

Abstract
The aim of this paper was to provide an overview of the current state of the art on research into the emission of biogenic volatile organic compounds (BVOCs) from vegetation fires. Significant amounts of VOCs are emitted from vegetation fires, including several reactive compounds, the majority belonging to the isoprenoid family, which rapidly disappear in the plume to yield pollutants such as secondary organic aerosol and ozone. This makes determination of fire-induced BVOC emission difficult, particularly in areas where the ratio between VOCs and anthropogenic NOx is favourable to the production of ozone, such as Mediterranean areas and highly anthropic temperate (and fire-prone) regions of the Earth. Fire emissions affecting relatively pristine areas, such as the Amazon and the African savannah, are representative of emissions of undisturbed plant communities. We also examined expected BVOC emissions at different stages of fire development and combustion, from drying to flaming, and from heatwaves coming into contact with unburned vegetation at the edge of fires. We conclude that forest fires may dramatically change emission factors and the profile of emitted BVOCs, thereby influencing the chemistry and physics of the atmosphere, the physiology of plants and the evolution of plant communities within the ecosystem.
Keywords: biomass burning, BVOC, combustion phases, forest fires, isoprenoids, plant communities and functional types
Introduction
Vegetation fires (naturally occurring bushfires and wildfires, and prescribed fires) are one of the most powerful factors shaping plant ecosystems globally, through their effects on plant biodiversity, community ecology and evolution (Cochrane 2003; Pierce et al. 2004; Bowman et al. 2009; Keeley et al. 2011). Vegetation fires also have strong impacts on atmosphere composition and climate. Biomass burning is one of the largest sources of atmospheric CO2, reactive trace gases and aerosols (Crutzen & Andreae 1990; Andreae & Merlet 2001) (Fig. 1), and a major factor in atmospheric chemistry and physics, often determining the deterioration of air quality observed between rural–urban interfaces at regional and global scales (Cochrane 2003). Fires are also powerful drivers of abrupt changes in land surface albedo. Fire-induced changes in atmospheric composition and the reflective properties of land surfaces, alongside decreased transpiration caused by the fragmentation of vegetation stands, may in turn reduce cloud formation and precipitation processes. Overall, the positive radiative forcing induced by vegetation fires is expected to substantially contribute to climate change, whereas a warmer future with more frequent and extensive drought events would likely increase the occurrence of vegetation fires (Cochrane 2003; Stephens et al. 2013).
Figure 1.

Combustion stages (Table 3) and the related compounds emitted. BVOC, biogenic volatile organic compound; PAH, polyaromatic hydrocarbon; VOC, volatile organic compound.
Almost all plants, but particularly trees, produce and emit a wide range of non-methane hydrocarbons, termed biogenic volatile organic compounds (BVOCs), to be distinguished from volatile organic compounds emitted by other sources (VOCs) (Loreto & Centritto 2008). Volatile isoprenoids (isoprene, monoterpenes and sesquiterpenes) are the most important BVOCs emitted by undisturbed forest trees (Centritto et al. 2011b; Loreto et al. 2014). Isoprenoids play an important role protecting against both abiotic and biotic stress, as they enhance tolerance to heat and oxidative stress (Sharkey & Yeh 2001; Loreto & Schnitzler 2010) and have anti-herbivore functions (Holopainen & Gershenzon 2010). The formation and emission of isoprenoids is influenced by physiological, biochemical and physicochemical processes, plant growth stage, seasonality and environmental conditions (Sharkey & Yeh 2001; Niinemets et al. 2004). The capacity to produce and store isoprenoids is strongly interspecific. Some plants may store volatile isoprenoids in liquid pools and specialized compartments (i.e. monoterpene- and sesquiterpene-emitting species: conifers, medicinal and aromatic plants, Eucalyptus spp.) or as temporary pools in non-specialized leaf structures (isoprene and light-dependent monoterpene- and sesquiterpene-emitting species) (Sharkey & Yeh 2001; Grote & Niinemets 2008). Copious amounts of these compounds can be emitted in response to wounding and high temperatures (Loreto et al. 2006; Centritto et al. 2011a; Fares et al. 2011). Fires may therefore be responsible for episodes of massive BVOC emission from burning vegetation, in addition to the vegetation in close proximity to the fires. Furthermore, BVOCs are known to be flammable and to influencethe intrinsic flammability of vegetation (Owens et al. 1998). Fire-induced BVOC emissions may have dramatic negative impacts on the chemistry and physics of the atmosphere, because of their significant contribution to the formation of tropospheric ozone, photo-oxidants and secondary organic aerosols (SOA) (Finnlayson-Pitts & Pitts 2000).
In this review, we provide insights into the relationships between fires and BVOCs, and the implications for atmospheric chemistry. We overview the type and amount of BVOCs emitted from forest fires, and envision possible emission rates in relation to the type of burned ecosystem and the characteristics of the fire type, albeit within the current limitations of BVOC determination.
Carbon and Pollutant Emissions from Vegetation Fires at the Global Level
Although VOC emissions from vegetation fires have occurred since terrestrial vascular plants first appeared on Earth approximately 350–400 million years ago, and forests have been burned by humans for thousands of years to create pastures and agricultural land (Scholes et al. 2003), forest fires have only recently became an environmental problem. According to recent global estimates (Thonicke et al. 2010), non-methane VOCs produced by forest and savannah fires account for 24 Tg/year, a value 50% lower than previously estimated by the EDGAR inventory (Reimann & Lewis 2007). To place these emission levels in context, VOC emissions from vegetation fires are lower than VOC emissions associated with the production and use of fossil fuels (77.4 Tg/year), comparable with those arising from industrial processes (26.7 Tg/year) and biofuel combustion (30.0 Tg/year), but higher than the VOC emissions generated by waste management (2.7 TG/year) (Reimann & Lewis 2007). The sources of VOCs produced by fire are unevenly distributed over the Earth; because of the 5130 Tg of vegetation burned every year, the largest fraction comes from biomass combustion of tropical forests (1330 Tg/year) and savannah/grasslands (3160 Tg/year). Combustion of biomass from extra-tropical forests and agricultural waste contributes 640 and 540 Tg/year, respectively (Scholes et al. 2003). A substantial fraction of biomass (2900 Tg/year) is burned as fuel for food cooking and heating, as this is the only fuel available in poorest economies.
Combustion is a highly exothermic reaction in which gaseous, liquid and solid materials containing different amounts of carbon (generically termed as fuel) are oxidized at high temperatures. If the fuel contains only C and H, then CO2, water and NOx are formed. As seen from Table 1, the combustion of vegetation produces a higher number of compounds, depending on the type and physical state of the fuel; the content of C, O, H, N and S; and the maximum temperature reached. Efficient combustion results in higher emissions of CO2, NOx and water, and lower emissions of VOCs, CO, CH4 and carbon particles. Nonetheless, due to the different content of carbon and water, and the conditions under which combustion takes place, the emissions produced by burning forests, savannas and agricultural residues differs to those produced by the combustion of biofuels, fossil fuels and urban wastes.
Table 1.
Emission Factors in g kg−1 dry matter of greenhouse gases and atmospheric pollutants emitted from vegetation fires and coal burning (data elaborated from Andreae & Merlet 2001)
| Chemical species | Savannah and grassland | Tropical forest | Extra-tropical forest | Agricultural residues | Charcoal burning |
|---|---|---|---|---|---|
| Greenhouse gases | |||||
| CO2 | 1613 ± 95 | 1580 ± 90 | 1569 ± 131 | 1515 ± 177 | 2611 ± 241 |
| CH4 | 2.3 ± 0.9 | 6.8 ± 2.0 | 4.7 ± 1.9 | 2.7 | 6.2 ± 3.3 |
| N2O | 0.21 ± 0.10 | −0.2 | 0.26 ± 0.07 | 0.07 | (0.2) |
| Air pollutants | |||||
| CO | 65 ± 20 | 104 ± 20 | 107 ± 37 | 92 ± 84 | 200 ± 38 |
| NOx as NO | 3.9 ± 2.4 | 1.6 ± 0.7 | 3.0 ± 1.4 | 2.5 ± 1.0 | 3.9 |
| SO2 | 0.35 ± 0.16 | 0.57 ± 0.23 | 1 | (0.4) | 0.4 |
| VOC | 3.4 ± 1.0 | 8.1 ± 3.0 | 5.7 ± 4.6 | (7) | 2.7 ± 1.9 |
| Total particulate matter | 8.3 ± 3.2 | 6.5–10.5 | 17.6 ± 6.4 | 13 | (12) |
| PM2.5 | 5.4 ± 1.5 | 9.1 ± 1.5 | 13.0 ± 7.0 | 3.9 | (9) |
| Total carbon | 3.7 ± 1.3 | 6.6 ± 1.5 | 6.1–10.4 | 4.0 | 6.3 |
| Organic carbon | 3.4 ± 1.4 | 5.2 ± 1.5 | 8.6–9.7 | 3.3 | 4.8 |
| Black carbon | 0.48 ± 0.18 | 0.66 ± 0.31 | 0.56 ± 0.19 | 0.69 ± 0.13 | 1.5 |
| Other gases | |||||
| NH3 | 0.6–1.5 | (1.3) | 1.4 ± 0.8 | (1.3) | (1.3) |
| HCN | 0.025–0.031 | (0.15) | (0.15) | (0.15) | (0.15) |
Data in brackets are best guesses.
HCN, hydrogen cyanide; VOC, volatile organic compound.
The spatial distribution of vegetation fires on Earth can be assessed by remote sensing, because fire products, and their plumes, are clearly visible from satellites equipped with different optical sensors, such as TOMS, GOME, MODIS and SCHAMACHY (Tyndall et al. 2003). Carbon particles are particularly visible as they absorb and scatter the light at different wavelengths than mineral dust released by desert soil and sulphate particles produced by photochemical oxidation of sulphur compounds. Some other fire-produced chemical species, as formaldehyde, CO and NO2, are also visible (Tyndall et al. 2003). Figure 2 shows the global emissions of carbon, mineral dust and sulphate particles recorded on the 22nd of August 2005 by elaborating the data obtained by MODIS with the Global Aerosol Model developed by the Navy Aerosol Analysis and Prediction System (NAAPS). These data are produced every 4 h and are accessible at the web page http://www.nrlmry.navy.mil/aerosol/#aerosolobservations.
Figure 2.

Maps showing the concentrations on Earth of black carbon from fossil fuel and biomass combustion (blue), mineral dust released from desert storms (green) and sulphate aerosols from secondary oxidation reactions (yellow). Data refer to the 22nd of August 2005 and were generated by the NAAPS Global Aerosol Transport model using both satellite and ground based information.
Figure 2 provides a view of the spatial distribution of the carbon particles produced by sources of combustion over the Earth, demonstrating how far heavily polluted plumes can travel. The number of fires is indicated by the red dots, whereas the area affected by their combustion plumes is shown in blue. The relative importance of the combustion sources with respect to those generating mineral dust and photochemical production of sulphate particles can be estimated by comparing the blue surface area with that of green and yellow, respectively. It is evident that during the studied day, combustion was the most significant source of atmospheric particulates on Earth; the major contribution was from forest fires in the Amazon region, and savannah and forest fires in Central-Southern Africa. Plumes of particles hundreds of kilometres wide and thousands of kilometres long were generated by each fire, overlapping on large portions of the Earth. According to process-based models reproducing the dynamics of fires on Earth (Thonicke et al. 2010), BVOCs produced from vegetation combustion (24 Tg/year) are associated with the production of 8200, 448, 19, 70 and 15 Tg/year of CO2, CO, CH4, total suspended particulate matter (TPM) and NOX, respectively. Similar estimates were earlier made by Andreae & Merlet (2001).
Substantial volumes of fine particles with an aerodynamic diameter less than 10 or 2.5 μm are always produced by vegetation fires (Table 1). In addition to the causing severe health problems and removing VOCs, particles with an aerometric diameter smaller than 0.1 μm affect the Earth's climate by acting as cloud condensation nuclei (CCN) (Andreae & Crutzen 2009). Of specific interest to this review is the effect that CCN have on marine clouds inducing rainfall on forest areas as part of the regular air circulation patterns. When marine clouds mix with the plume generated by vegetation fires, their volume, temperature and turbulence increases, whereas the average diameter of their droplets decreases due to the injection of CCN (Ramanathan et al. 2001). These changes prevent water droplets of the cloud reaching the critical diameter required to precipitate, at least until the clouds reach an altitude where the temperature is sufficiently low to allow the droplets to grow (Ramanathan et al. 2001). Because of this process, rainfall moves away from burned areas, altering the hydrogeological cycle, exacerbating the harmful effects of forest fires and even triggering desertification processes (Ramanathan et al. 2001).
General Reactions of VOCs in Fire Plumes
The data in Fig. 2 show that the temperature and turbulence generated by fires and wind regimes make the combustion plume a dynamic system where different chemical processes occur as a function of the time and type of airsheds in which fire products are diluted. Photochemistry takes place because fire-induced VOC/NOx mixtures are exposed to UV light, or dispersed in airsheds in which photochemical reactions occurs. The chemical species driving photochemical processes in the atmosphere during daytime are OH radicals formed by the reaction between water and the photolysis products of ozone (Finnlayson-Pitts & Pitts 2000). OH radicals react rapidly with VOCs to form ozone and a series of pollutants termed as photochemical oxidants (Finnlayson-Pitts & Pitts 2000). The oxidation of VOCs by OH radicals produces peroxyalkylradicals that, by reacting with NO, form NO2, hydroperoxyradicals (HO2) and carbonyl compounds (aldehydes and ketones) (Finnlayson-Pitts & Pitts 2000). Among these products, HO2 reacts rapidly with NO to form NO2 and OH radicals, thus self-sustaining the oxidation of VOCs and other gases in the system. Carbonyls can photolyse or react with OH radicals producing peroxyalkylradicals that are injected into the system. From the oxidation of carbonyls CO, H2O2 and peroxyacylnitrates (PAN) are formed together with OH radicals (Finnlayson-Pitts & Pitts 2000). As a result of these reactions, ozone is produced because the repeated conversion of NO to NO2 changes the photostationary state of NOx described by the following equilibrium:
| (1) |
where JNO2 is the photolysis rate of NO2, KNO the reaction constant between NO and ozone, and [O3], [NO] and [NO3] are the concentrations of ozone, NO and NO2, respectively (Finnlayson-Pitts & Pitts 2000). At night, when the production of OH radicals ceases, NO3 radicals are formed by the reaction between NO2 and ozone. NO3 radicals also exhibit fast reactions with VOCs to form organic nitrates (Finnlayson-Pitts & Pitts 2000).
This complex chain of gas–phase reactions is strongly altered by the presence of particles within the system because heterogeneous processes can then take place, where VOCs and their oxidation products are removed from the gas phase either by adsorption into hydrophobic carbon particles or by partition into the water layer covering the hygroscopic ones. Both removal processes are quite selective because they depend on the vapour pressure, and the functional groups of the VOC molecules (Hoffmann & Warnke 2007). Because their solubility in water, many polar VOCs, such as alcohols, acids, phenols and hydroxyl-furans, are preferentially transferred into the water layer surrounding hygroscopic particles (Finnlayson-Pitts & Pitts 2000; Hoffmann & Warnke 2007). Fine particles also act as seeds for the formation of SOA from the oxidation products of some VOCs (Hoffmann & Warnke 2007). In combustion plumes, seed particles are mostly composed of carbon particles and ammonium sulphate and nitrate salts formed by the photochemical oxidation of sulphur and nitrogen compounds by OH radicals (Finnlayson-Pitts & Pitts 2000). These chemical processes account for the higher content of organic carbon than black carbon in particles generated by vegetation combustion. Analysis of particles generated by forest fires in the Amazon region showed that 40–75% of the organic fraction was soluble in water and was composed of mono-, di- and polycarboxylic acids, aldehydes and alcohols (De Cesari et al. 2006). The presence of particles, especially those composed of black carbon, also affects the night-time chemistry of VOCs, because nitrous acid and nitric acid are formed by the heterogeneous reaction of NO, NO2 and water with the surface of the carbon (Finnlayson-Pitts & Pitts 2000). Under certain conditions, this heterogeneous reaction may prevail over the gas-phase reaction leading to the formation of NO3 radicals.
In the photochemical reaction chain, because the reaction rates of VOCs with OH radicals can differ by two to three orders of magnitudes, the composition of VOCs plays a fundamental role in determining the formation rate of ozone and photochemical oxidants (Finnlayson-Pitts & Pitts 2000). The residence time of a VOC in the atmosphere is expressed in terms of atmospheric lifetime, τ, which is the time required to reduce its initial concentration in air, C°, to a value of C = C/e, where e = 2.718 is the Euler constant. For many VOCs, the daytime reaction with OH radicals is much faster than with other atmospheric oxidants, such as ozone, and also faster than other removal processes, such as wet and dry deposition, or diffusion through the boundaries (Finnlayson-Pitts & Pitts 2000). It can thus be assumed that in many instances the lifetime of numerous VOCs is given by: τ = τOH = 1/kOH*[OH], where kOH is the pseudo first-order kinetic reaction constant with OH radicals in molecules cm−3 s−1, and [OH] the radical concentration in molecules cm−3.
Emissions and Reactions of BVOCs in Different Fire Plume Regimes
BVOCs are highly reactive because they contain one or more double bonds in the molecule (Atkinson & Arey 1998). Many plant-derived VOCs are more reactive than the majority of alkenes produced by pyrolysis in the flame or internal combustion of fossil fuels. In particular, as they are emitted at high rates by many plant species, isoprene, mono- and sesquiterpenes play a fundamental role in regulating ozone production in the atmosphere. Present estimates indicate annual emission rates of isoprenoids to be in the range of ca. 1 Pg/year, which largely exceeds anthropogenic emission of VOCs (Guenther 1999; Guenther et al. 2012). Identical to many anthropogenic alkenes, isoprenoids react rapidly with ozone, as their τO3 = 1/kO3 * [O3] is comparable with τOH (Atkinson & Arey 1998). Ozonolysis of all alkenes, including double-bonded biogenic alcohols, such as 2-methyl-3-butenol, 3-cis-hexenol and linalool, leads to the formation of an unstable ozonide, which decomposes by breaking the —O—O—O— and —C—C— bonds to form two aldehydes, or ketones and two excited radicals known as Criegee's intermediates. Their decomposition generates the same products that are formed by the reaction with OH radicals (Atkinson & Arey 1998). Ozonolysis of monoterpenes produces acidic species that are able to form SOAs by condensation between themselves or through interaction with seed particles (Hoffmann & Warnke 2007).
However, not all BVOCs react with ozone at the same rates, and the potential of ozone and SOA production of BVOC mixtures is strongly dependent on their composition. In Table 2, the atmospheric lifetimes of selected BVOCs are reported using the average mixing ratios of OH radicals (ca. 0.04 pptv) and ozone (ca. 28.4 ppbv) in the atmosphere (Atkinson & Arey 1998; Calogirou et al. 1999). As shown, the τOH values of BVOCs are quite variable as they range from 30 min for terpinolene to 2.6 h for α-pinene and camphene. The same is true for the τO3 values ranging from 30 min for terpinolene to 18 d for camphene. The first oxidation products of BVOCs also have very different lifetimes. Isoprene, the most abundant isoprenoid emitted by plants, reacts as rapidly as many monoterpenes with OH radicals, but is significantly less reactive with ozone. Methanol is strongly emitted by plants during leaf development and is comparatively unreactive with a long lifetime, even when reacting with OH radicals. On the other hand, sesquiterpenes generally have very short atmospheric lifetimes and are particularly active in forming SOA (Hoffmann & Warnke 2007; Li et al. 2011). BVOCs are also among the most rapidly reacting VOCs at night, because their lifetimes when mixed with NO3 radicals can be even shorter than those mixed with OH radicals (Atkinson & Arey 1998). The reactivity of BVOCs with radical species and ozone are in general so varied, that some disappear in few hours, whereas others persist to survive long-range transport.
Table 2.
Chemical lifetime (τ) of VOCs and their main oxidation products, calculated for the average atmospheric mixing ratios of OH radicals (= 0.04 pptv) and ozone (=28.4 ppbv)
| Biogenic VOCs | Biogenic VOC oxidation products | ||||
|---|---|---|---|---|---|
| Isoprene | τOH | τO3 | τOH | τO3 | |
| 1.4 h | 1.3 d | Methacrolein | 4.1 h | 15 d | |
| Methyl vinyl ketone | 6.8 h | 3.6 d | |||
| 3-Methyl furan | 1.5 h | 19 h | |||
| Monoterpenes | τOH | τO3 | τOH | τO3 | |
| Limonene | 49 min | 2 h | Limononaldehyde | 1.26 h | 2 d |
| Terpinolene | 37 min | 13 min | 4-Methyl-3-cyclohexenone | 1.5 h | n.r. |
| α-Pinene | 2.6 h | 4.6 h | Pinonaldehyde | 2.9 h | >2.4 year |
| β-Pinene | 1.8 h | 1.1 d | Nopinone | 12 h | >4.5 year |
| Δ3-Carene | 1.6 h | 11 h | Caroaldehyde | 2.9 h | >2.3 year |
| Sabinene | 1.2 h | 4.6 h | Sabinaketone | 18.8 d | >0.9 year |
| Camphene | 2.6 h | 18 d | Camphenilone | 2.3 d | >4.5 year |
| Sesquiterpenes | τOH | τO3 | |||
| β-Caryophyllene | 42 min | 2 min | |||
| α-Humulene | 28 min | 2 min | |||
| α-Cedrene | 2.1 h | 14 h | |||
| α-Copaene | 1.5 h | 2.5 h | |||
| Alcohols | τOH | τO3 | τOH | τO3 | |
| 2-Methyl-3-buten-2-ol | 2.4 h | 1.7 d | 5-Methyl-5-vinyltetrahydrofuran-2-ol | 1.56 d | 8.7 d |
| cis-3-Hexen-1-ol | 1.3 h | 6.2 h | |||
| Linalool | 52 min | 55 min | |||
| 1,8-Cineole | 1.0 d | >110 d | |||
| Methanol | 12 d | >4.5 year | |||
Data from Atkinson & Arey (2003), Calogirou et al. (1999) and Carrasco et al. (2007).
n.r., not reactive; VOC, volatile organic compound.
The highly non-linear nature of photochemical processes explains the large differences in the reactivity of VOCs during the dispersion of the combustion plume. Laboratory experiments performed with different VOC/NOx mixtures (both expressed in ppmv) indicate that the highest production of ozone occurs when VOC/NOx ratios are less than 4 or higher than 15 (Finnlayson-Pitts & Pitts 2000). For VOC/NOx <4, hydrocarbon-limited conditions are established where low production of ozone takes place (Finnlayson-Pitts & Pitts 2000). Low ozone is also produced in a NOx-limited condition where VOC/NOx >15 (Finnlayson-Pitts & Pitts 2000). Reasonable values of BVOC emission factors (EFs) from vegetation fires can be derived from determinations performed in their dispersion plumes only if the VOC/NOx ratios do not allow substantial ozone production, and if the concentration of OH radicals does not exceed values reported in Table 2. Remote sensing data show that intense vegetation fires in the Amazon region and in Central-Southern Africa create, indeed, a hydrocarbon-limited VOC/NOx regime, leading to low ozone content and high levels of NO2 in the plume (Tyndall et al. 2003). These conditions are determined by the significant emission of NO from vegetation fires that rapidly convert atmospheric ozone into NO2. In the flame of vegetation fires, NO is mainly formed by the direct reaction between N2 and O2 or by the reduction of NOx species on char, with both processes occurring at temperatures >1200 °C. The generation of NO is associated with a diminished production of NO2 and nitrous acid (Burling et al. 2011).
In summary, provided that hydrocarbon-limited conditions are maintained inside the vegetation combustion plume, the photochemical consumption of BVOCs is limited and their concentrations closely follow those occurring in fires. Given the huge presence of carbon particles and the elevated NO2 content, formation of nitrous acid is likely to prevail at night, limiting the removal of BVOCs by reaction with NO3 radicals (Finnlayson-Pitts & Pitts 2000). However, the photolysis of nitrous acid at sunrise can produce levels of OH radicals sufficient to remove the most reactive BVOCs (Finnlayson-Pitts & Pitts 2000; Alvarado & Prinn 2009). The persistence of hydrocarbon-limited conditions in vegetation combustion plumes is also dependent on the airshed where the plume is dispersed. If the plume is diluted in relatively clean airsheds, such as those typically present over the oceans, a longer time period can be required for the transition from a hydrocarbon-limited conditions to those generating ozone (Helas et al. 1995). However, if the plume is diluted in airsheds containing high levels of ozone, such as those generated by photochemically polluted urban plumes, a rapid production of OH radicals takes place, depleting large fractions of the most reactive BVOCs from the plume (Helas et al. 1995). As shown by Akagi et al. (2012, 2013,), the increase of ozone is mirrored by a substantial depletion of BVOCs within the combustion plume. Given the high ozone levels (80–100 ppbv) reached during summer over the whole Mediterranean basin (Millan-Millan et al. 1998), it is extremely difficult for hydrocarbon-limited conditions to occur in the plumes of vegetation fires generated in Greece, Spain, Portugal, Italy and France. Similar considerations apply to fires in California and in the South East of the United States. In these areas, fire-originated BVOCs can dramatically increase the existing levels of ozone and photochemical oxidants, such as PAN, aldehydes and sulphate and nitrates in particles (Akagi et al. 2012).
Energetics and the Phases of Biomass Combustion and BVOC Formation
As dry biomass is composed of 38–50% cellulose, 23–32% hemicelluloses and 15–25% lignin (Van Loo & Koppejan 2007), a substantial portion of VOCs produced by vegetation fires will come from the pyrolysis of these complex hydrocarbons. Disregarding the trace amounts of N and S, the biomass can be classified as an organic fuel with an elemental composition of CnHnxOny, where x ranges from ca. 1.2 to 1.7 and y from ca. 0.45 to 0.9 (Van Loo & Koppejan 2007). As a fuel, dry biomass contains less carbon and more oxygen than coal, which has x and y values in the range 0.7–1.0 and 0.05–0.3, respectively. Biomass combustion generates less energy (16 000 kJ/kg) than charcoal (31 000 kJ/kg), because some of the energy is required to evaporate water and organic gases. Organic gases are evolved from lipids, phenolic compounds, isoprenoids, fatty acids, resin acids, steryl esters, sterol, aminoacids, proteins and waxes that are present in variable amounts in different plant species (Rowell et al. 2012). Monoterpenes and sesquiterpenes are particularly abundant in plant species that are equipped with specialized organs, such as glands or resin ducts, for example, in needles, barks and trunks of conifers, where these isoprenoids are stored in the form of liquid micelles (Fall 1999) and play defensive roles against insects and pathogens (Keeling & Bohlmann 2006).
The different phases of biomass combustion and its transformation into charcoal (Table 3) may help to elucidate the mechanisms underpinning the emission of different classes of VOCs (Fig. 1). Thermogravimetric data, collected under a flow of inert gas, indicate that up to a temperature of 200 °C the biomass becomes dry (distillation phase) by losing free water, together with the majority of temporary stored BVOCs and those stored in the permanent pools (De Lillis et al. 2009). During pyrolysis, which occurs at temperatures between 200 and 500 °C, partly oxidized products are emitted. This is also the phase where CO is emitted at the maximum rate (Andreae & Merlet 2001). In particular, between 230 and 250 °C, biopolymers undergo small structural modifications, termed as rectification, coupled with the release of acetic acid and methanol (Evans & Milne 2006). Between 250 and 280 °C, biopolymers and some cell structures start to decompose (torrefaction), and pyrogenic VOCs are emitted from the preferential rupturing of —C—O, —C = O bonds in comparison to —C—C— and —C = C— bonds (Evans & Milne 2006). Additionally, more water is released by the progressive dehydration of the —OH and —COOH functional groups during this phase. Emission of oxygenated VOCs and water continues in the devolatilization phase occurring between 300 and 500 °C. Above this temperature, the carbonization phase begins, where pyrogenic arenes, alkenes, alkanes, alkynes and polyaromatic hydrocarbons (PAH) are released (Evans & Milne 2006).
Table 3.
Major biomass combustion phases
| Precombustion phases | Ignition | Combustion phase | ||
|---|---|---|---|---|
| Distillation | Rectification | Torrefaction | Flaming | Smouldering |
| T < 200 °C | 230 < T < 250 °C | 250 < T < 300 °C | 500 < T < 1600 °C | T < 600 °C |
| Endothermic | Endothermic | Exothermic | Exothermic | Exothermic |
| The wood becomes dehydrated and evolves water vapour with perhaps traces of carbon dioxide, formic and acetic acids, glyoxal BVOCs stored in the storage compartments of the plant are evolved. The gases produced by very slow pyrolysis are not ignitable. | Wood pyrolysis remains slow. Water vapour, carbon dioxide, formic and acetic acids, glyoxal and possibly a little carbon monoxide are evolved together with substantial amounts of vapours of BVOCs. Thus far, the reactions are endothermic and the gaseous products are still not ignitable. | This phase occurs suddenly and exothermically when the mixture of gases copiously evolved in the hot zones of wood becomes combustible. Active pyrolysis of wood begins swiftly and the temperature mounts rapidly unless the heat evolved is dissipated. Combustible gases and vapours are mainly carbon monoxide, methane, formaldehyde, formic and acetic acids, methanol, and hydrogen. They are emitted with carbon dioxide and water vapour carrying with them BVOCs and droplets of highly inflammable tars that appear as smoke. The primary pyrolysis products formed inside the wood undergo further pyrolysis and react one with another before they escape. They are produced by glowing combustion between the hot charred, formed by torrefaction and partial carbonization of wood, with encapsulated oxygen. | Flaming combustion occurs entirely in the gas phase outside the wood because the rapidly emerging gases lack the necessary oxygen until they have sufficiently mixed with air in proportions between the lower and upper limits of flammability. Indeed, under suitable conditions flaming combustion may occur at a considerable distance from the wood. Self-sustaining diffusion flames from organic fuels burn at 1100 °C or above. One-half to two-thirds of the heat of wood combustion is liberated in flaming, the rest by glowing combustion of charred wood. In this phase most of the pyrogenic VOCs, such as alkenes, arenes and PAHs are also emitted together with BVOCs and pyrolysis gases. | It is analogous to the glowing phase as it occurs by the direct reaction of oxygen with the surface of carbon. It takes place on the surface of the fragmented piece of charcoal formed by combustion, but is sustained by the emission of pyrolysis gases formed inside the material. This stage continues until the temperature drops below the combustion threshold value, or until only non-combustible ash remains. It is the phase where CO reaches it maximum value. It produces substantial amounts of pyrogenic VOCs, such as alkenes, arenes and PAHs. |
BVOC, biogenic volatile organic compound; PAH, polyaromatic hydrocarbon; VOC, volatile organic compound.
In the presence of O2 and at temperatures lower than 500 °C, the same dry distillation steps that occur in charcoal production take place when burning natural vegetation (Crutzen & Andreae 1990; Andreae & Merlet 2001). As the heat is transmitted to vegetation through waves that are originated by variations in the temperature of the flame in the moving fires, different parts of the plant simultaneously undergo drying, rectification, torrefaction, devolatilization and carbonization, thus emitting different blends of VOCs and BVOCs. These processes also continue during the glowing combustion phase (Andreae & Merlet 2001) where emission of BVOCs and pyrogenic VOCs also produces char and tar, forming a flammable white smoke. Above 500 °C, the mixture of BVOCs, pyrogenic VOCs, char and tars becomes a flammable fuel, whose ignition produces a flaming combustion. Under these conditions, all woody parts of vegetation are burned, forming ashes, carbon particles and inorganic gases (CO2, CO, H2O, NOx and SO2) (Andreae & Merlet 2001). During flaming combustion, CO2 emission reaches its maximum value (Andreae & Merlet 2001). Finally, flaming combustion ceases when most of the volatiles are distilled by the woody fuel and consumed near the flame. In this stage, known as the glowing/smouldering phases, combustion is sustained by the reaction of O2 with the carbon in the char layer of the burned wood, so emitting partly oxidized products that are very similar to those emitted in the pyrolysis and distillation phases (Andreae & Merlet 2001).
Factors Limiting Forest Fires in Nature
Forest fire regimes (fire frequency, spread of fire through vegetation, behaviour and severity) are affected by many factors. Forest canopy and sub-canopy moisture levels (e.g. closed-canopy wetter forests versus open-canopy drier forests), understory density (amount of shrubs and small trees), litter characteristics (type, composition, loading and humidity), degree of forest fragmentation (land cover) and the occurrence of extensive drought stress and heatwaves are major factors affecting ignition and the spread of forest fires. Early literature (Van Wagner 1967; Rothermel 1976; Trabaud 1979; Chandler et al. 1983) intuitively shows that plant ignitability and flammability are primarily governed by leaf water content (LWC). Fuel moisture acts as a heat sink (part of the heat is used to evaporate water, as shown earlier), dilutes flammable volatiles and excludes oxygen from the combustion zone (Nelson 2001), thus acting as one of the most important parameters in the determination of the properties of fire ignition and propagation (Van Wagner 1967; Chandler et al. 1983). The moisture content of leaves and small twigs is determined by morphological and physiological mechanisms by which plants adapt to the availability of water (sclerophylly, transpiration rate).
Trabaud (1979) found two thresholds of LWC influencing flammability: below ∼32% of LWC, leaves of major Mediterranean maquis species ignited rapidly, whereas above a LWC threshold of ∼45% the flame was either consistently delayed or did not appear. Interestingly, in a more recent study, De Lillis et al. (2009), working on Mediterranean species (three Quercus species, Myrtus communis and Pinus halepensis) confirmed that above a LWC threshold of ∼45% flaming only occurs at very high temperatures, whereas below that threshold, the temperature of the flame was not affected by LWC. However, M. communis and P. halepensis, two evergreen species that possess high storing capacity of monoterpenes, but respectively emit isoprene and monoterpenes, ignited at rather high LWC (∼60–70%). The large amount of monoterpenes stored in the glandular reservoirs of M. communis and in the resin ducts of P. halepensis may have facilitated leaf ignition. Furthermore, highly inflammable isoprenoids released during the pre-flaming stages may therefore be ignited by a nearby flame so facilitating fire propagation. The relationship between isoprenoids and flammability, and isoprenoids release during fire-generated heatwaves, will be analysed in the following section.
Influence of Leaf Isoprenoids on Flammability
The influence of isoprenoid concentration on the ignition of living leaves was first assessed by Owens et al. (1998) in a study performed on two populations of Juniperus ashei. Foliar flammability was positively related to limonene (monoterpene) concentration and negatively related to the concentration of the bornyl acetate (sesquiterpenoid). Limonene is among the most common and abundant monoterpene compound produced by plants in the Mediterranean region, an area that is characterized by the most significant forest fire hazards in Europe. However, Alessio et al. (2008) experimentally generated different LWC and monoterpene concentrations in leaves of Q. ilex and P. halepensis, and found no significant relationship between monoterpene content and flammability. Furthermore, Q. ilex leaves reached the smoke and pyrolysis phases earlier than P. halepensis needles despite monoterpene concentrations being three orders of magnitude lower (Alessio et al. 2008). No overall differences were shown in flame temperature and ignition delay time between the leaves of the two species. Taken together, these results confirm that LWC is a more important factor in determining ignitability than monoterpenes. However, P. halepensis needles then burned more rapidly than Q. ilex leaves, which may indicate that monoterpenes in their liquid state favour combustion.
Interestingly, De Lillis et al. (2009) found a lack of correlation between isoprenoid concentration and the temperature of ignition, whereas they found higher isoprenoid emissions in species that ignited at lower temperatures. Page et al. (2012) also showed that isoprenoid (especially E-β-ocimene and tricyclene) emissions were correlated with the flammability of P. contorta leaves, which had been attacked during the previous year by mountain pine beetle inducing significant changes in the foliage LWC and chemical composition. These results confirm that the flammability of a leaf depended to large extent on its hydration status; but being as isoprenoids dissolve in water, a high LWC may also imply high isoprenoid emission rates and/or concentrations (Fig. 3). If this is the case, then fresh tissues may ignite at higher hydration levels. De Lillis et al. (2009) also observed that the smoke appearance temperature rose with increasing foliar isoprenoid concentrations. This may again be a consequence of the tight association between isoprenoids and LWC. This effect was particularly evident in conifers where the resin ducts/glandular reservoirs in which large isoprenoid pools are tightly sealed are only broken when temperatures reach very high values.
Figure 3.
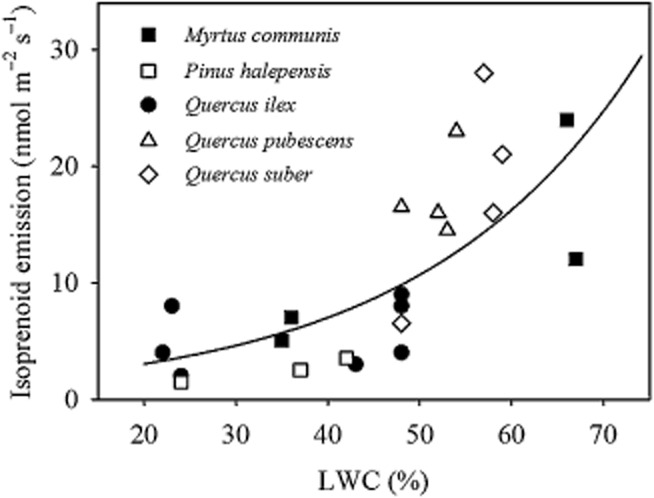
Relationship between isoprenoid emission and leaf water content (LWC) (redrawn from De Lillis et al. 2009).
Results obtained from the analysis of live leaves are still not fully definitive; the different experimental techniques utilized and the lack of a standardized methodology may account for the large variability found in the few published results. Isoprenoids are highly volatile and significant volumes are emitted as leaves are exposed to high temperatures. Thus, their intrinsic flammability may be masked by the dynamics of a fire event, because prior to leaf ignition the isoprenoid pool may be already depleted when the leaf was exposed to pre-heating and pre-ignition temperatures (i.e. distillation and then pyrolysis phases, see above) (Greenberg et al. 2006). Conversely, when leaves are directly and suddenly exposed to flame, high isoprenoid contents may facilitate flammability. These considerations raise the necessity of the development of standard methodologies for future experiments on leaf flammability.
Whether BVOCs also influence litter flammability is another important issue, as litter is among the most flammable biomass in forests. Litter is generally characterized by a significantly lower water content than fresh leaves. Therefore, the impact of water in determining flammability must be lower, and if BVOC pools are persist, their impact on flammability might be higher. A relationship between flammability and isoprenoid content was evident in the leaf litter of six different Pinus and Cistus species (Ormeño et al. 2009). As expected, the flammability of leaves with similar morphologies increased alongside isoprenoid concentrations. Notably, a higher flammability was observed in species with higher sesquiterpene (i.e. α-humulene, β-caryophyllene and caryophyllene oxide) contents than in those with higher pools of monoterpenes (i.e. α-pinene and β-pinene) (Ormeño et al. 2009).
BVOCs Emitted during Fire-Generated Heatwaves
The composition and emission of BVOCs varies significantly as temperature increases during the different stages of combustion (Greenberg et al. 2006) (Fig. 4). However, there is little available evidence regarding the effects of fire-generated heatwaves on the surrounding plants. These plants in close proximity to the fire-front experience a wide combination of pre-flaming temperatures, but do not experience direct contact with flame. Zhao et al. (2012) detected the emission of seven different monoterpenes from P. sylvestris needles exposed to 200 °C, whereas in ambient air conditions (air temperature of 28–30 °C) the emission blend was composed of eight monoterpenes and two sesquiterpenes. Furthermore, Greenberg et al. (2006) showed that the BVOC blend emitted during the distillation (temperatures up to 200 °C) and endothermic pyrolysis (temperatures between 200 and 300 °C) phases of combustion differed significantly to the BVOC blend emitted during flaming combustion, whereas it was relatively similar to that emitted during smouldering combustion. Water, stored VOCs and methanol were mostly emitted during these phases associated with the occurrence of a forest fire-generated heatwave.
Figure 4.

Comparison between isoprenoid emission, shown as percentage of total isoprenoid emission, from Eucalyptus citriodora and Quercus ilex leaves measured at 30 °C (upper panels) and during a progressive increase in temperature from 30 to 90 °C (bottom panels) (Centritto et al. unpublished results). Other = sum isoprenoid with an emission rate lower than 3% of the total emission.
Alessio et al. (2004) hypothesized that the effects of heat generated by a nearby fire on BVOC emission would depend on the negative impact on photosynthesis in light-dependent isoprenoid-emitting species, and on the rupture of reservoir integrity in plants storing BVOCs in specialized compartments. Indeed, exposure of leaves to 50 °C for 5 min concurrently inhibited photosynthesis and isoprenoid emissions in three Quercus species. However, as photosynthesis recovered with time, isoprenoid emission was either restored, or even increased relative to the pre-stress level. Identical results were found by Brilli et al. (2013), in a study of isoprene emission from Eucalyptus citriodora leaves in response to increased temperatures of up to 50 °C under different levels of water availability. Despite the presence of large glandular pools of isoprenoids in Eucalyptus leaves (which are absent in Quercus leaves), a full recovery of photosynthesis and isoprenoid emission was found, possibly due to the exposure time to 50 °C being limited to only 15 min. Data in Fig. 4 show that at 90 °C a substantial release of BVOCs from leaves occurs in both Eucalyptus and Quercus. In both species new compounds, such as isopregol, β-caryophyllene and τ-elemene were released alongside those emitted by unstressed leaves, thus again illustrating that the impact of a heatwave is somewhat similar in both species with and without permanent storage pools of isoprenoids. Although the experimental temperature was relatively mild in comparison with a real fire heatwave, and the process was mainly evaporative, the impact of the heatwave on isoprenoid composition was relatively dramatic. It is likely that at high temperatures BVOCs confined inside different leaf compartments are transported to the surface and released in the air by water vapour. The build-up of water pressure inside storage organs and glands can also disrupt their integrity and inducing the rapid release of BVOCs. Many of these compounds will contribute to the flammable mixture of air and BVOCs, thus initiating the flaming process.
A rather different dynamic is observed when a real heatwave is produced by a fire that reaches significantly higher temperatures (Fig. 5). Schinus molle saplings, planted adjacent to grass plots used to simulate a prescribed fire, were subjected to a heatwave that reached temperature of about 300 °C for approximately 30 s. Four hours after the fire event, the photosynthesis rates of leaves without detectable visible damage reached negative values, and isoprenoid emissions dropped dramatically to 7–28% of the pre-fire values. In the following 2 d, BVOC emission was almost zero, despite photosynthesis reaching values approximately two times higher than pre-stress levels. This experiment (Centritto, unpublished results) may indicate that, after the heatwave, the integrity of the BVOC storage pools was permanently damaged, and that photoassimilates were instead allocated to repair tissues within the plant, rather than being used for the synthesis of secondary metabolites.
Figure 5.
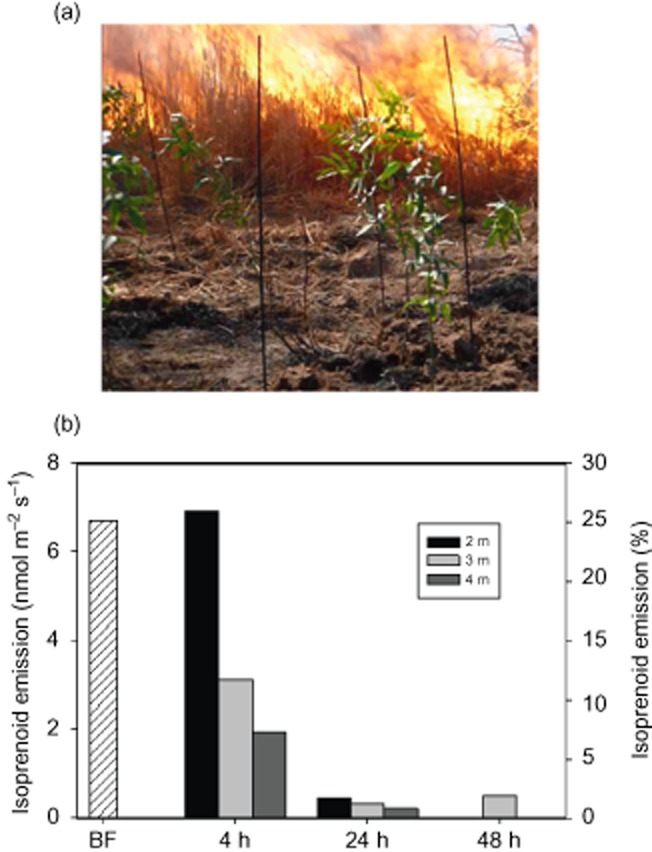
(a) Schinus molle saplings subjected to a heatwave generated by a grassland prescribed fire. The saplings were planted together with thermocouple rods at a distance of 2, 3 and 4 m from the edge the experimental plots. (b) Isoprenoid emissions from S. molle leaves before the fire event (BF), and isoprenoids emitted at the distance of 2, 3 and 4 m from the experimental plots 4, 24 and 48 h after the fire event, shown as a percentage of the BF emissions (Centritto et al. unpublished results).
Measurements of VOCs and BVOCs from Fires
In combustion facilities, the different phases of vegetation combustion can be clearly identified, as previously shown. In these cases, emitted compounds can be measured concurrently with the weight losses of the biomass and the temperature of the furnace (Crutzen & Andreae 1990; Ciccioli et al. 2001; Cerqueira et al. 2013), providing precise carbon balance information. However, in large forest or savannah fires, the phases of combustion are super-imposed over one another, and may last several days, especially in the case of smouldering. Therefore, only integrated values of VOC emissions can be used when referring to natural fires.
As the seminal paper of Andreae & Merlet (2001), many studies have focused on the determination of the EF of VOCs measuring their content in vegetation combustion plumes (Urbanski et al. 2009; Akagi et al. 2011). Airborne platforms equipped with a complex suite of fast sensors for the determination of VOCs and the other pollutants (such as Proton Transfer-Mass Spectrometry PTR-MS and different types of Fourier-Transform FTIR instruments) are generally used in this type of investigations, occasionally complementing real-time measurements with GC-MS analyses performed on air samples collected from canisters (Simpson et al. 2011; Akagi et al. 2013). By flying through the combustion plume, the mixing ratios of VOCs and other gases are measured inside and outside the plume, together with the concentration of particles. The gradients of VOCs are, then, normalized with respect to most abundant and least reactive gases released by vegetation combustion, CO2 and CO, and emission ratios (ERs) determined (Andreae & Merlet 2001). From these data the EF of VOCs per g kg−1 of dry biomass burned are calculated following Urbanski et al. (2009):
| (2) |
where MW is the molecular weight of the VOC, ER = ΔCVOC/ΔCCO2 and [%CF]Biomass is the percentage fraction of carbon in the dry vegetation, ranging from 0.45 to 0.55 (Urbanski et al. 2009). Information on the prevailing combustion conditions inside the plume are obtained by the modified combustion efficiency (MCE), defined by the ratio ΔCCO2/(ΔCCO2 + ΔCCO). This ratio ranges from near 0.80 for smouldering combustion to 0.99 for pure flaming combustion (Akagi et al. 2011; Simpson et al. 2011).
Data from different airborne campaigns present a very complex picture of VOCs emitted by vegetation combustion. The EF of VOCs emitted from vegetation combustion in various parts of the world have been recently reviewed (Urbanski et al. 2009; Akagi et al. 2011). Using the available data, EFs have been estimated for the burning of vegetation in savannas and tropical, boreal, temperate and extratropical forests (here considered as a subclass of boreal ecosystems developed at lower latitudes), in addition to the combustion of crop residues and pasture maintenance (Table 4). By separating these data into chemical classes (Ciccioli et al. 2001), the reactivity of VOCs with double bonds, or different functional groups, is highlighted (Table 4). Moreover, simple alkenes, in the most part derived from the pyrolysis of larger molecules, are separated from dienes (such as isoprene), trienes and larger terpenes, all of which are biological in origin.
Table 4.
Emission rates in g kg−1 dry matter of VOCs and other compounds proposed by Akagi et al. (2011) for modelling emissions from vegetation combustion
| Compound emitted | Tropical forests | Savannah | Crop residues | Boreal forests | Temperate forest |
|---|---|---|---|---|---|
| Carbon dioxide (CO2) | 1643 (58) | 1686 (38) | 1585 (100) | 1489 (121) | 1637 (71) |
| Carbon monoxide (CO) | 93 (27) | 63 (17) | 102 (33) | 127 (45) | 89 (32) |
| Nitrogen oxides (NOx as NO) | 2.55 (1.40) | 3.9 (0.80) | 3.11 (1.57) | 0.90 (0.69) | 2.51 (1.02) |
| Nitrous acid (HONO) | 1.18 (0.20) | – | 0.16 (0.07) | 0.52 (0.15) | 0.52 (0.15) |
| Sulphur dioxide (SO2) | 0.40 (0.19) | 0.48 (0.27) | – | – | – |
| Ammonia (NH3) | 1.33 (1.21) | 0.52 (0.35) | 2.17 (1.27) | 2.72 (2.32) | 0.78 (0.82) |
| Hydrogen cyanide (HCN) | 0.42 (0.26) | 0.41 (0.15) | 0.29 (0.38) | 1.52 (0.82) | 0.73 (0.19) |
| Methane (CH4) | 5.07 (1.98) | 1.94 (0.85) | 5.82 (3.56) | 5.96 (3.14) | 3.92 (2.39) |
| Alkanes | |||||
| Ethane (C2H6) | 0.71 (0.28) | 0.66 (0.41) | 0.91 (0.49) | 1.79 (1.14) | 1.12 (0.67) |
| Propane (C3H8) | 0.126 (0.060) | 0.10 (0.067) | 0.28 (0.15) | 0.44 | 0.26 (0.11) |
| n-Butane (C4H10) | 0.038 (0.023) | 0.016 (0.013) | 0.072 (0.036) | 0.12 | 0.083 (0.10) |
| i-Butane (C4H10) | 0.011 (0.009) | 0.0043 (0.0027) | 0.025 (0.013) | 0.042 | – |
| n-Pentane (C5H12) | 8.03 × 10−3 | 0.0032 (0.0032) | 0.025 (0.012) | 0.085 | – |
| i-Pentane (C5H12) | 0.010 (0.010) | 0.0022 (0.0032) | 0.020 (0.012) | 0.038 | – |
| Cyclopentane (C5H10) | – | – | 0.0019 (0.0012) | – | – |
| 2 + 3-Methylpentane (C6H14) | – | – | – | 0.036 | – |
| n-Hexane (C6H14) | 0.01 | 0.013 (0.0074) | – | 0.055 | – |
| Alkenes | |||||
| Ethylene (C2H4) | 1.06 (0.37) | 0.82 (0.35) | 1.46 (0.59) | 1.42 (0.43) | 1.12 (0.35) |
| Propylene (C3H6) | 0.64 (0.43) | 0.79 (0.56) | 0.68 (0.37) | 1.13 (0.60) | 0.95 (0.54) |
| 1-Butene (C4H8) | 0.079 (0.024) | 0.043 (0.022) | 0.134 (0.060) | 0.16 | – |
| i-Butene (C4H8) | 0.11 (0.051) | 0.024 (0.0051) | 0.117 (0.060) | 0.11 | – |
| trans-2-Butene (C4H8) | 0.029 (0.013) | 0.011 (0.0055) | 0.057 (0.030) | 0.04 | – |
| cis-2-Butene (C4H8) | 0.024 (0.010) | 0.0084 (0.0043) | 0.043 (0.023) | 0.03 | – |
| trans-2-Pentene (C5H10) | 3.30 × 10−3 | 0.0045 (0.0028) | – | – | – |
| cis-2-Pentene (C5H10) | 1.90 × 10−3 | 0.0025 (0.0018) | – | – | – |
| 3-Methyl-1-butene (C5H10) | 3.80 × 10−3 | 0.0051 (0.0034) | – | – | – |
| 2-Methyl-2-butene (C5H10) | 4.00 × 10−3 | 0.0048 (0.0035) | – | – | – |
| 2-Methyl-1-butene (C5H10) | 4.40 × 10−3 | 0.0059 (0.0037) | – | – | – |
| 2-Methyl-1-pentene (C6H12) | 2.80 × 10−3 | 0.0035 (0.0021) | – | – | – |
| Dienes trienes and terpenes | |||||
| Propadiene (C3H4) | 0.016 (0.006) | 0.012 (0.005) | – | – | – |
| 1,3-Butadiene (C4H6) | 0.039 | 0.052 (0.028) | 0.151 (0.072) | 0.14 | – |
| Isoprene (C5H8) | 0.13 (0.056) | 0.039 (0.027) | 0.38 (0.16) | 0.15 | – |
| α-Pinene (C10H16) | – | – | – | 1.64 | – |
| β-Pinene (C10H16) | – | – | – | 1.45 | – |
| Alkynes | |||||
| Acetylene (C2H2) | 0.44 (0.35) | 0.24 (0.10) | 0.27 (0.08) | 0.18 (0.10) | 0.29 (0.10) |
| Propyne (C3H4) | – | – | – | 0.059 | – |
| Arenes and substituted arenes | |||||
| Benzene (C6H6) | 0.39 (0.16) | 0.20 (0.084) | 0.15 (0.04) | 1.11 | – |
| Toluene (C7H8) | 0.26 (0.13) | 0.080 (0.058) | 0.19 (0.06) | 0.48 | – |
| Xylenes (C8H10) | 0.11 (0.082) | 0.014 (0.024) | – | 0.18 | – |
| Ethylbenzene (C8H10) | 0.050 (0.036) | 0.006 (0.010) | – | 0.051 | – |
| n-Propylbenzene (C9H12) | – | – | – | 0.018 | – |
| 3-Ethyltoluene (C9H12) | – | – | – | 0.024 | – |
| 2-Ethyltoluene (C9H12) | – | – | – | 0.011 | – |
| 4-Ethyltoluene (C9H12) | – | – | – | 0.015 | – |
| 1,2,3-Trimethylbenzene (C9H12) | – | – | – | 0.051 | – |
| 1,2,4-Trimethylbenzene (C9H12) | – | – | – | 0.03 | – |
| 1,3,5-Trimethylbenzene (C9H12) | – | – | – | 5.86 × 10−3 | – |
| Phenol (C6H5OH) | 0.45 (0.088) | 0.52 (0.36) | 0.52 (0.14) | 2.96 | 0.33 (0.38) |
| Aldehydes | |||||
| Formaldehyde (HCHO) | 1.73 (1.22) | 0.73 (0.62) | 2.08 (0.84) | 1.86 (1.26) | 2.27 (1.13) |
| Glycolaldehyde (C2H4O2) | 2.84 | 0.81 (0.38) | 2.01 (0.38) | 0.77 | 0.25 (0.45) |
| Acetaldehyde (C2H4O) | 1.55 (0.75) | 0.57 (0.30) | 1.24 (0.28) | – | – |
| Acrolein (C3H4O) | 0.65 (0.23) | – | – | – | – |
| Propanal (C3H6O) | 0.10 (0.026) | – | – | – | – |
| Methyl propanal (C4H8O) | 0.18 (0.075) | – | – | – | – |
| Methacrolein (C4H6O) | 0.15 (0.045) | – | – | 0.087 | – |
| Crotonaldehyde (C4H6O) | 0.24 (0.068) | – | – | – | – |
| Hexanal (C6H12O) | 0.01 (0.005) | – | – | – | – |
| Furaldehydes | 0.29 (0.0010) | – | – | – | – |
| Ketones | |||||
| Acetone (C3H6O) | 0.63 (0.17) | 0.16 (0.13) | 0.45 (0.07) | 0.75 | – |
| Acetol (C3H6O2) | 1.13 (0.12) | 0.45 (0.24) | 3.77 (0.91) | – | – |
| Methyl ethyl ketone (C4H8O) | 0.50 (0.21) | – | – | 0.22 | – |
| Methyl vinyl ketone (C4H6O) | 0.39 (0.11) | – | – | 0.2 | – |
| 2,3-Butanedione (C4H6O2) | 0.73 (0.22) | – | – | – | – |
| 2-Pentanone (C5H10O) | 0.08 (0.024) | – | – | – | – |
| 3-Pentanone (C5H10O | 0.03 (0.011) | – | – | – | – |
| Alcohols | |||||
| Methanol (CH3OH) | 2.43 (0.80) | 1.18 (0.41) | 3.29 (1.38) | 2.82 (1.62) | 1.93 (1.38) |
| Ethanol (C2H5OH) | – | – | – | 0.055 | – |
| Carboxylic acids | |||||
| Formic acid (HCO2H) | 0.79 (0.66) | 0.21 (0.096) | 1.00 (0.49) | 0.57 (0.46) | 0.35 (0.33) |
| Acetic acid (CH3CO2H) | 3.05 (0.90) | 3.55 (1.47) | 5.59 (2.55) | 4.41 (2.66) | 1.97 (1.66) |
| Esters | |||||
| Methyl vinyl ether (C3H6O) | – | 0.16 (0.045) | 0.08 (0.01) | – | – |
| Heterocyclic aromatics 5 rings | |||||
| Furan (C4H4O) | 0.41 (0.10) | 0.17 (0.058) | 0.11 (0.04) | 0.80 (0.50) | 0.20 (0.21) |
| 3-Methylfuran (C5H6O) | 0.59 (0.20) | – | – | – | – |
| 2-Methylfuran (C5H6O) | 0.08 (0.028) | – | – | – | – |
| Other substituted furans | 1.21 (0.016) | – | – | – | – |
| Pyrrole (C4H5N) | 0.12 (0.038) | – | – | – | – |
| Alkyl nitriles | |||||
| Acetonitrile (CH3CN) | 0.41 (0.10) | 0.11 (0.058) | 0.21 (0.06) | 0.61 | – |
| Propenenitrile (C3H3N) | 0.04 (0.01) | 0.051 (0.022) | 0.03 (0.002) | – | – |
| Propanenitrile (C3H5N) | 0.09 | 0.031 (0.014) | 0.06 (0.002) | – | – |
| Sulphur compounds | |||||
| Dimethyl sulphide (C2H6S) | 1.35 × 10−3 | 0.0013 (0.0011) | – | 4.65 × 10−3 | – |
| Carbonyl Sulphide (OCS) | 0.025 | – | – | 0.46 (0.47) | – |
| Alkyl halides | |||||
| Chloromethane (CH3Cl) | 0.053 (0.038) | 0.055 (0.036) | – | 0.059 | – |
| Trichloromethane (CHCl3) | 2.94 × 10−4 | 0.012 (0.020) | – | – | – |
Data in brackets are the observed variations.
VOC, volatile organic compound.
The data presented in Table 4 are only to be considered for general use as they come from relatively few campaigns in specific ecosystems. Essentially, data for tropical forests and savannah fires are derived from measurements in the Amazon region and in Central and Southern part of Africa where numerous flights have been performed in the last decade. The Boreal forest data set was collected from a few flights made in North America. The data set relating to the combustion of crop residues is equally scant. The temperate forest data set is particularly limited (Urbanski et al. 2009). In many instances, little or no information is provided regarding the type of ecosystem burned and the nature of the plants living in them. Despite these limitations, the data contained within Table 4 show that VOCs coming from the distillation and pyrolysis of plant biopolymers and wall cell components (such as carboxylic acids, aldehydes, ketones, alcohols and furans) are common to all vegetation types. Among them, acetaldehyde, methanol and acetic acid are by far the most abundant. Also common, but less abundant, are pyrogenic compounds such as alkanes, alkenes, dienes and arenes; however, many of these compounds, such as 1,3-butadiene and benzene, that are very toxic to humans are also non-specific to forest fires as they are also emitted by fossil fuel combustion (Ciccioli et al. 1999).
Among BVOCs, isoprene is the most abundant during the combustion of tropical forests of Brazil, whereas two monoterpenes (α-pinene and β-pinene) are found to be abundant in the emission of fires generated in Boreal forests of North America. Isoprene release is relatively low during the combustion of African savannah ecosystems, which also does not emit other terpenes. Oxygenated compounds with more than three carbon atoms are dominant in the emissions of tropical forest fires, whereas reduced carbon components dominate the fraction emitted by boreal forest fires.
Matching BVOC Emissions from Forest Fires with Functional Plant Types
Some of the data presented in Table 4 may bear similarities to BVOCs produced by the plants present in the investigated ecosystems. Monoterpenes below detection limits observed in the emissions of Brazilian forest fires might indicate that the Amazon forest ecosystem is dominated by trees that behave as poor monoterpene emitters. Indeed, tower-based and airborne determinations performed in Manaus clearly showed that monoterpene fluxes were one order of magnitude lower than those of isoprene (Kuhn et al. 2007), and mostly originated from plants without the ability to permanently store monoterpenes (Kuhn et al. 2004). In fact, isoprene emissions from Amazonian ecosystems are smaller than those measured in deciduous forests of the United States. These results were consistent with a screening study that showed that only ca. 30% of the plants that composed the Amazon forest emit isoprene (Harley et al. 2004).
Emissions from boreal forest fires (Simpson et al. 2011) reflected that the majority of trees in Northern Canada were conifers containing high levels of monoterpenes in their needles and bark. The observed blend of α-pinene and β-pinene originated from the combustion of several conifers, such as black, white and red spruce, tamarack and balsam fir. The data from savannah fires, which showed low emission rates, are also consistent with the type of vegetation, as the savannah ecosystem is characterized by the Graminaceae, which are known to be poor isoprenoid emitters (Kesselmeier & Staudt 1999).
A suite of 204 compounds was identified in the combustion emissions under laboratory conditions of P. pinea branches and needles during the flaming and smouldering phases (Ciccioli et al. 2001). As only GS-MS was used for the measurement of VOCs, some of the more volatile compounds, such as ethane, ethylene, propane, methanol, formaldehyde and glycolaldehyde, were not detected or quantified. Nonetheless, this early study provided the first evidence of the emission of sesquiterpene from the combustion of conifers, which was later confirmed by PTR-MS determinations (Warneke et al. 2011; Yokelson et al. 2013). Given the high number of emitted compounds, we only report in Table 5 the isoprenoid emissions of P. pinea, aiming to show that many isoprenoids produced during the early distillation phases of combustion (see Fig. 4) survive to flaming and, to a lesser extent, smouldering. It is worth noting that the isoprenoid profile displayed in Table 5 is different to that reported for conifers growing in South Carolina by Yokelson et al. (2013) and by Akagi et al. (2013). Overall, it seems that while the EFs of many pyrogenic VOCs can be similar in trees from temperate ecosystems in different parts of the world, BVOC fingerprints are different and effectively characterize plant species and ecosystems. Large differences in the BVOC profiles can be expected from the combustion of deciduous and conifer trees in temperate regions. For example, the emissions from forest fires are likely to be characterized by the compounds emitted by the dominant species; for example, sabinene is the major compound emitted by Fagus sylvatica that covers large portions of forests in Europe (Dindorf et al. 2006), 1,8-Cineol characterizes emissions from E. globulus forests in Australia, and 3-methyl-2-butenol is emitted in large volumes by the P. ponderosa of North America (Schade et al. 2000).
Table 5.
Emission rates mg kg−1 of terpenes emitted by the combustion of Pinus pinea (adapted from Ciccioli et al. 2001)
| Compound | Flaming | Smouldering |
|---|---|---|
| Tricyclene | 2.3 ± 0.3 | 0.5 ± 0.1 |
| Thujene | 2.2 ± 0.1 | 0.3 ± 0.0 |
| α-Pinene | 67.2 ± 2.6 | 6.3 ± 0.0 |
| Camphene | 10.4 ± 1.2 | 2.6 ± 0.2 |
| Sabinene | 1.9 ± 1.0 | – ± – |
| β-Pinene | 13.3 ± 1.6 | 0.7 ± 0.2 |
| Myrcene | 7.4 ± 0.5 | 0.6 ± 0.1 |
| α-Phellandrene | 6.4 ± 0.9 | 0.8 ± 0.2 |
| Δ3-Carene | 1.5 ± – | – ± – |
| α-Terpinene | 16.0 ± 3.4 | 1.9 ± 0.8 |
| p-Cymene | 79.3 ± 21.5 | 23.4 ± 2.1 |
| 1,8-Cineol | 415.1 ± 18.6 | 48.9 ± 4.5 |
| Limonene | 107.6 ± 9.2 | 17.5 ± 0.3 |
| γ-Terpinene | 0.9 ± – | – ± – |
| Terpinolene | 1.3 ± – | – ± – |
| Allocymene | 1.7 ± 0.6 | 0.4 ± 0.1 |
| Sesquiterpene* | 43.1 ± 9.3 | 15.6 ± 1.2 |
| Sesquiterpene* | 62.6 ± 3.0 | 14.4 ± 0.7 |
| Aromadendrene | 219.9 ± 133.6 | 67.9 ± 32.0 |
| Sesquiterpene* | 89.7 ± 108.7 | 49.3 ± 22.1 |
| Total | 1164.7 ± 318.0 | 253.0 ± 65.0 |
Compounds separated but not identified.
Despite many ecosystems in temperate regions (such as the Mediterranean basin, the south-eastern part of the United States, California and Australia), host large numbers of isoprene and monoterpene emitters (Kesselmeier & Staudt 1999; Loreto et al. 2014), none, or relatively few, isoprenoids or oxygenated BVOCs have been measured in the emission of temperate forest fires in Europe. Failure to observe BVOC emissions in temperate forests is probably due to the chemistry of the plume rather than the effect of fire on plant physiology. As highlighted in a previous section, BVOCs cannot be measured easily when oxidation reactions occur at the optimal ratios between VOCs and NOx (Akagi et al. 2012, 2013,). Moreover, BVOC depletion in the combustion plume might reflect their selective transfer from a gas to a solid phase, and their subsequent removal by deposition (de Gouw et al. 2006). This latter effect can be particularly apparent in some polar BVOCs, such as linalool and 1,8-cineol, that are strongly partitioned in the water layer of hygroscopic particles. Also due to the lower volatility of sesquiterpenes, they can be selectively removed by adsorption on carbon particles, as also shown by the analysis of particles emitted during the combustion of different wood types (Fine et al. 2004).
Conclusions
Although the complexity of BVOC production in plants has been long recognized (Kesselmeier & Staudt 1999; Loreto et al. 2014), the signature of BVOCs emitted by forest fires has only recently been recognized (Akagi et al. 2012; Yokelson et al. 2013). Indeed, we have acknowledged unforeseen problems in the determination of BVOCs emitted by vegetation fires, mainly due to the complex chemical reactions and transition phases occurring inside combustion plumes. Nevertheless, forest fires may dramatically change the EFs of BVOCs, therefore influencing the chemistry and physics of the atmosphere, the physiology of plants (Loreto & Schnitzler 2010), communication between plants and other organisms within the ecosystem (Trowbridge & Stoy 2013), and the evolution of plant communities in the ecosystem (Fineschi et al. 2013).
Acknowledgments
This work was supported by the European Commission (FP7-KBBE-2012-6 Development of improved perennial non-food biomass and bioproduct crops for water stressed environments, WATBIO, contract 311929), by the European Science Foundation scientific programme ‘EuroVol-Ecology of Plant Volatiles, from Molecules to the Globe’, and by the Ministero dell'Istruzione, dell'Università e della Ricerca of Italy – Programmi di Ricerca Scientifica di Rilevante Interesse Nazionale ‘Going to the root of plant productivity: how the rhizosphere interacts with the aboveground armament for indirect and direct defense against abiotic and biotic stressors (PRO-ROOT)’.
References
- Akagi SK, Yokelson RJ, Wiedinmyer C, Alvarado MJ, Reid JS, Karl T. Wennberg PO. Emission factors for open and domestic biomass burning for use in atmospheric models. Atmospheric Chemistry and Physics. 2011;11:4039–4072. [Google Scholar]
- Akagi SK, Craven JS, Taylor JW, McMeeking GR, Yokelson R, Burling IR. Weise DR. Evolution of trace gases and particles emitted by a chaparral fire in California. Atmospheric Chemistry and Physics. 2012;12:1397–1421. [Google Scholar]
- Akagi SK, Yokelson RJ, Burling IR, Meinardi S, Simpson I, Blake DR. Weise DR. Measurements of reactive trace gases and variable O3 formation rates in some South Carolina biomass burning plumes. Atmospheric Chemistry and Physics. 2013;13:1141–1165. [Google Scholar]
- Alessio GA, De Lillis M, Fanelli M, Pinelli P. Loreto F. Direct and indirect impacts of fire on isoprenoid emissions from Mediterranean vegetation. Functional Ecology. 2004;18:3357–3364. [Google Scholar]
- Alessio GA, Peñuelas J, De Lillis M. Llusia J. Implications of foliar terpene content and hydration on leaf flammability of Quercus ilex and Pinus halepensis. Plant Biology. 2008;10:123–128. doi: 10.1111/j.1438-8677.2007.00011.x. [DOI] [PubMed] [Google Scholar]
- Alvarado MJ. Prinn RG. Formation of ozone and growth of aerosols in young smoke plumes from biomass burning: 1. Lagrangian parcel studies. Journal of Geophysical Research. 2009;114:D09306. [Google Scholar]
- Andreae MO. Crutzen PJ. Atmospheric aerosols biogeochemical sources and role in atmospheric chemistry. Science. 1997;276:1052–1058. [Google Scholar]
- Andreae MO. Merlet P. Emission of trace gases and aerosols from biomass burning. Global Biogeochemical Cycles. 2001;15:955–966. [Google Scholar]
- Atkinson R. Arey J. Atmospheric chemistry of biogenic organic compounds. Accounts of Chemical Research. 1998;31:574–583. [Google Scholar]
- Atkinson R. Arey J. Gas-phase chemistry of biogenic volatile organic compounds: a review. Atmospheric Environment. 2003;37(Suppl. 2):S197–S219. [Google Scholar]
- Bowman DMJS, Balch JK, Artaxo P, Bond WJ, Carlson JM, Cochrane MA. Pyne SJ. Fire in the earth system. Science. 2009;324:481–484. doi: 10.1126/science.1163886. [DOI] [PubMed] [Google Scholar]
- Brilli F, Tsonev T, Mahmood T, Velikova V, Loreto F. Centritto M. Ultradian variation of isoprene emission, photosynthesis, mesophyll conductance and optimum temperature sensitivity for isoprene emission in water-stressed Eucalyptus citriodora saplings. Journal of Experimental Botany. 2013;64:519–528. doi: 10.1093/jxb/ers353. [DOI] [PubMed] [Google Scholar]
- Burling IR, Yokelson RJ, Akagi SK, Urbanski SP, Wold CE, Griffith DWT. Weise DR. Airborne and ground-based measurements of the trace gases and particles emitted by prescribed fires in the United States. Atmospheric Chemistry and Physics. 2011;11:12197–12216. [Google Scholar]
- Calogirou A, Larsen BR. Kotzias D. Gas phase oxidation products: a review. Atmospheric Environment. 1999;33:1428–1439. [Google Scholar]
- Carrasco N, Picquet-Varrault B. Doussin JF. Kinetic and product study of the gas-phase reaction of sabinaketone with OH radical. International Journal of Chemical Kinetics. 2007;39:415–421. [Google Scholar]
- Centritto M, Brilli F, Fodale R. Loreto F. Different sensitivity of isoprene emission, respiration, and photosynthesis to high growth temperature coupled with drought stress in black poplar (Populus nigra. Tree Physiology. 2011a;31:275–286. doi: 10.1093/treephys/tpq112. [DOI] [PubMed] [Google Scholar]
- Centritto M, Tognetti R, Leitgeb E, Střelcová K. Cohen S. Above ground processes: anticipating climate change influences. In: Schleppi P, editor; Bredemeier M, Cohen S, Godbold DL, Lode E, Pichler V, Schleppi P, editors. Forest Management and the Water Cycle: An Ecosystem-Based Approach. Dordrecht: Springer; 2011b. pp. 31–64. Ecological Studies 212. [Google Scholar]
- Cerqueira M, Gomes L, Tarelho L. Pio C. Formaldehyde and acetaldehyde emissions from residential wood combustion in Portugal. Atmospheric Environment. 2013;72:171–176. [Google Scholar]
- Chandler P, Cheney P, Thomas L, Trabaud L. Williams D. Fire in Forestry. Vol. 1. New York: Wiley; 1983. [Google Scholar]
- Ciccioli P, Brancaleoni E. Frattoni M. Reactive hydrocarbons in the atmosphere at urban and regional scales. In: Hewitt CN, editor; Reactive Hydrocarbons in the Atmosphere. San Diego, CA: Academic Press; 1999. pp. 159–207. [Google Scholar]
- Ciccioli P, Brancaleoni E, Frattoni M, Cecinato A. Pinciarelli L. Determination of volatile organic compounds (VOC) emitted from biomass burning of Mediterranean vegetation species by GC-MS. Analytical Letters. 2001;34:937–955. [Google Scholar]
- Cochrane MA. Fire science for rainforests. Nature. 2003;421:913–919. doi: 10.1038/nature01437. [DOI] [PubMed] [Google Scholar]
- Crutzen PJ. Andreae MO. Biomass burning in the tropics: impact on atmospheric chemistry and biogeochemical cycles. Science. 1990;250:1669–1678. doi: 10.1126/science.250.4988.1669. [DOI] [PubMed] [Google Scholar]
- De Cesari S, Fuzzi S, Facchini MC, Mircea M, Emblico L, Cavalli F. Artaxo P. Characterization of the organic composition of aerosols from Rondonia, Brazil, during the LBA-SMOCC 2002 experiment and its representation through model compounds. Atmospheric Chemistry and Physics. 2006;6:375–402. [Google Scholar]
- De Lillis M, Bianco PM. Loreto F. The influence of leaf water content and isoprenoids on flammability of some Mediterranean woody species. International Journal of Wildland Fire. 2009;18:203–212. [Google Scholar]
- Dindorf T, Kuhn U, Ganzeveld L, Schebeske G, Ciccioli P, Holzke C. Kesselmeier J. Significant light and temperature dependent monoterpene emissions from European beech (Fagus sylvatica L.) and their potential impact on the European volatile organic compound budget. Journal of Geophysical Research. 2006;111:D16305. [Google Scholar]
- Evans RJ. Milne TA. Molecular characterization of the pyrolysis of biomass, 1. Fundamentals. Energy and Fuels: An American Chemical Society Journal. 1987;1:123–137. [Google Scholar]
- Fall R. Biogenic emissions of volatile organic compounds from higher plants. In: Hewitt CN, editor. Reactive Hydrocarbons in the Atmosphere. San Diego, CA: Academic Press; 1999. pp. 41–86. [Google Scholar]
- Fares S, Mahmood T, Liu S, Loreto F. Centritto M. Influence of growth temperature and measuring temperature on isoprene emission, diffusive limitations of photosynthesis and respiration in hybrid poplars. Atmospheric Environment. 2011;45:155–161. [Google Scholar]
- Fine FM, Cass GR. Simoneit BRT. Chemical characterization of fine particle emissions from the wood stove combustion of prevalent United States tree species. Environmental Engineering Science. 2004;6:705–721. [Google Scholar]
- Fineschi S, Loreto F, Staudt M. Penuelas J. Diversification of volatile isoprenoid emissions from trees: evolutionary and ecological perspectives. In: Monson RK, editor; Niinemets U, Monson RK, editors. Biology, Controls and Models of Tree Volatile Organic Compound Emissions. London: Springer; 2013. pp. 1–20. Tree Physiology 5. [Google Scholar]
- Finnlayson-Pitts BJ. Pitts JN., Jr . Chemistry of the Upper and Lower Atmosphere. San Diego, CA: Academic Press; 2000. [Google Scholar]
- de Gouw JA, Warneke C, Stohl A, Wollny AG, Brock CA, Cooper OR. Lueb A. Volatile organic compounds composition of merged and aged forest fire plumes from Alaska and western Canada. Journal of Geophysical Research. 2006;111:D10303. [Google Scholar]
- Greenberg J, Friedli H, Guenther AB, Hanson D, Harley P. Karl T. Volatile organic emissions from the distillation and pyrolysis of vegetation. Atmospheric Chemistry and Physics. 2006;6:81–91. [Google Scholar]
- Grote R. Niinemets Ü. Modeling volatile isoprenoid emissions – a story with split ends. Plant Biology. 2008;10:8–28. doi: 10.1055/s-2007-964975. [DOI] [PubMed] [Google Scholar]
- Guenther A. Modeling biogenic organic compound emissions to the atmosphere. In: Hewitt CN, editor. Reactive Hydrocarbons in the Atmosphere. San Diego, CA: Academic Press; 1999. pp. 97–118. [Google Scholar]
- Guenther A, Jiang X, Heald CL, Sakulyanontvittaya T, Duhl T, Emmons LK. Wang X. The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions. Geoscientific Model Development. 2012;5:1471–1492. [Google Scholar]
- Harley P, Vascocellos P, Vierling L, De Pinheiro CC, Greenberg J, Guetnher A. Malhi YR. Variation in potential for isoprene emissions among neotropical forest sites. Global Change Biology. 2004;10:1–21. [Google Scholar]
- Helas G, Lobert J, Scharffe D, Schafer L, Goldhammer J, Bauset J. Andreae MO. Ozone production due to emissions from vegetation burning. Journal of Atmospheric Chemistry. 1995;22:163–174. [Google Scholar]
- Hoffmann T. Warnke J. Organic aerosols. In: Koppmann R, editor; Volatile Organic Compounds in the Atmosphere. Oxford: Blackwell Publishing; 2007. pp. 342–387. [Google Scholar]
- Holopainen JK. Gershenzon J. Multiple stress factors and the emission of plant VOCs. Trends in Plant Science. 2010;15:176–184. doi: 10.1016/j.tplants.2010.01.006. [DOI] [PubMed] [Google Scholar]
- Keeley JE, Pausas JG, Rundel PW, Bond WJ. Bradstock RA. Fire as an evolutionary pressure shaping plant traits. Trends in Plant Science. 2011;16:406–411. doi: 10.1016/j.tplants.2011.04.002. [DOI] [PubMed] [Google Scholar]
- Keeling CI. Bohlmann J. Genes, enzymes and chemicals of terpenoid diversity in the constitutive and induced defense of conifers against insects and pathogens. New Phytologist. 2006;170:657–675. doi: 10.1111/j.1469-8137.2006.01716.x. [DOI] [PubMed] [Google Scholar]
- Kesselmeier J. Staudt M. Biogenic volatile organic compounds (VOC): an overview on emission, physiology and ecology. Journal of Atmospheric Chemistry. 1999;33:23–88. [Google Scholar]
- Kuhn U, Rottenberger S, Biesenthal T, Wolf A, Schebeske G, Ciccioli P. Kesselmeier J. Seasonal differences in isoprene and light-dependent monoterpene emission by Amazonian tree species. Global Change Biology. 2004;10:663–682. [Google Scholar]
- Kuhn U, Andreae MO, Ammann C, Araujo AC, Brancaleoni E, Ciccioli P. Kesselmeier J. Isoprene and monoterpene fluxes from Central Amazonian rainforest inferred from tower-based and airborne measurements, and implications on the atmospheric chemistry and the local carbon budget. Atmospheric Chemistry and Physics. 2007;7:2855–2879. [Google Scholar]
- Li YJ, Chen Q, Guzman MI, Chan CK. Martin ST. Second-generation products contribute substantially to the particle-phase organic material produced by β-caryophyllene ozonolysis. Atmospheric Chemistry and Physics. 2011;11:121–132. [Google Scholar]
- Loreto F. Centritto M. Leaf carbon assimilation in a water-limited world. Plant Biosystems. 2008;142:154–161. [Google Scholar]
- Loreto F. Schnitzler J-P. Abiotic stresses and induced BVOCs. Trends in Plant Science. 2010;15:154–166. doi: 10.1016/j.tplants.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Loreto F, Barta C, Brilli F. Nogues I. On the induction of volatile organic compound emissions by plants as consequence of wounding or fluctuations of light and temperature. Plant, Cell and Environment. 2006;29:1820–1828. doi: 10.1111/j.1365-3040.2006.01561.x. [DOI] [PubMed] [Google Scholar]
- Loreto F, Bagnoli F, Calfapietra C, Cafasso D, De Lillis M, Filibeck G. Ricotta C. Isoprenoid emission in hygrophyte and xerophyte European woody flora: ecological and evolutionary implications. Global Ecology and Biogeography. 2014;23:334–345. [Google Scholar]
- Millan-Millan M, Salvador R, Mantella E. Artinano A. Meteorology of photochemical air pollution in Southern Europe: experimental results from EC research projects. Atmospheric Environment. 1998;30:2583–2593. [Google Scholar]
- Nelson RM., Jr . Water relations of forest fuels. In: Miyanishi K, editor; Johnson EA, Miyanishi K, editors. Forest Fires Behaviour and Ecological Effects. San Diego: Academic Press; 2001. pp. 79–149. [Google Scholar]
- Niinemets Ü, Loreto F. Reichstein M. Physiological and physicochemical controls on foliar volatile organic compound emissions. Trends in Plant Science. 2004;9:180–186. doi: 10.1016/j.tplants.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Ormeño E, Céspedes B, Sánchez IA, Velasco-García A, Moreno JM, Fernandez C. Baldy V. The relationship between terpenes and flammability of leaf litter. Forest Ecology and Management. 2009;257:471–482. [Google Scholar]
- Owens MK, Lin CD, Taylor CA. Whisenant SG. Seasonal patterns of plant flammability and monoterpenoid content in Juniperus ashei. Journal of Chemical Ecology. 1998;24:2115–2129. [Google Scholar]
- Page WG, Jenkins MJ. Runyon JB. Mountain pine beetle attack alters the chemistry and flammability of lodgepole pine foliage. Canadian Journal of Forest Research. 2012;42:1631–1647. [Google Scholar]
- Pierce JL, Meyer GA. Jull AJT. Fire-induced erosion and millennial-scale climate change in northern ponderosa pine forests. Nature. 2004;432:87–90. doi: 10.1038/nature03058. [DOI] [PubMed] [Google Scholar]
- Ramanathan V, Crutzen PJ, Kiehl JT. Rosenfeld D. Aerosols, climate, and the hydrological cycle. Science. 2001;294:2119–2124. doi: 10.1126/science.1064034. [DOI] [PubMed] [Google Scholar]
- Reimann S. Lewis CA. Anthropogenic VOCs. In: Koppmann R, editor; Volatile Organic Compounds in the Atmosphere. Oxford: Blackwell Publishing; 2007. pp. 33–70. [Google Scholar]
- Rothermel RC. Forest fires and the chemistry of forest fuels. In: Tillman DA, editor; Shafizadeh F, Karkanan KV, Tillman DA, editors. Thermal Use and Properties of Carbohydrates and Lignins. New York: Academic Press; 1976. pp. 245–259. [Google Scholar]
- Rowell RM, Pettersen R. Tshabalala MA. Cell wall chemistry. In: Rowell RM, editor; Handbook of Wood Chemistry and Wood Composites. London: CRC Press; 2012. pp. 35–74. [Google Scholar]
- Schade GW, Goldstein AH, Gray DW. Lerdau MT. Canopy and leaf level 2-methyl-3-buten-2-ol fluxes from a ponderosa pine plantation. Atmospheric Environment. 2000;34:3535–3544. [Google Scholar]
- Scholes MC, Matrai PA, Andreae MG, Smith KA. Manning MR. Biosphere-atmosphere interaction. In: Pszenny AAP, editor; Brasseur GP, Prinn RN, Pszenny AAp, editors. Atmospheric Chemistry in a Changing World. Berlin: Springer-Verlag; 2003. pp. 39–71. [Google Scholar]
- Sharkey TD. Yeh S. Isoprene emission from plants. Annual Review of Plant Physiology and Plant Molecular Biology. 2001;52:407–436. doi: 10.1146/annurev.arplant.52.1.407. [DOI] [PubMed] [Google Scholar]
- Simpson IJ, Akagi SK, Barletta B, Blake NJ, Choi Y, Diskin GS. Blake DR. Boreal forest fire emissions in fresh Canadian smoke plumes: C1-C10 volatile organic compounds (VOCs), CO2, CO, NO2, NO, HCN and CH3CN. Atmospheric Chemistry and Physics. 2011;11:6445–6463. [Google Scholar]
- Stephens SL, Agee JK, Fulé PZ, North MP, Romme WH, Swetnam TW. Turner MG. Managing forests and fire in changing climates. Science. 2013;342:41–42. doi: 10.1126/science.1240294. [DOI] [PubMed] [Google Scholar]
- Thonicke K, Spessa A, Prentice IC, Harrison SP, Dong L. Carmona-Moreno C. The influence of vegetation, fire spread and fire behavior on biomass burning and trace gas emissions: results from a process-based model. Biogeosciences. 2010;7:1991–2011. [Google Scholar]
- Trabaud L. Etude du comportement de feu dans la garrigue de chêne kèrmes à partir des températures et des vitesses de propagation. Annales des Sciences Forestieres. 1979;36:13–55. [Google Scholar]
- Trowbridge AM. Stoy CP. BVOC-mediated plant-herbivore interactions. In: Monson RK, editor; Niinemets U, Monson RK, editors. Biology, Controls and Models of Tree Volatile Organic Compound Emissions. London: Springer; 2013. pp. 21–46. Tree Physiology 5. [Google Scholar]
- Tyndall GS, Winker DM, Anderson TL, Eisele FL, Apel EC, Brenninkmeijer CAM. Wexler AS. Advances in laboratory and field Measurements. In: Pszenny AAP, editor; Brasseur GP, Prinn RN, Pszenny AAP, editors. Atmospheric Chemistry in a Changing World. Berlin: Springer-Verlag; 2003. pp. 157–184. [Google Scholar]
- Urbanski SP, Hao WM. Baker S. Chemical composition of wildland fire emissions. In: Andersen C, editor; Bytnerowicz A, Arbaugh M, Riebau A, Andersen C, editors. Developments in Environmental Science. The Netherlands: Elsevier; 2009. pp. 79–107. [Google Scholar]
- Van Loo S. Koppejan J. The Handbook of Biomass Combustion and Co-firing. London, UK: Earthscan Publ. Limited; 2007. [Google Scholar]
- Van Wagner CE. Calculations of forest fire spread by flame radiation. 1967. Canadian Department of Forestry, Report 1185.
- Warneke C, Roberts JM, Veresa P, Gilman J, Kustera WC, Burling I. de Gouw JA. VOC identification and inter-comparison from laboratory biomass burning using PTR-MS and PIT-MS. International Journal of Mass Spectrometry. 2011;303:6–14. [Google Scholar]
- Yokelson RJ, Burling IR, Gilman JB, Warneke C, Stockwell CE, de Gouw J. Weise DR. Coupling field and laboratory measurements to estimate the emission factors of identified and unidentified trace gases for prescribed fires. Atmospheric Chemistry and Physics. 2013;13:89–116. [Google Scholar]
- Zhao FJ, Shu LF. Wang QH. Terpenoid emissions from heated needles of Pinus sylvestris and their potential influences on forest fires. Acta Ecologica Sinica. 2012;32:33–37. [Google Scholar]