

Abstract
Inferring invasion routes and identifying reservoirs of diversity of plant pathogens are essential in proposing new strategies for their control. Magnaporthe oryzae, the fungus responsible for rice blast disease, has invaded all rice growing areas. Virulent genotypes regularly (re)emerge, causing rapid resistance breakdowns. However, the world-wide genetic subdivision of M. oryzae populations on rice and its past history of invasion have never been elucidated.
In order to investigate the centers of diversity, origin and migration of M. oryzae on rice, we analyzed the genetic diversity of 55 populations from 15 countries.
Three genetic clusters were identified world-wide. Asia was the center of diversity and the origin of most migrations to other continents. In Asia, two centers of diversity were revealed in the Himalayan foothills: South China–Laos–North Thailand, and western Nepal. Sexual reproduction persisted only in the South China–Laos–North Thailand region, which was identified as the putative center of origin of all M. oryzae populations on rice.
Our results suggest a scenario of early evolution of M. oryzae on rice that matches the past history of rice domestication. This study confirms that crop domestication may have considerable influence on the pestification process of natural enemies.
Keywords: center of origin, domestication, Magnaporthe, migration, population, rice
Introduction
Increasing globalization of trade and climatic changes enhance the probability of (re-) emergence of invasive pests. These latter are usually difficult to manage without using pesticides, and represent unprecedented economic and food safety risks (Estoup & Guillemaud, 2010). Understanding how past events shaped the contemporary genetic diversity of pathogen populations helps in predicting their future change (Lawson Handley et al., 2011). Pathogen species of domesticated organisms have likely adapted to their human-disturbed environments during the domestication process, enabling them to invade new habitats with similar characteristics (Lee & Gelembiuk, 2008; Hufbauer et al., 2012). The homogenization of cultivated landscapes over different geographical areas has enhanced the invasion capacity of crop pathogens, by minimizing the magnitude of the evolutionary response required to adapt to new environments (Stukenbrock & McDonald, 2008; Estoup & Guillemaud, 2010; Guillemaud et al., 2011). Plant pathogenic fungi, which represent major threats for several crops (Fisher et al., 2012), are outstanding examples of pests whose evolutionary potential has been shaped by ‘Anthropogenically induced adaptation to invade’ (Hufbauer et al., 2012). During the last decade, this has been exemplified for several emergent or re-emergent fungal pathogens (Banke & McDonald, 2005; Brunner et al., 2007; Gomez-Alpizar et al., 2007; Stukenbrock et al., 2007; Gladieux et al., 2008; Stukenbrock & McDonald, 2008).
Magnaporthe oryzae (Mo) is the Ascomycete fungus responsible for the most damaging rice disease world-wide: blast. This model species for the study of host–pathogen interactions (Valent, 1990; Dean et al., 2012) is a major threat to food security (Pennisi, 2010). Disease control is mainly genetic, but complete resistance genes of rice varieties are frequently overcome following the emergence of virulent blast strains. The molecular mechanisms of virulence acquisition have been documented (Dai et al., 2010; Takahashi et al., 2010; Chuma et al., 2011; Kanzaki et al., 2012; Cesari et al., 2013), but how such strains emerge and spread among and between populations remains misunderstood. Rice (Oryza sativa; Os), one of the host plants of Mo, probably originated from two independent domestication events involving wild rice O. rufipogon c. 7000 yr bp. These events occurred in two different regions that represent centers of diversification of cultivated rice, and resulted in two subspecies: Os ssp. japonica domesticated in southern China (Cheng et al., 2003; Londo et al., 2006; Fuller et al., 2009; Huang et al., 2012) and Os ssp. indica domesticated south of the Himalayas (likely eastern India, Myanmar or Thailand). However, the early history of Mo on cultivated rice, and especially the impact of rice domestication on Mo pestification, is still debated. How far human-mediated movements of infected materials are involved in inter-continental migration, especially between Asia and other continents, has seldom been investigated (Tharreau et al., 2009). Previous studies on blast populations give fragmentary information regarding these key questions. A single origin of Mo on cultivated rice has been suggested due to a single acquisition of pathogenicity (Shull & Hamer, 1994), possibly following a host shift from strains attacking foxtail millets (Setaria spp), probably in South China c. 10 000 yr ago where both rice and foxtail millet were domesticated and co-cultivated (Couch et al., 2005). While Mo reproduces asexually in most areas (Zeigler, 1998; Saleh et al., 2012), the presence of recombining populations is suspected in northeastern India (Zeigler, 1998; Kumar et al., 1999) and evidenced in Yunnan province, China (Saleh et al., 2012). Because sexual reproduction is believed to be ancestral in species that reproduce both sexually and asexually (Schurko & Logsdon, 2008), this geographic distribution also supports the hypothesis of Asia as the center of origin of the species. Finally, numerous studies have described the genetic diversity of Mo in different parts of the world using various genetic markers. Reviewing such studies, Zeigler (1998) concluded that the genetic diversity of Mo was higher in the area encompassing South, East and Southeast Asia than in other regions. More than 50 clonal lineages per country were characterized in India (Kumar et al., 1999), China (Chen et al., 2006) and Thailand (Zeigler, 1998), and 2–10 lineages were detected in Japan, (Don et al., 1999a), Korea (Park et al., 2003, 2008), the Philippines (Chen et al., 1995; Zeigler et al., 1995) and Vietnam (Don et al., 1999b). Outside of China, India and Thailand, by contrast, fewer (4–17) lineages were detected in Europe (Roumen et al., 1997; Piotti et al., 2005), Iran (Javan-Nikkah et al., 2004), USA (Levy et al., 1991; Xia et al., 1993, 2000; Correll et al., 2009), Argentina (Consolo et al., 2008), Colombia (Levy et al., 1993; Zeigler, 1998), Cuba (Fuentes et al., 2003) and West Africa (Takan et al., 2012). Besides these local studies, few attempts have been made to describe the population structure of Mo at a more global scale (Soubabère et al., 2000). In the most recent one, Tharreau et al. (2009) analyzed the genetic diversity of a world-wide collection of strains, and depicted a world-wide genetic structure of three clusters, Asian strains being scattered in the three clusters.
In sum these studies suggest that the origin of Mo strains pathogenic on rice may be in Asia and that most of the genetic diversity observed around the world is represented in this region. Thus, it is tempting to hypothesize that Asia may also be the center from which Mo dispersed towards the rest of the world. However, previous studies were limited to one country and were based on collections of strains maximizing diversity, not on population sampling. Testing these hypotheses requires population sampling covering native and secondary areas, coupled with analyses of population subdivision and genetic diversity without a priori on the population genetic structure. Such study – still lacking for Mo – is crucial to elucidate the routes and modalities of introduction, and would contribute to our understanding of how the pathogen emerged and spread, providing important clues for control methods to limit migrations of virulent strains and to improve the management of resistant varieties. Besides confirming the preliminary results of Tharreau et al. (2009) with appropriate population sampling and additional methods, the aim of the present work was therefore to address several questions about the origin, population structure and migration routes of world-wide populations of Mo. Using populations from different continents (Asia, Europe, the Americas and Africa), we asked: What are the main genetic groups in world-wide Mo populations? Can we localize one (or several) center(s) of genetic diversity? Based on the reproductive mode in the populations analyzed, can we infer the putative center of origin of the pathogen? Can we localize the geographic origin(s) of Mo migrations throughout the world?
Materials and Methods
Sampling
We used 55 world-wide population samples of Mo rice strains isolated between 2000 and 2009 (1372 strains in total; Table 1). A population was composed of strains collected in the same field on the same variety. In two cases (CH1 and MD1), different samples were collected at the same place but over two consecutive years; we grouped them as a single population after having verified that they were not genetically differentiated based on FST estimated from microsatellite markers. These 55 populations represented all continents (but without West African populations) with 423, 422, 136 and 391 strains from Asia, Europe, the Americas and Africa, respectively. Fungal strains were obtained after monospore isolation, as previously described by Silué & Nottéghem (1990) and stored as described by Valent et al. (1986).
Table 1.
Geographic origin and basic information on genetic diversity of 55 population samples of Magnaporthe oryzae
| Area | Country | Sample | N | Hn.b. | Na | Np | MLG | G : N |  |
|---|---|---|---|---|---|---|---|---|---|
| Asia | China | CH1 | 107 | 0.629 | 6.9 | 1.6 | 82 | 77% | 0.069 |
| – | – | CH2 | 38 | 0.522 | 3.4 | 0.2 | 21 | 55% | 0.292 |
| – | – | CH3 | 23 | 0.499 | 3.7 | 0.1 | 11 | 48% | 0.189 |
| – | – | CH4 | 25 | 0.051 | 1.3 | 0 | 4 | 16% | 0.209 |
| – | – | CH5 | 28 | 0.541 | 3.8 | 0 | 19 | 68% | 0.094 |
| – | – | CH6 | 30 | 0.253 | 2.6 | 0 | 7 | 23% | 0.368 |
| – | – | CH7 | 14 | 0.256 | 2.1 | 0 | 9 | 64% | 0.150 |
| – | Indonesia | ID1 | 20 | 0.277 | 2.3 | 0.1 | 10 | 50% | 0.135 |
| – | – | ID2 | 19 | 0.089 | 1.4 | 0.1 | 4 | 21% | 0.095 |
| – | – | ID3 | 16 | 0.114 | 1.7 | 0 | 6 | 38% | 0.053 |
| – | Laos | LA1 | 15 | 0.569 | 3.4 | 0.4 | 12 | 80% | 0.154 |
| – | – | LA2 | 9 | 0.518 | 3.5 | 0.2 | 8 | 89% | 0.073 |
| – | Nepal | NP1 | 31 | 0.168 | 2.6 | 0.1 | 11 | 35% | 0.295 |
| – | – | NP2 | 15 | 0.403 | 2.8 | 0.3 | 6 | 40% | 0.330 |
| – | – | NP3 | 6 | 0.491 | 2.4 | 0.1 | 4 | 67% | 0.651 |
| – | Thailand | TH | 27 | 0.460 | 3.6 | 0.1 | 18 | 67% | 0.166 |
| Europe/MB | France | FR1 | 23 | 0.124 | 1.7 | 0 | 9 | 39% | 0.050 |
| – | – | FR2 | 17 | 0.163 | 1.9 | 0 | 9 | 53% | −0.010 |
| – | – | FR3 | 18 | 0.112 | 1.6 | 0 | 6 | 33% | 0.113 |
| – | – | FR4 | 17 | 0.110 | 1.6 | 0 | 8 | 47% | 0.009 |
| – | – | FR5 | 22 | 0.019 | 1.2 | 0.1 | 3 | 14% | nd |
| – | – | FR6 | 37 | 0.317 | 2.3 | 0.1 | 8 | 22% | 0.398 |
| – | – | FR7 | 15 | 0.024 | 1.1 | 0 | 2 | 13% | nd |
| – | Greece | GR1 | 10 | 0.246 | 1.7 | 0 | 6 | 60% | 0.261 |
| – | – | GR2 | 10 | 0.183 | 1.8 | 0 | 6 | 60% | 0.144 |
| – | – | GR3 | 10 | 0.225 | 1.7 | 0 | 4 | 40% | 0.456 |
| – | – | GR4 | 9 | 0.102 | 1.5 | 0 | 3 | 33% | nd |
| – | – | GR5 | 9 | 0.167 | 1.6 | 0 | 3 | 33% | nd |
| – | – | GR6 | 10 | 0.190 | 2 | 0 | 4 | 40% | 0.544 |
| – | – | GR7 | 9 | 0.115 | 1.3 | 0 | 3 | 33% | nd |
| – | Hungary | HN1 | 7 | 0.070 | 1.2 | 0 | 3 | 43% | nd |
| – | – | HN2 | 3 | 0.053 | 1.1 | 0 | 2 | 67% | nd |
| – | – | HN3 | 7 | 0.101 | 1.3 | 0 | 4 | 57% | −0.055 |
| – | Morocco | MC | 15 | 0.169 | 1.9 | 0.1 | 8 | 53% | 0.097 |
| – | Spain | SP1 | 12 | 0.260 | 2 | 0 | 6 | 50% | 0.344 |
| – | – | SP2 | 31 | 0.204 | 2.1 | 0 | 11 | 35% | 0.245 |
| – | – | SP3 | 11 | 0.258 | 1.7 | 0 | 5 | 45% | 0.545 |
| – | – | SP4 | 22 | 0.278 | 2.1 | 0 | 9 | 41% | 0.399 |
| – | – | SP5 | 13 | 0.039 | 1.2 | 0 | 3 | 23% | nd |
| – | – | SP6 | 9 | 0.052 | 1.1 | 0 | 2 | 22% | nd |
| – | – | SP7 | 10 | 0.044 | 1.1 | 0 | 2 | 20% | nd |
| – | – | SP8 | 18 | 0.052 | 1.1 | 0 | 2 | 11% | nd |
| – | – | SP9 | 29 | 0.057 | 1.3 | 0.1 | 4 | 14% | −0.046 |
| – | Turkey | TR | 19 | 0.048 | 1.3 | 0 | 4 | 21% | −0.079 |
| Americas | Colombia | CL1 | 17 | 0.083 | 1.4 | 0 | 2 | 12% | nd |
| – | – | CL2 | 31 | 0.052 | 1.4 | 0.1 | 5 | 16% | −0.057 |
| – | French Guyana | GY | 12 | 0.219 | 1.9 | 0.2 | 3 | 25% | nd |
| – | USA | USA1 | 37 | 0.558 | 3.2 | 0.1 | 14 | 38% | 0.469 |
| – | – | USA2 | 39 | 0.018 | 1.3 | 0 | 3 | 8% | 0.280 |
| Africa | Madagascar | MD1 | 264 | 0.039 | 3.7 | 0.3 | 24 | 9% | 0.191 |
| – | – | MD2 | 27 | 0.169 | 2 | 0 | 10 | 37% | 0.047 |
| – | – | MD3 | 37 | 0.115 | 2.2 | 0 | 12 | 32% | 0.061 |
| – | – | MD4 | 15 | 0.000 | 1 | 0 | 1 | 7% | nd |
| – | – | MD5 | 23 | 0.193 | 2.4 | 0 | 11 | 48% | 0.022 |
| – | – | MD6 | 25 | 0.074 | 1.7 | 0 | 7 | 28% | −0.007 |
The first three columns give the area of origin (MB, Mediterranean Basin), the country of origin, and the name of the samples. N, sample size; Hn.b, unbiased gene diversity; Na, mean number of alleles per locus; Np, mean number of private alleles per locus; G, number of multilocus genotypes (MLG); G : N, proportion of unique MLG;  , multilocus linkage disequilibrium (
, multilocus linkage disequilibrium (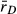 was not calculated (nd) when the number of MLGs was below 4).
was not calculated (nd) when the number of MLGs was below 4).
Determination of mating type and fertility
The mating type and female fertility of 600 strains were determined by in vitro crosses as described by Nottéghem & Silué (1992). Mating in Mo requires strains of opposite type and at least one of the strains must be female-fertile (able to produce perithecia). Crosses were performed by confronting the tested strain to female-fertile strains for which the mating type is known (reference strains). Mat1 reference strains were IN1, TH12, CH999 and CH1003. Mat2 reference strains were GY11, TH16, CH997 and CH1019. Tested strains were classified as Mat1 when inducing or forming perithecia with a Mat2 reference strain (and conversely). Tested strains were classified as female-fertile when forming perithecia with reference strains. For 175 additional strains, the mating type was determined by PCR amplification with the primers specific of Mat1 and of Mat2 (Xu & Hamer, 1995). In those cases, female fertility was not assessed.
DNA extraction and microsatellites amplification
DNA extraction was performed following a CIAA procedure (Adreit et al., 2007). All strains were genotyped with 10 microsatellites markers (Supporting Information Table S1) previously developed (Kaye et al., 2003; Adreit et al., 2007). Amplifications and allele size determination were performed as previously described (Saleh et al., 2012).
Indices of genetic diversity and linkage disequilibrium in populations
For each population, the mean number of alleles per locus Na, and the unbiased gene diversity Hn.b. (Nei, 1987) were calculated using Genetix v4.05 (Belkhir et al., 2004). We calculated the mean number of private alleles (Np) as the number of alleles that were present only in one population, averaged over the ten markers. The number of multilocus genotypes (MLG) and the index of association  were calculated using Multilocus v1.3 (Agapow & Burt, 2001). The proportion of unique MLG in each population was calculated as the G : N ratio (G, number of MLG; N, sample size).
were calculated using Multilocus v1.3 (Agapow & Burt, 2001). The proportion of unique MLG in each population was calculated as the G : N ratio (G, number of MLG; N, sample size).
Clustering and assignment analyses
Clustering methods were used to estimate the number of genetic groups that best explained the data. We used the Discriminant Analysis of Principal Components (DAPC; Jombart et al., 2010) that does not require any assumption on the biology of the organism, especially regarding panmixia. The DAPC was conducted using the adegenet package (v1.3-1) for the R software (v2.13.1; Vienna, Austria). We used the K-means procedure implemented in the function find.cluster to infer K, the optimal number of clusters, and let K vary between 1 and 60. K was determined using the Bayesian Information Criterion (BIC): if the function BIC = f(K) was U-shaped, then K was the abscissa of the minimum of this function; otherwise it corresponds to the point where the BIC decay rate abruptly changed (K then being the value after which the change in BIC was negligible; Jombart et al., 2010).
We also used the Structure Bayesian method (Pritchard et al., 2000; Falush et al., 2003). The basic assumption underlying this method is that the analyzed population can be theoretically subdivided into panmictic clusters. However, the method is supposed to be robust to departure from panmixia, and has given relevant results also in clonal or autogamous organisms (Garris et al., 2005; Bahri et al., 2009). We used the model with correlated allele frequencies and allowing admixture. Structure was run for K ranging from one to 32 with 10 replicates for each value of K. For each run, an 80 000-step Monte Carlo Markov Chain (MCMC) was performed after a 20 000 steps burn-in period. No a priori information was used on the assignments of individuals. We determined Ke, the optimal number of clusters, according to Evanno et al. (2005). Results were also checked for Ke−1 and Ke + 1. Individuals were assigned to a cluster if their probability of ancestry in this cluster was over the empirical cut-off of 0.7.
Unbiased gene diversity Hn.b and the mean number of private alleles Np were assessed for each genetic cluster as described above.
Genetic differentiation and genetic distances between populations or clusters
Pairwise FST (Weir & Cockerham, 1984) was calculated between clusters and the null hypothesis FST = 0 was tested using exact tests implemented in Genepop v4 (Raymond & Rousset, 1995). For the clusters inferred in Asia, the DA chord genetic distance calculated between all pairs of clusters was used to build an unrooted neighbor-joining tree using Populations v1.2.3.1 (O. Langella, http://bioinformatics.org/∼tryphon/populations/). We visualized the number of alleles shared between Asian clusters using a Venn diagram (package Venn.diagramm of the R software). At the world-wide scale, we calculated the number of MLG shared between different countries. We verified that these MLGs were real clones by calculating Psex, the probability that a genotype arose in several individuals within a population by independent reproduction events (Parks & Werth, 1993; Tibayrenc et al., 1990; Arnaud-Haond et al., 2005) using MLGsim (Stenberg et al., 2003). Identical MLGs with significantly low Psex values may be considered as belonging to the same clonal lineage. The program performs simulations of populations under random mating to assess the significance of Psex values.
An unrooted neighbor-joining tree of the world-wide populations was built from the pairwise DA distance (when the sample size was higher than 6).
Correlations between clusters assignments and biological features
Chi-squared tests were performed to assess if cluster assignment was independent from the rice subspecies (indica or japonica) the strains were collected on, the strain mating type (Mat1 and Mat2) and the female fertility status (female-fertile or female-sterile).
Migration capacities of M. oryzae in Asia
We evaluated spatial autocorrelation at different spatial scales in Asia using SPAGeDi v1.3 (Hardy & Vekemans, 2002). We calculated Moran's index, I, for all pairs of Asian individuals (either globally or by genetic cluster), in different distances classes. I ranges between −1 (negative spatial autocorrelation) and 1 (positive spatial autocorrelation). Distances classes were manually selected, class limits being positioned at breakpoints in the range of pairwise distances. The significance of I values was assessed by performing 1000 permutations. Linear regressions of I against distance (or its logarithm), as well as the significance of the regression slopes, were also estimated; the intrapopulation classes (i.e. individuals with identical geographic coordinates) were not considered in these analyses.
Results
Genetic structure and genetic diversity of world-wide populations
In order to infer the centers of diversity of Magnaporthe oryzae (Mo), the first stage was to study the genetic structure over all populations. We first inferred population subdivision at the global scale and evaluated genetic differentiation between genetic groups using FST. Then we studied the distribution of genetic diversity with regards to genetic structure and geography, within populations and within clusters.
We genotyped 1372 strains from 55 population samples of Mo rice strains from 15 countries (Table 1) with 10 microsatellite markers. In the DAPC, the 40 principal components retained explained more than 90% of the observed variance. The DAPC segregated the individuals into three genetic clusters (A, B, C). The Structure analysis also resulted in three clusters, the 10 replicates being 100% reproducible. The assignments of individuals to the three clusters were identical with DAPC and Structure (Fig. S1), except for four individuals (one from CH5, one from TH and two from SP1). With Structure, only 16 (among which 14 Asian strains) showed admixture signals (mixed ancestry in at least two clusters), but the DAPC assigned these 16 individuals to a single cluster.
The three clusters were highly differentiated (FST = 0.41, 0.44 and 0.65 between clusters A and B, A and C, and B and C, respectively) when compared to the average differentiation in phytopathogenic fungi (FST = 0.2 ± 0.05; Giraud et al., 2008).
The observed subdivision was highly associated with the geographic origin of the strains (Fig.1). All individuals from Europe/Mediterranean Basin belonged to cluster B and all individuals from Madagascar and Indonesia belonged to cluster C. Individuals from South America belonged to cluster C except for two Guyanese strains assigned to cluster A. Individuals from population USA1 were assigned to two clusters (A and B). Conversely, the three clusters described at the world-wide scale were all represented in Asia. Moreover, Asia was the only region in which the three clusters were represented in the same populations (CH2, CH3, CH5, CH6, NP2). Cluster A was over-represented in Asia (235 strains over 265).
Fig 1.

Proportion of strains belonging to the three clusters inferred using Discriminant Analysis of Principal Components (DAPC) in 55 world-wide samples of Magnaporthe oryzae. Cluster A, red; cluster B, green; cluster C, blue. The inset gives the coordinates of the individuals on the first two axes of the Principal Components Analysis.
We also looked at genetic diversity within clusters, within populations and within clusters × populations combinations.
Gene diversity (Hn.b.) calculated over all individuals within clusters, was at least two times higher in cluster A than in the other clusters (Hn.b. = 0.68, 0.32 and 0.23 for clusters A, B and C, respectively). The mean number of private alleles (Np) was five times higher in cluster A compared to the other clusters (Np = 5, 0.7 and 1 for clusters A, B and C, respectively).
Hn.b. and Np were also calculated within each population (Fig.2, black bars). In populations from Asia the Hn.b. mean value reached 0.38 ± 0.19 SD, whereas it was only 0.14 ± 0.09, 0.19 ± 0.22 and 0.10 ± 0.07 in populations from Europe/Mediterranean Basin, the Americas and Madagascar, respectively. Similarly, the mean Np value was 0.19 ± 0.38 in Asia but only 0.01 ± 0.04, 0.08 ± 0.08 and 0.05 ± 0.12 in Europe/Mediterranean Basin, the Americas and Madagascar, respectively. The lowest mean values of Hn.b and Np among Asian countries were found in Indonesia (Hn.b = 0.16 ± 0.10 and Np = 0.07 ± 0.06). Because sample sizes were highly different between some populations, and because gene diversity is known to be dependent on sample size (Leberg, 2002), we randomly sampled ten individuals in each of the 55 populations for which sample size was higher than ten, and recalculated Hn.b. and Np in these 55 re-samplings (with five replicates of this re-sampling procedure). This confirmed that gene diversity was higher in Asian populations (Table S2).
Fig 2.

Measures of gene diversity (Hn.b., upper panel) and mean number of private alleles (Np, lower panel) in samples (black bars) and in subsamples of Magnaporthe oryzae individuals from the same genetic cluster (colored bars). Population names are given in decreasing order of total Hn.b.. Red bars, subsample of individuals assigned to cluster A; green bars, subsample of individuals assigned to cluster B; blue bars, subsample of individuals assigned to cluster C. Asian samples and subsamples are shaded in gray. Hn.b. and Np were calculated only if the number of strains was higher than 6.
Hence, the higher genetic diversity observed in Asian populations had two explanations: some Asian populations encompassed individuals from the three clusters (e.g. CH2 and CH5), and others mainly encompassed individuals from the most diverse cluster, A (e.g. CH1 and LA1). To make the difference between these two potential causes, within each population we grouped strains belonging to the same cluster and calculated Hn.b. and Np on these subsamples (Fig.2, colored bars). To get reliable values, this was performed only on subsamples containing more than six individuals. The subsamples presenting the highest values for Hn.b. and Np belonged to cluster A. Only one subsample from outside Asia (USA1) had a gene diversity comparable to Asian subsamples but its number of private alleles was much lower. The most diverse subsamples belonging to cluster B and C also originated from Asia.
Hence, Asia was the best candidate to be, or include, the center of diversity of Mo compared to the other continents. To localize this center more precisely, we analyzed the genetic structure and the distribution of genetic diversity inside Asia.
Localization of the centers of diversity in Asia
At the Asian scale, we also inferred genetic structure using assignments methods. We evaluated FST between genetic groups. We then studied the distribution of genetic diversity with regards to genetic structure and geography: within populations and within clusters.
The DAPC performed on the 423 Asian individuals revealed that the region was organized in four genetic clusters (numbered from 1 to 4; Fig.3). The subdivision inferred using Structure was congruent with this result, and individual assignments to the four clusters were identical among the 10 replicates. 386 individuals could be assigned to a single cluster, the 37 remaining individuals showing admixture signal. The DAPC achieved to assign these 37 individuals to a single group. Individual assignments were identical with DAPC and with Structure, except for 12 individuals (six from CH1, three from LA1 and three from TH). Nine out of these 12 were assigned to cluster 1 by Structure and to cluster 4 by DAPC, or assigned to cluster 4 by Structure and to cluster 1 by DAPC.
Fig 3.

Discriminant Analysis of Principal Components (DAPC) on the 423 Asian strains of Magnaporthe oryzae. Cluster 1, yellow; cluster 2, green; cluster 3, blue; cluster 4, red. Samples are labelled according to their country of origin (CH, China; ID, Indonesia; LA, Laos; NP, Nepal; TH, Thailand). The inset gives the coordinates of the individuals on the first two axes of the Principal Components Analysis. Genotypes of strains from outside of Asia are represented in black: squares for Europe/Mediterranean basin, triangles for Madagascar, discs for Americas.
Each cluster was significantly differentiated from the others: pairwise FST values were always higher than 0.2, the lowest FST being between clusters 1 and 4 (FST = 0.44 between clusters 1 and 2; 0.49 between clusters 1 and 3; 0.27 between clusters 1 and 4; 0.63 between clusters 2 and 3; 0.48 between clusters 2 and 4; and 0.38 between clusters 3 and 4; P-values of Fisher's exact tests as implemented in Genepop v4 were below 10−5 for all pair of clusters, allowing rejection of the null hypothesis of no differentiation). The pairwise DA chord distance calculated among the four Asian clusters confirmed that clusters 1 and 4 were more closely related to each other than to clusters 2 and 3 (Fig.4a).
Fig 4.

Unrooted neighbor-joining trees based on the DA chord distance between Magnaporthe oryzae clusters or samples (a) between pairs of Asian clusters, and (b) between pairs of world-wide samples for which sample size was higher than 6.
All strains assigned to clusters 1 and 4 but one were assigned to cluster A in the world-wide analysis. Identically, the Asian cluster 2 mostly overlapped with the world-wide cluster B, and the Asian cluster 3 mostly overlapped with the world-wide cluster C. Therefore, the three-cluster structure depicted in Asia through the world-wide analysis was in accordance with the four-cluster structure obtained through the analysis of the Asian dataset alone. In Asia, different combinations of clusters could be observed in one geographic area (Fig.3). The populations from Yunnan province (South China), Laos and Thailand (CH1, LA1, LA2 and TH) shared a similar structure with individuals assigned essentially to cluster 4, and to a lower extent to cluster 1. The genetic composition of the other populations from Yunnan (CH3, CH4, CH5 and CH6) varied from one another, but the four clusters were detected there. The samples from Hunan province, China (CH2 and CH7), and the Indonesian populations were composed mainly of individuals from cluster 3. The Nepalese populations encompassed mainly individuals from cluster 1 (NP1 and NP3) or from cluster 3 (NP2).
Consistent with the three-cluster structure at the global scale, gene diversity was higher in clusters 1 and 4 than in clusters 2 and 3 (Hn.b: 0.50, 0.61, 0.26 and 0.26, respectively; Table 2). This also held for allelic diversity (Na: 7.9 and 7.7 in clusters 1 and 4, respectively; 3.6 and 5.5 in clusters 2 and 3, respectively), and for the mean number of private alleles per locus (Np: 1.9 and 1.4 in clusters 1 and 4, respectively; 0.4 and 0.3 in clusters 2 and 3, respectively). Hence, clusters 1 and 4 were more genetically diverse than clusters 2 and 3.
Table 2.
Genetic diversity within each of the four clusters of Magnaporthe oryzae Asian strains inferred using Discriminant Analysis of Principal Components (DAPC)
| Cluster | N | Hn.b | Na | Np | MLG | G : N |  |
|---|---|---|---|---|---|---|---|
| 1 | 103 | 0.504 | 7.9 | 1.9 | 55 | 53% | 0.097 |
| 2 | 72 | 0.256 | 3.6 | 0. 4 | 21 | 29% | 0.182 |
| 3 | 131 | 0.259 | 5.5 | 0. 3 | 49 | 37% | 0.078 |
| 4 | 124 | 0.610 | 7.7 | 1.4 | 96 | 77% | 0.030 |
Number of individuals (N), unbiased gene diversity (Hn.b.), mean number of alleles per locus (Na), mean number of private alleles per locus (Np), number of multilocus genotypes (MLG), clonal richness (G : N), and multilocus linkage disequilibrium ( ).
).
The only difference between genetic subdivision at the worldwide and Asian scale was the split of world-wide cluster A into Asian clusters 1 and 4. Therefore, the calculation of gene diversity and mean number of private alleles in the clusters × populations combination at the Asian scale (Table S3), gave results similar to those obtained at the global scale (Fig.2, colored bars). Populations exhibiting the highest genetic diversity were those composed of individuals mainly assigned to cluster 1 and/or 4.
Two geographic areas were identified to be composed of populations showing this feature: a first region comprising South China (Yunnan, CH1), Laos (LA1, LA2) and Thailand (TH) where cluster 4 dominated, and a second region in Western Nepal (NP1 and NP3) where cluster 1 dominated. So, these results allowed us to define these two regions as two centers of diversity.
Localization of the center of origin
We then wondered if one of these centers of diversity could correspond to the center of origin of Mo populations pathogenic on rice.
In order to test whether clusters 2 and 3 could originate from clusters 1 or 4, we compared the number of shared alleles between clusters (Fig. S2). We reasoned that any derived cluster should share more alleles with its cluster of origin than with other clusters. The number of shared alleles between clusters 2 and 1 was similar to the number shared between 2 and 4 (27 and 28, respectively). The number of shared alleles between clusters 3 and 1 was identical to the number shared between 3 and 4 (29). Thus, following this method, we could not figure out if clusters 2 and 3 are derived from cluster 1 or 4.
In organisms that can reproduce both sexually and asexually, the ability to reproduce sexually is believed to be an ancestral state that can be lost in certain conditions. In this case, sexual reproduction is expected in the center of origin rather than in introduced areas (Leslie & Klein, 1996). So, we looked for genetic and biological evidence of sexual reproduction in the four Asian clusters.
Footprints of recombination accompanying potential sexual reproduction were searched by measuring G : N, the proportion of multilocus genotypes (MLG) discriminated, and 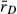 , the multilocus linkage disequilibrium. In Asia, the average G : N in populations was 54% ± 23% SD (China: 50% ± 23%; Indonesia: 36% ± 15%; Laos: 84% ± 6%; Nepal: 57% ± 24% and Thailand: 67%; Table 1). The mean value of
, the multilocus linkage disequilibrium. In Asia, the average G : N in populations was 54% ± 23% SD (China: 50% ± 23%; Indonesia: 36% ± 15%; Laos: 84% ± 6%; Nepal: 57% ± 24% and Thailand: 67%; Table 1). The mean value of  was 0.21 ± 0.15 SD (China: 0.20 ± 0.11; Indonesia: 0.09 ± 0.04; Laos: 0.11 ± 0.06; Nepal: 0.43 ± 0.20 and Thailand: 0.17; Table 1). The proportion of unique MLG was the highest in cluster 4 (G : N, 77%), intermediate in cluster 1 (G : N, 53%) and lowest in clusters 2 and 3 (G : N, 29% and 37%, respectively; Table 2). Multilocus linkage disequilibrium was lowest in cluster 4 (
was 0.21 ± 0.15 SD (China: 0.20 ± 0.11; Indonesia: 0.09 ± 0.04; Laos: 0.11 ± 0.06; Nepal: 0.43 ± 0.20 and Thailand: 0.17; Table 1). The proportion of unique MLG was the highest in cluster 4 (G : N, 77%), intermediate in cluster 1 (G : N, 53%) and lowest in clusters 2 and 3 (G : N, 29% and 37%, respectively; Table 2). Multilocus linkage disequilibrium was lowest in cluster 4 (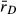 : 0.030), intermediate in clusters 1 and 3 (
: 0.030), intermediate in clusters 1 and 3 ( : 0.097 and 0.078, respectively) and highest in cluster 2 (
: 0.097 and 0.078, respectively) and highest in cluster 2 ( : 0.182; Table 2).
: 0.182; Table 2).
Biological evidence of sexual reproduction was searched for by measuring the proportions of both mating types and of female-fertile strains within populations, both being required for sexual reproduction. Overall in Asia, the distribution of mating types (χ2 = 330, P = 3.2 × 10−71, df = 3) and of female-fertile strains (χ2 = 294, P = 2.0 × 10−64, df = 3) significantly depended on the cluster of origin. The frequency of Mat1 and Mat2 strains in clusters 1 and 4 was relatively balanced and not significantly different from a random distribution (Table 3). On the contrary, Mat1 strains were over-represented in cluster 2 and Mat2 strains were over-represented in cluster 3 (Table 3). Similarly, the proportion of female-fertile strains was highly different within the four clusters. Interestingly, the percentage of female-fertile strains was highest in clusters 4 (76%), intermediate in cluster 1 (42%), and lowest in clusters 2 and 3 (4% and 11%, respectively).
Table 3.
Distribution of Magnaporthe oryzae individuals in genetic clusters at different scales as a function of the mating type and female fertility
| Mat1 | Mat2 | Female-fertile | Female-sterile | ||||
|---|---|---|---|---|---|---|---|
| (a) Asian scale | |||||||
| 1 | 36 (34.1) | 33 (34.9) | 69 | 1 | 27 (27.0) | 38 (38.0) | 65 |
| 2 | 61 (34.1) | 8 (34.9) | 69 | 2 | 3 (28.6) | 66 (40.4) | 69 |
| 3 | 22 (38.0) | 55 (39.0) | 77 | 3 | 5 (18.7) | 40 (26.3) | 45 |
| 4 | 44 (56.8) | 71 (58.2) | 115 | 4 | 87 (47.7) | 28 (67.3) | 115 |
| 163 | 167 | 330 | 122 | 172 | 294 | ||
| (b) Global scale | |||||||
| A | 92 (88.7) | 132 (135.3) | 224 | A | 116 (46.5) | 111 (180.5) | 227 |
| B | 195 (80.0) | 7 (122.0) | 202 | B | 3 (33.0) | 158 (128.0) | 161 |
| C | 20 (138.3) | 329 (210.8) | 349 | C | 4 (43.5) | 208 (168.5) | 212 |
| 307 | 468 | 775 | 123 | 477 | 600 | ||
Values in brackets are expected values for an independent assortment calculated on the overall frequencies of the different characters.
Altogether, the low genotypic diversity, the high linkage disequilibrium, the dominance of one mating type and the very low proportion of female-fertile strains in clusters 2 and 3 suggested a low probability that sexual reproduction occurred in these groups. Although Cluster 1 presented a higher genotypic diversity and balanced proportions of Mat1 and Mat2 strains, the average proportion of female-fertile strains and the high linkage disequilibrium were not in agreement with the expectations of sexual reproduction. However, the high genotypic diversity, the low linkage disequilibrium, the balanced proportions of Mat1 and Mat2 strains and the high proportions of female-fertile strains in cluster 4 were consistent with footprints of sexual reproduction in this genetic group. Hence, the region where cluster 4 dominates – that is, the region comprising South China (Yunnan), Laos and North Thailand – should be considered as a putative center of origin of Mo populations pathogenic on rice.
Localizations of the centers of migration
All clusters are present together only in Asia, and hence we hypothesized that Asia could be the center of origin of world-wide migrations. To test this hypothesis, we wondered if strains from populations outside Asia could be genetically related to the Asian clusters. We calculated the coordinates of the 949 nonAsian strains as supplementary individuals in the DAPC performed on the 423 Asian strains using the function pred.sup (package Adegenet, R software). All European/Mediterranean strains but one were assigned to cluster 2 (inset of Fig.3). All South American and the Madagascan strains were assigned to cluster 3. The North American strains were assigned to clusters 1, 2 and 4. These results show that all strains from outside Asia can be related to one of the Asian genetic groups. This points to Asia as the center of dispersion of Mo. We further tested this hypothesis by analyzing the shared MLG between different countries.
Twenty MLG were shared between different populations within and between countries. Shared MLG between countries belonged to cluster B and to cluster C (Fig.5). No MLG were shared between countries within cluster A. Most of the shared MLG were found between countries of the same region: within Europe (five between France and Spain, five between France and Greece, two between Spain and Greece, one between France and Turkey) and within Asia (two between China and Indonesia, two between China and Nepal). Interestingly, several MLG were also shared between geographically distant countries, and especially between Asian countries and nonAsian ones (one between China and Spain, two between China and Hungary, two between China and Colombia, one between Indonesia and Madagascar, three between Indonesia and Colombia, one between Thailand and French Guyana). Only two MLG were shared between countries of different regions outside Asia: between Spain and USA. To validate these results, we tested the resolution power of the markers to discriminate clones, that can be affected in clonal organisms (Arnaud-Haond et al., 2005, 2007). For MLGs that were both shared between at least two different populations and repeated within each population, we calculated Psex within each population (the probability that repeated genotypes originate from distinct reproductive events). All the Psex values were highly significant (Table S4), indicating that all the MLG shared between countries were real clones.
Fig 5.

Magnaporthe oryzae multilocus genotypes (MLG) shared between countries. The width of the line is proportional to the number of shared MLG. Green lines and blue lines represent MLG that belong to cluster B and cluster C, respectively. CH, China; CL, Columbia; FR, France; GR, Greece; GY, French Guyana; HN, Hungary; ID, Indonesia; MD, Madagascar; NP, Nepal; SP, Spain; TH, Thailand; TR, Turkey; USA, United States of America.
These results suggest long intercontinental migrations from Asia towards the other regions of the world, as well as intracontinental migrations. Furthermore, none of the MLG shared between continents were assigned to cluster A. In addition, only few strains assigned to cluster A were found out of Asia. This suggests that most of these migrations did not originate from the most ancestral genotypes, belonging to cluster A, but from more recent genotypes belonging to clusters B and C.
In order to further address the migration capacities of Mo in Asia, we evaluated the spatial autocorrelation in this region using Moran's index I. When considering all Asian individuals (Fig.6a), significant positive spatial autocorrelation was observed only for the intrapopulation class (i.e. between pairs of individuals from the same spatial location). For all non-null distance classes, I was never significantly different from 0, indicating a random spatial pattern whatever the geographic scale considered. We obtained similar results when separating individuals by genetic cluster (Fig.6b), except for cluster C in which a weak but significantly positive spatial auto-correlation was observed in the distance class (0–300 km) (I = 0.16, P = 0.045, one-sided test). We never found any significant linear regression of I against distance (or its logarithm).
Fig 6.

Spatial autocorrelation between Magnaporthe oryzae individuals in Asia. Moran's index I was calculated between pairs of Asian individuals for different distance classes, (a) for all Asian individuals; (b) in each of the four Asian genetic clusters. Abscissas of points are calculated as the mean pairwise distance of all pairs of individuals in the class considered. The points situated on abscissa 0 correspond to intrapopulation classes (individuals from the same spatial location). The red dotted lines represent upper and lower bounds of the 95% confidence interval, assessed after 1000 permutations. Vertical gray lines represent upper bounds of distance classes, manually chosen after the distribution of pairwise geographic distances.
Genetic distance between populations
At the world-wide scale, the neighbor-joining tree based on DA chord distance between pairs of populations (except the HN2 sample which size was considered too small) showed that populations were grouped according to their mosaic composition in the different genetic clusters (Fig.4b). One clade grouped Asian LA, TH, CH1, NP1 and NP3 populations from the two centers of diversity (the first three being also from the putative center of origin). Another clade grouped Asian populations CH2, CH3, CH5 and NP2 which have a similar mosaic composition, with populations mostly or completely composed of individuals assigned to the Asian cluster 3 (CH7 and ID populations) or to world-wide cluster C (GY, CL and MD populations). The third clade grouped populations mainly composed of individuals belonging to Asian cluster 2 (CH4 and CH6) and to world-wide cluster B (populations from Europe/Mediterranean Basin).
Discussion
Phylogeographic studies on different phytopathogenic fungi have shown a variety of situations regarding the co-localization of centers of origin, diversity and migration (Robert et al., 2012). Here, we provided evidence that the center of origin of Mo on cultivated rice colocalizes with one of the two centers of diversity in South-East Asia but not with the center of dispersal towards the rest of the world.
The centers of diversity of Mo match the centers of domestication of rice in Asia
The highest genetic diversity of Mo was found in Asia, at the regional scale (whole South-East Asia) as well as at the population scale, except for Indonesian populations. Allelic and gene diversities, and the number of private alleles were (respectively) two, three and four times higher in Asian populations than in populations from Europe/Mediterranean Basin, the Americas or Madagascar. Asian strains formed four genetic clusters that did not strictly match a single Asian country or region. Rather, Asian populations were composed of ‘mosaics’ of these clusters, whose composition corresponded roughly to the geography. Such a mosaic structure, described for example for the fungus Venturia inequalis in its area of origin (Gladieux et al., 2008), shows that regional groups have a common but complex evolutionary origin involving mixing between several pre-existing genetic groups. Here, some populations were mostly composed of one cluster, and others comprised several clusters. The geographic distribution of the two most diverse clusters (1 and 4) determined two centers of diversity, one covering Yunnan province (China), Laos and Thailand, and the other located in western Nepal. Interestingly, these two centers of diversity match the putative domestication areas of rice, localized in South China and northeastern India (Londo et al., 2006; Huang et al., 2012).
The center of origin of Mo matches one of its centers of diversity
Assuming that the ancestral reproductive character is sexuality rather than clonality (Schurko & Logsdon, 2008) and that asexual fungal crop pathogens might still reproduce sexually near their center of origin (Leslie & Klein, 1996), we inferred the center of origin of Mo by localizing those areas where footprints of sexual reproduction are detected. In Mo, sexual reproduction was previously inferred in India (Kumar et al., 1999) and evidenced in South China (Saleh et al., 2012). Here, genetic and biological evidences of past or present sexual reproduction designate the region comprising South China (Yunnan), Laos and North Thailand as the putative center of origin of Mo strains pathogenic on rice. Following Huang et al. (2012), this area matches the initial center of domestication of rice Os var. japonica. This result agrees with the formerly proposed hypothesis of a single origin of Mo strains pathogenic to cultivated rice in China following a host shift (Shull & Hamer, 1994; Couch et al., 2005). As for other plant pathogens, the center of origin of Mo rice strains corresponds to one center of domestication of its host.
Bridgehead effect in Asia: intercontinental migrations of Mo originated from secondary Asian areas
Tharreau et al. (2009) suggested the occurrence of intercontinental migrations of Mo. Our results confirmed that all secondary areas outside Asia actually had an Asian origin (Figs 5). At the global scale we found three clusters consistent with the four clusters found in Asia. The most diverse cluster (A) did not disperse much outside Asia, whereas the two others (B and C) were found extensively world-wide. Genetic diversity, especially the number of alleles shared between clusters, indicated that world-wide clusters B and C originated from Asian clusters 2 and 3, respectively. This illustrates a bridgehead effect in Asia; that is, that the secondary sources of long-range migrations are different from the centers of diversity and from the center of origin of blast (Lombaert et al., 2010).
Our results also illustrate different invasion histories of secondary areas. In the European/Mediterranean Basin populations, all individuals but one belonged to a single world-wide cluster (B). Interestingly, the 69 Asian individuals also assigned to this cluster came mostly from two Chinese populations: 25 from CH4 and 26 from CH6. Moreover, we found common MLG between Hungarian strains and strains from CH3 and CH4 populations. There were also common MLG between Spanish strains and CH4 strains. Therefore, Yunnan, where CH3, CH4 and CH6 populations were collected, could likely represent the source of European/Mediterranean populations. A single introduction in the European/Mediterranean area is supported by the low genetic diversity observed in this region and by the fact that only one mating type (Mat1) is present there. The fungus was probably subsequently dispersed throughout Europe from a single, still undetermined entry.
Indonesian and Chinese populations CH2 and CH7 had common MLGs, showing possible exchange between the two regions. Madagascan populations had a single origin and belonged to the same cluster as the Chinese population CH7 and Indonesian populations. In addition, a common MLG was detected between Madagascar and Indonesia. So, either the Indonesian and Madagascan populations originated from the same genetic pool independently, or the Madagascan strains migrated from Indonesia. We favour the second hypothesis because the diversity observed in Madagascar is lower than in Indonesia, and because it matches the history of human migrations in these areas. The first human groups arrived in Madagascar from Indonesia at least 2000 yr ago (Hurles et al., 2005), and may have carried with them blast-infected rice seeds.
Populations from Colombia and French Guyana belonged to the same cluster (C), and shared common MLGs with Asian populations: CL1 with CH2 and CH7 from Hunan province (China) and with ID1 and ID2 from Indonesia, and GY (especially the most common MLG of this population) with the Thailand population. So, Mo strains from South America may have different origins: Colombian populations might have originated either directly from western China or from Indonesia. In French Guyana, strains might have been introduced from Thailand or Vietnam, probably through recent migrations of H'Mongs.
The two North American populations obviously had different origins. All USA2 strains belonged to cluster B, like European strains, with two MLG shared with Spanish strains. So, this population might have originated either from the same Chinese genetic pool that migrated towards Europe, or directly from Europe itself. The USA1 population likely resulted from multiple introductions from different gene pools because it gathered strains belonging to two clusters (A and B): this suggests either two introductions from Asia, or one from Asia and one from Europe. Similar patterns of multiple introductions have already been described for other phytopathogenic fungi (Dutech et al., 2010; Montarry et al., 2010; González-Varela et al., 2011; Robert et al., 2012).
The world-wide organization of genetic diversity agrees with stochastic human-driven migrations outside Asia. Inside Asia, spatial autocorrelation analyses did not reveal any significant deviation from random spatial pattern. Hence, inside Asia, whatever the distance range considered (even at the intraregional geographic scale, i.e. up to 300 km), and whatever the genetic origin of individuals, migration events were also probably purely stochastic, and closely linked to movements of human groups or to seed exchanges. This agrees with previous knowledge on the short-distance natural migration capacities of M. oryzae (a few meters; D. Tharreau & J. L. Nottéghem, pers. comm.).
Selection by the host might explain the differentiation of the secondary Asian centers
Our results support the hypothesis, also proposed by Levy et al. (1991) and Zeigler (1998), that selection by rice contributed to shaping the genetic structure of Mo populations. The higher diversity observed in clusters 1 and 4 may be explained by the higher diversity of the host in the Asian areas where clusters 1 and 4 are found. Indeed, both clusters encompassed mainly (179/226) strains collected on rice grown in upland conditions, where many traditional and diverse varieties were maintained (at least in the Asian regions sampled). Either the host diversity maintained directly pathogen genotypic diversity by selection, or indirectly by maintaining sexual reproduction.
We also found that membership to a particular genetic cluster was significantly associated with the prevalence of varieties of indica or japonica type in the area sampled. In Asia, cluster 3 was significantly over-represented in regions where indica rice is prevalent (Table S5a), and conversely all individuals but one from cluster 4 were sampled in regions were japonica rice is prevalent. World-wide, cluster B is prevalent in areas where japonica varieties – especially temperate – are grown (Table S5b), while cluster C is over-represented in areas where indica varieties are grown. Pathogenic specialization of blast on the different rice subspecies, suggested by Bonman et al. (1990), could explain this distribution. This specialization, that may not be strict and remains to be demonstrated for the clusters we identified, could be the result of host-pathogen coevolution. Indeed, indica and japonica rice subspecies were domesticated independently in two Asian areas, and our results show that population subdivision of blast in Asia matches this domestication process. Thus, following rice domestication, Mo possibly adapted independently to these two subspecies leading to differentiation in two clusters (B and C). This subdivision was maintained when these clusters spread into different countries because Mo was probably introduced with the rice varieties it was adapted to, and because different rice subspecies are used in different agrosystems, limiting the possibility of cross-adaptation between strains of the B cluster on indica rice varieties and of A cluster on japonica.
Altogether, our results suggest that Mo could have evolved as a major pathogen on cultivated rice through a host-tracking process, following a host shift from an unknown plant towards wild rice. Host-tracking – that is, the coevolution of the host and the pathogen during domestication (Stukenbrock & McDonald, 2008) – implies that both partners have the same center of origin, and has been suggested as an emerging mechanism for several important pathogenic fungi (Banke & McDonald, 2005; Gomez-Alpizar et al., 2007; Raboin et al., 2007; Gladieux et al., 2008; Robert et al., 2012).
Sexual reproduction was probably lost during the differentiation of secondary Asian centers and intercontinental migrations
This study supports the hypothesis suggested by Levy et al. (1991) and Zeigler (1998) that Mo populations outside Asia derived recently from a limited set of founders. In addition, we found a nonrandom distribution of mating types and of female-fertile strains in the different clusters (χ2 = 441, P = 1.7 × 10−96, df = 2 and χ2 = 600, P = 5.1 × 10−131, df = 3, respectively; Table 3b), confirming the clonal structure of all nonAsian populations already demonstrated by Saleh et al. (2012). Within cluster A, we observed balanced proportions of the two mating types, and of female-fertile/female-sterile strains. Cluster B gathered almost only Mat1 strains (175/180), and cluster C almost only Mat2 strains (280/293). Cluster A gathered 94% of the total number of female-fertile strains, these strains being rare both within the two other clusters (2/97 in cluster B, 2/173 in cluster C). Furthermore our results suggest that the clonal populations found outside Asia likely originated from clonal Asian populations that pre-existed before world-wide migrations. Indeed, in Asian groups 2 and 3 mating type ratio is biased towards Mat1 and Mat2, respectively. Moreover, the frequency of female-fertile strains is low in these groups (4% and 11%, respectively). So, sexual reproduction was probably lost in these groups compared to groups 1 and 4 from which they likely derived. Following our ‘out of Asia’ dispersal scenario, the genetic groups B and C likely originated from groups 2 and 3, respectively. The absence of sexual reproduction in all areas outside Asia may thus be explained by migrations from source populations that were already exclusively clonal.
Conclusion
Our study provides new insights on the native areas, diversity reservoirs and invasion routes of rice blast. We showed that several independent events of intercontinental migrations occurred which are likely linked with the transportation of infected materials. In a context of intense global exchanges, knowledge about these past events should lead to increased vigilance on the risk of introductions of new genotypes of the pathogen through the exchanges of rice seeds. Our work also exemplifies the role of plant domestication in shaping the population structure of plant pathogens. For the Mo/rice pathosystem, the independent domestication of indica and japonica rice subgroups led to the appearance of two genetic groups of the pathogen. Such a structure, probably accompanied by a specialization of the pathogen on the different rice subspecies, could be exploited to develop new strategies of deployment of resistance genes. Wild species have been proposed as a source of resistance genes to improve related crop species (Izawa & Shimamoto, 1996; Brar & Khush, 1997). A similar strategy could be used in the case of subspecies. It is likely that some so called major resistance genes, and defense mechanisms involving several genes, are specific for each rice subspecies. By introducing these genes in the other subspecies, the pathogen population would then be confronted by genes that it had never met before.
Acknowledgments
D.S. was supported by CIRAD and INRA grants. We thank Agropolis Fondation (‘Rice blast networking’ project), the European Community (Residiv ICA4-CT-2000-30021) and ANR (Genblast ANR-06-ERA-PG-013, Emerfundis ANR-Biodiv-07) for financial support. We thank the labex ‘Centre Méditerranéen de l'Environnement et de la Biodiversité’ for access to molecular genetic analysis technical facilities. We thank the following colleagues for their help in sharing or collecting strains: Ying Shen (CNRRI, China), Peng Xu (YAAS, China), Li Chengyun (YAU, China), Fernando Correa (CIAT, Colombia), Dimitris Katsantonis (Cereal Institute, Greece), Santoso (RIR, Indonesia), Somphet Phengchanh (NAFRI, Laos), Dodelys Andrianatsimialona (FOFIFA, Madagascar), Gopal Prasad Parajuli (NARC, Nepal), Maria del Mar Catala (IRTA, Spain), Mercedes Castejon-Muñoz (ITFAP, Spain), Manuel Aguilar-Portero (ITFAP, Spain), Ramon Carreres (IVIA, Spain), Jose-Maria Osca (UPV, Spain), Surëk Halil (TARI, Turkey) and Jim Correll (U. Arkansas, USA).
Supporting Information
Additional supporting information may be found in the online version of this article.
Genetic structure of worldwide individuals of Magnaporthe oryzae: comparison between the DAPC and Structure methods.
Fig. S2 Number of shared and specific alleles calculated over 10 microsatellites for the four Magnaporthe oryzae clusters identified in Asia.
Characteristics of the 10 microsatellites used for Magnaporthe oryzae genotyping
Table S2 Unbiased gene diversity and number of private alleles in random subsamples performed in 46 Magnaporthe oryzae worldwide populations
Table S3 Gene diversity and mean number of private alleles in subsamples of Magnaporthe oryzae individuals from the same genetic cluster in Asia
Table S4 Information on Magnaporthe oryzae multilocus genotypes repeated within populations and shared between populations
Table S5 Distribution of Magnaporthe oryzae individuals in clusters according to the type of rice culture
References
- Adreit H, Santoso , Andriantsimialona D, Utami DW, Nottéghem JL, Lebrun MH, Tharreau D. Microsatellite markers for population studies of the rice blast fungus, Magnaporthe grisea. Molecular Ecology Notes. 2007;7:667–670. [Google Scholar]
- Agapow PM, Burt A. Indices of multilocus linkage disequilibrium. Molecular Ecology Notes. 2001;1:101–102. [Google Scholar]
- Arnaud-Haond S, Alberto F, Teixeira S, Procaccini G, Serrao EA, Duarte CM. Assessing genetic diversity in clonal organisms: low diversity or low resolution? Combining power and cost efficiency in selecting markers. Journal of Heredity. 2005;96:434–440. doi: 10.1093/jhered/esi043. [DOI] [PubMed] [Google Scholar]
- Arnaud-Haond S, Duarte CM, Alberto F, Serrao EA. Standardizing methods to address clonality in population studies. Molecular Ecology. 2007;16:5115–5139. doi: 10.1111/j.1365-294X.2007.03535.x. [DOI] [PubMed] [Google Scholar]
- Bahri B, Leconte M, Ouffroukh A, De Vallavieille-Pope C, Enjalbert J. Geographic limits of a clonal population of wheat yellow rust in the Mediterranean region. Molecular Ecology. 2009;18:4165–4179. doi: 10.1111/j.1365-294X.2009.04267.x. [DOI] [PubMed] [Google Scholar]
- Banke S, McDonald BA. Migration patterns among global populations of the pathogenic fungus Mycosphaerella graminicola. Molecular Ecology. 2005;14:1881–1896. doi: 10.1111/j.1365-294X.2005.02536.x. [DOI] [PubMed] [Google Scholar]
- Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F. GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Montpellier, France: Laboratoire Génome, Populations, Interactions; 1996–2004. [Google Scholar]
- Bonman JM, Mew TW, Koganezawa H, Vergel de Dios-Mew TI, Vera-Cruz CM, Medella ES, Kim KK, Nottéghem JL, Glaszmann JC. Resistance to key diseases in sub-specific groups of rice. Phytopathology. 1990;80:999. (Abstract) [Google Scholar]
- Brar DS, Khush GS. Alien introgression in rice. Plant Molecular Biology. 1997;35:35–47. [PubMed] [Google Scholar]
- Brunner PC, Schuerch S, McDonald BA. The origin and colonization history of the barley scald pathogen Rhynchosporium secalis. Journal of Evolutionary Biology. 2007;20:1311–1321. doi: 10.1111/j.1420-9101.2007.01347.x. [DOI] [PubMed] [Google Scholar]
- Cesari S, Thilliez G, Ribot C, Chalvon V, Michel C, Jauneau A, Rivas S, Alaux L, Kanzaki H, Okuyama Y, et al. The rice resistance protein pair RGA4/RGA5 recognizes the Magnaporthe oryzae effectors AVR-Pia and AVR1-CO39 by direct binding. Plant Cell. 2013;25:1463–1481. doi: 10.1105/tpc.112.107201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DH, Zeigler RS, Leung H, Nelson RJ. Population structure of Pyricularia grisea at two screening sites in the Philippines. Phytopathology. 1995;85:1011–1020. [Google Scholar]
- Chen QH, Wang YC, Zheng XB. Genetic diversity of Magnaporthe grisea in China as revealed by DNA fingerprint haplotypes and pathotypes. Journal of Phytopathology. 2006;154:361–369. [Google Scholar]
- Cheng CY, Motohashi R, Tsuchimoto S, Fukuta Y, Ohtsubo H, Ohtsubo E. Polyphyletic origin of cultivated rice: based on the interspersion pattern of SINEs. Molecular Biology and Evolution. 2003;20:67–75. doi: 10.1093/molbev/msg004. [DOI] [PubMed] [Google Scholar]
- Chuma I, Isobe C, Hotta Y, Ibaragi K, Futamata N, Kusaba M, Yoshida K, Terauchi R, Fujita Y, Nakayashiki H, et al. Multiple translocation of the AVR-Pita effector gene among chromosomes of the rice blast fungus Magnaporthe oryzae and related species. PLoS Pathogens. 2011;7:e1002147. doi: 10.1371/journal.ppat.1002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consolo VF, Cordo CA, Salerno GL. DNA fingerprint and pathotype diversity of Pyricularia oryzae populations from Argentina. Australasian Plant Pathology. 2008;37:357–364. [Google Scholar]
- Correll JC, Boza EJ, Seyran E, Cartwright RD, Jia Y, Lee FN. Examination of the rice blast pathogen population diversity in Arkansas, USA – Stable or unstable? In: Wang GL, Valent B, editors. Advances in genetics, genomics and control of rice blast disease. Dordrecht, the Netherlands: Springer; 2009. pp. 217–228. [Google Scholar]
- Couch BC, Fudal I, Lebrun MH, Tharreau D, Valent B, van Kim P, Nottéghem JL, Kohn L. Origins of host-specific populations of the blast pathogen Magnaporthe oryzae in crop domestication with subsequent expansion of pandemic clones on rice and weeds of rice. Genetics. 2005;170:613–630. doi: 10.1534/genetics.105.041780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Jia Y, Correll J, Wang X, Wang Y. Diversification and evolution of the avirulence gene AVR-Pita1 in field isolates of Magnaporthe oryzae. Fungal Genetics and Biology. 2010;47:973–980. doi: 10.1016/j.fgb.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Dean R, Van Kan JA, Pretorius ZA, Hammond-Kosack KE, Di Pietro A, Spanu PD, Rudd JJ, Dickman M, Kahmann R, Ellis J, et al. The top 10 fungal pathogens in molecular plant pathology. Molecular Plant Pathology. 2012;13:414–430. doi: 10.1111/j.1364-3703.2011.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Don LD, Kusaba M, Urashima AS, Tosa Y, Nakayashiki H, Mayama S. Population structure of the rice blast fungus in Japan examined by DNA fingerprinting. Annals of the Phytopathological Society of Japan. 1999a;65:15–24. [Google Scholar]
- Don LD, Tosa Y, Nakayashiki H, Mayama S. Population structure of the rice blast fungus in Vietnam. Annals of the Phytopathological Society of Japan. 1999b;65:475–479. [Google Scholar]
- Dutech C, Fabreguettes O, Capdevielle X, Robin C. Multiple introductions of divergent genetic lineages in an invasive fungal pathogen, Cryphonectria parasitica, in France. Heredity. 2010;105:220–228. doi: 10.1038/hdy.2009.164. [DOI] [PubMed] [Google Scholar]
- Estoup A, Guillemaud T. Reconstructing routes of invasion using genetic data: why, how and so what? Molecular Ecology. 2010;19:4113–4130. doi: 10.1111/j.1365-294X.2010.04773.x. [DOI] [PubMed] [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Molecular Ecology. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard J. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL, Gurr SJ. Emerging fungal threats to animal, plant and ecosystem health. Nature. 2012;484:186–194. doi: 10.1038/nature10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes JL, Correa-Victoria FJ, Escobar F, Mora L, Duque MC, Deus JE, Cornide MT. Genetic diversity analysis of the rice blast pathogen population at two locations in Cuba. Biotecnología Aplicada. 2003;20:14–19. [Google Scholar]
- Fuller DQ, Qin L, Zheng Y, Zhao Z, Chen X, Hosoya LA, Sun GP. The domestication process and domestication rate in rice: spikelet bases from the Lower Yangtze. Science. 2009;323:1607–1610. doi: 10.1126/science.1166605. [DOI] [PubMed] [Google Scholar]
- Garris AJ, Tai TH, Coburn J, Kresovich S, McCouch S. Genetic structure and diversity in Oryza sativa L. Genetics. 2005;169:1631–1638. doi: 10.1534/genetics.104.035642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud T, Enjalbert J, Fournier E, Delmote F, Dutech C. Population genetics of fungal diseases of plants. Parasite. 2008;15:449–454. doi: 10.1051/parasite/2008153449. [DOI] [PubMed] [Google Scholar]
- Gladieux P, Zhang XG, Afoufa-Bastien D, Valdebenito Sanhueza RM, Sbaghi M, Le Cam B. On the origin and spread of the scab disease of apple: out of Central Asia. PLoS One. 2008;1:e1455. doi: 10.1371/journal.pone.0001455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Alpizar L, Carbone I, Ristaino JB. An Andean origin of Phytophthora infestans inferred from mitochondrial and nuclear gene genealogies. Proceedings of the National Academy of Sciences, USA. 2007;104:3306–3311. doi: 10.1073/pnas.0611479104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Varela G, González AJ, Milgroom MG. Clonal population structure and introductions of the chestnut blight fungus, Cryphonectria parasitica, in Asturias, northern Spain. European Journal of Plant Pathology. 2011;131:67–69. [Google Scholar]
- Guillemaud T, Ciosi M, Lombaert E, Estoup A. Biological invasions in agricultural settings: insights from evolutionary biology and population genetics. Comptes Rendus Biologies. 2011;334:237–246. doi: 10.1016/j.crvi.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Hardy OJ, Vekemans X. SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Molecular Ecology Notes. 2002;2:618–620. [Google Scholar]
- Huang X, Kurata N, Wei X, Wang ZX, Wang A, Zhao Y, Liu K, Lu H, Li W, Guo Y, et al. A map of rice genome variation reveals the origin of cultivated rice. Nature. 2012;490:497–503. doi: 10.1038/nature11532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufbauer RA, Facon B, Ravigné V, Turgeon J, Foucaud J, Lee CE, Rey O, Estoup A. Anthropogenically induced adaptation to invade (AIAI): contemporary adaptation to human-altered habitats within the native range can promote invasions. Evolutionary Applications. 2012;5:89–101. doi: 10.1111/j.1752-4571.2011.00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurles ME, Sykes BC, Jobling MA, Forster P. The dual origin of the Malagasy in Island Southeast Asia and East Africa: evidence from maternal and paternal lineages. American Journal of Human Genetics. 2005;76:894–901. doi: 10.1086/430051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa T, Shimamoto K. Becoming a model plant: the importance of rice to plant science. Trends in Plant Science. 1996;1:95–99. [Google Scholar]
- Javan-Nikkah M, McDonald BA, Banke S, Hedjaroude GA. Genetic structure of Iranian Pyricularia grisea populations based on re-PCR fingerprinting. European Journal of Phytopathology. 2004;110:909–919. [Google Scholar]
- Jombart T, Devillard S, Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genetics. 2010;11:94. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzaki H, Yoshida K, Saitoh H, Fujisaki K, Hirabuchi A, Allaux L, Fournier E, Tharreau D, Terauchi R. Arms race co-evolution of Magnaporthe oryzae AVR-Pik and rice Pik genes driven by their physical interactions. Plant Journal. 2012;72:894–907. doi: 10.1111/j.1365-313X.2012.05110.x. [DOI] [PubMed] [Google Scholar]
- Kaye C, Milazzo J, Rozenfeld S, Lebrun MH, Tharreau D. The development of simple sequence repeat markers for Magnaporthe grisea and their integration into an established genetic linkage map. Fungal Genetics and Biology. 2003;40:207–214. doi: 10.1016/j.fgb.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Kumar J, Nelson RJ, Zeigler RS. Population structure and dynamics of Magnaporthe grisea in the Indian Himalayas. Genetics. 1999;152:971–984. doi: 10.1093/genetics/152.3.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson Handley LJ, Estoup A, Evans DM, Thomas CE, Lombaert E, Facon B, Aebi A, Roy HE. Ecological genetics of invasive alien species. BioControl. 2011;56:409–428. [Google Scholar]
- Leberg PL. Estimating allelic richness: effects of sample size and bottlenecks. Molecular Ecology. 2002;11:2445–2449. doi: 10.1046/j.1365-294x.2002.01612.x. [DOI] [PubMed] [Google Scholar]
- Lee CE, Gelembiuk GW. Evolutionary origins of invasive populations. Evolutionary Applications. 2008;1:427–448. doi: 10.1111/j.1752-4571.2008.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie JF, Klein KK. Female fertility and mating type effects on effective population size and evolution in filamentous fungi. Genetics. 1996;144:557–567. doi: 10.1093/genetics/144.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy M, Correa FJ, Zeigler RS, Xu S, Hamer JE. Genetic diversity of the rice blast fungus in a disease nursery in Colombia. Phytopathology. 1993;83:1427–1433. [Google Scholar]
- Levy M, Romao J, Marchetti MA, Hamer JE. DNA fingerprinting with a dispersed repeated sequence resolves pathotype diversity in the rice blast fungus. Plant Cell. 1991;3:95–102. doi: 10.1105/tpc.3.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombaert E, Guillemaud T, Cornuet J-M, Malausa T, Facon B, Estoup A. Bridgehead effect in the worldwide invasion of the biocontrol Harlequin ladybird. PLoS One. 2010;5:e9743. doi: 10.1371/journal.pone.0009743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londo JP, Chiang YC, Hung KH, Chiang TY, Schaal BA. Phylogeography of Asian wild rice, Oryza rufipogon, reveals multiple independent domestications of cultivated rice, Oryza sativa. Proceedings of the National Academy of Sciences, USA. 2006;103:9578–9583. doi: 10.1073/pnas.0603152103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montarry J, Andrivon D, Glais I, Corbiere R, Mialdea G, Delmotte F. Microsatellite markers reveal two admixed genetic groups and an ongoing displacement within the French population of the invasive plant pathogen Phytophthora infestans. Molecular Ecology. 2010;19:1965–1977. doi: 10.1111/j.1365-294X.2010.04619.x. [DOI] [PubMed] [Google Scholar]
- Nei M. Molecular evolutionary genetics. New York, NY, USA: Columbia University Press; 1987. [Google Scholar]
- Nottéghem JL, Silué D. Distribution of the mating type alleles in Magnaporthe grisea populations pathogenic on rice. Phytopathology. 1992;82:421–424. [Google Scholar]
- Park SY, Milgroom MG, Han SS, Kang S, Lee YH. Diversity of pathotypes and DNA fingerprint haplotypes in populations of Magnaporthe grisea in Korea over two decades. Phytopathology. 2003;93:1378–1385. doi: 10.1094/PHYTO.2003.93.11.1378. [DOI] [PubMed] [Google Scholar]
- Park SY, Milgroom MG, Han SS, Kang S, Lee YH. Genetic differentiation of Magnaporthe oryzae populations from scouting plots and commercial rice fields in Korea. Phytopathology. 2008;98:436–442. doi: 10.1094/PHYTO-98-4-0436. [DOI] [PubMed] [Google Scholar]
- Parks JC, Werth CR. A study of spatial features of clones in a population of bracken fern, Pteridium aquilinumDennstaedtiaceae. American Journal of Botany. 1993;80:537–544. doi: 10.1002/j.1537-2197.1993.tb13837.x. [DOI] [PubMed] [Google Scholar]
- Pennisi E. Armed and dangerous. Science. 2010;327:804–805. doi: 10.1126/science.327.5967.804. [DOI] [PubMed] [Google Scholar]
- Piotti E, Rigano MM, Rodino D, Rodolfi M, Castiglione S, Picco AM, Sala F. Genetic structure of Pyricularia grisea (Cooke) Sacc. Isolates from Italian paddy fields. Journal of Phytopathology. 2005;153:80–86. [Google Scholar]
- Pritchard JK, Stephens P, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raboin LM, Selvi A, Oliveira KM, Paulet F, Calatayud C, Zapater MF, Brottier P, Luzaran R, Garsmeur O, Carlier J, et al. Evidence for the dispersal of a unique lineage from Asia to America and Africa in the sugarcane fungal pathogen Ustilago scitaminea. Fungal Genetics and Biology. 2007;44:64–76. doi: 10.1016/j.fgb.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Raymond M, Rousset F. Genepop (version-1.2) – Population-genetics software for exact tests and ecumenicism. Journal of Heredity. 1995;86:248–249. [Google Scholar]
- Robert S, Ravigne V, Zapater MF, Abadie C, Carlier J. Contrasting introduction scenarios among continents in the worldwide invasion of the banana fungal pathogen Mycosphaerella fijiensis. Molecular Ecology. 2012;21:1098–1114. doi: 10.1111/j.1365-294X.2011.05432.x. [DOI] [PubMed] [Google Scholar]
- Roumen E, Levy M, Nottéghem JL. Characterisation of the European pathogen population of Magnaporthe grisea by DNA fingerprinting and pathotype analysis. European Journal of Plant Pathology. 1997;103:363–371. [Google Scholar]
- Saleh D, Xu P, Shen Y, Li CY, Adreit H, Milazzo J, Ravigné V, Bazin E, Nottéghem JL, Fournier E, et al. Sex at the origin: an Asian population of the rice blast fungus Magnaporthe oryzae reproduces sexually. Molecular Ecology. 2012;21:1330–1344. doi: 10.1111/j.1365-294X.2012.05469.x. [DOI] [PubMed] [Google Scholar]
- Schurko AM, Logsdon JM., Jr Using a meiosis detection toolkit to investigate ancient asexual “scandals” and the evolution of sex. BioEssays. 2008;30:579–589. doi: 10.1002/bies.20764. [DOI] [PubMed] [Google Scholar]
- Shull V, Hamer J. Genome structure and variability in Pyricularia grisea. In: Zeigler RS, Leong SA, Teng P, editors. Rice blast disease. Wallingford, UK: CAB International; 1994. pp. 65–86. [Google Scholar]
- Silué D, Nottéghem JL. Production of perithecia of Magnaporthe oryzae on rice plants. Mycological Research. 1990;94:1151–1152. [Google Scholar]
- Soubabère O, Dioh W, Lebrun MH, Nottéghem JL, Tharreau D. Comparative continental variation of the rice blast fungus using Sequence Characterized Amplified Region markers. In: Tharreau D, Lebrun MH, Talbot NJ, Nottéghem JL, editors. Advances in rice blast research. Dordrecht, the Netherlands: Kluwer Academic Press; 2000. pp. 209–213. [Google Scholar]
- Stenberg P, Lundmark M, Saura A. MLGSim: a program for detecting clones using a simulation approach. Molecular Ecology Notes. 2003;3:329–331. [Google Scholar]
- Stukenbrock EH, Banke S, Javan-Nikkhah M, McDonald BA. Origin and domestication of the fungal wheat pathogen Mycosphaerella graminicola via sympatric speciation. Molecular Biology and Evolution. 2007;24:398–411. doi: 10.1093/molbev/msl169. [DOI] [PubMed] [Google Scholar]
- Stukenbrock EH, McDonald BA. The origins of plant pathogens in agro-ecosystems. Annual Review of Phytopathology. 2008;46:75–100. doi: 10.1146/annurev.phyto.010708.154114. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Ashizawa T, Hirayae K, Moriwaki J, Sone T, Sonoda R, Noguchi MT, Nagashima S, Ishikawa K, Arai M. One of two major paralogs of AVR-Pita1 is functional in Japanese rice blast isolates. Phytopathology. 2010;100:612–618. doi: 10.1094/PHYTO-100-6-0612. [DOI] [PubMed] [Google Scholar]
- Takan JP, Chipili J, Muthumeenakshi S, Talbot NJ, Manyasa EO, Bandyopadhyay R, Sere Y, Nutsugah SK, Talhinhas P, Hossain M, et al. Magnaporthe oryzae populations adapted to finger millet and rice exhibit distinctive patterns of genetic diversity, sexuality and host interaction. Molecular Biotechnologies. 2012;50:145–158. doi: 10.1007/s12033-011-9429-z. [DOI] [PubMed] [Google Scholar]
- Tharreau D, Fudal I, Andriantsimialona D, Santoso , Utami D, Fournier E, Lebrun MH, Nottéghem JL. World population structure and migration of the rice blast fungus, Magnaporthe oryzae. In: Wang GL, Valent B, editors. Advances in genetics, genomics and control of rice blast disease. Dordrecht, the Netherlands: Springer; 2009. pp. 209–215. [Google Scholar]
- Tibayrenc M, Kjellberg F, Ayala FJ. A clonal theory of parasitic protozoa: the population structures of Entomœba, Giardia, Leishmania, Nægleria, Plasmodium, Trichomonas, and Trypanosoma and their medical and taxonomical consequences. Proceedings of the Natural Academy of Sciences, USA. 1990;87:2414–2418. doi: 10.1073/pnas.87.7.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valent B. Rice blast as a model system for plant pathology. Phytopathology. 1990;80:33–36. [Google Scholar]
- Valent B, Crawford MS, Weaver CG, Chumley FG. Genetic studies of fertility and pathogenicity in Magnaporthe griseaPyricularia oryzae. Iowa State Journal of Research. 1986;60:569–594. [Google Scholar]
- Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- Xia JQ, Correll JC, Lee FN, Rhoads DD, Marchetti MA. DNA fingerprint to examine variation in the Magnaporthe griseaPyricularia grisea) population in two rice fields in Arkansas. Phytopathology. 1993;83:1029–1035. [Google Scholar]
- Xia JQ, Correll J, Lee FN, Ross WJ. Regional population diversity of Pyricularia grisea in Arkansas and the influence of host selection. Plant Disease. 2000;84:877–884. doi: 10.1094/PDIS.2000.84.8.877. [DOI] [PubMed] [Google Scholar]
- Xu JR, Hamer JE. Assessment of Magnaporthe grisea mating type by spore PCR. Fungal Genetics Newsletter. 1995;42:80–81. [Google Scholar]
- Zeigler RS. Recombination in Magnaporthe grisea. Annual Review of Phytopathology. 1998;36:249–275. doi: 10.1146/annurev.phyto.36.1.249. [DOI] [PubMed] [Google Scholar]
- Zeigler RS, Cuoc LX, Scott RP, Bernardo MA, Chen DH, Valent B, Nelson RJ. The relationship between lineage and virulence in Pyricularia grisea in the Philippines. Phytopathology. 1995;85:443–451. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genetic structure of worldwide individuals of Magnaporthe oryzae: comparison between the DAPC and Structure methods.
Fig. S2 Number of shared and specific alleles calculated over 10 microsatellites for the four Magnaporthe oryzae clusters identified in Asia.
Characteristics of the 10 microsatellites used for Magnaporthe oryzae genotyping
Table S2 Unbiased gene diversity and number of private alleles in random subsamples performed in 46 Magnaporthe oryzae worldwide populations
Table S3 Gene diversity and mean number of private alleles in subsamples of Magnaporthe oryzae individuals from the same genetic cluster in Asia
Table S4 Information on Magnaporthe oryzae multilocus genotypes repeated within populations and shared between populations
Table S5 Distribution of Magnaporthe oryzae individuals in clusters according to the type of rice culture