

Abstract
The rising tide of antibacterial resistance and the lack of a diverse, vibrant pipeline of novel antibacterial agents is a global crisis that impairs our ability to treat life-threatening infections. The recent introduction of a tiered approach to the regulatory framework in this area offers one path to resolving some of the challenges. By drawing heavily on the predictive power of the related sciences of pharmacokinetics and pharmacodynamics, smaller, focused clinical trial programs have become possible for agents that might not otherwise have been possible to progress. There are limitations to these pathways, and they are not easy to implement, but making reliable noninferiority-based approaches available is critical to reinvigorating the global antibiotic pipeline. With the recognition of these ideas by key regulatory authorities in recent guidance, the next challenges in this area will focus on interpretive breakpoints, the extent of data in the prescribing information, ensuring that multiple agents can be progressed, and the challenge of the antibiotic business model.
Keywords: regulatory framework, antibacterial drug development, antibacterial resistance
Introduction
The crisis of rising rates of antibacterial resistance in the face of a relatively thin global pipeline threatens our ability to continue to deliver adequate health care. The Centers for Disease Control and Prevention in the United States recently estimated that 2 million infections with resistant bacteria occur each year in the United States and that 23,000 people die as result.1 Similar rates of disease burden have been described for Europe.2
The limited pipeline of new agents has been raising alarm for years, with a recent review by the Infectious Diseases Society of America (IDSA)3 noting “some progress in development of new antibacterial drugs that target infections caused by resistant Gram-negative bacteria, but progress remains alarmingly elusive.” This conclusion was challenged by a 2013 review of antibiotic approvals and withdrawals in which the authors concluded that drug approval rates are down in many classes of drugs.4 These authors further noted that antibiotics have sometimes been withdrawn after approval for reasons ranging from safety-related issues to lack of clinical or commercial significance and that, as a consequence, policies seeking to remedy the lack of antibiotic innovation should focus on the quality of new agents, not just their quantity. We, however, find this view to be overly simplified—from our perspective within industry, the situation is indeed alarming, with corporate support for anti-infective programs remaining sparse and fragile. While it is easy to call for greater quality, the practical truth is that development timelines are long and the true quality of any given agent cannot be readily known until some years into its life cycle. If there were a tool that could predict quality for a newly discovered compound based solely on preclinical testing, it would certainly be in widespread use.
Identifying a path to solving this problem depends on an accurate assessment of the core drivers.5,6 From our standpoint within the pharmaceutical industry, it is helpful to view the challenges via a tripartite framework. First, antibacterial discovery is difficult. Relatively high drug concentrations are often needed for efficacy, and thus finding agents that kill bacteria without host toxicity is difficult. Second, antibacterial development is difficult. As will be discussed in detail below, the traditional regulatory pathway's requirement for large phase III programs is a challenge if we wish to have new agents developed in advance of widespread resistance. Third, the economics of antibacterial agents are difficult. Antibacterial agents have tended to be modestly priced and are used only briefly. When taken with the community need to limit their use, the net effect for a developer is a limited return on investment, which has often been cited as a substantial driver of reduced effort in this area.7
Each of these challenges requires a different approach to its resolution. The focus of this paper is on changes to the regulatory environment, but the other areas are also being addressed in parallel. Improved discovery success is being sought via collaborative efforts such as the New Drugs For Bad Bugs (ND4BB) project sponsored in Europe under the aegis of the Innovative Medicines Initiative project (http://www.imi.europa.eu/). Research support is also being made available in the United States via grants from the National Institute of Allergy and Infectious Diseases (NIAID) and the Biomedical Advanced Research and Development Authority (BARDA). Generation of ideas for possible changes in the business model for antibiotics will be the focus of an ND4BB-sponsored project (see below).
The tiered framework
The core challenge with the traditional framework for approving antibacterial agents is that it rests on the assumption that pairs of relatively large (usually >750 total subjects per study) phase III studies can be conducted for the pathogen(s) of interest at the body site(s) of interest. This approach has worked well in the past, but has become problematic as the development goal for new antibiotics has focused increasingly on specific problem pathogens (e.g., Pseudomonas aeruginosa) or on agents targeted to address specific types of emerging resistance. In these scenarios, enrolling such large study sizes may be impossible or impractical.
As a specific example, consider the goal of developing a narrow-spectrum agent focused on P. aeruginosa. Using reasonably standard assumptions, the study might be sized based on an endpoint with a failure rate of about 20%, a noninferiority margin of 10%, and a power of 90%. Such a study would require ∼672 evaluable cases (336/arm). The problem is that the rate of positive cultures for P. aeruginosa is relatively low—rates of 22% for nosocomial pneumonia, 11% for complicated intra-abdominal infection, and 3% for complicated urinary tract infection are typical for recent trials.8–11 If evaluable cases require a positive culture for this organism, then from 3064 (nosocomial pneumonia) to 22,466 subjects are required to be enrolled. That even the smallest of these goals is not feasible is demonstrated by a recent nosocomial pneumonia trial that took nearly 5 years to enroll ∼1200 patients.12
To address this challenge, new ways of working will be required. As one possible approach, Alemayehu and colleagues called in 2012 for a move toward a graduated approval process.13 As a further example of innovative approaches to this problem, we recently worked as part of a cross-industry group to propose a comprehensive regulatory framework based on four tiers and a totality of the evidence approach (Fig. 1) in which there is greater reliance on preclinical data and the combination of animal and human pharmacokinetic and pharmacodynamic (PK–PD) data that are tightly integrated with a focused clinical program.14 In this model, Tier A is the traditional approach of two large phase III studies and Tier D encompasses the so-called “animal rule” used for advance registration for agents for biothreat pathogens where the current number of human cases is zero.
Figure 1.
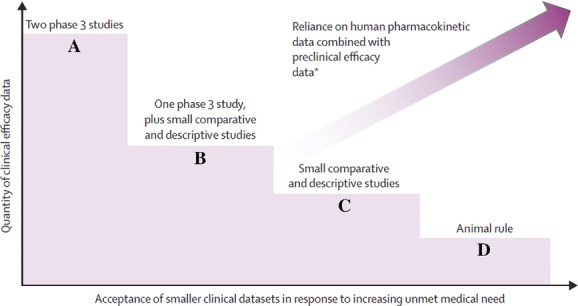
Overview of the tiered regulatory framework. Figure reprinted with permission from Ref. 14.
Tiers B and C provide intermediate steps between Tiers A and D (Fig. 1 and Table 1). Tier B is well suited to agents with a reasonably broad spectrum of pathogen coverage and provides for registration based on a single phase III trial plus small pathogen-focused studies. Tier C is well suited for agents with a narrow spectrum of activity and is based solely on small pathogen-focused studies.
Table 1.
Examples of drugs well suited for Tier B versus C development
| Attribute | Tier B | Tier C |
|---|---|---|
| Example spectrum | Broad with MDR pathogen coverage | Narrow MDR pathogen coverage |
| Example target pathogen | MDR Enterobacteriaceae (also covers if non-MDR) | Pseudomonas aeruginosa only |
| Challenge in studying the MDR pathogen in large numbers? | Yes | Yes |
| Detailed insight into: | ||
| Microbiology including mechanism of action and resistance? | Yes | Yes |
| Animal models that mimic human disease? | Yes | Yes |
| Exposure response in animals? | Yes | Yes |
| Detailed PK–PD justification of dose selection in humansa | Yes | Yes |
| Can do “standard” P3 study versus susceptible organisms? | Yesb | No |
| Randomized comparative data generated? | Yes (single body site, vs. standard comparator) | Yes (multiple body sites, vs. BATc) |
| Able to do “usual strength” statistical inference testing? | Yes, but only in the standard P3 study | No |
| Pooling of data across infection sites proposed? | Yes | Yes |
| Reliance on a totality-of-evidence approach?d | High | Even higher |
MDR, multidrug resistant.
The mechanism of action is understood, animal models are available that reasonably mimic human disease at relevant sites, an exposure–response relationship in the animal studies informs human dose with an adequate safety margin, and PK is known in healthy volunteers and relevant patient groups.
This provides relevant efficacy data if MDR pathogens have same susceptibility to a new agent as do non-MDR pathogens.
BAT, best available therapy, standardized insofar as possible.
All drug reviews consider the totality of evidence, but the reliance on such things as PK–PD predictions and pooled responses across sites will be very high here.
Development under any of the tiers begins with a detailed exploration of the PK–PD properties of the new agent.15–17 Recent regulatory approvals across all categories of agents have increasingly incorporated PK–PD insights.18 For antibacterial agents developed under Tiers B–D, this work takes on additional significance, as it will provide a key efficacy argument for the overall registration package. Instead of using two phase III studies to provide empiric evidence of causality,19 preclinical work must provide clarity on the mechanism of action (MOA) plus PK–PD data showing strong evidence that a proper dosage regimen has been selected. This PK–PD package should include data showing (1) a clear understanding of the microbiologic mode of action, presence or lack of cross-resistance with other agents, and appropriate methods of in vitro susceptibility testing; (2) the exposure curve(s) associated with response in several animal models of infection; (3) the presence of a consistent exposure-minimal inhibitory concentration (MIC) response relationship for the agent; and (4) that the selected dosage regimen produces in patients the exposure required to be reasonably likely to be active.
The last of these requirements is noteworthy—the argument needs to rest on PK data from patients with the target syndrome (or with metabolic disturbances similar to those of patients with the target syndrome) plus a demonstration that most (usually taken as ≥∼90%) of subjects will reliably achieve that exposure relative to the expected range of MICs. The data supporting this argument would follow from progressively building a full PK model based on experience with the agent, first in healthy volunteers and then in patients from phase II and III studies. The overall goal is to ensure a good understanding of the covariates influencing exposure.
With the importance of the PK–PD package understood, Tiers B and C can be usefully compared (Table 1). A hypothetical Tier B program is built around two core studies. The first study would be a single standard phase III noninferiority study at a single body site versus a standard comparator. Because of the use of a standard comparator, this study is not expected to enroll any multidrug-resistant (MDR) pathogens, and indeed study procedures must include active steps to avoid enrolling subjects infected with pathogens likely resistant to the comparator. The value of this study lies rather in using the broad activity of the Tier B agent to permit simple implementation of a standard trial that has low technical risk. By providing data on the responses of seriously ill patients infected with susceptible strains of the organism, important insight is gained into the safety and efficacy of the agent at the tested dosage(s). If the test agent shows good efficacy and also lacks cross-resistance with other agents, then it is entirely appropriate to predict therapeutic response when the agent is used against MDR pathogens that retain susceptibility to the new agent. The second Tier B study would be a small study focused on MDR pathogens that are susceptible to the Tier B agent. Optimally, this study would compare randomization versus the best available therapy (BAT).
By comparison, a Tier C program is built around a single small study focused on MDR pathogens that are susceptible to the Tier C agent. Even though it is small and not powered for standard inferential statistical testing, this study gains substantial value if it is possible for it to be a randomized study of the new agent versus the BAT.
Both Tier B and C programs could usefully be supported by the provision of an open-label salvage study that accrues patients for whom there is no BAT.
Finally, both programs rely heavily on a totality-of-evidence approach plus a willingness to attempt to pool data across multiple body sites.
The role of observational control data, whether collected retrospectively or prospectively, has been debated at some length. We originally proposed inclusion of such data in a registration program, but such data have many limitations—most notably, it is very difficult to demonstrate comparability of the external control cohort to those subjects in the prospective Tier B or Tier C studies. Approaches to using nonrandomized data have been described,20–22 but these approaches all require assumptions that may not be universally supported. A full solution to this problem has not yet emerged and will require further work. If at all possible, it is desirable to attempt to provide the small but randomized comparison from the MDR-focused study that is at the heart of the Tier B and C types of programs.
Strengths
These approaches should provide substantial flexibility for developers and enable the design of programs that might otherwise never even be attempted. Enthusiasm for these approaches has been high to date, and we have been encouraged by the number of new entrants into the antimicrobial development arena.23
These approaches are also consistent with the steadily increasing power of the newer diagnostic tools to quickly and accurately identify the causative pathogen. As discussed in a recent policy paper from the IDSA,24 the availability of improved diagnostic tests should improve health care and facilitate good antibiotic stewardship.
Limitations
There are, however, a number of limitations that must be recognized by all parties. Due to the small size of these data sets, significant heterogeneity of the patient populations is almost certain to be seen. This, in turn, could lead to the appearance of safety or efficacy signals that are positive or negative with regard to the new agent. Typical subset analyses (e.g., response by region of the world, response by age of subjects) will result in very small groups that have even higher risks of confounding results. There is no specific remedy for this issue other than to focus analyses on the totality of the evidence.
On a related theme, the total size of the safety database will be relatively small with these programs. The required size of the safety database will vary with the presence or absence of safety signals in the clinical program and will also need to be balanced against the level of unmet need.
Enrollment into the key randomized MDR pathogen studies can be surprisingly difficult. MDR pathogens appear in clusters, and sites that have had problems with such pathogens are often appropriately engaged in active infection-control processes that reduce case rates even as a trial is initiated. Furthermore, these patients tend to be relatively complex, and recent experience has suggested that challenges with both identifying a suitable BAT regimen and obtaining informed consent can reduce accrual rates.
The issue of the appropriate use of new and existing antibiotics often comes into discussion at the time of introduction of a new agent. It is important to acknowledge this challenge, but also to realize that resolving the challenge requires a perspective much broader than that of the approval process. Full resolution of the challenge of appropriate use will require a coordinated approach to the entire antibiotic business model.
The challenge of superiority: great when it works, but not a reliable path for the long term
As an alternative to the above approaches, the idea of registration based on superiority designs is often suggested.25–27 Although it might be the case that some measure of superiority might be shown versus external historical controls, such comparisons have limitations, as discussed above, in that it may be difficult to show comparability of those subjects to those treated with the test agent.
With respect to demonstration of superiority in a prospective, randomized study, it is important to realize that design features of such studies (most notably, that control patients must receive a therapy expected to be active) make superiority unlikely except in exceptional circumstances. Here, we agree with the recent statement by the European Medicines Agency (EMA)28 that “it is not expected to be feasible to demonstrate superiority for the new agent over BAT based on the usual endpoints that would be applied to each type of infection.”
Although it might currently be possible to envision superiority on secondary endpoints such as relative tolerability (e.g., colistin may be needed as part of current comparison regimens and its nephrotoxicity might provide an avenue to a demonstration of superiority), this approach will become decreasingly feasible as new agents are developed. It is thus important that a reliable and feasible pathway is available that does not require demonstration of superiority but that focuses instead on noninferiority-based approaches (even if limited in size and not powered for inferential testing) that do not require the new agent's activity to be greater than that of a previously approved therapy. It is only by providing such reliable noninferiority-based approaches that we can ensure that we have multiple therapeutic options with different MOAs, routes of elimination, and toxicity profiles.
A further challenge is that current debates on the validity of endpoints that appear to involve physician judgment have led to criticism of more traditional endpoints such as overall response at test of cure.29 Differences in opinion on what constitutes of a useful endpoint have strong advocates on both sides of the debate.28,30
One possible approach to at least a partial demonstration of superiority or validation of novel endpoints might be found in post hoc pharmacometric analyses of clinical trial data.31 When adequate information is available from study subjects to permit estimation of likely drug exposure in relation to isolated MIC, the resulting analysis of the relationship of the key PD exposure MIC variable versus response may permit demonstration of a within-study trend based on the sometimes large dynamic range of the exposure MIC variable (Fig. 2). This is tantamount to a dose–response study and can provide powerful support for efficacy or a novel endpoint.
Figure 2.
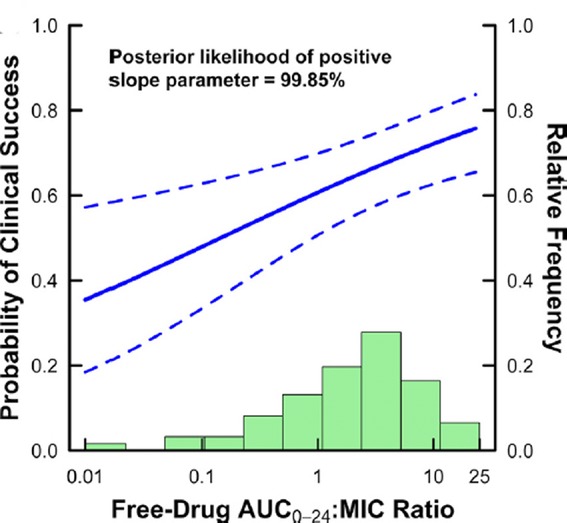
Pharmacometric approach to estimating the placebo effect size. Shown is an approach to estimating the size of the placebo treatment effect from actual clinical trial data. In the analysis, clinical success is analyzed versus observed isolate MIC and estimated drug exposure measured as 24-h area under the curve (AUC0–24). The right-hand y-axis corresponds to the frequency histogram of observed AUC0–24/MIC ratios. The left-hand y-axis shows observed clinical response as an estimate logistic regression function (solid line) and its 95% confidence bounds (dotted lines). The placebo treatment effect size can be estimated by examining the success rate when the AUC0–24/MIC approaches zero. In this case, the success rate with placebo is estimated at slightly less than 40%. Figure reprinted with permission from Ref. 31.
Regulatory status
The EMA released their updated thinking on this topic in a document dated October 24, 2013.28 This document provides a section focused on circumstances in which limited clinical data may be accepted and describes pathways that are effectively identical to the Tier B and C concepts discussed above. The document even proposes that a Tier C–like approach built entirely around external controls could be envisioned in extreme circumstances. The guidance indicates that agency views are still evolving, but suggests possible indication wording along these lines for agents studied in small, comparative MDR-focused studies: (1) “For the treatment of infections due to (genus or species) in patients with limited treatment options,” and (2) “For the treatment of infections due to (some types of) pathogens in patients with limited treatment options.”
In addition, it suggests that Section 4.2 of the Summary of Product Characteristics would contain the statement: “It is recommended that [agent name] should be used to treat patients that have limited treatment options only after consultation with a physician with appropriate experience in the management of infectious diseases.”
The EMA addendum also addresses the use of the data from the standard phase III study in a Tier B–like program. If such a study is done, standard indication wording (e.g., “Drug X is indicated for the treatment [in adults or other specific population] with [name of standard body site infection]” could result. Granting of this wording requires that (1) the development program has included a standard study of the test agent for infections at a single body site, (2) the study has used the usual noninferiority margin recommended for the type of infection under study and level of alpha, (3) noninferiority is convincingly demonstrated in that study for the test product compared to the active comparator, and (4) the indication for use in patients with limited therapeutic options is considered to be soundly supported.
Current thinking from the U.S. Food and Drug Administration (FDA) was summarized in the July 2013 release of a draft guidance document entitled “Antibacterial therapies for patients with unmet medical need for the treatment of serious bacterial diseases.”26 Structured in a question-and-answer format, the document provides outlines for several development programs, but these all hinge significantly on the presumption that some demonstration of superiority will be possible. One example includes the idea of using different statistical parameters in noninferiority-based programs, but it is not clear from the test that meeting noninferiority criteria alone would be adequate for registration in such a situation. However, the document also signals significant flexibility from the agency, ongoing evolution of agency thinking, and a willingness to entertain further discussion. Product-specific contacts with the agency have suggested that the FDA is willing to support the concepts underpinning the ideas of Tiers B and C as endorsed by the EMA.
The FDA document suggests that label wording for agents approved using programs built around these designs might be as follows: “Drug X is indicated, in [approved patient population], for the treatment of [HABP/VABP, cIAI, ABSSSI, CABP, cUTI (include as appropriate)] caused by the following susceptible microorganism(s): [list the genus and species of the bacterial pathogen(s)]. Drug X has been approved for use in patients with [HABP/VABP, cIAI, ABSSSI, CABP, cUTI (include as appropriate)] where limited or no alternative therapies are available. The safety and effectiveness of Drug X have not been established beyond this patient population. This indication is based on (summarize the limitations of available data that supported the approval).” This proposed labeling concept implies a willingness to consider data from multiple body sites and to use the labeling information as way to describe the limitations of the data to the prescriber.
Issues requiring further work
Interpretive breakpoints
Just as PK–PD data can be used to provide important, supportive information for new antibiotic approval, it can be harnessed to tackle another challenging area involved with antibiotic development, approval, and use: interpretative susceptibility breakpoints.32 Over the past 25 years, PK–PD methodologies have evolved to allow for generation of sound scientific information that can aid in our understanding of the relationship between drug exposure and effect. Of particular importance, PK–PD data can provide additional insight in cases of resistant pathogens, which are often excluded from traditional registration trials, and organisms with MICs at the edge of the MIC distribution curve, which are almost always underrepresented.
In 2009, the FDA provided a draft guidance to industry for the development, analysis, and presentation of microbiologic data for new systemic antibiotics, and described how provisional breakpoints are established using PK–PD data before initiation of phase III clinical trials.33 An organism is considered susceptible if the compound reaches sufficient concentration at the infection site and is likely to inhibit growth, whereas an organism is considered resistant when the concentration a compound usually achieves at the site is not likely to inhibit growth. Finally, an organism is considered intermediate in situations where a higher drug dosage can be used and/or is physically concentrated at the relevant body site. In recent reviews, both U.S.34 and E.U. authors35 have extensively discussed the rationale and the approaches taken.
The preference in the past has been to use clinical data from the phase III trials to set the final interpretative breakpoints. The breakpoint has typically been only as high as the highest MIC isolate recovered from a successfully treated patient in the trial. All organisms with MICs beyond this upper edge are considered nonsusceptible, a category which clinicians generally interpret as being equivalent to being in the resistant category. This approach is conservative in that it prevents the occurrence of the “very major error” category of interpretive errors in which the clinical microbiology laboratory (based on the MIC determination of the isolated pathogen) reports that the antibiotic is effective when it actually is not.
However, the challenge is that very few patients in a trial have MICs at the upper edge of the MIC distribution curve, and the rarity of high MIC isolates will increase as trial programs shrink. Thus, this approach has the corresponding weakness of causing “major errors” (incorrectly identifying a susceptible pathogen as resistant) to occur because as noted the category “nonsusceptible” is in practice interpreted as “resistant.” Unfortunately, at a time of increasing antibiotic resistance and fewer new antibiotics available in the clinic, society ends up having possible highly effective therapeutic options unavailable at the very time we need them the most.
To address these concerns, susceptibility breakpoints should be set based on a combination of clinical and PK–PD data. Furthermore, susceptibility breakpoints should be considered provisional and be updated as insights accrue. Thus, breakpoints for a number of the cephalosporin antibiotics, established years before extended spectrum β-lactamases were recognized and became widespread clinically, were lowered by the Clinical and Laboratory Standards Institute (CLSI) and the FDA.36,37 Finally, use of the interpretive category “susceptible dose-dependent” may facilitate communication to the provider of the importance of the use of the maximal drug dosage in settings of reduced susceptibility that fall short of complete resistance.38
On-label versus scientifically (medically) appropriate use
The approved labeling of a prescription drug is the primary mechanism through which regulatory agencies and drug manufacturers communicate essential, science-based information to healthcare professionals. As an example of this, the FDA notes that “the primary purpose of the package insert is to provide practitioners with the essential information they need to prescribe the drug safely and effectively for the care of patients.”39 Thus, it is critical to include in the label a complete and accurate explanation of a prescription drug to facilitate safe and effective prescribing.
In the case of antibacterial agents, the need to include all available and scientifically valid data about a product is even more critical, as important clinical situations often do not appear (nor may they ever appear) in the “Indications and usage” section of the label. For example, infections due to resistant or less common pathogens might occur in settings that can never be studied outside very isolated cases. Alternatively, a health-care professional might be confronted with a patient with an infection at a less-studied body site. In these situations, physicians must make an educated judgment regarding whether or not technically off-label usage is appropriate in the particular situation. To do this, the health-care provider must rely on available data, their clinical experience with the drug, and the principles of infectious disease in general (including the utility of preclinical testing to predict antibiotic effects at body sites) in order to make informed judgments in their practice of medicine. In the United States, it has been determined that the FDA must not interfere with such judgment. See, for example, Weaver v. Reagen, 886 F.2d 194, 198–99 (8th Cir. 1989), which concluded, “FDA[-]approved indications were not intended to limit or interfere with the practice of medicine nor to preclude physicians from using their best judgment in the interest of the patient.” A supporting example is found in Proposed New Drug, Antibiotic, and Biologic Drug Regulations, 48 Fed. Reg. 26720, 26733 (proposed June 9, 1983) which states, “Once a drug product has been approved for marketing, a physician may … prescribe the drug for uses not included in the drug's approved labeling.” In fact, United States v. Caronia, 703 F.3d 149 (2nd Cir. 2012) notes that courts and the FDA have long “recognized the proprietary and potential public value of unapproved or off-label drug use.” The importance of off-label use is further reinforced in an FDA draft guidance that states that “[o]ff-label uses or treatment regimens may be important and may even constitute a medically recognized standard of care.”40
To support this type of informed, optimal clinical decision making, the label of an antibacterial agent should thus include all available data (PK, PD, clinical data, etc.), as well as describe any limitations of those data. Such information contributes to the choice of an appropriate antibiotic in a particular setting and permits the treating physician to develop additional insights about the uses of a given new agent. This approach also informs physicians about potential inappropriate use of an agent (e.g., with data for lack of penetration to a given site) and use in situations with limited data (e.g., less-studied sites of infection).
Agency concerns that including all available data in the label could mislead treating physicians about appropriate treatment may be mitigated by clearly communicating areas of residual uncertainty, perhaps by using a disclaimer. For example, current FDA labeling regulations regarding inclusion of certain in vitro data for anti-infective drugs require that such data be preceded by a disclaimer concerning their clinical significance (21 US Code of Federal Regulations Section 201.57(c)(13)(ii)(A)). A similar approach may be appropriate for other types of data. Language describing the data would be carefully negotiated between the agency and the sponsor, as is the rest of the labeling. Finally, promotion of the drug would be regulated following the usual models so that these additional data do not form a basis for or unduly influence promotional claims.
The need for multiple agents to be progressed
To prevent antimicrobial resistance from repeatedly becoming a major public health threat after each new antibiotic launch, it is important that we have a diverse, vibrant, and sustained pipeline of new agents. The IDSA has even made this part of their call to arms, calling for 10 new agents by 2020 in their “10 × 20” campaign (http://www.idsociety.org/10x20/).
A tension arises when one or more new agents have been approved. As both the documents from the EMA28 and the FDA26 focus on facilitating the development of new agents via smaller programs when there is an unmet need, does the approval of one or two new agents eliminate that unmet need?
We do not think so, and both agencies acknowledge this in their commentary. The EMA comments that “second (or sequential) agent that may address the same types of pathogens could also be considered for an initial approval based on limited data taking into account that having a choice of agents available has several obvious benefits.”28 The FDA gives an example of one of those benefits when it notes that “The approval of more than one therapy addresses an emerging or anticipated public health need, such as a drug shortage or the development of antimicrobial resistance.”26
Reimbursement
Although the societal value of antibiotics has been estimated to be in the range of $60–300 trillion for the United States alone,41 antibiotic developers face a well-described set of difficult economic challenges.5,42 As a specific example, Sharma and Towse modeled the entire antibiotic lifecycle for an average new agent, estimating the net present value (NPV) of this work at –€38m on average.42 A significant amount of the negative return is the result of the way that value is eroded by the impact of time discounting over the lengthy life cycle (33 years in the Sharma–Towse model) of a typical drug. Understanding this is critical when understanding the limited impact of so-called pull incentives. In the modeling of Sharma and Towse, it was not found to be possible for such approaches to move the model into positive territory. A subsequent paper explored this in some detail and demonstrated that push incentives (funding provided early in the R&D cycle) could be as much as 95% smaller than pull incentives with comparable economic effects.43
A variety of approaches to antibiotic business models are now being discussed. At the most fundamental level, these discussions all focus on the challenge of attracting the kind of long-term capital and patient investors required to successfully rebuild the global pipeline of antibacterial agents. All companies face pressure from shareholders. However, an important comparison for future investment in antibiotics in the years ahead will be whether companies investing in this area will be able to command the kinds of significant financial returns that are experienced with some biotechnology companies focused on orphan disorders, selected forms of cancer, multiple sclerosis, and other infectious diseases, such as hepatitis C. In this light, the possibility of premium pricing for well-targeted antibacterial agents is one often-discussed example. Given the increasing availability of diagnostic tools that enable earlier and more specific identification of the infecting pathogen, premium pricing may make sense to both the payer and the drug sponsor. As a specific study of this, Spellberg and Rex44 modeled the value of a narrow-spectrum agent focused on resistant Acinetobacter. Using a QALY (quality-adjusted life-year) approach and a cost per course of therapy of $10,000, the model estimated that net costs per life year saved were $1908 in the United States and $6319 globally, with costs per QALY estimated at $3180 in the United States and $10,531 globally. These estimates were robust in a variety of sensitivity analyses, with costs per QALY remaining below typical benchmark values of $50,000/QALY even when the price per course of therapy approached $30,000.
Significant further work is required in this area. Key questions to be resolved include definitions of appropriate antibiotic stewardship (we lack a common language for these ideas and we also lack ready measures of progress toward the ill-defined goal of better stewardship), best approaches to implementing stewardship on a broad scale (e.g., what lessons have been learned from the implementation in 2010 of such a program in California for general acute care hospitals (http://www.cdph.ca.gov/programs/hai/Pages/AntimicrobialStewardshipProgramInitiative.aspx)), and how government agencies in the United States and across Europe can help stimulate this important market (e.g., exemptions from European health technology assessments and support of an expansion of Centers for Medicare & Medicaid Services (CMS) add-on payment for antibiotics). In addition, it may be possible for governments and other stakeholders to find ways to dissociate antibiotic utilization (or sales volume) from reward to innovators. The interwoven nature of these challenges has led to a recent proposal as part of IMI Call 9 that this area be approached via a multistakeholder consortium within the ND4BB project (http://www.imi.europa.eu/content/9th-call-2013). This project is currently entitled “Driving reinvestment in R&D and responsible use of antimicrobials” and is anticipated to launch during 2014.
Conclusions
The threat of antimicrobial resistance and the incredibly thin global development pipeline is an issue for us all. It is encouraging to see the significant attention being paid to this area and the changes that are underway to easy the challenges of discovery, development, and economics. We are far from having a resolution to the current gap in our therapeutic armamentarium, but we agree with the recent observation by Pucci and Bush23 that “there is at least some hope that we may be able to tackle at least some of our current problems with new agents in the current pipeline.”
Acknowledgments
The authors gratefully acknowledge and are thankful for conversations over time both with their coauthors on Reference 14 (Jeff Alder, Robert Meyer, Aaron Dane, Ian Friedland, Charles Knirsch, Wendy R. Sanhai, John Tomayko, Cindy Lancaster, and Jennifer Jackson) and leadership at both the FDA and the EMA.
Conflicts of interest
The authors are employees of their respective pharmaceutical companies. In addition, JHR is a non-executive director of, shareholder of, and consultant to F2G Pharmaceuticals and also a consultant to Advent Life Sciences, an investor in F2G Pharmaceuticals.
References
- 1.Anonymous. US Dept HHS, Centers for Disease Control and Prevention. Antibiotic resistance threats in the United States, 2013. 2013. http://www.cdc.gov/drugresistance/threat-report-2013/. Accessed April 13, 2014.
- 2.Anonymous. The bacterial challenge: time to reach. European CDC/European Medicines Agency Joint Technical Report. 2009. http://www.ecdc.europa.eu/en/publications/_layouts/forms/Publication_DispForm.aspx?ID=199&List=4f55ad51-4aed-4d32-b960-af70113dbb90. Accessed April 13, 2014.
- 3.Boucher HW, Talbot GH, Benjamin DK, Jr, et al. 10 x ‘20 progress–development of new drugs active against Gram-negative bacilli: an update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2013;56:1685–1694. doi: 10.1093/cid/cit152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Outterson K, Powers JH, Seoane-Vazquez E, et al. Approval and withdrawal of new antibiotics and other antiinfectives in the U.S., 1980–2009. J. Law Med. Ethics. 2013;41:688–696. doi: 10.1111/jlme.12079. [DOI] [PubMed] [Google Scholar]
- 5.Morel CM. Mossialos E. Stoking the antibiotic pipeline. BMJ. 2010;340:1115–1118. doi: 10.1136/bmj.c2115. [DOI] [PubMed] [Google Scholar]
- 6.Spellberg B, Blaser M, Guidos RJ, et al. Combating antimicrobial resistance: policy recommendations to save lives. Clin. Infect. Dis. 2011;52(Suppl 5):S397–428. doi: 10.1093/cid/cir153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Projan SJ. Why is big Pharma getting out of antibacterial drug discovery? Curr. Opin. Microbiol. 2003;6:427–430. doi: 10.1016/j.mib.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 8.Chastre J, Wunderink R, Prokocimer P, et al. Efficacy and safety of intravenous infusion of doripenem versus imipenem in ventilator-associated pneumonia: a multicenter, randomized study. Crit. Care Med. 2008;36:1089–1096. doi: 10.1097/CCM.0b013e3181691b99. [DOI] [PubMed] [Google Scholar]
- 9.Brun-Buisson C, Sollet JP, Schweich H, et al. Treatment of ventilator-associated pneumonia with piperacillin-tazobactam/amikacin versus ceftazidime/amikacin: a multicenter, randomized controlled trial. Clin. Infect. Dis. 1998;26:346–354. doi: 10.1086/516294. [DOI] [PubMed] [Google Scholar]
- 10.Lucasti C, Jasovich A, Umeh O, et al. Efficacy and tolerability of IV doripenem versus meropenem in adults with complicated intra-abdominal infection: a phase III, prospective, multicenter, randomized, double-blind, noninferiority study. Clin. Ther. 2008;30:868–883. doi: 10.1016/j.clinthera.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 11.Naber KG, Llorens L, Kaniga K, et al. Intravenous doripenem at 500 milligrams versus levofloxacin at 250 milligrams, with an option to switch to oral therapy, for treatment of complicated lower urinary tract infection and pyelonephritis. Antimicrob. Agents Chemother. 2009;53:3782–3792. doi: 10.1128/AAC.00837-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wunderink RG, Niederman MS, Kollef MH, et al. Linezolid in methicillin-resistant Staphylococcus aureus nosocomial pneumonia: a randomized, controlled study. Clin. Infect. Dis. 2012;54:621–629. doi: 10.1093/cid/cir895. [DOI] [PubMed] [Google Scholar]
- 13.Alemayehu D, Quinn J, Cook J, et al. A paradigm shift in drug development for treatment of rare multidrug-resistant gram-negative pathogens. Clin. Infect. Dis. 2012;55:562–567. doi: 10.1093/cid/cis503. [DOI] [PubMed] [Google Scholar]
- 14.Rex JH, Eisenstein BI, Alder J, et al. A comprehensive regulatory framework to address the unmet need for new antibacterial treatments. Lancet Infect. Dis. 2013;13:269–275. doi: 10.1016/S1473-3099(12)70293-1. [DOI] [PubMed] [Google Scholar]
- 15.Ambrose PG, Bhavnani SM, Ellis-Grosse EJ. Drusano GL. Pharmacokinetic-pharmacodynamic considerations in the design of hospital-acquired or ventilator-associated bacterial pneumonia studies: look before you leap. Clin. Infect. Dis. 2010;51:S103–S110. doi: 10.1086/653057. [DOI] [PubMed] [Google Scholar]
- 16.Ambrose PG. Use of pharmacokinetics and pharmacodynamics in a failure analysis of community-acquired pneumonia: implications for future clinical trial study design. Clin. Infect. Dis. 2008;47:S225–S231. doi: 10.1086/591427. [DOI] [PubMed] [Google Scholar]
- 17.Drusano GL. Pharmacokinetics and pharmacodynamics of antimicrobials. Clin. Infect. Dis. 2007;45:S89–S95. doi: 10.1086/518137. [DOI] [PubMed] [Google Scholar]
- 18.Lee JY, Garnett CE, Gobburu JV, et al. Impact of pharmacometric analyses on new drug approval and labelling decisions: a review of 198 submissions between 2000 and 2008. Clin. Pharmacokinet. 2011;50:627–635. doi: 10.2165/11593210-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 19.Peck CC, Rubin DB. Sheiner LB. Hypothesis: a single clinical trial plus causal evidence of effectiveness is sufficient for drug approval. Clin. Pharmacol. Ther. 2003;73:481–490. doi: 10.1016/S0009-9236(03)00018-3. [DOI] [PubMed] [Google Scholar]
- 20.Shadish WR, Galindo R, Wong VC, et al. A randomized experiment comparing random and cutoff-based assignment. Psychol. Methods. 2011;16:179–191. doi: 10.1037/a0023345. [DOI] [PubMed] [Google Scholar]
- 21.Gamalo MA, Tiwari RC. Lavange LM. Bayesian approach to the design and analysis of non-inferiority trials for anti-infective products. Pharm Stat. 2014;13:25–40. doi: 10.1002/pst.1588. [DOI] [PubMed] [Google Scholar]
- 22.Viele K, Berry S, Neuenschwander B, et al. Use of historical control data for assessing treatment effects in clinical trials. Pharm Stat. 2014;13:41–54. doi: 10.1002/pst.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pucci MJ. Bush K. Investigational antimicrobial agents of 2013. Clin. Microbiol. Rev. 2013;26:792–821. doi: 10.1128/CMR.00033-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caliendo AM, Gilbert DN, Ginocchio CC, et al. Better tests, better care: improved diagnostics for infectious diseases. Clin. Infect. Dis. 2013;57(Suppl 3):S139–S170. doi: 10.1093/cid/cit578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Infectious Diseases Society of America. White paper: recommendations on the conduct of superiority and organism-specific clinical trials of antibacterial agents for the treatment of infections caused by drug-resistant bacterial pathogens. Clin. Infect. Dis. 2012;55:1031–1046. doi: 10.1093/cid/cis688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anonymous. Guidance for industry: antibacterial therapies for patients with unmet medical need for the treatment of serious bacterial diseases. 2013. US Dept HHS, FDA, CDER, CBER. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm. Accessed April 13, 2014.
- 27.Shlaes DM. Spellberg B. Overcoming the challenges to developing new antibiotics. Curr. Opin. Pharmacol. 2012;12:522–526. doi: 10.1016/j.coph.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 28.Committee for Medicinal Products for Human Use (CHMP) European Medicines Agency; 2013. Addendum to the guideline on the evaluation of medicinal products indicated for treatment of bacterial infections. EMA/CHMP/351889/2013. [Google Scholar]
- 29.Talbot GH, Powers JH, Fleming TR, et al. Progress on developing endpoints for registrational clinical trials of community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections: update from the Biomarkers Consortium of the Foundation for the National Institutes of Health. Clin. Infect. Dis. 2012;55:1114–1121. doi: 10.1093/cid/cis566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Powers JH. Editorial commentary: asking the right questions: morbidity, mortality, and measuring what's important in unbiased evaluations of antimicrobials. Clin. Infect. Dis. 2012;54:1710–1713. doi: 10.1093/cid/cis274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ambrose PG, Hammel JP, Bhavnani SM, et al. Frequentist and Bayesian pharmacometric-based approaches to facilitate critically needed new antibiotic development: overcoming lies, damn lies, and statistics. Antimicrob. Agents Chemother. 2012;56:1466–1470. doi: 10.1128/AAC.01743-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dudley MN. Ambrose PG. Pharmacodynamics in the study of drug resistance and establishing in vitro susceptibility breakpoints: ready for prime time. Curr. Opin. Microbiol. 2000;3:515–521. doi: 10.1016/s1369-5274(00)00132-6. [DOI] [PubMed] [Google Scholar]
- 33.Anonymous. Guidance for industry: microbiological data for systemic antibacterial drug products—development, analysis, and presentation. 2009. US Dept HHS, FDA, CDER and CBER, September. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm064980.htm.
- 34.Dudley MN, Ambrose PG. Jones RN. Commentary: revised susceptibility breakpoints: fear, loathing and good science. Pediatr. Infect. Dis. J. 2013;32:970–971. doi: 10.1097/INF.0b013e3182a1b8cd. [DOI] [PubMed] [Google Scholar]
- 35.Mouton JW, Brown DF, Apfalter P, et al. The role of pharmacokinetics/pharmacodynamics in setting clinical MIC breakpoints: the EUCAST approach. Clin. Microbiol. Infect. 2012;18:E37–45. doi: 10.1111/j.1469-0691.2011.03752.x. [DOI] [PubMed] [Google Scholar]
- 36.Turnidge JD. Cefazolin and enterobacteriaceae: rationale for revised susceptibility testing breakpoints. Clin. Infect. Dis. 2011;52:917–924. doi: 10.1093/cid/cir031. [DOI] [PubMed] [Google Scholar]
- 37.Dudley MN, Ambrose PG, Bhavnani SM, et al. Background and rationale for revised clinical and laboratory standards institute interpretive criteria (Breakpoints) for Enterobacteriaceae and Pseudomonas aeruginosa: I. Cephalosporins and Aztreonam. Clin. Infect. Dis. 2013;56:1301–1309. doi: 10.1093/cid/cit017. [DOI] [PubMed] [Google Scholar]
- 38.Rex JH, Pfaller MA, Galgiani JN, et al. Development of interpretive breakpoints for antifungal susceptibility testing: conceptual framework and analysis of in vitro-in vivo correlation data for fluconazole, itraconazole, and Candida infections. Clin. Infect. Dis. 1997;24:235–247. doi: 10.1093/clinids/24.2.235. [DOI] [PubMed] [Google Scholar]
- 39.Anonymous. 65 Fed. Reg. 81082, 81083. 2000. US Dept HHS, FDA.
- 40.Anonymous. Good reprint practices for the distribution of medical journal articles and medical or scientific reference publications on unapproved new uses of approved drugs and approved or cleared medical devices. 2009. US Dept HHS, FDA, CDER, CBER. http://www.fda.gov/regulatoryinformation/guidances/ucm125126.htm Accessed April 13, 2014.
- 41.Hollis A. Ahmed Z. Preserving antibiotics, rationally. N. Engl. J. Med. 2013;369:2474–2476. doi: 10.1056/NEJMp1311479. [DOI] [PubMed] [Google Scholar]
- 42.Sharma P. Towse A. New drugs to tackle antimicrobial resistance. London: Office of Health Economics; 2010. http://www.ohe.org/publications/article/new-drugs-to-tackle-antimicrobial-resistance-analysis-of-eu-policy-options-21.cfm. Accessed April 13, 2014. [Google Scholar]
- 43.Spellberg B, Sharma P. Rex JH. The critical impact of time discounting on economic incentives to overcome the antibiotic market failure. Nat. Rev. Drug Discov. 2012;11:168. doi: 10.1038/nrd3560-c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spellberg B. Rex JH. The value of single-pathogen antibacterial agents. Nat. Rev. Drug Discov. 2013;12:963. doi: 10.1038/nrd3957-c1. [DOI] [PMC free article] [PubMed] [Google Scholar]