

Abstract
Numerous empirical studies have reported lack of migration–drift equilibrium in wild populations. Determining the causes of nonequilibrium population structure is challenging because different evolutionary processes acting at a variety of spatiotemporal scales can produce similar patterns. Studies of contemporary populations in northern latitudes suggest that nonequilibrium population structure is probably caused by recent colonization of the region after the last Pleistocene ice age ended ∼13 000 years ago. The chum salmon's (Oncorhynchus keta) range was fragmented by dramatic environmental changes during the Pleistocene. We investigated the population structure of chum salmon on the North Alaska Peninsula (NAP) and, using both empirical data and simulations, evaluated the effects of colonization timing and founder population heterogeneity on patterns of genetic differentiation. We screened 161 single nucleotide polymorphisms and found evidence of nonequilibrium population structure when the slope of the isolation-by-distance relationship was examined at incremental spatial scales. In addition, simulations suggested that this pattern closely matched models of recent colonization of the NAP by secondary contact. Our results agree with geological and archaeological data indicating that the NAP was a dynamic landscape that may have been more recently colonized than during the last deglaciation because of dramatic changes in coastal hydrology over the last several thousand years.
Keywords: chum salmon, glaciation, isolation by distance, population genetics, single nucleotide polymorphisms
Introduction
A central goal of population genetics is to describe the spatial distribution of population structure and understand how it is influenced by environmental and demographic conditions. In wild populations, allele frequencies change in response to natural selection, genetic drift, mutation and gene flow. However, these different evolutionary forces can produce similar genetic patterns, making it difficult to infer which processes are responsible for observed population structure unless demographic or historical landscape data are available (Strand et al. 2012). Many species have locally restricted patterns of dispersal and gene flow (hereafter referred to as migration), mediated both by dispersal ability and the surrounding environment. Understanding migration patterns is pivotal to interpretations of hierarchical population structure and selection (Meirmans 2012).
A commonly tested hypothesis is that migration is spatially limited and occurs more frequently between proximate populations. If this relationship exists over many generations, then genetic differentiation is expected to be positively correlated with the geographical distance separating populations, a pattern known as isolation by distance (IBD; Wright 1943; Kimura & Weiss 1964). The geographical extent over which IBD is present depends on the spatial arrangement of populations, the rate of migration, and elapsed time. Theoretical models show that in a one-dimensional array of ideal populations (equal size, equal migration rates, no selection), the distance over which IBD is present depends on 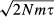 , where τ = generations since separation, m = migration rate and N = effective population size (Slatkin 1993). IBD forms first between adjacent populations (Kimura & Weiss 1964) and extends over larger spatial scales as time passes. When this migration pattern remains stable over time, populations may reach migration–drift equilibrium and the same IBD slope will be observed over all geographical distances. In two-dimensional IBD systems, genetic differentiation is linearly correlated with the log of geographical distance (Slatkin 1993) and therefore decreases asymptotically when plotted against linear geographical distance. Nonequilibrium one-dimensional and equilibrium two-dimensional systems therefore show similar patterns of IBD.
, where τ = generations since separation, m = migration rate and N = effective population size (Slatkin 1993). IBD forms first between adjacent populations (Kimura & Weiss 1964) and extends over larger spatial scales as time passes. When this migration pattern remains stable over time, populations may reach migration–drift equilibrium and the same IBD slope will be observed over all geographical distances. In two-dimensional IBD systems, genetic differentiation is linearly correlated with the log of geographical distance (Slatkin 1993) and therefore decreases asymptotically when plotted against linear geographical distance. Nonequilibrium one-dimensional and equilibrium two-dimensional systems therefore show similar patterns of IBD.
Numerous empirical studies have reported lack of migration–drift equilibrium in wild populations (Poissant et al. 2005; Bradbury & Bentzen 2007). It is straightforward to identify nonequilibrium population structure (Hutchison & Templeton 1999), but distinguishing bet-ween the causes of nonequilibrium population structure is challenging because different evolutionary processes acting at various spatiotemporal scales can produce similar patterns. For example, abrupt genetic differentiation over small spatial scales and nonequilibrium population structure is characteristic of both populations that were historically allopatric (e.g. secondary contact, Swenson & Howard 2005; Duvernell et al. 2008; Hewitt 2011) and populations with contemporary barriers to migration, either temporal (Ramstad et al. 2004) or physical (Koizumi et al. 2006). Similarly, allele frequency gradients produced by natural selection across an ecotone (Endler 1977) can strongly resemble gradients formed by secondary contact (Strand et al. 2012) or restricted migration (Vasemägi 2006).
Studies of contemporary populations in northern latitudes suggest that, in many cases, nonequilibrium population structure is caused by relatively recent colonization of the region after the Pleistocene ice age. The Pleistocene glacial advances reached their largest extent during the last glacial maximum, approximately 25 000 years before present (ybp); this period was characterized by colder temperatures, reduced precipitation and lower sea levels than contemporary conditions (Clark et al. 2009). The reduction in sea level and encroachment of ice sheets onto the continental shelf restricted available habitat for coastal species occupying subpolar latitudes. These climatic changes could have affected population structure in two ways (Bernatchez & Dodson 1991). First, populations persisting in separate glacial refugia might have diverged into genetically distinct lineages due to geographical isolation. Alternatively, changes in hydrology might have facilitated gene flow between previously isolated populations. Previous studies of aquatic organisms (Oncorhynchus kisutch, Smith et al. 2001; Salvelinus sp., Redenbach & Taylor 2002; Prosopium coulteri, Witt et al. 2011) have reported population structure that was probably affected by climatic changes of the Pleistocene.
Chum salmon (Oncorhynchus keta) is widely distributed across the northern Pacific Rim (Salo 1991). During the last glacial maximum, its range was fragmented by changes in sea level, the emergence of the Bering Land Bridge and presence of ice sheets along the coast (Pewé 1975; Dyke et al. 2002). In addition, geological evidence indicates that within the last two thousand years, sudden changes in relative sea level altered Alaska's coastal hydrology (Jordan 2001) and may have isolated populations of chum salmon spawning in coastal streams.
The strong homing behaviour of salmon to their natal stream has been well documented (reviewed in Quinn 2005), and numerous studies contribute to our understanding of chum salmon population structure (Wilmot et al. 1994; Seeb & Crane 1999; Sato et al. 2004; Beacham et al. 2009; Seeb et al. 2011). Populations generally cluster in large-scale geographical groups. However, the north coast of the Alaska Peninsula (NAP) contains a major phylogeographical break for the species, and allozyme studies (Wilmot et al. 1994; Seeb & Crane 1999) demonstrated that populations sampled from the western NAP are similar to populations from the southern range of chum salmon in North America, while populations from the eastern NAP are similar to populations from the northern range of the species. This abrupt genetic differentiation over a small geographical area is unusual for chum salmon, and it has been suggested (Seeb & Crane 1999) that the NAP is a secondary contact zone between lineages that survived the Pleistocene ice age in allopatric glacial refugia.
Theory predicts that genetic differences between populations in secondary contact will decay as a function of migration and time since initial contact (Endler 1977), assuming that there are no selective barriers to hybridization (but see Fraser et al. (2011) for a review of fitness differences among allopatric salmonids). However, secondary contact zones can persist over many generations when maintained by a barrier to migration (Barton & Hewitt 1985). We investigated patterns of population structure and IBD among summer-spawning chum salmon in the putative secondary contact zone of the NAP using empirical data. In addition, we evaluated the effects of colonization timing and founder population heterogeneity on IBD patterns using simulated data. Because most NAP streams are short, the system can be considered one dimensional (Fig.1). We screened 161 single nucleotide polymorphisms (SNPs) to explore patterns of genetic differentiation, and empirical data were subsequently compared with simulations of two different colonization scenarios of the NAP: (i) two lineages meeting in a secondary contact zone and, (ii) colonization by a single genetic lineage. The simulation procedure allowed us to estimate the time required for idealized populations to reach migration–drift equilibrium and test the hypothesis that population structure on the NAP is caused by secondary contact.
Fig 1.
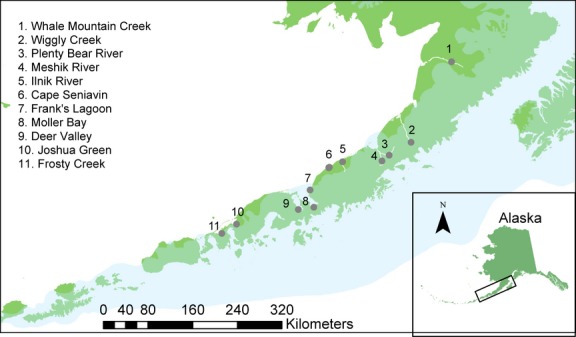
Map of the Alaska Peninsula, indicating where spawning chum salmon were sampled. Landmasses are depicted in green, and the translucent blue overlay represents the Cordilleran Ice Sheet c. 25 000 ybp. The inset displays the state of Alaska, and the study area is outlined in a black box.
Materials and methods
Empirical data
Samples were collected from 2001 to 2010 by the Alaska Department of Fish and Game (ADFG). Approximately 95 sexually mature salmon were sampled from each of 11 different locations on the NAP that probably represent distinct breeding populations and whose individuals return to freshwater spawning grounds in the summer months (Fig.1 and Table1). In 2001 and 2002 (Table1), salmon were captured by beach seine, and heart tissue was collected and frozen in liquid nitrogen until transferred to long-term storage at -80 °C. From 2008 onward, individual salmon were captured by beach seine, held momentarily while the axillary process on the ventral fin was removed, and subsequently released. Axillary processes were stored in 95% ethanol.
Table 1.
Sampling locations and associated collection information
| Location number | Location name | Latitude | Longitude | Collection dates | N |
|---|---|---|---|---|---|
| 1 | Whale Mountain Creek | 58.2549 | −156.5817 | August 2010 | 95 |
| 2 | Wiggly Creek | 56.9846 | −157.6637 | August 2009 | 95 |
| 3 | Plenty Bear River | 56.7070 | −158.2994 | August 2009 | 50 |
| 4 | Meshik River | 56.6100 | −158.5014 | August 2009 | 95 |
| 5 | Ilnik River | 56.4390 | −160.0142 | July 2002 | 50 |
| 6a | Cape Seniavin | 56.4381 | −160.0143 | August 2001 | 55 |
| 6b | 56.4402 | −160.0109 | August 2009 | 21 | |
| 6c | 56.4334 | −160.0250 | August 2010 | 30 | |
| 7 | Frank's Lagoon | 56.0449 | −160.5066 | July 2010 | 95 |
| 8 | Moller Bay | 55.7779 | −160.3461 | August 2009 | 95 |
| 9 | Deer Valley | 55.7160 | −160.7860 | August 2008 | 95 |
| 10 | Joshua Green | 55.3717 | −162.4746 | August 2009 | 95 |
| 11 | Frosty Creek | 55.1947 | −162.8583 | August 2009 | 95 |
DNA was extracted from 966 individuals using the DNeasy 96 Tissue Kit (QIAGEN, Valencia, CA). A total of 192 SNPs (Table S1, Supporting information) were assayed using 5′ nuclease reactions (Seeb et al. 2009) on the BioMark 96.96 Dynamic Array (Fluidigm, San Francisco, CA). Eighty-four loci were previously described (Smith et al. 2005a,b; Elfstrom et al. 2007; Seeb et al. 2011), and novel assays for 108 loci were developed specifically for this project (Tables S1 and S2, Supporting information). As a quality control measure, 8% of the samples were genotyped again to ensure that genotypes were accurate and reproducible.
Allele frequencies and the expected heterozygosity of each locus were calculated in GENALEX 6.4 (Peakall & Smouse 2006). We tested for homogeneity of allele frequencies in the Cape Seniavin collections sampled in different years using a test for genic differentiation in the program GENEPOP (Raymond & Rousset 1995; Rousset 2008). If no statistically significant difference (α = 0.01) over all loci was found between collection years, individuals sampled from the same geographical location were pooled (Table1) to achieve a larger sampling size as recommended by Waples (1990).
Deviations from Hardy–Weinberg equilibrium (HWE) at each population were detected using exact tests (α = 0.01) and the MC algorithm in GENEPOP 4.0; tests were performed across all loci using the default parameters. We examined patterns of linkage disequilibria between all locus pairs using exact tests in GENEPOP 4.0. We used the program LDNE (Waples & Do 2008) to estimate the effective population size (Ne) of each sample based on the magnitude of linkage disequilibria.
Genetic differentiation between populations was estimated using the Weir & Cockerham (1984) FST statistic in GENEPOP 4.0. Tests for statistical significance of pairwise population differentiation were conducted in ARLEQUIN 3.5 using 1000 permutations (Excoffier & Lischer 2010). Patterns of differentiation were explored with principal components analysis (PCA) based on individual genotypes using the ADEGENET package (Jombart 2008) in R (R Development Core Team 2008).
The shortest waterway distance between sampling locations was estimated using a least cost path analysis in ARCGIS ver. 10 (ESRI, Inc). Linearized pairwise FST estimates (FST/(1-FST)) (Rousset 1997) were regressed to waterway distance to test for IBD. The statistical significance of IBD was evaluated using a Mantel test with 1000 permutations in the ISOLDE subroutine of GENEPOP. A linear array of populations in migration–drift equilibrium is expected to have a constant and monotonic IBD slope over all spatial scales (Hutchison & Templeton 1999). We explored changes in IBD slope over spatial scales (Bradbury & Bentzen 2007) in two different ways: estimates of linearized FST were iteratively regressed to (i) increase waterway distances (0-50, 0-100, 0-150 km) and (ii) increase step distance along a linear array. Adjacent populations were defined as having a step distance of one.
Simulated data
We explored how long it would take idealized populations to reach migration–drift equilibrium using the program EASYPOP (Balloux 2001) to model genotypic data for 161 loci in twelve populations. Each population had an Ne of 1000 randomly mating, diploid individuals, with even sex ratios. Every locus had two possible allelic states. Alleles were randomly assigned to the initial populations, and free recombination was allowed between all loci. Each locus mutated at a rate of 10−8, which approximates the mutation rate of neutral SNPs (Brumfield et al. 2003). Simulated individuals were subsequently allowed to migrate along a linear array of populations by following either a (i) secondary contact or (ii) single lineage model of colonization.
Secondary contact simulations
A hierarchical island model (Carmelli & Cavalli-Sforza 1976; Sawyer & Felsenstein 1983) with two groups was used to generate two differentiated lineages. Each group consisted of six populations, and individuals migrated to any population with a uniform migration rate of m = 0.05. Migration between groups was not allowed. We ran the model for 6250 generations or approximately 25 000 years [chum salmon commonly reach sexual maturity at 3–4 years of age (Beacham & Murray 1987)] to simulate allopatric isolation between two genetic lineages during the Pleistocene ice age.
Subsequently, we simulated secondary contact along a linear habitat using a one-dimensional stepping stone model of migration (Kimura & Weiss 1964). Individuals were allowed to migrate to adjacent populations with a uniform migration rate of m = 0.05, which is within the estimated range of straying rates between natal streams for adult chum salmon (reviewed in Johnson et al. 1997). Populations at either end of the one-dimensional array only exchanged migrants with one adjacent population. The one-dimensional stepping stone model is an appropriate approximation of our study region because sampled streams on the NAP are hydrologically separated and flow north from the interior of the peninsula to the Bering Sea. In addition, our sampling locations are ordered along a transect from east to west (Fig.1). We ran the one-dimensional stepping stone model for different number of generations (100, 250, 500 and 1000 generations) to observe how IBD developed over time. Once simulations were finished, data were tested for IBD and migration–drift equilibrium as described for the empirical data.
Single lineage simulations
A genetically homogeneous lineage was simulated by allowing individuals to migrate to any other population (n = 11) with a uniform migration rate of m = 0.05 for 6250 generations (∼25 000 years). Subsequently, all populations were ordered along a one-dimensional stepping stone (as described above), and data were simulated for 100, 250, 500 or 1000 generations. Once simulations were finished, data were tested for IBD and migration–drift equilibrium as described for the empirical data.
Results
Empirical data
Repeated genotyping of individuals at all SNPs indicated that genotyping errors occurred at a frequency of 0.05%. A few individuals from each population were missing more than 15% of genotypes (indicating poor-quality DNA) and were removed from the data set, leaving 912 of 966 individuals for the remainder of the analyses. Monomorphic SNPs and SNPs with an average minor allele frequency <0.01 across all populations were discarded (Table S1, Supporting information). No significant temporal differentiation across all SNPs (α = 0.01) was found for the Cape Seniavin collections taken from different years (2001, 2009, 2010), and individuals were pooled to achieve a larger sample size for that sampling location.
We conducted 1772 tests to screen for deviations from HWE and found nine tests (0.5%) to be statistically significant (α = 0.01); however, deviations from HWE were inconsistent across SNPs or populations. As the number of significant tests was smaller than what was expected by chance alone (1%), we did not reject the null hypothesis of HWE in these populations.
We tested for linkage disequilibrium by conducting 207 717 tests between pairs of SNPs and found 2182 (1.0%) to be statistically significant at α = 0.01. Sixteen SNP pairs were in linkage disequilibrium in almost all populations, and each SNP pair was ascertained from the same sequence (Table S2, Supporting information). We retained only one SNP from each pair for the remainder of the analyses to avoid redundant data. After accounting for linked markers and low minor allele frequency, 161 nuclear SNPs were available for use in the downstream analyses (Table S1, Supporting information). The median estimate of Ne was 822, and values ranged from 333 to 7286 (Table2).
Table 2.
Estimates of the effective population size (Ne) and 95% confidence intervals of Ne in empirical populations. Individuals from Cape Seniavin were pooled as described in the Materials and methods section, and parameters were estimated using the program LDNE (Waples & Do 2008)
| Location number | Location name | Estimated Ne | 95% CI of Ne |
|---|---|---|---|
| 1 | Whale Mountain Creek | 822 | 471–2879 |
| 2 | Wiggly Creek | 868 | 452–7384 |
| 3 | Plenty Bear River | 7286 | 605–infinite |
| 4 | Meshik River | 457 | 318–786 |
| 5 | Ilnik River | 333 | 202–868 |
| 6 | Cape Seniavin | 437 | 290–842 |
| 7 | Frank's Lagoon | 339 | 254–499 |
| 8 | Moller Bay | 617 | 386–1438 |
| 9 | Deer Valley | 3324 | 798–infinite |
| 10 | Joshua Green | 3211 | 775–infinite |
| 11 | Frosty Creek | 1360 | 584–infinite |
Several SNPs (Oke_ccd16-77, Oke_FANK1-96, Oke_ras1-249, Oke_ROA1-209, Oke_serpin 140, Oke_TCTA-202, Oke_thic-84, Oke_Tf-278, Oke_u200-385, Oke_U504-228, Oke_U509-219) were characterized by abrupt and coincident changes in allele frequency (Fig.2) between the Meshik River and Ilnik River populations. The change in allele frequency at these SNPs was >0.30 in all cases, while the distance between the two sampling sites was only ∼100 waterway km (Fig.1).
Fig 2.
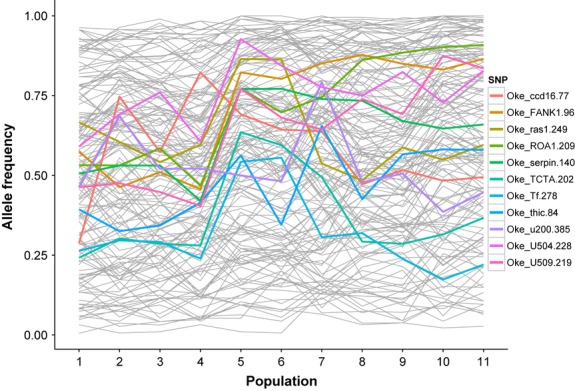
Allele frequency trends over all of the SNPs examined in this study. SNPs with allele frequency differences >0.30 between adjacent populations have been highlighted in colour. Populations are ordered from east to west on the Alaska Peninsula, as indicated in Table2.
We estimated a global FST of 0.04. Pairwise FST values ranged from 0.002 to 0.08 (Table3), and all pairwise comparisons of populations were statistically significant (α = 0.05). PCA of individual genotypic data allowed us to visualize differentiation among and between populations; we observed variation among individuals taken from the same sampling location, but recognizable groups of individuals were evident (Fig.3). Individuals from Whale Mountain Creek, Wiggly Creek, Plenty Bear River and Meshik River (sampling locations 1–4 in Table1) were grouped together and were clearly distinct from individuals from Joshua Green and Frosty Creek (sampling locations 10 and 11). These findings indicate that individuals sampled from the eastern base of the NAP are genetically distinct from individuals sampled from the western tip of the NAP.
Table 3.
Pairwise comparisons from the empirical data set; each population was represented by the location number designated in Table2. The pairwise distance between populations (km) is shown above the diagonal. Pairwise FST is shown below the diagonal; all comparisons were statistically significant at α = 0.05
| Population | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | * | 225 | 278 | 273 | 296 | 314 | 372 | 409 | 411 | 522 | 551 |
| 2 | 0.018 | * | 163 | 158 | 188 | 214 | 275 | 312 | 314 | 434 | 464 |
| 3 | 0.010 | 0.007 | * | 35 | 107 | 132 | 194 | 231 | 233 | 352 | 382 |
| 4 | 0.014 | 0.012 | 0.009 | * | 102 | 128 | 189 | 226 | 228 | 348 | 377 |
| 5 | 0.034 | 0.039 | 0.030 | 0.038 | * | 34 | 95 | 133 | 134 | 254 | 284 |
| 6 | 0.037 | 0.034 | 0.031 | 0.040 | 0.002 | * | 61 | 99 | 100 | 220 | 250 |
| 7 | 0.041 | 0.044 | 0.042 | 0.043 | 0.020 | 0.027 | * | 37 | 40 | 166 | 195 |
| 8 | 0.038 | 0.048 | 0.045 | 0.044 | 0.027 | 0.033 | 0.024 | * | 50 | 187 | 217 |
| 9 | 0.036 | 0.046 | 0.040 | 0.044 | 0.023 | 0.030 | 0.025 | 0.003 | * | 175 | 205 |
| 10 | 0.072 | 0.081 | 0.080 | 0.073 | 0.047 | 0.058 | 0.045 | 0.026 | 0.027 | * | 41 |
| 11 | 0.066 | 0.070 | 0.069 | 0.066 | 0.039 | 0.047 | 0.036 | 0.020 | 0.020 | 0.006 | * |
Fig 3.
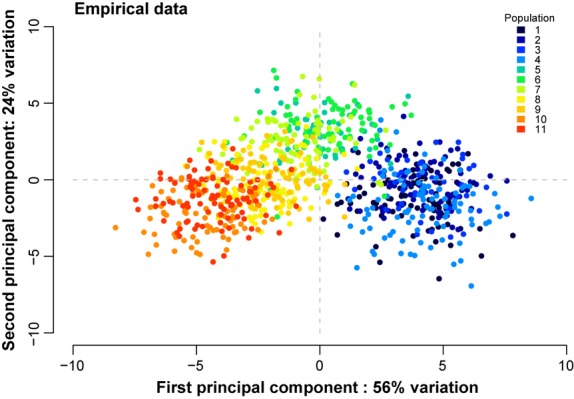
PCA of individuals based on the allele frequencies over all SNPs. Each dot is an individual, and colours represent the different populations sampled. Populations are numbered as in Table2, moving from east to west along the Alaska Peninsula.
IBD was examined over all populations, and a significant correlation between genetic and waterway distances was found (Fig.4A, P < 0.0001, r2 = 0.53) based on Mantel tests. However, the IBD slope was highly variable over increasing spatial scales when FST/1-FST was iteratively regressed against the waterway distance separating populations (Fig.4B) as well as against increasing step distance along the NAP (Fig.4C). As chum spawning habitat can be considered a one-dimensional space, these results indicate that chum salmon populations from the NAP were not in migration–drift equilibrium.
Fig 4.

IBD and IBD slope analyses of the empirical data. (A) Regression of linearized FST to the waterway distance separating populations. (B) IBD slope calculated by including pairwise comparisons at increasing distance intervals between populations. (C) IBD slope calculated by including pairwise comparisons at increasing step distance intervals between populations along a linear array.
Simulated data
Secondary contact simulations
Populations simulated with 100 generations of secondary contact were highly differentiated with regard to pairwise FST, and this differentiation was strongly correlated with the step distance separating populations (Fig.5A). However, this relationship was highly nonlinear (Fig.5B); IBD slope was not uniform, but rather increased over increasing spatial scales until reaching a maximum. Populations simulated with 250 and 500 generations of secondary contact were characterized by smaller pairwise FST values (Fig.5A) because multiple generations of migration homogenized allele frequency differences between the two lineages. However, at 250 and 500 generations of secondary contact, the IBD slope varied over increasing spatial scales (Fig.5B), indicating that populations had not reached migration–drift equilibrium.
Fig 5.
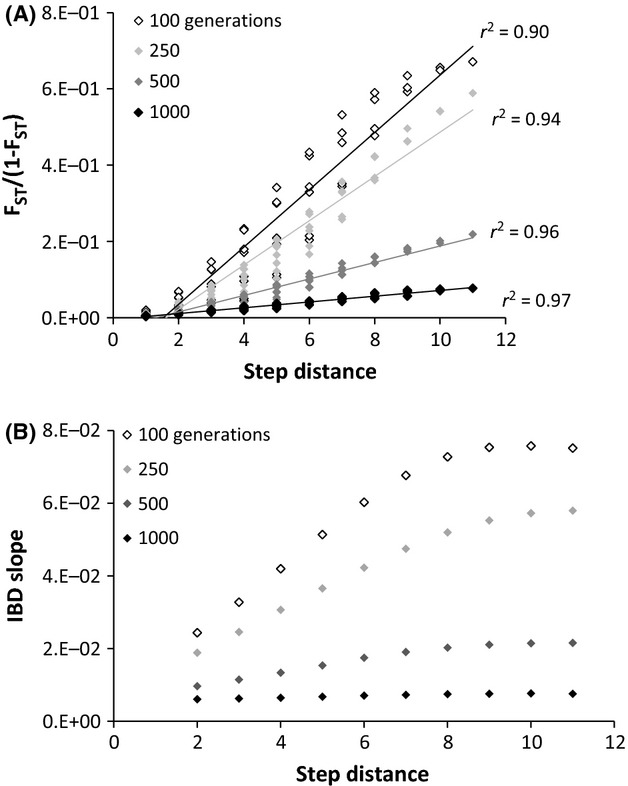
IBD and IBD slope analyses of data simulated by the secondary contact model of colonization. (A) Regression of linearized FST to the step distance separating populations. (B) IBD slope calculated by including pairwise comparisons at increasing step distance intervals between populations along a linear array.
After 1000 generations of secondary contact, populations exhibited nearly perfect correlation between linearized FST and step distance (r2 = 0.97, P < 0.0001). In addition, populations simulated with 1000 generations of secondary contact had uniform IBD slopes at increasing spatial scales (Fig.5B), indicating that they were approaching migration–drift equilibrium. Thus, when populations originating from allopatric lineages were connected by migration for 1000 generations, it was impossible to detect that secondary contact had occurred by examining the IBD slope.
Single lineage simulations
We found statistically significant correlations between increasing geographical and genetic distances for all simulations, regardless of the number of generations (100, 250, 500 and 1000 generations) spent in a one-dimensional stepping stone model of migration (Fig.6A). However, the IBD slope decreased with increasing distance for populations simulated with 100 and 250 generations (Fig.6B). The IBD slope became more constant as the number of generations spent in one-dimensional stepping stone model increased, indicating that populations approached migration–drift equilibrium.
Fig 6.
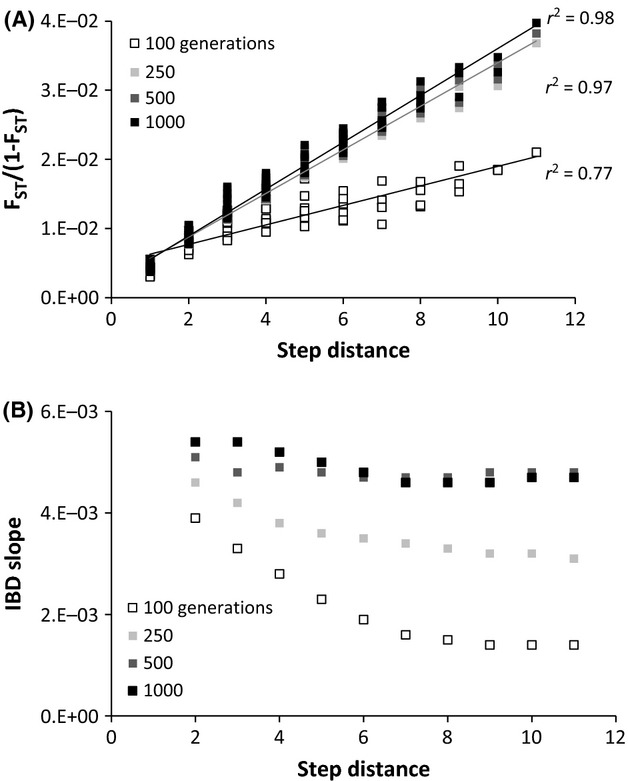
IBD and IBD slope analyses of data simulated by the single lineage model of colonization. (A) Regression of linearized FST to the step distance separating populations. (B) IBD slope calculated by including pairwise comparisons at increasing step distance intervals between populations along a linear array.
Discussion
Population structure in empirical data
Chum salmon is a species generally characterized by low levels of population structure. For example, summer-spawning chum salmon from western Alaska rivers that are thousands of kilometres apart have a regional FST of only 0.004, as estimated using a subset of SNPs analysed in this study (Seeb et al. 2011). In comparison, summer-spawning chum salmon originating from different rivers on the NAP exhibit strong population structure. Genetic differentiation was greatest when comparing populations from the eastern and western ends of the NAP. Although sampling locations spanned a linear distance of only ∼500 km, the overall FST of these populations was 0.036. These findings indicate that the NAP is a relative hot spot of genetic differentiation for chum salmon, and our results agree with previous studies using other genetic markers to describe the population structure of the species in Alaska (Seeb & Crane 1999; Smith & Seeb 2008). These interesting patterns of genetic differentiation could be explained in two different ways: (i) present-day migration is restricted in contemporary populations, or (ii) populations retain signatures of historical isolation and secondary contact.
Migration patterns are influenced by geographical features of the surrounding landscape (Castric et al. 2001; Koizumi et al. 2006); for example, physical barriers such as dams and waterfalls restrict migration between aquatic populations and contribute to increased population structure. We are not aware of any contemporary physical barriers to migration in our study system; streams on the NAP flow north to the Bering Sea along relatively flat terrain, and there are no dams in the sampled streams. Thus, we think it is unlikely that contemporary barriers to migration have caused the high genetic differentiation observed between chum salmon populations on the NAP.
Rivers on the NAP run approximately parallel to each other, and all except two of the waterways sampled in our study (Plenty Bear Creek and Meshik River) are hydrologically separated (Fig.1). It is possible that migration between populations on the NAP is less than other parts of the species' range where rivers share a common outlet or confluence. However, previous research (Olsen et al. 2008) examining the population structure of chum salmon spawning in parallel waterways in Norton Sound, Alaska, found very low levels of genetic differentiation. Instead, they discovered that gene flow between populations decreased as the hydrographic complexity and environmental heterogeneity of the river networks increased. Furthermore, chum salmon populations spawning in short, coastal streams on the South Alaska Peninsula exhibit low population structure and are much more homogeneous than populations spawning on the NAP (Petrou et al. 2013). This comparison between geographical regions suggests that contemporary river topography might be a secondary driver of population structure on the NAP.
Temporal differences in spawning time can be a barrier to migration between populations, a phenomenon contributing to ‘isolation by time’ (Hendry & Day 2005). However, our samples were sexually mature chum salmon collected over only a month (late July to late August), so it is unlikely that temporal differences in spawning date generated the observed patterns of genetic differentiation.
A previous study (Seeb & Crane 1999) suggested that the strong genetic differentiation between spawning populations on the NAP was probably caused by secondary contact between two genetic lineages that recolonized Alaska after the Pleistocene ice age ended. It was postulated that these lineages probably originated from allopatric refugia that existed in the northern (Beringia) and southern (Cascadia) part of the contemporary species’ range. Secondary contact zones are often characterized by steep gradients in genetic or morphological characteristics (Endler 1977; Barton & Hewitt 1985) that degrade over time when populations are connected through migration. Eleven SNPs used in this study (Oke_ccd16-77, Oke_FANK1-96, Oke_ras1-249, Oke_ROA1-209, Oke_serpin 140, Oke_TCTA-202, Oke_thic-84, Oke_Tf-278, Oke_u200-385, Oke_U504-228 and Oke_U509-219) showed abrupt and coincident changes in allele frequency. These loci had a maximum difference of 0.37 in allele frequency between chum salmon populations in the Meshik and Ilnik rivers, adjacent spawning locations separated by ∼100 waterway km (Fig.1). The observed sharp differences in allele frequency are an unusual pattern that is consistent with hypotheses of restricted migration, secondary contact and/or natural selection at these SNPs. Unfortunately, we could not test hypotheses regarding divergent selection at these SNPs because existing methods to detect outlier loci (Beaumont & Nichols 1996; Excoffier et al. 2009) assume an island model of migration and are not appropriate for analysing populations that exhibit IBD (Meirmans 2012). Indeed, in exploratory tests of divergent selection using the methods of Antao et al. (2008) and Foll & Gaggiotti (2008), we found that data simulated under a neutral model of evolution by the secondary contact and single lineage simulations produced a large number of false positives.
Incomplete sampling of populations can also produce abrupt allele frequency gradients, and patchy sampling of individuals across a study region can lead to signals of population differentiation that are not representative of biological processes (Schwartz & McKelvey 2009). This pattern is more pronounced when there is a strong correlation between genetic and geographical distances. The authors suggest sampling on a fine scale relative to the species’ life history. As our sampling targeted spawning populations that were tens to hundreds of kilometres apart, it is unlikely that we failed to sample important intermediate populations. As a result, we suggest that the relatively large genetic differentiation that was observed on the NAP is likely due to historical factors rather than incomplete sampling.
Comparison of IBD slope in empirical and simulated data
Species with spatially restricted migration should exhibit IBD if enough time has passed for populations to approach migration–drift equilibrium (Slatkin 1993). Theoretical models (Malécot 1955) have shown that at migration–drift equilibrium, populations following a stepping stone model of migration exhibit a positive and monotonic relationship between increasing pairwise genetic differentiation and geographical distance. The equilibrium hypothesis can be rejected for populations with spatially restricted migration if: (i) there is no significant correlation between genetic and geographical distances (no IBD), and/or (ii) IBD slope varies over the spatial scale examined (Hutchison & Templeton 1999).
In simulations depicting recent colonization (100–250 generations ago), populations from both the secondary contact and single lineage models were characterized by IBD slopes that varied over increasing spatial scales (Figs5B and 6B), indicating a lack of migration–drift equilibrium. However, the two colonization scenarios had different IBD slope patterns. In the secondary contact model, IBD slopes were smallest at small step distances and increased over larger spatial scales (Fig.5B). This pattern was present because small step distances encompassed the secondary contact zone; as a result, there was comparatively high variation in pairwise FST and a weak IBD relationship at small spatial scales. The opposite pattern was observed in the single lineage model; IBD slope was largest at small step distances and decreased over larger spatial scales (Fig.6B). This pattern is consistent with theoretical models describing how IBD develops in a stepping stone model consisting of genetically homogeneous founder populations (Hutchison & Templeton 1999): migration initially occurs more frequently between adjacent populations, causing allele frequencies to be correlated at small spatial scales.
In agreement with previous research (Slatkin 1993; Hutchison & Templeton 1999; Bradbury & Bentzen 2007), we found that the IBD slope became more stable with increasing time since colonization. One thousand generations after colonization, both the secondary contact (Fig.5) and single lineage (Fig.6) simulations were characterized by positive and constant IBD slope over all spatial scales, indicating that populations were in migration–drift equilibrium. Our simulations suggest that if the NAP were colonized by chum salmon more than 1000 generations ago (∼4000 years), then neutrally evolving populations should be approaching migration–drift equilibrium.
We investigated whether empirical populations on the NAP were in migration–drift equilibrium by analysing patterns of IBD slope at increasing spatial scales. When IBD slope was partitioned by increasing bin sizes (Fig.4B), it fluctuated greatly over increasing spatial scales, indicating that despite the relatively robust overall correlation between genetic and geographical distance, populations were not in migration–drift equilibrium. However, the resulting IBD slope pattern resembled neither the secondary contact nor single lineage simulations because empirical populations were not evenly spaced across the NAP and increasing distance bins contained haphazard pairwise comparisons that did not mirror the hierarchical population structure of the data. To obtain a more analogous comparison between the empirical and simulated data, we also analysed IBD slope by increasing step distance along a one-dimensional stepping stone (as in the simulated data). In this case, the empirical IBD slope strongly resembled the pattern from the secondary contact simulation (Figs4C and 5B). We suggest that these results provide additional evidence for secondary contact as the underlying colonization history of the NAP by chum salmon. Sharp genetic breaks at some SNPs (Fig.2) further support the notion of a secondary contact and suggest the area between Meshik and Ilnik River as the contact zone.
Several assumptions were made in the simulations that may not reflect the biological reality of our study system. We assumed that all simulated populations have the same effective population size (Ne = 1000) and migration rate (m = 0.05). Given the remote location of our sampling sites, few estimates of spawning adult abundance exist. The only available data (report of escapement across river weirs, Murphy & Hartill 2009) indicate that abundance of sexually mature adults in regional spawning streams ranges from several hundreds to thousands of individuals, with yearly variation. Although Ne and m probably varied among populations, these estimates of abundance are similar to genetic estimates of Ne (Table2) and the Ne used in our models.
Geological and archaeological evidence support recent colonization
Our simulations suggest that one can distinguish between two different colonization hypotheses (secondary contact vs. single lineage) using contemporary genetic data, but only if <1000 generations have elapsed since colonization. The Cordilleran Ice Sheet receded from the Alaska Peninsula approximately 16 000 ybp (Mann & Peteet 1994). Radiocarbon dating of a willow leaf (11 530 ± 100 14C ybp) and peat (10 830 ± 60 ybp) from the region indicates that vegetation had been locally established by the early Holocene (Dochat 1997). Given that the Pleistocene ice ages ended more than 4000 chum salmon generations ago, it may seem unlikely that any signal of historical isolation could be detected in contemporary populations.
However, spatial patterns of population structure are influenced by migration patterns that may vary over time. Relatively recent changes in the coastal geography of the Alaska Peninsula could have impacted the timing of colonization of the region's streams by chum salmon. Holocene coastal geography of the Alaska Peninsula was very different from the present, and sea level records provide information that helps constrain temporal estimates of salmon colonization in the region. Prior to 10 000 ybp, the relative sea level was 25 m higher than today due to rapid eustatic sea level rise following deglaciation (Jordan 2001). The NAP is a broad coastal plain where elevation ranges from 0 to 150 m above sea level, so the availability of freshwater habitats would have been severely limited at this time (Jordan 2001); lack of coastal freshwater streams makes it unlikely that chum salmon populations spawned in the region.
Over the next several thousand years, relative sea level slowly dropped (Fig.7) as isostatic rebound became relatively more influential than sea level rise in the region (Jordan 2001). This geological change was accompanied by the emergence of the coastal plain and stabilization of freshwater environments. An archaeological excavation (Maschner 1999) on the NAP unearthed a village in Moffet Lagoon that contained salmon remains dating to ∼3950 ybp. It is unclear whether these salmon were fished from the marine environment or from freshwater spawning sites on the NAP, but these artefacts might evidence that salmon occupied regional watersheds at this time.
Fig 7.
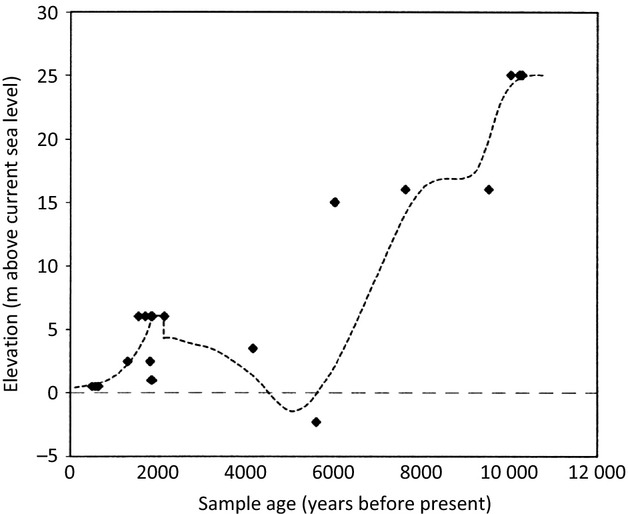
Postglacial sea level trend estimated from shorelines on the western Alaska Peninsula. Reprinted from Quaternary Science Reviews, Volume 20, J. Jordan, Late Quaternary sea level change in Southern Beringia: postglacial emergence of the western Alaska Peninsula, 509–523, Copyright (2001), with permission from Elsevier.
The coastal geography and ecology of the NAP were disrupted by an earthquake that occurred ∼2100 ybp (Fig.7), which caused sudden subsidence and rapid rise in relative sea level (Jordan 2001). An increase of 2 m in relative sea level would have inundated coastal salmon spawning habitats with salt water and probably displaced or extirpated local chum salmon populations. Interestingly, there is also a gap in the archaeological record during this period. Although there is evidence of human settlement on the Alaska Peninsula as early as 5000 ybp, no human artefacts are dated from 2500 to 2200 ybp. In addition, villages appear to have been destroyed or abandoned at this time (Maschner 1999). This gap in the archaeological record has been attributed to the sudden increase in sea level and subsequent degradation of coastal resources. Archaeological evidence indicates that it was not until 1250 ybp that settlements were constructed close to streams supporting contemporary populations of pink (O. gorbuscha) and chum salmon, and salmon fishing intensified again (Maschner 1999).
Both geological and archaeological data suggest that the contemporary ecology of coastal habitats on the NAP was established relatively recently. Chum salmon populations have probably been using present-day coastal spawning habitats on the NAP for ∼300 to 400 generations. Our simulations predict that populations would not be in migration–drift equilibrium if this amount of time had passed. In addition, our analyses of IBD in the empirical data indicated that contemporary chum salmon populations on the NAP were not in migration–drift equilibrium. This finding, in conjunction with the geological and archaeological evidence, leads us to conclude that the nonequilibrium population structure is caused by the recent colonization of a very dynamic coastal environment by chum salmon. Furthermore, the observed nonequilibrium IBD slope patterns most closely resemble simulated data from the secondary contact model of colonization. This research has shown that using information from contemporary genetic data, simulation models and historical landscape information can improve our ability to interpret existing patterns of population structure. By referring to geological and archaeological data, we were able to identify recent landscape rearrangements that probably disrupted migration between populations and contributed to contemporary patterns of nonequilibrium population structure.
Acknowledgments
We thank colleagues from the ADFG (Nick DeCovich, Lisa Fox, Penny Crane, Mark Witteveen, and Judy Berger) who conducted field sampling and organized data sharing. Carita Pascal gave indispensable laboratory support and guidance. This research was partially funded by the Alaska Sustainable Salmon Fund under Study # 45919 from NOAA, US Department of Commerce, administered by the ADFG. The statements, findings, conclusions and recommendations are those of the authors and do not necessarily reflect the views of the NOAA, the US Department of Commerce or the ADFG. Additional funding was provided by the Gordon and Betty Moore Foundation and the State of Alaska. A National Science Foundation GK-12 graduate teaching fellowship through the Ocean and Coastal Interdisciplinary Science Program (OACIS, proposal number DGE-0742559) provided ELP with additional funding during the final year of study.
L.W.S. and E.L.P. designed this study with input from R.S.W., L.H. and J.E.S. Samples were contributed by W.D.T. J.E.S. coordinated SNP development. E.L.P. and D.G.U. analysed the data. E.L.P. wrote the paper with contributions from all co-authors.
Data accessibility
Novel SNPs developed for this project are available in GenBank (dbSNP) under Accession nos ss538825712–ss538825890. Individual SNP genotypes are available in the DRYAD data depository (doi:10.5061/dryad.sg573).
Supporting Information
Additional supporting information may be found in the online version of this article.
List of SNP loci used in genotyping.
Sequence and primer data associated with each novel SNP developed during this study.
References
- Antao T, Lopes A, Lopes R, Beja-Pereira A, Luikart G. LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics. 2008;9:323. doi: 10.1186/1471-2105-9-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balloux F. Easypop (Version 1.7): a computer program for population genetics simulations. Journal of Heredity. 2001;92:301–302. doi: 10.1093/jhered/92.3.301. [DOI] [PubMed] [Google Scholar]
- Barton NH, Hewitt GM. Analysis of hybrid zones. Annual Review of Ecology and Systematics. 1985;16:113–148. [Google Scholar]
- Beacham TD, Murray CB. Adaptive variation in body size, age, morphology, egg size, and developmental biology of chum salmon (Oncorhynchus keta) in British Columbia. Canadian Journal of Fisheries and Aquatic Sciences. 1987;44:244–261. [Google Scholar]
- Beacham TD, Candy JR, Wallace C, et al. Microsatellite stock identification of chum salmon on a Pacific Rim basis. North American Journal of Fisheries Management. 2009;29:1757–1776. [Google Scholar]
- Beaumont MA, Nichols RA. Evaluating loci for use in the genetic analysis of population structure. Proceedings of the Royal Society of London Series B: Biological Sciences. 1996;263:1619–1626. [Google Scholar]
- Bernatchez L, Dodson JJ. Phylogeographic structure in mitochondrial DNA of the lake whitefish (Coregonus clupeaformis) and its relation to Pleistocene glaciations. Evolution. 1991;45:1016–1035. doi: 10.1111/j.1558-5646.1991.tb04367.x. [DOI] [PubMed] [Google Scholar]
- Bradbury IR, Bentzen P. Non-linear genetic isolation by distance: implications for dispersal estimation in anadromous and marine fish populations. Marine Ecology Progress Series. 2007;340:245–257. [Google Scholar]
- Brumfield RT, Beerli P, Nickerson DA, Edwards SV. The utility of single nucleotide polymorphisms in inferences of population history. Trends in Ecology and Evolution. 2003;18:249–256. [Google Scholar]
- Carmelli D, Cavalli-Sforza LL. Some models of population structure and evolution. Theoretical Population Biology. 1976;9:329–359. doi: 10.1016/0040-5809(76)90052-6. [DOI] [PubMed] [Google Scholar]
- Castric V, Bonney F, Bernatchez L. Landscape structure and hierarchical genetic diversity in the brook charr, Salvelinus fontinalis. Evolution. 2001;55:1016–1028. doi: 10.1554/0014-3820(2001)055[1016:lsahgd]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Clark PU, Dyke AS, Shakun JD, et al. The Last Glacial Maximum. Science. 2009;325:710–714. doi: 10.1126/science.1172873. [DOI] [PubMed] [Google Scholar]
- Dochat TM. Quaternary stratigraphy and geomorphology of the Cold Bay region of the Alaska Peninsula: A basis for paleoenvironmental reconstruction. Ph.D. Madison, WI: Thesis, Department of Geology, University of Wisconsin-Madison; 1997. [Google Scholar]
- Duvernell DD, Lindmeier JB, Faust KE, Whitehead A. Relative influences of historical and contemporary forces shaping the distribution of genetic variation in the Atlantic killifish, Fundulus heteroclitus. Molecular Ecology. 2008;17:1344–1360. doi: 10.1111/j.1365-294X.2007.03648.x. [DOI] [PubMed] [Google Scholar]
- Dyke AS, Andrews JT, Clark PU, et al. The Laurentide and Innuitian ice sheets during the Last Glacial Maximum. Quaternary Science Reviews. 2002;21:9–31. [Google Scholar]
- Elfstrom CM, Smith CT, Seeb LW. Thirty-eight single nucleotide polymorphism markers for high-throughput genotyping of chum salmon. Molecular Ecology Notes. 2007;7:1211–1215. [Google Scholar]
- Endler JA. Geographic Variation, Speciation, and Clines. Princeton: Princeton University Press; 1977. [PubMed] [Google Scholar]
- Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Hofer T, Foll M. Detecting loci under selection in a hierarchically structured population. Heredity. 2009;103:285–298. doi: 10.1038/hdy.2009.74. [DOI] [PubMed] [Google Scholar]
- Foll M, Gaggiotti O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics. 2008;180:977–993. doi: 10.1534/genetics.108.092221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser DJ, Weir LK, Bernatchez L, Hansen MM, Taylor EB. Extent and scale of local adaptation in salmonid fishes: review and meta-analysis. Heredity. 2011;106:404–420. doi: 10.1038/hdy.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendry AP, Day T. Population structure attributable to reproductive time: isolation by time and adaptation by time. Molecular Ecology. 2005;14:901–916. doi: 10.1111/j.1365-294X.2005.02480.x. [DOI] [PubMed] [Google Scholar]
- Hewitt G. Quaternary phylogeography: the roots of hybrid zones. Genetica. 2011;139:617–638. doi: 10.1007/s10709-011-9547-3. [DOI] [PubMed] [Google Scholar]
- Hutchison DW, Templeton AR. Correlation of pairwise genetic and geographic distance measures: inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution. 1999;53:1898–1914. doi: 10.1111/j.1558-5646.1999.tb04571.x. [DOI] [PubMed] [Google Scholar]
- Johnson OW, Grant WS, Kope RG, et al. Status Review of Chum Salmon from Washington, Oregon, and California. Seattle: U.S. Dept. of Commerce, National Oceanic and Atmospheric Administration, National Marine Fisheries Service; 1997. [Google Scholar]
- Jombart T. Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24:1403–1405. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- Jordan J. Late Quaternary sea level change in Southern Beringia: postglacial emergence of the Western Alaska Peninsula. Quaternary Science Reviews. 2001;20:509–523. [Google Scholar]
- Kimura M, Weiss GH. The stepping stone model of population structure and the decrease of genetic correlation with distance. Genetics. 1964;49:561–576. doi: 10.1093/genetics/49.4.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi I, Yamamoto S, Maekawa K. Decomposed pairwise regression analysis of genetic and geographic distances reveals a metapopulation structure of stream-dwelling Dolly Varden charr. Molecular Ecology. 2006;15:3175–3189. doi: 10.1111/j.1365-294X.2006.03019.x. [DOI] [PubMed] [Google Scholar]
- Malécot G. The decrease of relationship with distance. Cold Spring Harbor Symposia on Quantitative Biology. 1955;20:52–53. [Google Scholar]
- Mann DH, Peteet DM. Extent and Timing of the Last Glacial Maximum in Southwestern Alaska. Quaternary Research. 1994;42:136–148. [Google Scholar]
- Maschner HDG. Prologue to the prehistory of the lower Alaska Peninsula. Arctic Anthropology. 1999;36:84–102. [Google Scholar]
- Meirmans PG. The trouble with isolation by distance. Molecular Ecology. 2012;21:2839–2846. doi: 10.1111/j.1365-294X.2012.05578.x. [DOI] [PubMed] [Google Scholar]
- Murphy RL, Hartill TG. The North Alaska Peninsula Salmon Report to the Alaska Board of Fisheries, 2010. Anchorage: Alaska Department of Fish and Game; 2009. [Google Scholar]
- Olsen JB, Flannery BG, Beacham TD, et al. The influence of hydrographic structure and seasonal run timing on genetic diversity and isolation-by-distance in chum salmon (Oncorhynchus keta. Canadian Journal of Fisheries and Aquatic Sciences. 2008;65:2026–2042. [Google Scholar]
- Peakall R, Smouse P. Genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes. 2006;6:288–295. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrou EL, Seeb JE, Witteveen MJ, Seeb LW. Fine-scale sampling reveals distinct isolation by distance patterns in salmonid populations occupying a glacially dynamic environment. Conservation Genetics. 2013 [Google Scholar]
- Pewé TL. Quaternary Geology of Alaska. Washington, D.C: U.S. Government Printing Office; 1975. [Google Scholar]
- Poissant J, Knight TW, Ferguson MM. Nonequilibrium conditions following landscape rearrangement: the relative contribution of past and current hydrological landscapes on the genetic structure of a stream-dwelling fish. Molecular Ecology. 2005;14:1321–1331. doi: 10.1111/j.1365-294X.2005.02500.x. [DOI] [PubMed] [Google Scholar]
- Quinn TP. The Ecology and Behavior of Pacific Salmon and Trout. Seattle: University of Washington Press; 2005. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2008. ISBN 3-900051-07-0. Available at: http://www.R-project.org. [Google Scholar]
- Ramstad KM, Woody CA, Sage GK, Allendorf FW. Founding events influence genetic population structure of sockeye salmon (Oncorhynchus nerka) in Lake Clark, Alaska. Molecular Ecology. 2004;13:277–290. doi: 10.1046/j.1365-294x.2003.2062.x. [DOI] [PubMed] [Google Scholar]
- Raymond M, Rousset F. Genepop version 1.2: population genetics software for exact tests and ecumenicism. Journal of Heredity. 1995;86:248–249. [Google Scholar]
- Redenbach Z, Taylor EB. Evidence for historical introgression along a contact zone between two species of char (PISCES:SALMONIDAE) in northwestern North America. Evolution. 2002;56:1021–1035. doi: 10.1111/j.0014-3820.2002.tb01413.x. [DOI] [PubMed] [Google Scholar]
- Rousset F. Genetic differentiation and estimation of gene flow from F-Statistics under isolation by distance. Genetics. 1997;145:1219–1228. doi: 10.1093/genetics/145.4.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset F. Genepop'007: a complete reimplementation of the Genepop software for Windows and Linux. Molecular Ecology Resources. 2008;8:103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- Salo EO. Life history of chum salmon (Oncorhynchus keta. In: Groot C, Margolis L, editors. Pacific Salmon Life Histories. Vancouver: University of British Columbia Press; 1991. pp. 231–309. [Google Scholar]
- Sato S, Kojima H, Ando J, et al. Genetic population structure of chum salmon in the Pacific Rim inferred from mitochondrial DNA sequence variation. Environmental Biology of Fishes. 2004;69:37–50. [Google Scholar]
- Sawyer S, Felsenstein J. Isolation by distance in a hierarchically clustered population. Journal of Applied Probability. 1983;20:1–10. [Google Scholar]
- Schwartz M, McKelvey K. Why sampling scheme matters: the effect of sampling scheme on landscape genetic results. Conservation Genetics. 2009;10:441–452. [Google Scholar]
- Seeb LW, Crane PA. High genetic heterogeneity in chum salmon in Western Alaska, the contact zone between northern and southern lineages. Transactions of the American Fisheries Society. 1999;128:58–87. [Google Scholar]
- Seeb JE, Pascal CE, Ramakrishnan R, Seeb LW. SNP genotyping by the 5′-nuclease reaction: advances in high throughput genotyping with non-model organisms. In: Komar A, editor. Methods in Molecular Biology, Single Nucleotide Polymorphisms. New York: Humana Press; 2009. pp. 277–292. [DOI] [PubMed] [Google Scholar]
- Seeb LW, Templin WD, Sato S, et al. Single nucleotide polymorphisms across a species’ range: implications for conservation studies of Pacific salmon. Molecular Ecology Resources. 2011;11:195–217. doi: 10.1111/j.1755-0998.2010.02966.x. [DOI] [PubMed] [Google Scholar]
- Slatkin M. Isolation by distance in equilibrium and non-equilibrium populations. Evolution. 1993;47:264–279. doi: 10.1111/j.1558-5646.1993.tb01215.x. [DOI] [PubMed] [Google Scholar]
- Smith CT, Seeb LW. Number of alleles as a predictor of the relative assignment accuracy of short tandem repeat (STR) and single-nucleotide-polymorphism (SNP) baselines for chum salmon. Transactions of the American Fisheries Society. 2008;137:751–762. [Google Scholar]
- Smith CT, Nelson RJ, Wood CC, Koop BF. Glacial biogeography of North American coho salmon (Oncorhynchus kisutch. Molecular Ecology. 2001;10:2775–2785. doi: 10.1046/j.1365-294x.2001.t01-1-01405.x. [DOI] [PubMed] [Google Scholar]
- Smith CT, Baker J, Park L, et al. Characterization of 13 single nucleotide polymorphism markers for chum salmon. Molecular Ecology Notes. 2005a;5:259–262. [Google Scholar]
- Smith CT, Elfstrom CM, Seeb LW, Seeb JE. Use of sequence data from rainbow trout and Atlantic salmon for SNP detection in Pacific salmon. Molecular Ecology. 2005b;14:4193–4203. doi: 10.1111/j.1365-294X.2005.02731.x. [DOI] [PubMed] [Google Scholar]
- Strand AE, Williams LM, Oleksiak MF, Sotka EE. Can diversifying selection be distinguished from history in geographic clines? A population genomic study of killifish (Fundulus heteroclitus. PLoS ONE. 2012;7:e45138. doi: 10.1371/journal.pone.0045138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson N G, Howard D J. Clustering of contact zones, hybrid zones, and phylogeographic breaks in North America. The American Naturalist. 2005;166:581–591. doi: 10.1086/491688. [DOI] [PubMed] [Google Scholar]
- Vasemägi A. The adaptive hypothesis of clinal variation revisited: single-locus clines as a result of spatially restricted gene flow. Genetics. 2006;173:2411–2414. doi: 10.1534/genetics.106.059881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples RS. Temporal changes of allele frequency in Pacific salmon: implications for mixed-stock fishery analysis. Canadian Journal of Fisheries and Aquatic Sciences. 1990;47:968–976. [Google Scholar]
- Waples RS, Do C. LDNE: a program for estimating effective population size from data on linkage disequilibrium. Molecular Ecology Resources. 2008;8:753–756. doi: 10.1111/j.1755-0998.2007.02061.x. [DOI] [PubMed] [Google Scholar]
- Weir B, Cockerham C. Estimating F-Statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- Wilmot RL, Everett RJ, Spearman WJ, et al. Genetic stock structure of western Alaska chum salmon and a comparison with Russian Far East stocks. Canadian Journal of Fisheries and Aquatic Sciences. 1994;51:84–94. [Google Scholar]
- Witt JDS, Zemlak RJ, Taylor EB. Phylogeography and the origins of range disjunctions in a north temperate fish, the pygmy whitefish (Prosopium coulterii), inferred from mitochondrial and nuclear DNA sequence analysis. Journal of Biogeography. 2011;38:1557–1569. [Google Scholar]
- Wright S. Isolation by distance. Genetics. 1943;28:114–138. doi: 10.1093/genetics/28.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of SNP loci used in genotyping.
Sequence and primer data associated with each novel SNP developed during this study.
Data Availability Statement
Novel SNPs developed for this project are available in GenBank (dbSNP) under Accession nos ss538825712–ss538825890. Individual SNP genotypes are available in the DRYAD data depository (doi:10.5061/dryad.sg573).