

Abstract
Chemical mutagenesis is routinely used to create large numbers of rare mutations in plant and animal populations, which can be subsequently subjected to selection for beneficial traits and phenotypes that enable the characterization of gene functions. Several next-generation sequencing (NGS)-based target enrichment methods have been developed for the detection of mutations in target DNA regions. However, most of these methods aim to sequence a large number of target regions from a small number of individuals. Here, we demonstrate an effective and affordable strategy for the discovery of rare mutations in a large sodium azide-induced mutant rice population (F2). The integration of multiplex, semi-nested PCR combined with NGS library construction allowed for the amplification of multiple target DNA fragments for sequencing. The 8 × 8 × 8 tridimensional DNA sample pooling strategy enabled us to obtain DNA sequences of 512 individuals while only sequencing 24 samples. A stepwise filtering procedure was then elaborated to eliminate most of the false positives expected to arise through sequencing error, and the application of a simple Student's t-test against position-prone error allowed for the discovery of 16 mutations from 36 enriched targeted DNA fragments of 1024 mutagenized rice plants, all without any false calls.
Keywords: NGS, induced mutation detection, large samples pooling, multiplexed target enrichment
Introduction
Genetic variations that are caused by radiation, chemical mutagens and errors in the process of DNA replication are the basis of genetic diversity. Chemical mutagens, such as ethyl methanesulfonate (EMS), ethylnitrosourea (ENU) and sodium azide, have been widely used in animal and plant mutagenesis (Natarajan, 2005). The alkylating agents, for example EMS, can alkylate guanine bases, leading to the misplacing of a thymine residue over a cytosine residue opposite to the O-6-ethyl guanine by DNA polymerase during replication. Also, sodium azide was believed to be metabolized by the cells to form the mutagenic agent, presumably azidoalanine (Owais and Kleinhofs, 1988), which preferentially create A to G and T to C mutations (Cooper et al., 2008; Olsen et al., 1993; Suzuki et al., 2008; Till et al., 2007). The high frequency of point mutations in the chemical-induced mutagenized population often leads to a large phenotypic variation. A reverse genetic approach, namely target-induced local lesions in genome (TILLING), was developed to identify mutations from this type of mutated population (McCallum et al., 2000). Basically, point mutations of the heteroduplex DNA are recognized and nicked by Cel-I endonuclease (Yang et al., 2000), resulting in a mixture of two shorter DNA fragments in addition to the original PCR products (Colbert et al., 2001). High-performance liquid chromatography (Caldwell et al., 2004), polyacrylamide gel electrophoresis (PAGE) (Raghavan et al., 2007), capillary electrophoresis (Suzuki et al., 2008) and matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) (Van Den Boom and Ehrich, 2007) have been applied to effectively separate and detect those nicked DNA fragments from highly mutated mutant populations (Wang et al., 2010). TILLING has been applied for target gene functional analysis in all major crops such as rice (Leung et al., 2001), barley (Caldwell et al., 2004), wheat (Slade et al., 2005), maize (Till et al., 2004), pea (Triques et al., 2007) and soybean (Cooper et al., 2008). But, detection of point mutations from a mutant population consisting of several thousand M2 individuals by TILLING technology is labour-intensive and time-consuming.
The next-generation sequencing (NGS) technology enables scientists to obtain huge amount of DNA sequence data from a single experiment, providing a potential way to directly detect point mutations in a highly mixed DNA samples in a large-scale population. As NGS remains too costly to allow for routine whole-genome sequencing of large numbers of individuals within a given species (Mamanova et al., 2010), the focus has been to develop target enrichment methods to allow sequencing to be directed at a small number of specific genomic regions. Such strategies are relevant for assessing levels of genetic variation and for developing diagnostic marker assays, where nontarget genomic regions are irrelevant (O'Roak et al., 2012). The three leading NGS target enrichment strategies involved (i) a PCR-based approach, (ii) the use of molecular inversion probes (MIP) (O'Roak et al., 2012) and (iii) microarray-based capture (Nijman et al., 2010). Both the MIP- and microarray-based methods are particularly well suited to the parallel sequencing of large numbers of target DNA fragments, but given the complexity of the enrichment process and the need to construct an NGS library, these methods are not cost- and/or labour-efficient enough to deal with the sort of sample numbers needed for the detection of rare mutations. Instead, a TILLING by sequencing strategy has been proposed, in which NGS was applied to amplicons obtained from a template of pooled DNA samples (Tsai et al., 2011). Such a PCR-based approach is difficult to multiplex, and the many cycles of PCR required to initially achieve the amplification of the target DNA sequences, then subsequently constructing the NGS library, risk the introduction of many artefactual nucleotide changes, leading to an unacceptably high background error rate. Moreover, the uneven pooling strategy of the tridimensional pool (12 × 16 × 16) lowers the comparability of bulked samples from different dimensions, which resulted in the unresolved false-negative problem.
We present here a strategy in which multiplexed semi-nested PCR is combined with NGS library construction for the sequencing of amplicons derived from tridimensional, evenly pooled DNA samples (Figure 1). The application of semi-nested PCR, which is similar to a nested PCR (Bej et al., 1991; Haqqi et al., 1988) except that one of the primers in the first PCR is reused in the second PCR, ensures high specificity and efficiency in target enrichment. Integration of semi-nested PCR with NGS library construction allows for a reduction in the number of PCR cycles, thereby greatly diminishing the background mutation rate. A stepwise filtering procedure was then elaborated to eliminate most of the false positives expected to arise through sequencing error while including most of the genuine mutations, and a simple Student's t-test was applied to further eliminate position-prone errors. The even number of samples pooled in each bulk ensured the comparability of bulks from different dimensions, which enhanced the accuracy of mutation calling. As a result of this novel strategy, the detection of rare mutations via direct sequencing became both cost-effective and easily manageable in terms of the complexity of the data handling procedures.
Figure 1.
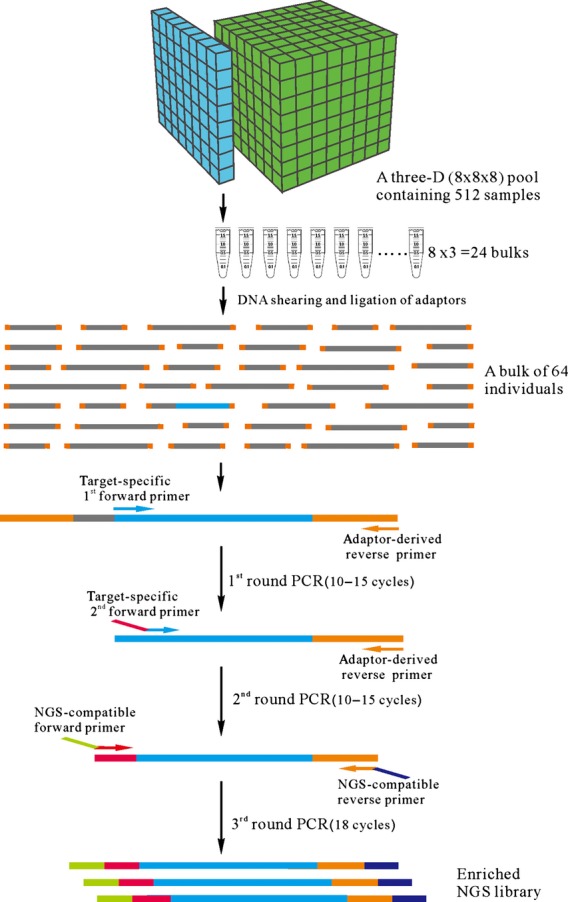
Schematic illustration of the semi-nested PCR-based multiple target enrichment method, based on tridimensional pooling of DNA. Two independent sets of 512 DNA samples were arranged in the form of 8 × 8 × 8 arrays, from each of which 24 DNA bulks, each comprising 64 individuals, were created by pooling equimolar amounts of DNA taken from the samples arrayed in each plane in all three orthogonal directions. The DNA was then sheared, ligated to adaptors and amplified by three rounds of PCR. The first PCR was based on a target-specific forward primer and an adapter-derived reverse primer. The second PCR was based on a target-specific nested primer with a 5′ overhang and the nested adapter-derived reverse primer. The overhang and the adapter that anchored the NGS-compatible forward and reverse primers were used for the third round PCR. Bar codes were embedded within the adaptors. The resulting NGS library consisted of a population of fragments of variable length, each having one fixed and one variable end.
Results
Multiplex enrichment of target DNA from pooled samples
In a pilot experiment, a model template was generated by mixing DNA extracted from five known mutants (C4151T, C7973T, C9835T, C9880T and G9818A) for the rice gene Os08g12740 with DNA from wild type cv. Zhonghua 11 in a ratio of 1:63, which simulated a mutant allele frequency of 1/64 where the mutation was in the homozygous state or1/128 if heterozygous. The subpools were enriched for the targets by a semi-nested PCR based on a variable number (3, 5, 7, 10 or 13) of target-specific primers (Table S1). When NGS was applied to these five libraries, a bar coding method (Table S1) showed that of the 2.04 Gbp of sequence generated, 19.21% related to C4151T, 18.71% to C7973T, 22.75% to C9835T, 22.82% to C9880T and 16.50% to G9818A. Overall, 54–72% of the high-quality reads are mapped to the targeted sequences (Figure 2a). Encouragingly, the semi-nested PCRs involving ten or fewer primers achieved an even enrichment of all of the target DNA fragments (Figure 2a). The highest on-target ratio (72%) was achieved by the ten primer reaction. With 13 primers, only 11 of the target fragments were enriched, and an uneven enrichment of the target fragments was observed (varying from 3% to 20%). An uneven depth coverage pattern (‘cliff’ shape), which resulted from overlapping reads derived from both the fixed and random amplified ends, featured in nearly all of the enriched regions (Figure 2b). The length of enriched target fragments ranged from 208 to 431 bp, when a minimum sequencing depth of 512 was applied. In a 10-plex semi-nested PCR, 3563 bp of the target DNA fragments were amplified (Figure 2b). This model experiment demonstrated that combining semi-nested PCR with NGS library construction provided an effective means of enriching for multiple target DNA fragments.
Figure 2.

Summary of the pilot experiment. (a) The number of enriched target fragments and the enrichment efficiency. The pie chart at the top shows the total number of reads and the maximum and minimum proportions of reads in the five enriched libraries produced by varying the number of target-specific semi-nested PCR primers. The red line indicates the number of enriched target fragments when 3–13 target-specific semi-nested PCR primers were used to amplify templates of the corresponding subpools. The histogram in cyan indicates the ratio of on-target sequences. (b) Sequencing depth of the enriched target DNA fragments. (c) A box plot showing the rate of both sequencing errors and genuine mutations in the five subpools. The mean rate of each nucleotide substitution type is shown above the box plot. (d) The sequencing error rate in the vicinity of GGT/ACC trinucleotides. (e) The detected base substitution rates of C to T or G to A plotted against sequencing depth. (f) The background sequencing error rate plotted against sequencing depth. The dotted lines indicate expected allele frequencies of 15.6 × 10−3 and 7.8 × 10−3 in a pooled sample comprising, respectively, a single homozygous and a single heterozygous mutant along with 63 wild-type individuals.
Reducing the number of PCR cycles, for the most part, resulted in a reduction in sequencing error (0.19–0.62 × 10−3, see Figure 2c), as compared to the rate (0.08–1.3 × 10−3) observed in a rice TILLING population in a previous study (Tsai et al., 2011). To our surprise, the GGT/ACC motif appeared to be particularly prone to sequencing error (6.21 × 10−3 and 13.53 × 10−3, respectively) (Figure 2d). As Illumina HiSeq2000-based sequencing uses the same laser wavelength to excite the fluorophore attached to both G and T (Minoche et al., 2011), this may simply be a contamination artefact. The estimated mean frequencies of the known heterozygous and homozygous mutations (5.37–17.32 × 10−3) were close to their expected values of 7.81 and 15.63 × 10−3 (Figure 2c). Where the sequencing depth was particularly high (i.e. >10 000), all five mutations could be unambiguously distinguished (Figure 2e). It was not possible to discriminate between genuine mutations and sequencing errors at sequencing depths <10 000, even though the true mutation rate was about one order of magnitude higher than the average sequencing error rate (Figure 2f).
Estimation of the mutation rate variation
To discriminate between genuine mutations and sequencing errors, the variation of the mutation rate was analysed based on the data from the pilot experiment. The minimum mutation rate (MiMR, see Experimental procedures section) was proposed to distinguish the genuine mutations from sequencing errors. This criterion was estimated utilizing the genuine mutation data from the pilot experiment. The two heterozygous mutations, C7973T and C9880T, were each sequenced five times, which gave five m/n values (Figure 2e). The respective mean m/n values of C7973T and C9880T were unbiased estimators of X; therefore, they were used to calculate the σX (0.0010). The five m/n values of C7973T and C9880T, respectively, were used to calculate their own σm/n (Figure 3). This quantity was assumed to be independent of X, so the two separate σm/n estimates were then averaged, giving σm/n = 2.8 × 10−4. The probability density function of the normal distributions of both X and m/n is presented in Figure 3. The sample size of X was eight (two in the same subpool were obviously higher than expected; therefore, they were excluded from the calculation); therefore, we used t= = 0.025, df = 7 to calculate the one-tail leftborder of X (3.7 × 10−3). The one-tail left border representing 97.5% of m/n lies at 3.02 × 10−3. This value was considered as the MiMR threshold, indicating that any position with an m/n below this value was due to sequencing error. Theoretically, 95% (97.5% × 97.5%) of the genuine mutations will remain following the application of this threshold filter.
Figure 3.
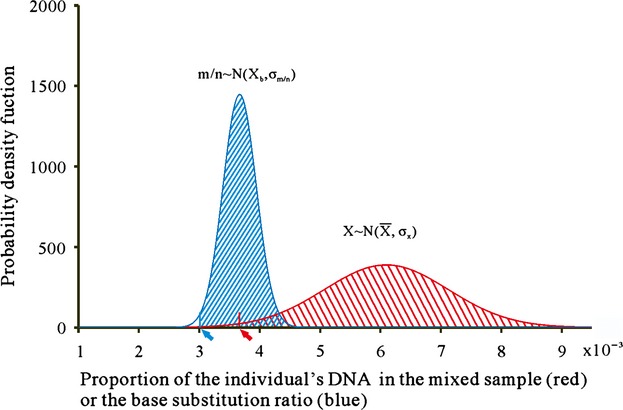
The probability density function of the normal distribution of DNA proportions of genuine mutations in the bulked samples (red area) and the ratio of base substitution numbers in total sequencing depth (the m/n ratio, blue area). The red and blue arrows indicate the one-tail left-hand borders equivalent to 97.5% of the DNA proportion and the m/n ratio, respectively.
Sequencing the enriched target DNA from extensively pooled tridimensional samples
In total, 70 468 200 high-quality reads (6.95 Gbp) were obtained from the 48 bulked templates from two pools (Pool A and Pool B), derived from 1024 M2 individuals of a rice sodium azide-induced mutated population. The mean proportion of on-target reads across all 48 bulks was 57.2%, rising to 73.9% in one case (Table S3); thus, the enrichment achieved was similar to that attained in the pilot experiment (Figure 2a). The average length of the 36 target fragments was 146 bp, ranging from 27 to 289 bp in Pool A and from 11 to 292 bp in Pool B (Figure S1a,b, Tables S4 and S5). The average length of the screened region was 2603 bp for each individual in Pool A and 3162 bp in Pool B (Tables S4 and S5). The number of enriched target fragments in the 24 bulks of Pool A was 732 (84.7%) and 785 (90.9%) for Pool B (Figure S1a,b). The total enriched fragment length of the 512 M2 progeny was 1.33 Mbp in Pool A and 1.62 Mbp in Pool B. The average sequencing depth was 7002 in Pool A and 8402 in Pool B (see Figure S1c,d).
Application of multiple criteria for rare mutation discovery
As the mutations induced by sodium azide are typically C to T or G to A (Qi et al., 2006), our focus was directed onto the 117 602 Cs and 120 148 Gs which were mapped to the reference sequences (Figure 4a; Tables S6 and S7). The set of criteria, a minimum sequencing depth (MiSD) of 512 and a minimum mutation rate (MiMR) of 3.02 × 10−3, were applied sequentially, which succeeded in excluding most of the likely sequencing errors (Figure 4a). What remained after the application of the 1X1Y1Z criterion, that is, a candidate mutation was defined by having exactly one base position's mutation rates above 3.02 × 10−3 in each of tridimensional bulks, was a set of 36 Cs and 45 Gs, corresponding to 12 C to T and 15 G to A candidate mutations. Application of a Student's t-test enabled the elimination of position-prone errors at the same base position and resulted in a set of 16 highly likely candidate mutations (Figure 4a). Sanger sequencing confirmed that the identified 16 candidates were all the genuine mutations and that the selected 12 false mutations which were eliminated by the t-test were indeed false positives (Figure 4b; Table S8).
Figure 4.
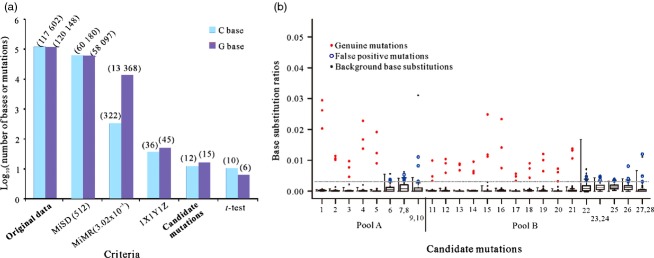
The putative and confirmed candidates detected in the tridimensional pooled DNA samples. (a) The log10 of the number of presumptive mutations remaining after each filtering step (original numbers shown above the relevant column). (b) Validation of the candidate mutations. The grey dotted lines indicate the one-tail left-hand border, equivalent to 95% of the ‘true mutations’ base substitution rate (3.02 × 10−3). The confirmed genuine mutations are marked by red dots. Sequencing errors are marked by blue circles.
We used a very stringent criterion of the 1X1Y1Z to define the candidate mutations. It is possible that more than one genuine mutation is included at the same base position in a bulk which contains 64 highly mutated genomic DNA samples from F2 individuals, or a few extra-high error rates may have been undistinguishable from genuine mutations. When a maximum cumulative bulk data (MaCBD) of 4, 5 and 6 were applied, three more genuine mutations were identified, but 12 false-positive mutations were included (Table 1). Usage of higher MaCBD values more than three results in the substantial increase of false-positive ratios to 62% and 46%. The exclusion of these three genuine mutations by the 1X1Y1Z criterion was due to the extra-high error rates of the same base positions in other bulks.
Table 1.
The number of genuine mutations identified using different MaCBD values
| MaCBD | Pool A | Pool B | ||||
|---|---|---|---|---|---|---|
| Number of genuine mutations | Number of false-positive mutations | False-positive rate (%) | Number of genuine mutations | Number of false-positive mutations | False-positive rate (%) | |
| 4 | 6 | 4 | 40 | 12 | 3 | 20 |
| 5 | 6 | 6 | 50 | 13 | 6 | 32 |
| 6 | 6 | 10 | 62 | 13 | 11 | 46 |
MaCBD, Maximum cumulative bulk data.
Discussion
The strategy that we used for rare mutation discovery relies on three key attributes. Firstly, the combination of multiplex semi-nested PCR with NGS library construction allowed a 12-plex amplification of target DNA fragments and greatly reduced the total number of PCR cycles. This strategy reduces not only the effort required in the experimental process, but also resulted in the reduction in the background mutation rate. Secondly, the application of a 8 × 8 × 8 tridimensional pooling strategy allowed us to simultaneously screen 512 individuals within each 3-D pool while only actually needing to sequence 24 bulked samples. Thirdly, the development of multiple criteria and the stepwise filtering strategy that gradually increases filtering stringency enabled us to eliminate most of the false positives expected to arise through sequencing errors. We were thus able to unambiguously identify genuine rare mutations.
Multiplexing PCR often results in uneven and unspecific amplification of the targets, which may be due to the different efficiencies in primer binding and extension. Our method reduces the differences of primer binding and increases the amplification specificity by introducing semi-nested PCR. Furthermore, the method minimizes the extension difficulty by fragmenting the template. Without additional instrumentation or procedures, we achieved multiplexing of at least 11 primers with minimum 5% to maximum 16% proportion of enriched targets and enriched 3563-bp target DNA fragments by a single 10-plex nested PCR following with NGS library construction reaction (Figure 2b). The multiplex semi-nested PCR strategy used in this study generated relative short enriched target DNA fragments (208–431 bp). This method is particularly well suited to targeting exon sequences of genes with many introns, which can greatly increase chances of screening coding sequences. However, the length of a screened region is determined by the shortest amplified fragments of the tridimensional (X, Y and Z) pools. Indeed, in our mutation identification experiments, much shorter target fragments (2603 bp for each individual in Pool A and 3162 bp in Pool B by three 12-plex PCR enrichments) were screened. To further increase the screening efficiency, techniques for obtaining longer enriched fragments in all tridimensional pools are required.
Recently, two studies (Chen et al., 2013; Tsai et al., 2011) have reported practical efforts in sequencing extensively pooled samples. In these two papers, single targeted DNA fragment was amplified by PCR, and the amplicons were then used for NGS library construction. The singleplex PCR strategy mainly involves eight steps, that is, PCR amplification of targeted DNA fragments, amplicons purification, quantification and fragmentation, adaptor ligation following column purification, one-round PCR and a gel purification. The strategy used in this study integrates target selection into NGS library construction and includes nine major steps: genomic DNA fragmentation, adaptor ligation, three rounds of PCR, column purification of the ligation and the first two PCR products, and gel purification of the third PCR product. Reducing the number of purification and PCR will simplify the experiment procedure and reduce the cost.
Previous works (Chen et al., 2013; and Tsai et al., 2011) used 3-D pooling strategy in rather complex ways. Chen et al. (2013) claimed that they could identify each singleton's pooling pattern without any further experiments. However, there are actually 192 pairs of singletons in their pooling design that have the same pooling pattern (due to the symmetrical positions of each pair in their Pooling 3), which cannot be distinguished from the sequencing result alone. Moreover, the number of mixed samples in each pool did not reach the full pooling capacity; that is, for a 12 × 12 × 12 tridimensional pool, there were only 96 samples in each bulk (if orthogonal pooling was used, there would be 12 × 12 = 144 samples in each pool). Therefore, the number of possible patterns of singletons is less than 10 × 10 × 10 = 1000 < 384 × 3 = 1152 < 12 × 12 × 12 =1728 (pooling of 96 samples in each bulk resulted in less pooling patterns than 10 × 10 = 100 samples in each bulk). This is less than the 243 claimed by Chen et al., which might be the reason why they cannot actually distinguish every singleton's pooling pattern with their method. It is worth noting that full pooling capacity was not achieved in the Tsai et al. method either. They screened 768 samples by sequencing 44 bulked samples (12 + 16 + 16 = 44), using 12 × 16 × 16 tridimensional pooling. In contrast, with our simple 8 × 8 × 8 orthogonal pooling strategy, we sequenced 48 bulked samples to screen 1024 samples of the population, with 9% more bulked samples and 33% more samples screened overall.
The major challenge in detection of rare mutations from large populations is to distinguish genuine mutations from sequencing errors. Therefore, the model for sequencing errors is extraordinarily important for reducing false positives. As pointed out by many previous researches, the sequence context of a given base position can greatly influence the sequencing error rate of that site. However, the algorithm in Chen et al. (2013) did not take sequence context into account (the noise data for the null model was drawn randomly from a pool of all positions' noise data), which resulted in low repeatability of the variant detection (two experimental repeats, one detected 138 putative mutations, the other 161, with a total of 118 overlapped detected putative mutations). Tsai et al. (2011) and Missirian et al. (2011) took the sequence context into account and developed a complex algorithm, but due to their uneven pooling strategies, bulked samples from different dimensions were not comparable, which resulted in an unresolved false-negative problem. In contrast to these particular failings, our stepwise filtering of the data took the advantage of our even number of multidimensional pooling, by comparing the predicted mutation's base substitution rate to all the other 21 predicted noises' sequencing error rates at the same base position (hence fixed the influence of the sequence context) using a simple Student's t-test. This eliminated all of the false-positive predictions from the former filtering step.
In this study, five genuine mutations were identified through direct sequencing of 1.33 Mbp enriched target DNA fragments among the 512 M2 progeny in Pool A, and 11 genuine mutations were identified in Pool B (1.62 Mbp enriched target sequences) of a sodium azide-induced rice mutant population, equivalent to a mutation rate of one per 267 and 147 Kbp, respectively. This represents a higher mutation frequency than the one mutation per 500 Kbp reported elsewhere (Till et al., 2007) and showed that our NGS method is likely more effective/sensitive than traditional TILLING.
Compared with most of the current target re-sequencing strategies (such as Rain-dance PCR (Tewhey et al., 2009), array-based hybridization (Nijman et al., 2010) and molecular inverse probe (O'Roak et al., 2012)) that aim at parallel sequencing of tens of thousands of target regions in a small number of individuals, our method is particularly suitable for sequencing of less than a hundred target DNA regions in thousands of individuals. As such, it is particularly well suited to applications such as the identification of target gene mutants from ten thousand individuals in chemical-induced mutant populations of plants or animals. It is important to note that the extensive tridimensional pooling strategy is better used in populations containing random rare mutations, such as the chemical-induced plant F2 populations. For applications focusing on populations containing inherited rare mutations, such as the detection of disease rare mutations in large human populations, it is recommended to use a more limited pooling strategy that bulks only a few samples, and re-confirmation of rare mutations in the individual samples is required (Comai et al., 2004; Till et al., 2006).
Reduction in sequencing error rate is particularly important for rare mutation detection in the extensively pooled samples. Lowering the sequencing error rate will enable the detection of mutations more accurately and may henceforth allow us to use an even more extensively pooled tridimensional strategy (e.g. 12 × 12 × 12) for sequencing more individuals. Lowering the sequencing error rate would also reduce the required sequencing depth. Coupling our method with the recently proposed duplex sequencing strategy (Schmitt et al., 2013), which eliminates most of the point mutations induced in PCRs, would achieve more cost-effective/labour-effective methods for rare mutation detection.
In summary, through thorough consideration of the critical factors that affect the accuracy of rare mutation detection and carefully designed pooling and multiplexed enrichment strategies, our method achieved a higher efficiency in rare mutation detection in large populations as compared to existing methods. The reductions in cost and improvements in dimensionality (number of individuals screened) will be particularly attractive to research groups who are seeking ways to economically employ and benefit from NGS methods.
Experimental procedures
Generation of rice mutant population
Batches of 25 g of mature caryopses of rice (Oryza sativa L. ssp. japonica) cultivar Zhonghua 11 (obtained from the Chinese Academy of Agricultural Science, Beijing, China) were imbibed in water and then immersed in 2 mm sodium azide for 6 h. The germinating caryopses were rinsed three times for 5 min in water and left to soak for a further 13 h at 30–35 °C, before being planted in a field at Shangzhuang in Beijing. DNA was isolated from each self-fertile individual (M2 plant), using a standard CTAB extraction method.
Establishment of pooled DNA samples
In the pilot experiment, a 3 μL aliquot of 200 ng/μL DNA extracted from each of the five known Os08g12740 mutants (C4151T, C7973T, C9835T, C9880T, G9818A) was mixed with 177 μL of 200 ng/μL cv. Zhonghua 11 DNA. A 30 μL aliquot of this subpool DNA was ligated to adaptors and amplified sequentially using appropriate primers (Table S1).
A tridimensional pooling strategy (as shown in Figure 1), where 512 samples were arrayed as a cube (8 × 8 × 8 = 512), was applied in this study. The three dimensions, or directions, were designated as X, Y and Z, respectively. Each sample in this pool was represented as a unique coordinate, (Xi, Yj, Zk) [i, j, k ∈ (1, 2, 3, …, 8)]. The samples located on the same plane were mixed together; for example, samples (X1, Yj, Zk) [j,k ∈ (1,2,3,..8)] containing 64 individuals' DNA samples were bulked together and named as X1. As such, samples (Xi, Y1, Zk) [i, k ∈ (1,2,3,..8)] were pooled and named as Y1, and so on. For each tridimensional pool, there were 24 bulked samples in total. Each 8 bulked samples of the 24 comes from one of three directions (X1, X2, …, X8 for direction X, Y1, Y2, …, Y8 for direction Y and Z1, Z2,…, Z8 for direction Z). Therefore, each individual's DNA samples must be present once in each of the three directional bulked samples. For example, an individual (X3, Y5, Z6) would only be present in the bulked samples X3, Y5 and Z6. In this study, two unique tridimensional pools, each containing the DNA of 512 individuals, were established.
Multiple target enrichment and NGS library construction
A 6 μg aliquot of each of the five subpool DNA samples of the pilot experiment (or the 48 bulk DNA of the TILLING population) was fragmented by a 90 s exposure to sonication (40 kHz,80W), and then, the DNA was purified using a QIAquick PCR purification kit using protocols recommended by the manufacturer (Qiagen, Hilden, Germany). The fragments were blunted in a reaction mixture containing 20 μL of 50 ng/μL sonicated DNA, 55 μL ddH2O, 10 μL 10 × T4 DNA ligase buffer containing 10 mm ATP, 4 μL 10 mm dNTPs, 5 μL 3 U/μL T4 DNA polymerase, 1 μL 5 U/μL Klenow fragment and 5 μL 10 U/μL T4 PNK. The reaction mixtures were held at 20 °C for 30 min. The resulting DNA was purified using a Qiagen QIAquick PCR Purification kit using protocols recommended by the manufacturer. An adenine was added to the 3′ end of each fragment by mixing 32 μL of the blunting reaction mixture with 25 μL 10 × NEBuffer4, 10 μL 1 mm dATP and 3 μL 5 U/μL Klenow fragment. This mixture was held at 37 °C for 30 min. To prepare the adaptors for ligation, each oligomer (‘long’ and ‘short’, Tables S1 and S2) was diluted to 100 μm, and 25 μL of each dilution was added to 10 μL 10 × NEBuffer4 and 40 μL ddH2O. The DNA was denatured by holding at 95 °C for 2 min, after which the temperature was reduced by 1 °C and held for 36 s for each additional cycle until the temperature reached 24 °C, for the formation of double-stranded DNA. For the ligation reaction, 12 μL of the DNA fragment solution was mixed with 25 μL 2 × quick ligase buffer, 6 μL 25 μm treated adaptors, 5 μL T4 DNA quick ligase and 2 μL ddH2O and held at 20 °C for 15 min. The reaction was finally purified using a MinElute PCR Purification kit, using the protocols recommended by the manufacturer (Qiagen).
The primers for the first two PCRs were presented in an equimolar mixture made up to 2 μm with 1 × TE buffer. In the first round of PCR of the pilot experiment, the forward primers of each subpool consisted of different number of TSPs (target-specific primer, Table S2). The reverse primer was AP1 (adaptor-derived primer), which was used for each of the first round PCRs. In the second round PCRs of the pilot experiment, the forward primers for each subpool were similar to the first round PCR primers, with TSP2-1 instead of TSP 1-1, TSP2-2 instead of TSP1-2,…, TSP2-13 instead of TSP1-13. The reverse primer used in the second round was AP2. In the study of the mutagenized rice population, we used 12 primers in each multiplexed PCR. Therefore, the 36 TSPs were grouped in three sets, with each set containing 12 TSPs. The third round PCR primers were AP3F and AP3R, which were universal for all of the third round PCRs. For the first round PCRs, a 6 μL aliquot of adaptor-ligated DNA was added to 2 μL primer mixture, 0.4 μL of first step universal primer (AP1), 15 μL 2 × Phusion High Fidelity Polymerase PCR Master Mix and 6.6 μL ddH2O; the PCR program comprised 95 °C/60 s denaturation, followed by 12 cycles of 95 °C/20 s, 60 °C/60 s, 68 °C/120 s and an extension step of 68 °C/5 min. The amplicons were purified using a QIAquick PCR purification kit, using protocols recommended by the manufacturer. For the second round PCRs, the template was 6 μL of the amplicon produced by the first PCR, 3 μL primer mixture, 0.6 μL of the second step universal primer (AP2), 15 μL 2 × Phusion High Fidelity Polymerase PCR Master Mix and 5.4 μL ddH2O, and the PCR program comprised 95 °C/60 s denaturation, followed by 13 cycles of 95 °C/20 s, 60 °C/60 s, 68 °C/120 s and followed by an extension of 68 °C/5 min. The resulting amplicons were purified as above. For the third round PCRs, the template was 2 μL of the purified amplicon produced by the second round PCR, 0.6 μL 25 μm of AP3F and AP3R each, 15 μL 2 × Phusion High Fidelity Polymerase PCR Master Mix and 11.8 μL ddH2O. The PCR program comprised 98 °C/30 s denaturation, followed by 18 cycles of 98 °C/10 s, 65 °C/30 s, 72 °C/30 s and then by an extension of 72 °C/5 min. The amplicons were purified using a QIAquick Gel Extraction kit, using protocols recommended by the manufacturer, applying a size selection of 300–500 bp.
Next-generation sequencing and data processing
The resulting library was sequenced using an Illumina HiSeq 2000 platform (paired-end, 2 × 100 bp read length). Low-quality reads (phred quality score lower than 20, that is, error rate >1%) and reads including undefined nucleotides were rejected. The remaining reads were sorted according to the incorporated bar codes (Tables S1 and S2). Then, the bar code sequences were removed by a customized perl script, and the resulting sequences were aligned to the reference sequences of the relevant rice genes using Bowtie 2.0 (a software tool which aligns NGS data with reference sequences, http://bowtie-bio.sourceforge.net/bowtie2/index.shtml). The data files were converted into ‘bam’ format to allow sorting and pile-up analyses using the software package SAMtools (http://samtools.sourceforge.net/). A customized perl script was written to calculate sequencing depth and the number of base substitutions for all possible base substitutions at a given position.
Establishment of multiple criteria for rare mutation discovery
A set of criteria was established to distinguish between genuine mutations and those arising from sequencing errors. This set consisted of the following: (i) a minimum sequencing depth (MiSD), (ii) a minimum mutation rate (MiMR), (iii) the feature of the 3-D pool that a genuine mutation must be presented in each of the tridimensional bulks, X, Y and Z (defined as 1X 1Y 1Z) and (iv) a Student's t-test that was used to discriminate genuine mutations from position-prone sequencing errors. Our strategy for the selection of these criteria was to maximize the retention of genuine mutations through a stepwise exclusion of sequencing errors.
The MiSD threshold of 512
In a bulk of 64 DNA samples, a heterozygous allele is represented by a variant base in one of 128 of the DNA samples. To balance error-free base calling with the depth of sequencing, the aim was to identify the mutant sequence four times. This required a minimum sequencing depth of 128 × 4 = 512.
The MiMR threshold of 3.02 × 10−3
The data for each base position of one of the 48 bulks consisted of two major elements: the number of sequencing depth (the number of times for this base to be sequenced, here designated as n) and the number of base substitutions (here designated as m). Theoretically, for a heterozygous mutation site, the m/n ratio should be 1/128, equalling the proportion of the mutated individual's DNA. But in practice, there were biases in DNA mixing, which could cause a deviation to the mutation rate. For a heterozygous mutation site, if we designated the proportion of the mutated individual's DNA in the mixture to be X, then it was reasonable to assume that X follows a normal distribution (caused by DNA handling), that is,
and σX was unknown. After the DNA samples were mixed, X of each mutated individual was fixed, respectively, but the library construction step caused the mutation rate (m/n) to vary from X. Assuming m/n follows a normal distribution, then
As the library construction and sequencing process were conducted in parallel for each individual's DNA, it was logical to assume that σm/n stays constant for each individual. Estimation of σX, X and σm/n allowed us to calculate a minimum mutation rate under a certain confident level (in this case, 97.5% × 97.5% = 95%).
The 1X1Y1Z
A consequence of the three-dimensional pooling strategy was that a genuine mutation (as opposed to a sequencing error) should be represented once in each of the three-dimensional bulks, that is, one in bulk X, one in bulk Y and one in bulk Z, of the total 24 (8 Xs + 8 Ys + 8 Xs) bulks of the tridimensional pool. The 1X 1Y 1Z criterion is a high-stringency filter that allows only exactly matched positions to pass through, prior to further statistical analysis.
Student's t-test of significance
The DNA sequence context generally has certain influences on sequencing error rates. A few error-prone motifs, such as the GGT/ACC motif, that were identified in this study can be excluded in the analysis when it was predicted or identified. However, it is still hard to deal with a base position where the error rate is close to that of a genuine mutation. For each identified candidate mutation, a nucleic acid base position-based t-test was applied. The 24 m/n ratios of the tridimensional pool at this particular base position were grouped as that of three from candidate genuine mutations and 21 from randomized sequencing error. The t-test was introduced with the null hypothesis that one of the m/n ratios from the identified candidate mutation was due to a sequencing error. The one-tail right-hand border of 99% sequencing errors' m/n ratio was calculated for each of the candidate positions. Any m/n ratio that was smaller than this value was not considered to be significantly different from a sequencing error and was excluded, while whose base positions having mutation rates significantly different from the cut-off values were used to identify the candidate genuine mutations.
Acknowledgments
We thank Xuan Li, John Hugh Snyder and Saleha Bakht for discussion of the manuscript, Yan Yan, Yongzhen Sun and Haiyin Wang for the help on the statistics and the perl script.
Funding
This work was supported by the fund of National Transgenic Megaproject of China [2013ZX08009001] and the Key Project of Chinese National Programs for Fundamental Research and Development [2013CB127000]. Funding for Open Access Charge: [2013ZX08009001].
Supporting Information
Additional Supporting information may be found in the online version of this article:
Summary of the enriched products in Pools A and B.
Table S1 Adaptors and primers used in the pilot experiment.
Table S2 Adaptors and primers used in mutation discovery from the sodium azide-induced rice population.
Table S3 Ratio of on-target reads.
Table S4 Summary of the enriched fragment length of Pool A.
Table S5 Summary of the enriched fragment length of Pool B.
Table S6 Number of candidate mutations remaining after each filtering step in Pool A.
Table S7 Number of candidate mutations remaining after each filtering step in Pool B.
Table S8 The confirmed genuine and false-positive mutations by Sanger sequencing.
References
- Bej AK, Mahbubani MH, Atlas RM. Amplification of nucleic acids by polymerase chain reaction (PCR) and other methods and their applications. Crit. Rev. Biochem. Mol. Biol. 1991;26:301–334. doi: 10.3109/10409239109114071. [DOI] [PubMed] [Google Scholar]
- Caldwell DG, McCallum N, Shaw P, Muehlbauer GJ, Marshall DF, Waugh RA. Structured mutant population for forward and reverse genetics in Barley (Hordeum vulgare L.) Plant J. 2004;40:143–150. doi: 10.1111/j.1365-313X.2004.02190.x. [DOI] [PubMed] [Google Scholar]
- Chen CT, McDavid AN, Kahsai OJ, Zebari AS, Carlson CS. Efficient identification of rare variants in large populations: deep re-sequencing the CRP locus in the CARDIA study. Nucleic Acids Res. 2013;41:e85. doi: 10.1093/nar/gkt092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbert T, Till BJ, Tompa R, Reynolds S, Steine MN, Yeung AT, McCallum CM, Comai L, Henikoff S. High-ghroughput screening for induced point mutations. Plant Physiol. 2001;126:480–484. doi: 10.1104/pp.126.2.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comai L, Young K, Till BJ, Reynolds SH, Greene EA, Codomo CA, Enns LC, Johnson JE, Burtner C, Odden AR, Henikoff S. Efficient discovery of DNA polymorphisms in natural populations by Ecotilling. Plant J. 2004;37:778–786. doi: 10.1111/j.0960-7412.2003.01999.x. [DOI] [PubMed] [Google Scholar]
- Cooper JL, Till BJ, Laport RG, Darlow MC, Kleffner JM, Jamai A, El-Mellouki T, Liu S, Ritchie R, Nielsen N, Bilyeu KD, Meksem K, Comai L, Henikoff S. TILLING to detect induced mutations in soybean. BMC Plant Biol. 2008;8:9. doi: 10.1186/1471-2229-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haqqi TM, Sarkar G, David CS, Sommer SS. Specific amplification with PCR of a refractory segment of genomic DNA. Nucleic Acids Res. 1988;16:11844. doi: 10.1093/nar/16.24.11844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung H, Wu C, Baraoidan M, Bordeos A, Ramos M, Madamba S, Cabauatan P, Vera Cruz C, Portugal A, Reyes G, Bruskiewich R, Mclaren G, Lafitte R, Gregorio G, Bennett J, Brar D, Khush G, Schnable P, Wang G, Leach J. Deletion mutants for functional genomics: progress in phenotyping, sequence assignment, and database development. Rice Genet. 2001;4:239–251. [Google Scholar]
- Mamanova L, Coffey AJ, Scott CE, Kozarewa I, Turner EH, Kumar A, Howard E, Shendure J, Turner DJ. Target-enrichment strategies for next-generation sequencing. Nat. Methods. 2010;7:111–118. doi: 10.1038/nmeth.1419. [DOI] [PubMed] [Google Scholar]
- McCallum CM, Comai L, Greene EA, Henikoff S. Targeted screening for induced mutations. Nat. Biotechnol. 2000;18:455–457. doi: 10.1038/74542. [DOI] [PubMed] [Google Scholar]
- Minoche AE, Dohm JC, Himmelbauer H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and Genome Analyzer systems. Genome Biol. 2011;12:R112. doi: 10.1186/gb-2011-12-11-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missirian V, Comai L, Filkov V. Statistical mutation calling from sequenced overlapping DNA pools in TILLING experiments. BMC Bioinformatics. 2011;12:287. doi: 10.1186/1471-2105-12-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan AT. Chemical mutagenesis: from plants to human. Curr. Sci. India. 2005;89:2. [Google Scholar]
- Nijman IJ, Mokry M, Boxtel R, Toonen P, Bruijn E, Cuppen E. Mutation discovery by targeted genomic enrichment of multiplexed barcoded samples. Nat. Methods. 2010;7:913–915. doi: 10.1038/nmeth.1516. [DOI] [PubMed] [Google Scholar]
- Olsen O, Wang X, Von Wettstein D. Sodium azide mutagenesis: preferential generation of A·T→G·C transitions in the barley Ant18 gene. Proc. Natl Acad. Sci. USA. 1993;90:8043–8047. doi: 10.1073/pnas.90.17.8043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, Munson J, Hiatt JB, Turner EH, Levy R, O'Day DR, Krumm N, Coe BP, Martin BK, Borenstein E, Nickerson DA, Mefford HC, Doherty D, Akey JM, Bernier R, Eichler EE, Shendure J. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owais WM, Kleinhofs A. Metabolic activation of the mutagen azide in biological systems. Mutat. Res. 1988;197:313–323. doi: 10.1016/0027-5107(88)90101-7. [DOI] [PubMed] [Google Scholar]
- Qi X, Bakht S, Qin B, Leggett M, Hemmings A, Mellon F, Eagles J, Werck-Reichhart D, Schaller H, Lesot A, Melton R, Osbourn A. A different function for a number of an ancient and highly conserved cytochrome P450 family: from essential sterols to plant defense. Proc. Natl Acad. Sci. USA. 2006;103:18848–18853. doi: 10.1073/pnas.0607849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan C, Naredo MEB, Wang H, Atienza G, Liu B, Qiu F, McNally KL, Leung H. Rapid method for detecting SNPs on agarose gels and its application in candidate gene mapping. Mol. Breed. 2007;19:87–101. [Google Scholar]
- Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl Acad. Sci. USA. 2013;109:14508–14513. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade AJ, Fuerstenberg SI, Loeffler D, Steine MN, Facciotti D. A reverse genetic nontransgenic approach to wheat crop improvement by TILLING. Nat. Biotechnol. 2005;23:75–81. doi: 10.1038/nbt1043. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Eiguchi M, Kumamaru T, Satoh H, Matsusaka H, Moriguchi K, Nagato Y, Kurata N. MNU-induced mutant pools and high performance TILLING enable finding of any gene mutation in rice. Mol. Genet. Genom. 2008;279:213–223. doi: 10.1007/s00438-007-0293-2. [DOI] [PubMed] [Google Scholar]
- Tewhey R, Warner JB, Nakano M, Libby B, Medkova M, David PH, Kotsopoulos SK, Samuels ML, Hutchison JB, Larson JW, Topol EJ, Weiner MP, Harismendy O, Olson J, Link DR, Frazer KA. Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nat. Biotechnol. 2009;27:1025–1031. doi: 10.1038/nbt.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till BJ, Reynolds SH, Weil C, Springer N, Burtner C, Young K, Bowers E, Codomo CA, Enns LC, Odden AR, Greene EA, Comai L, Henikoff S. Discovery of induced point mutations in maize genes by TILLING. BMC Plant Biol. 2004;4:12. doi: 10.1186/1471-2229-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till BJ, Zerr T, Bowers E, Greene EA, Comai L, Henikoff S. High-throughput discovery of rare human nucleotide polymorphisms by Ecotilling. Nucleic Acids Res. 2006;34:e99. doi: 10.1093/nar/gkl479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till BJ, Cooper J, Tai TH, Colowit P, Greene EA, Henikoff S, Comai L. Discovery of chemically induced mutations in rice by TILLING. BMC Plant Biol. 2007;7:19. doi: 10.1186/1471-2229-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triques K, Sturbois B, Gallais S, Dalmais M, Chauvin S, Clepet C, Aubourg S, Rameau C, Caboche M, Bendahmane A. Characterization of Arabidopsis thaliana mismatch specific endonucleases: application to mutation discovery by TILLING in pea. Plant J. 2007;51:1116–1125. doi: 10.1111/j.1365-313X.2007.03201.x. [DOI] [PubMed] [Google Scholar]
- Tsai H, Howell T, Nitcher R, Missirian V, Watson B, Ngo KJ, Lieberman M, Fass J, Uauy C, Tran RK, Khan AA, Filkov V, Tai TH, Dubcovsky J, Comai L. Discovery of rare mutations in populations: TILLING by sequencing. Plant Physiol. 2011;156:1257–1268. doi: 10.1104/pp.110.169748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Boom D, Ehrich M. Discovery and identification of sequence polymorphism and mutations with MALDI-TOF MS. Methods Mol. Biol. 2007;366:287–306. doi: 10.1007/978-1-59745-030-0_16. [DOI] [PubMed] [Google Scholar]
- Wang T, Uauy C, Till B, Liu CM. TILLING and associated technologies. J. Integr. Plant Biol. 2010;52:1027–1030. doi: 10.1111/j.1744-7909.2010.00999.x. [DOI] [PubMed] [Google Scholar]
- Yang B, Wen X, Kodali NS, Oleykowski CA, Miller CG, Kulinsi J, Besack D, Yeung JA, Kowalski D, Yeung AT. Purification, cloning, and characterization of the CEL I nuclease. Biochemistry. 2000;39:3533–3541. doi: 10.1021/bi992376z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of the enriched products in Pools A and B.
Table S1 Adaptors and primers used in the pilot experiment.
Table S2 Adaptors and primers used in mutation discovery from the sodium azide-induced rice population.
Table S3 Ratio of on-target reads.
Table S4 Summary of the enriched fragment length of Pool A.
Table S5 Summary of the enriched fragment length of Pool B.
Table S6 Number of candidate mutations remaining after each filtering step in Pool A.
Table S7 Number of candidate mutations remaining after each filtering step in Pool B.
Table S8 The confirmed genuine and false-positive mutations by Sanger sequencing.