72°C for 13 min (for a long-extension PCR) or 80 sec (for a short-extension PCR), 35 cycles after an initial denaturation at 95°C for 1 min. For quantitative real-time PCR (qPCR) analysis, each PCR product was separately amplified from 50 ng of total DNA or 5 ng of purified mtDNA in a 10 μL reaction using SYBR Premix Ex Taq reagent. For deleted mtDNA molecules, qPCR was based on short-extension PCR. Accumulation of mtDNA deletions was assessed by determining the ratio to total mtDNA quantified with amplification of the D-loop using the primer pair D1 and D2. Other primer sets were designed to anneal to the COX1 gene and the ND4 gene for mtDNA, and the ApoB gene for nuclear DNA. Copy number was quantified by determining the ratio of total mtDNA to nuclear DNA. Real-time qPCR reactions were carried out in quadruplicate for all measurements. In every run, the quantities were calibrated using the standard curves with each control plasmid (R2 > 0.99), as described previously.32 Only a standard curve of mtDNA deletions was used to standardize the differences among runs and was calculated between Ct values and the logarithm of concentration of extracted mtDNA from old PolgD257A/D257A mice (>60 weeks old) within the linear range (Fig. S4B, R2 > 0.95).
In vivo mtDNA labeling and Southwestern blot analysis
Newly synthesized mtDNA molecules were labeled by 5-bromo-2-deoxyuridine (BrdU) treatment and detected by Southwestern blot analysis using anti-BrdU antibody. Eight-week-old, male C57BL/6J mice (CLEA Japan) were injected intraperitoneally with prewarmed 2′-Deoxyuridine, 5′-Triphosphate (dUTP) or BrdU (500 mg/kg body weight; 50 mg/mL; NACALAI TESQUE, Kyoto, Japan) dissolved in 0.1 mol/L NaOH and adjusted to neutral pH. mtDNA was sampled each hour from 0–12 h and at 24, 36, 48, 60, 72, 84, and 96 h after intraperitoneal injection. Southwestern blot analysis was developed using a method described previously.33 mtDNA was digested by incubation with the restriction endonuclease EagI (New England Biolabs, Beverly, MA) and transferred using the same method used for Southern blot analysis. After BrdU imaging with anti-BrdU antibody (Becton Dickinson Immunocytometry Systems, San Jose, CA), and an HRP-conjugated anti-mouse IgG secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) by Western Lightning™ Chemiluminescence Reagent (Perkin Elmer, Waltham, MA) and LAS-3000 (FUJIFILM, Tokyo, Japan), antibodies were stripped for Southern blot analysis. The relative levels of newly generated mtDNA after injection was quantified by determining the value of incorporation of BrdU into mtDNA based on the BrdU/mtDNA signal intensity ratio measured using Multi Gauge Ver 2.2 software (FUJIFILM).
Cell culture and immunofluorescence analysis
Primary neurons were isolated from mouse embryos at gestational day 17–18 and cultured as described previously.34 To detect localization of BrdU signals in the mitochondria as an indicator of newly synthesized mtDNA, neurons were cultured in the presence 10 μmol/L BrdU for 48 h after 7 days of incubation for differentiation. Neurons were fixed and immunocytochemically stained as reported previously.35 In some experiments, neurons were also cultured in the presence of 100 nmol/L MitoTracker Red CM-H2XRos for 1 h before fixation or were incubated with NeuroTrace Red Fluorescent Nissl Stain (Life Technologies, Carlsbad, CA) at room temperature for 20 min following the anti-BrdU staining. Images were acquired using a confocal microscope (FV1000; Olympus, Tokyo, Japan) from at least three independent culture preparation. Protocols to isolate neural stem cells and generate neurospheres from embryonic brain in vitro have been described previously.36
Statistical analysis
The results of quantitative experiments were analyzed by parametric or nonparametric tests after conducting a Kolmogorov–Smirnov test to confirm a normal distribution. A two-tailed test was used for all analysis. Statistical significance (P < 0.05) was determined using SPSS 18.0 software (SPSS, Chicago, IL) and KyPlot 4.0 (KyensLab, Tokyo, Japan).
Results
Behavioral phenotypes of Polg+/D257A mice
We examined 34 weeks old male mice carrying the heterozygous PolgD257A mutation (referred to as Polg+/D257A) using a conventional behavioral test battery. Homozygous PolgD257A-mutant mice (referred to as PolgD257A/D257A mice) were also assessed as a positive control group for mitochondrial dysfunction, apoptosis, and sarcopenia in skeletal muscle.25,26,28 As expected, PolgD257A/D257A mice showed significantly lower body weight than mice of other genotypes, poor performance in the rotarod test, and reduced spontaneous motor activity in the open field test, elevated plus maze, and Y-maze test (Fig. S1). In the rotarod test, not only PolgD257A/D257A mice but also Polg+/D257A mice showed significantly poor performance than Polg+/+ mice for a number of trials (Fig. 1). Polg+/D257A mice did not differ from Polg+/+ mice in other tests. The result indicates that the motor dysfunction caused by the heterozygous mutation in Polg is milder than that caused by the homozygous mutation.
Figure 1.
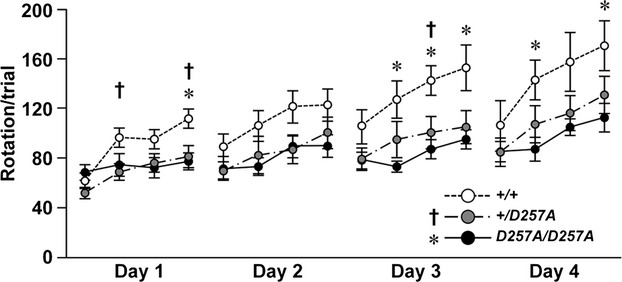
Rotarod test for evaluating motor function among Polg+/+ mice (n = 9), Polg+/D257A (n = 10), and PolgD257A/D257A (n = 10) mice. The general linear model repeated measures ANOVA followed by Tukey’s honestly significant difference (HSD) (mean rotations ± SEM) showed significant effects of genotype (P = 0.026) and days (P < 1.00 × 10−10), but no interaction between genotype and days (P = 0.082). Polg+/D257A mice generally showed lower performance than Polg+/+ mice (P = 0.077), which was similar to PolgD257A/D257A mice (Polg+/D257A mice vs. PolgD257A/D257A mice: P = 0.885, PolgD257A/D257A mice vs. Polg+/+ mice: P = 0.029). These results were supported by significant differences between Polg+/+ and Polg+/D257A mice (†P < 0.05), or PolgD257A/D257A mice (*P < 0.05) in analysis of each trial.
Accumulation of mtDNA deletions in the brain of Polg+/D257A mice
The motor dysfunction in Polg+/D257A mice prompted us to investigate whether mtDNA was impaired by the heterozygous mutation in Polg. Therefore, we evaluated mtDNA alterations in the brain of Polg+/D257A mice. Southern blot analysis of purified mtDNA using a probe for the control region (D-loop) (Fig. 2A and B) showed the presence of small mtDNAs corresponding to deletion mutants in the frontal lobe of Polg+/D257A mice. These small mtDNAs were observed in the frontal lobe of Polg+/D257A mice at 62 weeks of age upon overexposure (Fig. 2B). However, small mtDNAs were not detectable in Polg+/D257A mice at 24 weeks and in Polg+/+ mice at 62 weeks at the same experimental condition. Additionally, apparent multiple mtDNA signals were observed in PolgD257A/D257A mice at 62 weeks, subtle signals were observed at 24 weeks (Fig. 2A). Consistent with the results of previous studies, there was no detectable signal other than full-length mtDNA using probes for gene-encoding regions of mtDNA, even in PolgD257A/D257A mice (Fig. S2A). Accumulation of multiple mtDNA deletions in the frontal lobe of Polg+/D257A mice at 62 weeks was confirmed by PCR analyses (Fig. 2C–E). Long-range PCR amplification of mtDNA revealed the presence of variously sized mtDNA molecules with deletions in the brain of Polg+/D257A mice (Fig. 2C). Full-length mtDNA was observed, but there were fewer deletion molecules in Polg+/+ mice than in Polg+/D257A mice. Moreover, short-extension PCR was used to selectively amplify small mtDNA molecules to clarify the difference between Polg+/D257A and Polg+/+ mice (Fig. 2D). Using highly sensitive qPCR analysis (see Materials and Methods, Fig. S2B and C), significantly increased accumulation of mtDNA deletions was observed in Polg+/D257A mice (Fig. 2E). In frontal lobe of Polg+/D257A mice, there was a reduction in protein level of mtDNA-encoded subunits of Complex IV (Fig. S3).
Figure 2.
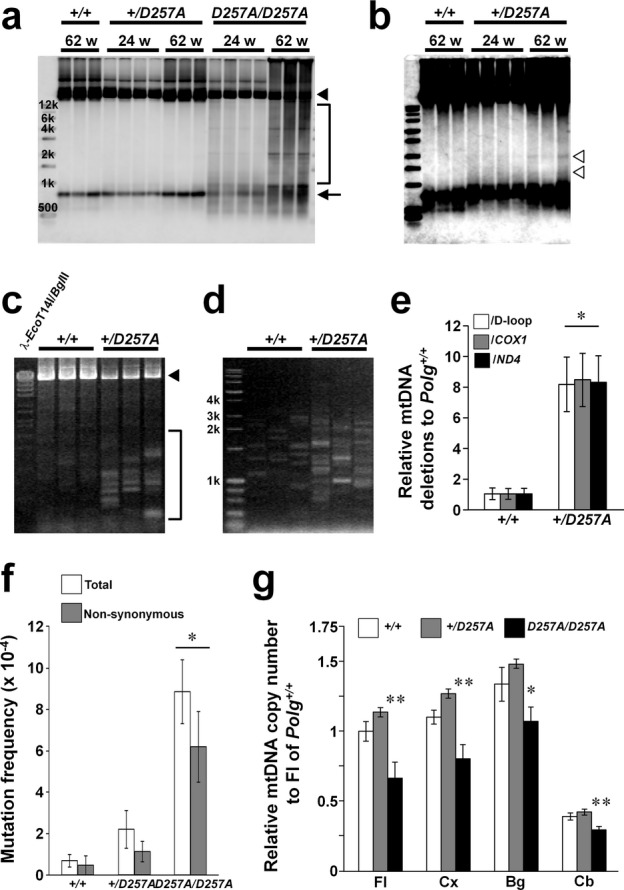
mtDNA analysis in the brain of mice. (A) Southern blot analysis of the frontal lobe mtDNA using a probe for the control region of mtDNA. Signals of full-length mtDNA (arrowhead), 7S DNA forming the displacement loop (D-loop; arrow), and mtDNA deletions (square bracket) can be observed. Signals for mtDNAs smaller than the full-length 16.5-kb signal were detected strongly in the frontal lobe of PolgD257A/D257A mice at 62 weeks and more subtly at 24 weeks. (B) Overexposed Southern blot analysis of mtDNA in the frontal lobe of Polg+/+ and Polg+/D257A mice in panel A. Modest mtDNA signals (open arrowheads) were detectable in Polg+/D257A mice, but not in Polg+/+ mice. (C) Long-range PCR of the frontal lobe mtDNA. Full-length mtDNA (arrow head) in all mice and deletion molecule size (square bracket) in Polg+/D257A mice (n = 3) were observed. (D) Short-extension PCR for selective amplification of deletion molecules. Amplicon-derived mtDNA deletions in the frontal lobe were more obvious in Polg+/D257A mice than Polg+/+ mice. (E) Quantitative analysis of accumulation of mtDNA deletions in the frontal lobe using qPCR. Accumulation levels were determined using the D-loop (white), COX1 (gray), or ND4 (black) region of mtDNA as a reference (n = 3, mean ± SD, *P < 0.02, t-test). (F) Quantitative analysis of mtDNA point mutation in the frontal lobe of mice at 62 weeks. Numbers of total mutations (white columns, Welch ANOVA followed by Dunnett’s T3 test) and nonsynonymous mutations (gray columns, one-way ANOVA followed by Tukey’s HSD) are indicated (n = 3, mean ± SD, *P < 0.025). (G) Analysis of mtDNA copy number in the frontal lobe, posterior cortex, basal ganglia, and cerebellum of mice: Polg+/+ mice (white columns), Polg+/D257A mice (gray columns), and PolgD257A/D257A mice (black columns). The vertical axis represents the relative value normalized to the average copy number of mtDNA in frontal lobe of Polg+/+ mice (n = 3, mean ± SD, **P < 0.01, *P < 0.05, Welch ANOVA followed by the Dunnett’s T3 test).
We also investigated mtDNA point mutations in frontal lobe. PolgD257A/D257A mice showed significant accumulation of point mutations compared to other mice. Although Polg+/D257A mice also showed a similar tendency of higher number of mtDNA point mutations than Polg+/+ mice, this difference was not statistically significant (Fig. 2F). In addition to point mutations, crucial single nucleotide insertion/deletion were found in six clones from PolgD257A/D257A mice. Moreover, PolgD257A/D257A mice showed a significantly reduced mtDNA copy number in frontal lobe, posterior cortex, basal ganglia, and cerebellum. No significant difference in mtDNA copy number was observed between Polg+/+ and Polg+/D257A mice (Fig. 2G). Thus, the formation and accumulation of mtDNA deletions, but not point mutations and mtDNA depletion, were detected in Polg+/D257A mice.
Age-dependent increase in mtDNA deletions is accelerated in muscle and the brain of Polg+/D257A mice
We investigated mtDNA deletion levels in detail in the brain, which was divided into five areas (frontal lobe, posterior cortex, cerebral cortex except for the frontal lobe, hippocampus, basal ganglia, cerebellum), and in other somatic tissues (heart, liver, kidney, and skeletal muscles) of Polg+/+ and Polg+/D257A mice at 48 weeks of age. Deletions in the heart were clearly detected in all Polg+/D257A mice by Southern blot analysis (Fig. 3A, upper panel). Deleted mtDNA signals in the frontal lobe, posterior cortex, and hippocampus of Polg+/D257A mice were detectable after overexposure in Southern blot analysis, same as is shown in Figure 2B, but were not detectable at all in Polg+/+ mice (Fig. 3A, lower panel). Consistent with the results of Southern blot analysis, significant differences were verified by short-extension qPCR analysis in the frontal lobe, posterior cortex, and hippocampus, and heart, but not in the cerebellum, liver, kidney, and skeletal muscles at 48 weeks (Fig. 3B). On the other hand, PolgD257A/D257A mice indeed showed much higher levels of mtDNA deletions in all tissues we tested (Fig. S4).
Figure 3.

Tissue-specific accumulation of mtDNA deletions in Polg+/D257A mice. (A) Southern blot analysis of accumulation in tissues of Polg+/D257A and Polg+/+ mice (n = 3) at 48 weeks using a probe containing D-loop, a representative result is shown. mtDNA deletions were detected in the heart of Polg+/D257A mice (open arrowheads) but not in Polg+/+ mice. In Polg+/D257A mice, slight signals of deletion molecules were detectable in the frontal lobe, posterior cortex, and hippocampus of only Polg+/D257A mice after overexposure (lower panel, arrowheads). (B) qPCR analysis to compare accumulation of mtDNA deletions in the frontal lobe (Fl), posterior cortex (Pc), hippocampus (Hp), basal ganglia (Bg), cerebellum (Cb), heart, liver (Liv), kidney (Kid), and skeletal muscles (Sk) of Polg+/D257A mice with that of Polg+/+ mice at 48 weeks old. Relative accumulation normalized to that in the frontal lobe of Polg+/+ mice is represented (n = 3, mean ± SD, **P < 0.01, *P < 0.05, t-test). Significant differences were observed among tissues of Polg+/D257A mice: Fl and heart > Liv, Kid, and Sk; heart > all brain areas (P < 0.05, Welch ANOVA followed by the Dunnett T3 test). (C) Analysis of age-dependent accumulation of mtDNA deletions in the frontal lobe and skeletal muscles of Polg+/D257A and Polg+/+ mice at 12, 24, 48, 60, 72, 84, and 101 weeks using qPCR. Values are those relative to the average of 12-week-old Polg+/+ mice in each tissue. Each circle indicates one mouse (n = 4; two males and two females). Results in other brain areas, heart, and liver are shown in Figure S5. A significant effect of genotype and a significant interaction between genotype and age upon increase in mtDNA deletions was observed in the brain and muscles (P < 0.001, ANCOVA with a factor of genotype and a covariate of age), but not for the liver (P > 0.2). In ANCOVA with a factor of tissues and a covariate of age in each genotype, difference among tissues and the interaction between tissue and age were significant (P < 0.001). Significant differences were detected at several ages, except for in the liver (*P < 0.05). Significant effects of age were observed in all tissues regardless of genotype (P < 1.00 × 10−5, ANCOVA with a factor of genotype and a covariate of age).
To understand the effect of age on accumulation of multiple mtDNA deletions, we measured mtDNA deletion levels in each brain region, muscles, and liver in Polg+/+ and Polg+/D257A mice of various ages (Figs. 3C and S5). mtDNA deletions accumulated with age in an exponential manner regardless of tissues or genotypes. However, there was a significant interaction between genotype and age; the heterozygous Polg mutation accelerated the age-dependent increase in mtDNA deletions in the brain and muscles, including the cerebellum and skeletal muscles, but not the liver. In contrast, mtDNA copy number was not affected by age or genotype, whereas a significant difference was found between tissues (Fig. S6). Although the increase in mtDNA deletion was not detected until 60 weeks in mtDNA analysis utilizing the entire skeletal muscle in hind limb, a part of skeletal muscle of hind limb of Polg+/D257A mice showed the significant enhancement of accumulation of mtDNA deletions at 37 weeks corresponding period of the rotarod test (Fig. S7). Furthermore, a significant decrease in complex IV (COX) activities was observed in skeletal muscles of Polg+/D257A mice at 96 weeks (Fig. S8) similar to that in cortex of Polg+/D257A mice in previous report.37
In neural stem cells (NSCs) cultured for 5 weeks from mouse embryos, no difference was observed between NSCs from Polg+/D257A mice and those from Polg+/+ mice, although NSCs from PolgD257A/D257A mice accumulated mtDNA deletions (Fig. S9). These results suggest that aging plays an important role in the accumulation of mtDNA deletions in the brain of Polg+/D257A mice.
mtDNA replicates more rapidly in the mouse brain than in the liver
Since mtDNA deletions and point mutations were accumulated in mouse brain which contains nondividing neurons, we estimated the probability that quiescent cells are constantly metabolizing mtDNA and accumulate mtDNA mutations during mtDNA synthesis. To verify the active mtDNA synthesis, we treated primary cultured neurons from wild-type mouse embryos with BrdU for 48 h. We showed that BrdU signals were localized in the mitochondria in the cell bodies and neurites, but not in the nuclei (Fig. 4A), in postmitotic neurons.
Figure 4.
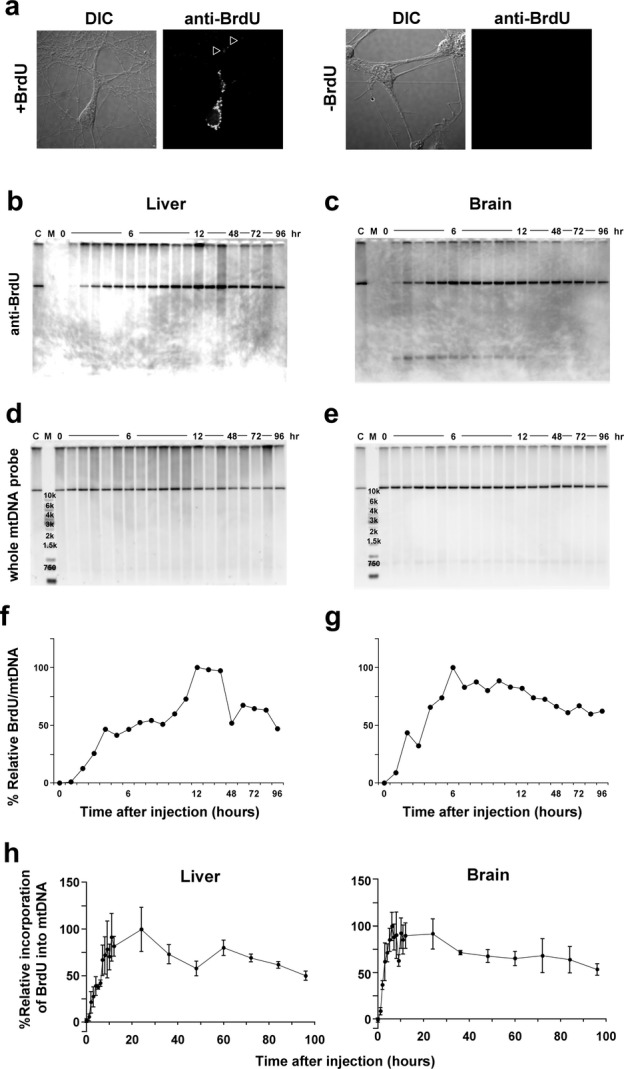
(A) Immunofluorescence analysis for the incorporation of BrdU in primary cultures of neurons prepared from mice at E17. After 9 days of culture, neurons exhibiting morphological differentiation with neurite elongations (left panels) were stained with an anti-BrdU antibody (right panels). Localization of BrdU was observed in the mitochondria of cell bodies and neurites (open arrow heads), but not in the nuclei of postmitotic neurons in the presence 10 μmol/L BrdU (lower panels), although no signal was observed in the absence of BrdU (upper panels). Assessment of newly generated mtDNA in the liver (B, D, and F) and brain (C, E, and G) using BrdU. Representative result is shown. (B and C) Southwestern blot analysis of purified mtDNA from the liver and posterior cortex after intraperitoneal injection of BrdU into adult mice. A control sample was applied to normalize the signals on each membrane (lane “C”). (D and E) Southern blot analysis using the same membrane from B and C. Analysis was performed from each injection independently (n = 3). (F and G) Quantification of incorporation of BrdU into mtDNA in the liver and cerebral cortex. The vertical axis represents the relative level of BrdU incorporation into mtDNA to the peak in each experiment. (H) Quantitative analysis of BrdU incorporation into mtDNA in the liver (upper graph) and cerebral cortex (lower graph). In ANCOVA in which a factor of tissue and a covariate of time was applied (n = 3, % mean ± SE), there was a significant effect of tissue (P < 0.05) and time (P < 0.001) as well as a significant interaction between region and time (P < 0.03). The time to reach the half of the maximum level of BrdU incorporation was shorter in the cerebral cortex (2–3 h) of the brain than in the liver (7–8 h).
Tissue-dependent difference in mtDNA synthesis rate may also contribute to the tissue-dependent accumulation of mtDNA deletions. Thus, we examined differences in mtDNA synthesis between the liver (Fig. 4B, D, and F) and brain (posterior cortex) (Fig. 4C, E, and G) by in vivo mtDNA labeling with BrdUin adult wild-type mice at 8 weeks of age. Incorporation of BrdU (Fig. 4B and C) into mtDNA (Fig. 4D and E) increased over time in the both tissues from an hour or two after the injection (Fig. 4F and G). A significant dependence was observed between tissues over time. The time to reach the half-maximal level of BrdU incorporation (T1/2) was shorter in the brain (posterior cortex) (Fig. 4G and H, ~4 h) than in the liver (Fig. 4F and H, 7–8 h).
Discussion
Despite clinical studies indicating that heterozygous POLG mutations cause CPEO, neuromuscular symptoms and accumulation of mtDNA deletions in Polg+/D257A mice have not been reported. In this study, we found that a heterozygous mutation of POLG could cause neuromuscular symptoms similar to those found in CPEO patients. Moreover, we showed that the D257A mutation in one allele of the Polg gene was sufficient to cause accumulation of multiple mtDNA deletions in the brain and muscles, but accumulation of mtDNA point mutations and mtDNA copy number reduction is absent or less prominent. It is still controversial whether mtDNA point mutations are increased in the Polg+/D257A mice.22,27 However, the present results suggest that mtDNA deletions are more prominent than point mutations. In addition to skeletal muscles in posterior crus, we cannot rule out a possibility that accumulations of mtDNA deletions in tissues other than skeletal muscles, such as the posterior cortex, basal ganglia, and heart, affect motor dysfunction of Polg+/D257A mice, because they already showed significant increase in mtDNA deletions at 24 weeks. In either case, the tissue-specific accumulation of mtDNA deletions was similar to CPEO patients and the worse rotarod performance of Polg+/D257A mice might be resulted from accumulation of mtDNA deletions. Thus, the Polg+/D257A mouse could be an animal model of adCPEO, in contrast with the PolgD257A/D257A mice that have the systemically premature-aging phenotype derived from excessive mtDNA alterations, including mtDNA depletion and point mutations.
Recently, Dai et al. showed that Polg+/D257A mice performed comparably in the rotarod test to control mice.37 There are several possible explanations for this discrepancy: protocol of rotarod test, analysis methods, genetic back ground, and housing environment. These differences between the previous report and our study could lead to this apparent discrepancy. Among them, housing environment of mice might have impact on the motor dysfunction because mice were kept in cages with a running wheel. Exercise might have become load for muscles. In addition, the genetic background of mice seems to be different: we backcrossed Polg+/D257A mice into C57BL/6JJcl before a conventional behavioral test battery, whereas Dai and colleagues intercrossed Polg+/D257A mice in a colony. Clinical heterogeneity of features and different age of onset is observed among patients even within a family of adCPEO carrying an identical mutation in POLG.8,10,11,18 This may also be explained by genetic background and environmental factors. We clearly demonstrated that aging is a critical determinant for accumulation of mtDNA deletions due to Polg mutation. However, other genetic or environmental risk factors associated with disease onset and progression remain unclear. The Polg+/D257A mouse will be useful for exploring and evaluating these risk factors.
There are two explanations for the tissue specificity of adCPEO symptoms due to a POLG mutation. First, the muscles and brain may show functional impairment at relatively low levels of mtDNA deletions, which does not cause dysfunction in other tissues. Second, the muscles and brain may accumulate more mtDNA deletions than other types of tissues. Our data suggest that the latter possibility is more likely as mtDNA deletions preferentially accumulated in the muscles and brain in Polg+/D257A mice. mtDNA deletions may preferentially accumulate in the muscles and brain because myocytes and neurons are not frequently replaced by newly generated cells.
The results of the present study analyzing mtDNA in Polg+/D257A mice suggest that normal Polg in one allele is sufficient to repair point mutations or to maintain the mtDNA copy number but does not inhibit the formation of deletions during mtDNA synthesis by PolgD257A.38,39 Previous reports did not detect the accumulation of mtDNA deletions in Polg+/D257A mice.22,23,26–29 This apparent discrepancy may be because different methods were used; Southern blot analysis with labeled whole mtDNA or a large variety of labeled oligonucleotides as a probe was used in previous studies, while the D-loop region that containing the replication origin for the heavy strand of mtDNA was used as a probe in this study. The presence of the D-loop in deleted mtDNAs suggests that the molecules are replicated and amplified, which explains why mtDNA deletions accumulate in an exponential manner along with aging. Indeed, D-loop retaining mtDNAs with deletions replicate more efficiently than full-length mtDNAs.40 Therefore, higher activity of mtDNA synthesis in the brain may contribute at least in part to tissue-specific accumulation of mtDNA deletions. Further studies are needed to understand how mtDNAs containing deletions replicate through the action of normal or mutant Polg or both in the brain subregions of Polg+/D257A mice and to clarify how pathological accumulation of mtDNA deletions cause chronic progression of neuromuscular symptoms of CPEO.
Acknowledgments
This work was supported by grants from the Laboratory for Molecular Dynamics of Mental Disorders, RIKEN Brain Science Institute (BSI), a Grant-in-Aid from the Japanese Ministry of Health and Labor, and Grants-in-Aid from the Japanese Ministry of Education, Culture, Sports, Science and Technology. S. F. is supported by Grants-in-Aid for young scientists from the Japanese Ministry of Education, Culture, Sports, Science and Technology, and Grants-in-Aid for young scientists from Shiga University of Medical Science. We are grateful to the staff at the Divisions of Animal Experiments and Common Instrumentations in Research Resources Center, RIKEN BSI, for technical assistance, Yoko Nakano for DNA extraction from mouse tissues, and the other members in our laboratory for various discussions.
Conflict of Interest
Dr. Kato received honorarium for manuscripts, lectures, or consultancy from Kyowa Hakko Kirin Co., Ltd., Eli Lilly Japan K.K., Otsuka Pharmaceutical Co., Ltd., GlaxoSmithKline K.K., Taisho Toyama Pharmaceutical Co., Ltd., Dainippon Sumitomo Pharma Co., Ltd., Meiji Seika Pharma Co., Ltd., Pfizer Japan Inc., Mochida Pharmaceutical Co., Ltd., Shionogi & Co., Ltd., Janssen Pharmaceutical K.K. and Astellas Pharma Inc., and grants from Takeda Pharmaceutical Co., Ltd. outside the submitted work. Dr. Prolla has a United States patent 7,126,040 and a founding owner and consultant for LifeGen Technologies, specializing in nutraceutical interventions in aging. Dr. Kujoth has a United States patent 7,126,040. Other authors declare no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Results of conventional behavioral test battery. Male mice at 34 weeks old were used for these analyses (mean ± SEM is shown in all graphs.). (A) Body weight. PolgD257A/D257A mice weighed less than Polg+/D257A and Polg+/+ mice (n = 10, *P < 0.05, one-way ANOVA followed by Tukey’s HSD). (B) Open field test. Total distance traveled per 5 min is shown. There was a significant main effect of genotype (P = 0.046) and bin of time (P = 0.00307) in the GLM repeated measures ANOVA. A significant interaction between genotype and the bin was observed (P = 1.01 × 10−5). In analysis of each 5 min, the total distance of PolgD257A/D257A mice was shorter than for Polg+/D257A and Polg+/+ mice (n = 10, *P < 0.05, one-way ANOVA followed by Tukey’s HSD). (C) A percentage of total time in center in an open field test. There was no significant difference among genotypes (n = 10, P = 0.817, one-way ANOVA). (D) Total distance traveled in elevated plus maze test. The total distance of PolgD257A/D257A mice was shorter than for Polg+/D257A and Polg+/+ mice (n = 10, *P < 0.05, one-way ANOVA followed by Tukey’s HSD). (E) A percentage of total time in the open arm of the elevated plus maze. There was no significant difference among genotypes (n = 10, P = 0.142, Kruskal–Wallis test). (F) A percentage of number of entries into the open arm in the elevated plus maze test. There was no significant difference among genotypes (n = 10, P = 0.066, one-way ANOVA). (G) Y-maze test. Numbers of entries into the arm are shown. PolgD257A/D257A mice showed fewer entries than Polg+/D257A and Polg+/+ mice (n > 9, *P < 0.05, one-way ANOVA followed by Tukey’s HSD). The low number of entries of PolgD257A/D257A mice (2.3 ± 0.65) was seemingly due to a lack of spontaneous behavior. Thus, the number of alternations does not make sense in the PolgD257A/D257A mice. No difference was found in the alternation task between Polg+/D257A and Polg+/+ mice (n = 8, P = 0.618, t-test). (H) Prepulse inhibition (PPI) test. Effect of prepulse on the auditory startle response was examined. There was a significant main effect of prepulse (P = 7.74 × 10−9) but not genotype (P = 0.171) with the GLM repeated measures ANOVA. There was no significant interaction between PPI and genotype (P = 0.469). (I) Passive avoidance test. The genotypes did not differ in time until entry into the black box in both training (P = 0.913, Kruskal–Wallis test) and the test (P = 0.795, Kruskal–Wallis test).
Figure S2. Methodology for analysis of accumulation of multiple mtDNA deletions. (A) Southern blot analysis of total mtDNA from frontal lobe using the same membrane as Figure 2C with a probe for cytochrome c oxidase subunit I (COX1) gene (left panel), for nicotinamide adenine dinucleotide dehydrogenase subunit 4 (ND4) genes (middle panel), or for whole mtDNA (right panel). Even in PolgD257A/D257A mice, there was no detectable signal other than full-length mtDNA. (B) For a quantitative real-time PCR (qPCR) analysis for the statistical comparison of the relative accumulation of mtDNA deletions, typical standard curves of serially diluted control mtDNA, which was extracted from the frontal lobe of aged PolgD257A/D257A mice and contains mtDNA deletions, as measured by qPCR analysis. Each circle indicates a single measurement (n = 4). Significantly high linearity in this range for the standard samples was assured in each PCR plate (R2 > 0.95). (C) Agarose gel electrophoresis analysis and identification of amplifications of multiple mtDNA deletions after real-time PCR. A dilution series of control mtDNA for a standard curve, mtDNA of the frontal lobe of Polg+/+ and Polg+/D257A mice are shown. No amplification was detected without mtDNA (lane N).
Figure S3. Western blot analysis for levels of the respiratory chain enzyme complex subunits in the frontal lobes of Polg+/+ and Polg+/D257A mice at 96 weeks of age. (A) Subunits of mitochondrial complex are shown as follows: complex I subunit Ndufb8 (CI), complex II subunit Sdhb (CII), complex III subunit Uqcrc2 (CIII), complex IV subunit mt-Co1 (CIV), and complex V subunit Atp5a1 (CV) (upper panel). β-actin was detected as a loading control in the same membrane after stripping antibodies (lower panel). (B) Densitometric quantification of the ratios of each mitochondrial complex subunit/β-actin immunoblot levels in Western blot analysis (n = 6, mean ± SE, *P < 0.05, t-test). One of mtDNA-encoded subunits of respiratory complex, mt-Co1, showed a significant decrease in protein level.
Figure S4. Tissue distribution of accumulation of mtDNA deletions in Polg+/+ mice at 48 weeks (n = 3), Polg+/D257A and PolgD257A/D257A mice at 24 weeks (n = 4). The qPCR analysis was applied to mtDNAs extracted from five areas of the brain (frontal lobe [Fl], posterior cortices [Cx], hippocampus [Hp], basal ganglia [Bg], and cerebellum [Cb]) and four somatic tissues (heart, liver [Liv], kidney [Kid], and skeletal muscle [Sk]). The vertical axis represents the relative value normalized to the average level of deletions in the frontal lobe of Polg+/+ mice (*P < 0.05, **P < 0.01, mean ± SD, Welch ANOVA followed by the Dunnett T3 test). In all areas and tissues, PolgD257A/D257A mice showed significantly higher accumulation of mtDNA deletions than did the other mice. In both PolgD257A/D257A and Polg+/D257A mice (P < 0.0001), but not in Polg+/+ mice (P = 0.332), a significant tissue-dependent variation in the level of the accumulation was found by Welch ANOVA. A multiple comparison by the Dunnett T3 test showed a significant tissue-dependent difference in accumulation in PolgD257A/D257A and Polg+/D257A mice, as follows. In PolgD257A/D257A mice: Fl, Cx, and heart > Cb, Liv, and Sk; Hp and Bg > Liv and Sk; Fl > Bg (P < 0.05). In Polg+/D257A mice: Fl and heart > Liv, Kid, and Sk; Fl > Bg and Cb (P < 0.05).
Figure S5. Analysis of age-dependent accumulation of mtDNA deletions in posterior cortices, hippocampus, basal ganglia, cerebellum, and heart of Polg+/D257A and Polg+/+ mice at 12, 24, 48, 60, 72, 84, 101 weeks using qPCR. Values are represented relative to the average of 12-week-old Polg+/+ mice in each tissue. Each circle indicates one mouse (n = 4; two males and two females). Significant differences were detected at several ages except for liver (*P < 0.05). Statistical analysis results including the frontal lobe and skeletal muscles are described in Figure 3C.
Figure S6. Effect of age, tissue, and genotype on mtDNA copy number, as measured by qPCR from total DNA. The mtDNA copy number was measured in eight regions: frontal lobe, other cortices, hippocampus, basal ganglia, cerebellum, heart, liver, and skeletal muscle. The copy number per cell was measured by qPCR for the D-loop of mtDNA using the ApoB gene as a reference for nuclear genome copy number. The vertical axis represents the relative value normalized to each ratio in the frontal lobe of Polg+/+ mice (n = 4, mean ± SD). The horizontal axis represents the age. A significant difference was found in mtDNA copy number among tissues (P < 1.0 × 10−10, ANCOVA), whereas age and genotype showed no significant effect (P > 0.05).
Figure S7. Analysis of region-dependent accumulation of mtDNA deletions in skeletal muscles from hind limbs of Polg+/D257A and Polg+/+ mice at 37 weeks using qPCR. Values are represented relative accumulation of mtDNA deletions to that in the thigh of Polg+/+ mice. Each circle indicates one mouse (n = 5; two males and three females), and each horizontal bar show the mean value. Significant differences were detected only in posterior crus (*P < 0.01, Mann–Whitney U test).
Figure S8. Analysis of complex IV (COX) enzyme activity in skeletal muscles of male Polg+/D257A and Polg+/+ mice at 96 weeks of age (n = 6, mean ± SE). The vertical axis represents absorbance at 550 nm (A) or the relative activities normalized to the average value of activities in Polg+/+ mice (B). There was a significant decrease in COX activities in skeletal muscles of Polg+/D257A mice (*P < 0.01, t-test).
Figure S9. Southern blot analysis of mtDNA in neural stem cell (NSC) cultured for 5 weeks from the mouse embryo at gestational day 14.5 (Polg+/+ mice: n = 2, Polg+/D257A mice: n = 3, PolgD257A/D257A mice: n = 3) and 16.5 (Polg+/+ mice: n = 2, Polg+/D257A mice: n = 2). NSC from PolgD257A/D257A mice at E14.5 accumulated mtDNA deletions. However, there was no detectable signal other than full-length mtDNA both in NSC from Polg+/D257A mice and that from Polg+/+ mice.
Table S1. PCR primers.
Data S1. Supplementary methods.
References
- DiMauro S, Davidzon G. Mitochondrial DNA and disease. Ann Med. 2005;37:222–232. doi: 10.1080/07853890510007368. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- Van Goethem G, Martin JJ, Van Broeckhoven C. Progressive external ophthalmoplegia characterized by multiple deletions of mitochondrial DNA: unraveling the pathogenesis of human mitochondrial DNA instability and the initiation of a genetic classification. Neuromolecular Med. 2003;3:129–146. doi: 10.1385/NMM:3:3:129. [DOI] [PubMed] [Google Scholar]
- Suomalainen A, Majander A, Haltia M, et al. Multiple deletions of mitochondrial DNA in several tissues of a patient with severe retarded depression and familial progressive external ophthalmoplegia. J Clin Invest. 1992;90:61–66. doi: 10.1172/JCI115856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melberg A, Arnell H, Dahl N, et al. Anticipation of autosomal dominant progressive external ophthalmoplegia with hypogonadism. Muscle Nerve. 1996;19:1561–1569. doi: 10.1002/(SICI)1097-4598(199612)19:12<1561::AID-MUS5>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Suomalainen A, Majander A, Wallin M, et al. Autosomal dominant progressive external ophthalmoplegia with multiple deletions of mtDNA: clinical, biochemical, and molecular genetic features of the 10q-linked disease. Neurology. 1997;48:1244–1253. doi: 10.1212/wnl.48.5.1244. [DOI] [PubMed] [Google Scholar]
- Moslemi AR, Melberg A, Holme E, Oldfors A. Autosomal dominant progressive external ophthalmoplegia: distribution of multiple mitochondrial DNA deletions. Neurology. 1999;53:79–84. doi: 10.1212/wnl.53.1.79. [DOI] [PubMed] [Google Scholar]
- Van Goethem G, Dermaut B, Lofgren A, et al. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- Siciliano G, Tessa A, Petrini S, et al. Autosomal dominant external ophthalmoplegia and bipolar affective disorder associated with a mutation in the ANT1 gene. Neuromuscul Disord. 2003;13:162–165. doi: 10.1016/s0960-8966(02)00221-3. [DOI] [PubMed] [Google Scholar]
- Luoma P, Melberg A, Rinne JO, et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase γ mutations: clinical and molecular genetic study. Lancet. 2004;364:875–882. doi: 10.1016/S0140-6736(04)16983-3. [DOI] [PubMed] [Google Scholar]
- Mancuso M, Filosto M, Bellan M, et al. POLG mutations causing ophthalmoplegia, sensorimotor polyneuropathy, ataxia, and deafness. Neurology. 2004;62:316–318. doi: 10.1212/wnl.62.2.316. [DOI] [PubMed] [Google Scholar]
- Kato T. Mitochondrial dysfunction as the molecular basis of bipolar disorder: therapeutic implications. CNS Drugs. 2007;21:1–11. doi: 10.2165/00023210-200721010-00001. [DOI] [PubMed] [Google Scholar]
- Smits BW, Fermont J, Delnooz CC, et al. Disease impact in chronic progressive external ophthalmoplegia: more than meets the eye. Neuromuscul Disord. 2011;21:272–278. doi: 10.1016/j.nmd.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Stumpf JD, Copeland WC. Mitochondrial DNA replication and disease: insights from DNA polymerase γ mutations. Cell Mol Life Sci. 2011;68:219–233. doi: 10.1007/s00018-010-0530-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozzo R, Hirano M, Fromenty B, et al. Multiple mtDNA deletions features in autosomal dominant and recessive diseases suggest distinct pathogeneses. Neurology. 1998;50:99–106. doi: 10.1212/wnl.50.1.99. [DOI] [PubMed] [Google Scholar]
- Horvath R, Hudson G, Ferrari G, et al. Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene. Brain. 2006;129:1674–1684. doi: 10.1093/brain/awl088. [DOI] [PubMed] [Google Scholar]
- Kollberg G, Jansson M, Pérez-Bercoff A, et al. Low frequency of mtDNA point mutations in patients with PEO associated with POLG1 mutations. Eur J Hum Genet. 2005;13:463–469. doi: 10.1038/sj.ejhg.5201341. [DOI] [PubMed] [Google Scholar]
- Hudson G, Schaefer AM, Taylor RW, et al. Mutation of the linker region of the polymerase γ-1 (POLG1) gene associated with progressive external ophthalmoplegia and Parkinsonism. Arch Neurol. 2007;64:553–557. doi: 10.1001/archneur.64.4.553. [DOI] [PubMed] [Google Scholar]
- Suomalainen A, Kaukonen J. Diseases caused by nuclear genes affecting mtDNA stability. Am J Med Genet. 2001;106:53–61. doi: 10.1002/ajmg.1379. [DOI] [PubMed] [Google Scholar]
- Spelbrink JN, Li FY, Tiranti V, et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- Longley MJ, Clark S, Yu Wai Man C, et al. Mutant POLG2 disrupts DNA polymerase γ subunits and causes progressive external ophthalmoplegia. Am J Hum Genet. 2006;78:1026–1034. doi: 10.1086/504303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, et al. Premature aging in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Edgar D, Shabalina I, Camara Y, et al. Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metab. 2009;10:131–138. doi: 10.1016/j.cmet.2009.06.010. [DOI] [PubMed] [Google Scholar]
- Hiona A, Sanz A, Kujoth GC, et al. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS One. 2010;5:e11468. doi: 10.1371/journal.pone.0011468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SL, Huang J, Edwards YJ, et al. The mtDNA mutation spectrum of the progeroid Polg mutator mouse includes abundant control region multimers. Cell Metab. 2010;12:675–682. doi: 10.1016/j.cmet.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameur A, Stewart JB, Freyer C, et al. Ultra-deep sequencing of mouse mitochondrial DNA: mutational patterns and their origins. PLoS Genet. 2011;7:e1002028. doi: 10.1371/journal.pgen.1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermulst M, Bielas JH, Kujoth GC, et al. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007;39:540–543. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- Vermulst M, Wanagat J, Kujoth GC, et al. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet. 2008;40:392–394. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y, Simon DK, Turnbull DM, et al. Do mtDNA deletions drive premature aging in mtDNA mutator mice? Aging Cell. 2009;8:502–506. doi: 10.1111/j.1474-9726.2009.00484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara T, Kubota M, Miyauchi T, et al. Mice with neuron-specific accumulation of mitochondrial DNA mutations show mood disorder-like phenotypes. Mol Psychiatry. 2006;11:577–593. doi: 10.1038/sj.mp.4001824. [DOI] [PubMed] [Google Scholar]
- Fuke S, Kubota-Sakashita M, Kasahara T, et al. Regional variation in mitochondrial DNA copy number in mouse brain. Biochim Biophys Acta. 2011;1807:270–274. doi: 10.1016/j.bbabio.2010.11.016. [DOI] [PubMed] [Google Scholar]
- Battersby BJ, Shoubridge EA. Selection of a mtDNA sequence variant in hepatocytes of heteroplasmic mice is not due to differences in respiratory chain function or efficiency of replication. Hum Mol Genet. 2001;10:2469–2479. doi: 10.1093/hmg/10.22.2469. [DOI] [PubMed] [Google Scholar]
- Hayashi A, Kasahara T, Iwamoto K, et al. The role of brain-derived neurotrophic factor (BDNF)-induced XBP1 splicing during brain development. J Biol Chem. 2007;282:34525–34534. doi: 10.1074/jbc.M704300200. [DOI] [PubMed] [Google Scholar]
- Magnusson J, Orth M, Lestienne P, et al. Replication of mitochondrial DNA occurs throughout the mitochondria of cultured human cells. Exp Cell Res. 2003;289:133–142. doi: 10.1016/s0014-4827(03)00249-0. [DOI] [PubMed] [Google Scholar]
- Hitoshi S, Alexon T, Tropepe V, et al. Notch pathway molecules are essential for the maintenance, but not for the generation, of mammalian neural stem cells. Genes Dev. 2002;16:846–858. doi: 10.1101/gad.975202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Kiselak T, Clark J, et al. Behavioral and metabolic characterization of heterozygous and homozygous POLG mutator mice. Mitochondrion. 2013;13:282–291. doi: 10.1016/j.mito.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey L, Cluett TJ, Reyes A, et al. Mice expressing an error-prone DNA polymerase in mitochondria display elevated replication pausing and chromosomal breakage at fragile sites of mitochondrial DNA. Nucleic Acid Res. 2009;37:2327–2335. doi: 10.1093/nar/gkp091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan KJ, Reeve AK, Samuels DC, et al. What causes mitochondrial DNA deletions in human cells? Nat Genet. 2008;40:275–279. doi: 10.1038/ng.f.94. [DOI] [PubMed] [Google Scholar]
- Diaz F, Bayona-Bafaluy MP, Rana M, et al. Human mitochondrial DNA with large deletions repopulates organelles faster than full-length genomes under relaxed copy number control. Nucleic Acids Res. 2002;30:4626–4633. doi: 10.1093/nar/gkf602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Results of conventional behavioral test battery. Male mice at 34 weeks old were used for these analyses (mean ± SEM is shown in all graphs.). (A) Body weight. PolgD257A/D257A mice weighed less than Polg+/D257A and Polg+/+ mice (n = 10, *P < 0.05, one-way ANOVA followed by Tukey’s HSD). (B) Open field test. Total distance traveled per 5 min is shown. There was a significant main effect of genotype (P = 0.046) and bin of time (P = 0.00307) in the GLM repeated measures ANOVA. A significant interaction between genotype and the bin was observed (P = 1.01 × 10−5). In analysis of each 5 min, the total distance of PolgD257A/D257A mice was shorter than for Polg+/D257A and Polg+/+ mice (n = 10, *P < 0.05, one-way ANOVA followed by Tukey’s HSD). (C) A percentage of total time in center in an open field test. There was no significant difference among genotypes (n = 10, P = 0.817, one-way ANOVA). (D) Total distance traveled in elevated plus maze test. The total distance of PolgD257A/D257A mice was shorter than for Polg+/D257A and Polg+/+ mice (n = 10, *P < 0.05, one-way ANOVA followed by Tukey’s HSD). (E) A percentage of total time in the open arm of the elevated plus maze. There was no significant difference among genotypes (n = 10, P = 0.142, Kruskal–Wallis test). (F) A percentage of number of entries into the open arm in the elevated plus maze test. There was no significant difference among genotypes (n = 10, P = 0.066, one-way ANOVA). (G) Y-maze test. Numbers of entries into the arm are shown. PolgD257A/D257A mice showed fewer entries than Polg+/D257A and Polg+/+ mice (n > 9, *P < 0.05, one-way ANOVA followed by Tukey’s HSD). The low number of entries of PolgD257A/D257A mice (2.3 ± 0.65) was seemingly due to a lack of spontaneous behavior. Thus, the number of alternations does not make sense in the PolgD257A/D257A mice. No difference was found in the alternation task between Polg+/D257A and Polg+/+ mice (n = 8, P = 0.618, t-test). (H) Prepulse inhibition (PPI) test. Effect of prepulse on the auditory startle response was examined. There was a significant main effect of prepulse (P = 7.74 × 10−9) but not genotype (P = 0.171) with the GLM repeated measures ANOVA. There was no significant interaction between PPI and genotype (P = 0.469). (I) Passive avoidance test. The genotypes did not differ in time until entry into the black box in both training (P = 0.913, Kruskal–Wallis test) and the test (P = 0.795, Kruskal–Wallis test).
Figure S2. Methodology for analysis of accumulation of multiple mtDNA deletions. (A) Southern blot analysis of total mtDNA from frontal lobe using the same membrane as Figure 2C with a probe for cytochrome c oxidase subunit I (COX1) gene (left panel), for nicotinamide adenine dinucleotide dehydrogenase subunit 4 (ND4) genes (middle panel), or for whole mtDNA (right panel). Even in PolgD257A/D257A mice, there was no detectable signal other than full-length mtDNA. (B) For a quantitative real-time PCR (qPCR) analysis for the statistical comparison of the relative accumulation of mtDNA deletions, typical standard curves of serially diluted control mtDNA, which was extracted from the frontal lobe of aged PolgD257A/D257A mice and contains mtDNA deletions, as measured by qPCR analysis. Each circle indicates a single measurement (n = 4). Significantly high linearity in this range for the standard samples was assured in each PCR plate (R2 > 0.95). (C) Agarose gel electrophoresis analysis and identification of amplifications of multiple mtDNA deletions after real-time PCR. A dilution series of control mtDNA for a standard curve, mtDNA of the frontal lobe of Polg+/+ and Polg+/D257A mice are shown. No amplification was detected without mtDNA (lane N).
Figure S3. Western blot analysis for levels of the respiratory chain enzyme complex subunits in the frontal lobes of Polg+/+ and Polg+/D257A mice at 96 weeks of age. (A) Subunits of mitochondrial complex are shown as follows: complex I subunit Ndufb8 (CI), complex II subunit Sdhb (CII), complex III subunit Uqcrc2 (CIII), complex IV subunit mt-Co1 (CIV), and complex V subunit Atp5a1 (CV) (upper panel). β-actin was detected as a loading control in the same membrane after stripping antibodies (lower panel). (B) Densitometric quantification of the ratios of each mitochondrial complex subunit/β-actin immunoblot levels in Western blot analysis (n = 6, mean ± SE, *P < 0.05, t-test). One of mtDNA-encoded subunits of respiratory complex, mt-Co1, showed a significant decrease in protein level.
Figure S4. Tissue distribution of accumulation of mtDNA deletions in Polg+/+ mice at 48 weeks (n = 3), Polg+/D257A and PolgD257A/D257A mice at 24 weeks (n = 4). The qPCR analysis was applied to mtDNAs extracted from five areas of the brain (frontal lobe [Fl], posterior cortices [Cx], hippocampus [Hp], basal ganglia [Bg], and cerebellum [Cb]) and four somatic tissues (heart, liver [Liv], kidney [Kid], and skeletal muscle [Sk]). The vertical axis represents the relative value normalized to the average level of deletions in the frontal lobe of Polg+/+ mice (*P < 0.05, **P < 0.01, mean ± SD, Welch ANOVA followed by the Dunnett T3 test). In all areas and tissues, PolgD257A/D257A mice showed significantly higher accumulation of mtDNA deletions than did the other mice. In both PolgD257A/D257A and Polg+/D257A mice (P < 0.0001), but not in Polg+/+ mice (P = 0.332), a significant tissue-dependent variation in the level of the accumulation was found by Welch ANOVA. A multiple comparison by the Dunnett T3 test showed a significant tissue-dependent difference in accumulation in PolgD257A/D257A and Polg+/D257A mice, as follows. In PolgD257A/D257A mice: Fl, Cx, and heart > Cb, Liv, and Sk; Hp and Bg > Liv and Sk; Fl > Bg (P < 0.05). In Polg+/D257A mice: Fl and heart > Liv, Kid, and Sk; Fl > Bg and Cb (P < 0.05).
Figure S5. Analysis of age-dependent accumulation of mtDNA deletions in posterior cortices, hippocampus, basal ganglia, cerebellum, and heart of Polg+/D257A and Polg+/+ mice at 12, 24, 48, 60, 72, 84, 101 weeks using qPCR. Values are represented relative to the average of 12-week-old Polg+/+ mice in each tissue. Each circle indicates one mouse (n = 4; two males and two females). Significant differences were detected at several ages except for liver (*P < 0.05). Statistical analysis results including the frontal lobe and skeletal muscles are described in Figure 3C.
Figure S6. Effect of age, tissue, and genotype on mtDNA copy number, as measured by qPCR from total DNA. The mtDNA copy number was measured in eight regions: frontal lobe, other cortices, hippocampus, basal ganglia, cerebellum, heart, liver, and skeletal muscle. The copy number per cell was measured by qPCR for the D-loop of mtDNA using the ApoB gene as a reference for nuclear genome copy number. The vertical axis represents the relative value normalized to each ratio in the frontal lobe of Polg+/+ mice (n = 4, mean ± SD). The horizontal axis represents the age. A significant difference was found in mtDNA copy number among tissues (P < 1.0 × 10−10, ANCOVA), whereas age and genotype showed no significant effect (P > 0.05).
Figure S7. Analysis of region-dependent accumulation of mtDNA deletions in skeletal muscles from hind limbs of Polg+/D257A and Polg+/+ mice at 37 weeks using qPCR. Values are represented relative accumulation of mtDNA deletions to that in the thigh of Polg+/+ mice. Each circle indicates one mouse (n = 5; two males and three females), and each horizontal bar show the mean value. Significant differences were detected only in posterior crus (*P < 0.01, Mann–Whitney U test).
Figure S8. Analysis of complex IV (COX) enzyme activity in skeletal muscles of male Polg+/D257A and Polg+/+ mice at 96 weeks of age (n = 6, mean ± SE). The vertical axis represents absorbance at 550 nm (A) or the relative activities normalized to the average value of activities in Polg+/+ mice (B). There was a significant decrease in COX activities in skeletal muscles of Polg+/D257A mice (*P < 0.01, t-test).
Figure S9. Southern blot analysis of mtDNA in neural stem cell (NSC) cultured for 5 weeks from the mouse embryo at gestational day 14.5 (Polg+/+ mice: n = 2, Polg+/D257A mice: n = 3, PolgD257A/D257A mice: n = 3) and 16.5 (Polg+/+ mice: n = 2, Polg+/D257A mice: n = 2). NSC from PolgD257A/D257A mice at E14.5 accumulated mtDNA deletions. However, there was no detectable signal other than full-length mtDNA both in NSC from Polg+/D257A mice and that from Polg+/+ mice.
Table S1. PCR primers.
Data S1. Supplementary methods.