

Abstract
Background
Hfq functions in post-transcriptional gene regulation in a wide range of bacteria, usually by promoting base pairing of mRNAs with trans-encoded sRNAs. It was previously shown that Hfq down-regulates Tn10 transposition by inhibiting IS10 transposase expression at the post-transcriptional level. This provided the first example of Hfq playing a role in DNA transposition and led us to ask if a related transposon, Tn5, is similarly regulated.
Results
We show that Hfq strongly suppresses Tn5 transposition in Escherichia coli by inhibiting IS50 transposase expression. However, in contrast to the situation for Tn10, Hfq primarily inhibits IS50 transposase transcription. As Hfq does not typically function directly in transcription, we searched for a transcription factor that also down-regulated IS50 transposase transcription and is itself under Hfq control. We show that Crp (cyclic AMP receptor protein) fits these criteria as: (1) disruption of the crp gene led to an increase in IS50 transposase expression and the magnitude of this increase was comparable to that observed for an hfq disruption; and (2) Crp expression decreased in hfq−. We also demonstrate that IS50 transposase expression and Tn5 transposition are induced by over-expression of the sRNA SgrS and link this response to glucose limitation.
Conclusions
Tn5 transposition is negatively regulated by Hfq primarily through inhibition of IS50 transposase transcription. Preliminary results support the possibility that this regulation is mediated through Crp. We also provide evidence that glucose limitation activates IS50 transposase transcription and transposition.
Electronic supplementary material
The online version of this article (doi:10.1186/s13100-014-0027-z) contains supplementary material, which is available to authorized users.
Keywords: Tn5/IS50, Hfq, Crp, SgrS, DNA transposition
Background
Transposase proteins catalyze the chemical steps in bacterial transposition reactions. It follows that the regulation of expression of these genes is a critical feature in dictating the transposition frequency of most transposons. In many instances, including Tn10/IS10 and Tn5/IS50, transposase gene promoters are inherently weak. In addition, DNA adenine methylase (DAM) limits initiation of IS10 and IS50 transposase gene transcription by methylating promoter elements [1,2]. These factors together make transcription initiation a limiting step in Tn10/IS10 and Tn5/IS50 transposition reactions [3,4]. There are also examples where translation of transposase transcripts is subject to both intrinsic and host levels of regulation. In the case of IS10 transposase, the ribosome binding site is inherently weak and the transposon encodes an antisense RNA that binds the translation initiation region (TIR), blocking ribosome binding [5,6]. There is also evidence that the ‘host’ protein Hfq helps mediate the pairing interaction between the antisense RNA and the IS10 transposase transcript [7,8].
Hfq is a global regulator of gene expression in bacteria. It typically functions at the post-transcriptional level, influencing translation initiation and/or transcript stability by catalyzing the pairing of small RNAs (sRNA) and their mRNA targets (Figure 1B and reviewed in [9]). In contrast to the many examples of Hfq acting in a post-transcriptional capacity to impact gene expression, there is (to our knowledge) only one example in the literature of Hfq acting at the level of transcription to influence gene expression. In the case of ribosomal proteins rpsO, rpsT and rpsB-tsf, Hfq was shown to increase transcript levels without influencing transcript stability. It was suggested that this is accomplished through Hfq binding to secondary structure elements in the respective transcripts that form early in the elongation phase of transcription and that this interaction reduces RNA polymerase pausing [10].
Figure 1.
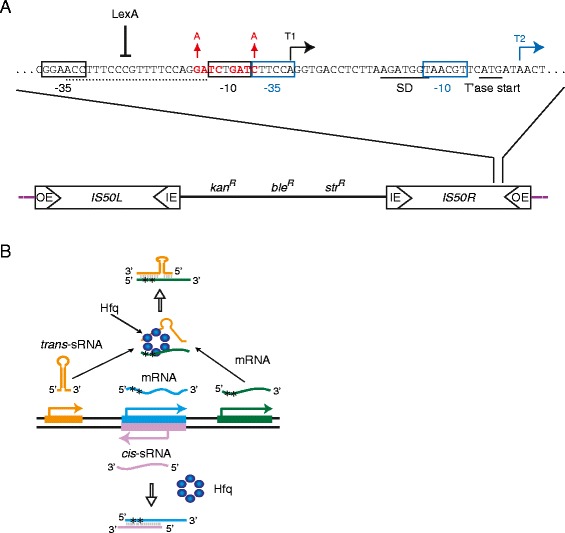
Tn5 / IS50 structure and gene expression. (A) The structure of Tn5 is shown along with transcription units within IS50-Right. There are two distinct promoters defined by -35/-10 regions that control transposase (black) and inhibitor (blue) expression. T1 is the transposase transcript and T2 is the inhibitor transcript. The Shine-Dalgarno sequence of T1 is also shown. Expression of T1 but not T2 is regulated by DAM methylation at two GATC sequences (red) and potentially LexA binding (dotted line defines a putative LexA binding site). Mutations in the dam sites used in this work are shown. kan R, ble R and str R are kanamycin, bleomycin and streptomycin resistance genes, respectively. (B) Post-transcriptional regulation by Hfq. Hfq (blue hexamer) is shown catalyzing the pairing of an sRNA with an mRNA. The sRNA can be either cis or trans encoded relative to its target mRNA. In both cases the sRNA is shown pairing to the translation initiation region of the mRNA (asterisks) and would block translation.
As noted above, Hfq has been implicated in the regulation of Tn10/IS10 transposition. Under conditions of hfq deficiency, a large increase in both Tn10/IS10 transposition (up to 80-fold) and transposase expression (up to 7-fold) were observed. The existing evidence is consistent with Hfq acting as a negative regulator of IS10 transposase expression by both antisense dependent and independent pathways. In support of the latter, it was found that hfq deficiency (or hfq−) had a significant impact on Tn10 transposition even when the level of antisense RNA was insufficient to impact on transposase expression (that is when Tn10 is present in single copy in the bacterial chromosome). In addition, there was a synergistic increase in transposase expression when both hfq and the antisense RNA were knocked out, implying that Hfq does not function exclusively in the same pathway as the antisense RNA [7].
Taking the above results into account, and considering that most bacterial transposition systems are not regulated by antisense RNAs, we wondered if Hfq might play a more general role in regulating transposition systems. In the current work, we tested this hypothesis by asking if Tn5 transposition is also regulated by Hfq. Like Tn10, Tn5 is a composite transposon (Figure 1A). The two transposons are closely related but Tn5 lacks an antisense RNA regulatory system and consequently if Hfq were to regulate this system at the post-transcriptional level, it is likely that a trans-encoded sRNA would play a role [11-13]. Tn5 does encode an inhibitor protein that limits Tn5/IS50 transposition by dimerizing with the transposase protein, forming an inactive complex [14]. Transposase and the inhibitor protein are expressed from overlapping promoters, P1 and P2 (color coded in Figure 1A), with the inhibitor transcript (T2) being expressed at a higher level than the transposase transcript (T1). T1 expression is down-regulated by DAM (reviewed in [15]). There is some evidence that P1 is also negatively regulated by LexA, an SOS-inducible transcriptional repressor [16]. However, there is little else known with regard to host proteins that influence either transposase transcription or translation.
In the current work, we show that both Tn5 transposition and IS50 transposase expression increase significantly in E. coli under conditions of hfq deficiency. However, unlike the situation in Tn10/IS10 transposition, the up-regulation of IS50 transposase expression appears mainly to be due to an increase in transposase gene transcription. As Hfq does not typically function directly in transcription, we looked at the possibility that Hfq regulates IS50 transposase expression by controlling the expression of a transcription factor. Towards this end, we provide evidence that Hfq acts in a regulatory network with Crp (cyclic AMP receptor protein) to down-regulate IS50 transposase transcription. Finally, we demonstrate that over-expression of an sRNA (SgrS) activates expression of the IS50 transposase gene specifically when cells are grown with glucose as the sole carbon source. Evidence is presented that this up-regulation is a consequence of glucose limitation, demonstrating that the IS50 transposase promoter (and Tn5 transposition) is responsive to the nutrient status of the cell.
Results
Hfq is a potent negative regulator of Tn5 transposition
We asked if Hfq regulates Tn5 transposition in E. coli by measuring the frequency of Tn5 transposition under conditions of hfq deficiency using the ‘mating out’ assay. In this assay, an F+ donor strain harboring a chromosomal copy of Tn5 was mated to an F− recipient strain and the mating efficiency and number of transposition events were measured by plating mating mixes on the appropriate selective media (see Methods). We show in Figure 2A that in one donor strain background (DBH179) Tn5 transposition increased by close to 75-fold under conditions of hfq deficiency. Note that we did not have a defective copy of Tn5 to act as a negative control in this experiment. In lieu of this, we carried out physical mapping on a sampling of colonies present on ‘hop’ plates to ensure that bona fide transposition events were being measured in both wt and hfq− strains (Additional file 1).
Figure 2.
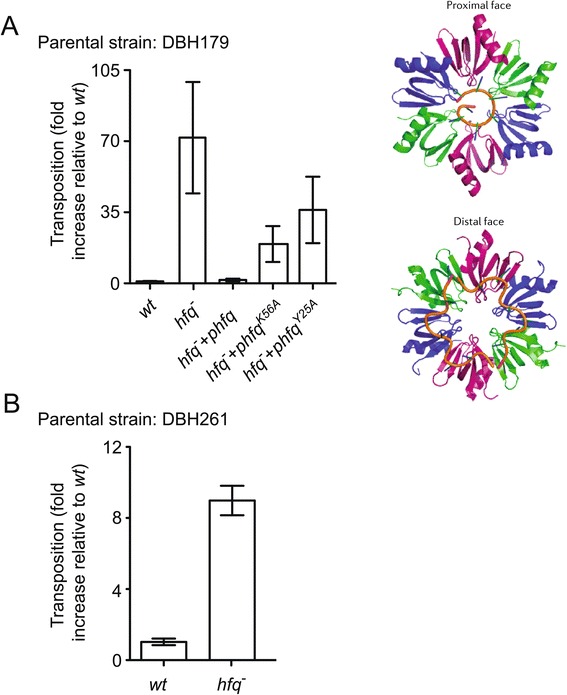
Frequencies of Tn5 transposition in hfq − versus wt strains of E. coli . (A) Tn5 transposition from the chromosome of DBH179 and derivatives (hfq − and dam −) was measured by the conjugal ‘mating out’ assay as described in Methods. For purposes of trans-complementation, strains contained an empty vector or a low-copy plasmid encoding either wild type hfq WT or mutant forms of hfq (K56A or Y25A) expressed from the hfq P3 promoter. The data was compiled from four independent experiments, each with at least three isolates of each strain. The average transposition frequency was 8.33 × 10−5 events per mL of mating mix for the wt strain (no ‘hfq plasmid’) and for purposes of comparison this value was set at 1 and all other values normalized to this. The illustration shows the structure of an Hfq hexamer with RNA (gold) bound either to the proximal or distal face [9]. The Y25A mutation inhibits RNA binding to the distal face and the K56A mutation inhibits RNA binding to the proximal face. Adapted from Nature Reviews: Microbiology [9] with permission from Macmillan Publishers. (B) Tn5 transposition from the chromosome of DBH261 and derivatives (hfq − and dam −) was measured as in (A). The data shown is from one experiment with five independent isolates of each strain. The average transposition frequency for the wt strain was 2.57 × 10−6 events per mL of mating mix. In (A) and (B) the error bars indicate standard error of the mean.
We also performed a complementation assay in the DBH179 strain background to further test that the increase in transposition reported above in hfq− was actually due to the absence of Hfq, as opposed to possible polar effects of the hfq disruption allele. Towards this end, we introduced hfq on a low-copy plasmid (pDH700) into the hfq− strain and measured Tn5 transposition as above. We observed nearly complete complementation by plasmid-borne hfq, as transposition was reduced approximately 45-fold relative to when no hfq was present (Figure 2A). Furthermore, plasmid-encoded variants of hfq, including K56A and Y25A, which are impaired for RNA-binding at the ‘proximal’ and ‘distal’ surface, respectively, failed to complement hfq deficiency [17]. This confirms that specific functions of Hfq, namely interaction with RNA via known RNA-binding surfaces, are required for effective repression of Tn5 transposition.
We also tested the impact of hfq deficiency on Tn5 transposition in a second donor strain background (DBH261) via the ‘mating out’ assay (Figure 2B). In this experiment hfq− also caused an increase in Tn5 transposition, although the magnitude of the effect was smaller (approximately 9-fold) than reported for the DBH179 strain background.
IS50 transposase expression increases in hfq− cells
We next asked if hfq status influenced IS50 transposase expression. In one approach, we measured transposase expression by constructing IS50-lacZ transcriptional and translational fusions (‘TCF’ and ‘TLF’, respectively; see Figure 3A for schematics), integrating these reporters into the chromosome of a lac−E. coli strain (DBH107), and then performing β-galactosidase assays. This was done for each reporter in isogenic strains that were either wt, dam− or hfq−. As expected for a promoter that is DAM-sensitive, transposase expression increased in the context of both transcriptional and translational fusions in the dam− strain relative to wt (approximately19- and 25-fold, respectively; Figure 3B). The increase in transposase expression for both constructs in dam− is indicative of expression coming predominantly from the P1 promoter [2]. Transposase expression in TCF and TLF constructs also increased in hfq− cells (11-fold and 7.4-fold, respectively), indicating that Hfq (or a factor under Hfq control) represses IS50 transposase expression. As the TCF encodes only 15 nucleotides of the transposase transcript (T1), it seemed most likely that up-regulation of transposase expression in hfq− was primarily due to enhanced transcription in both TCF and TLF constructs.
Figure 3.
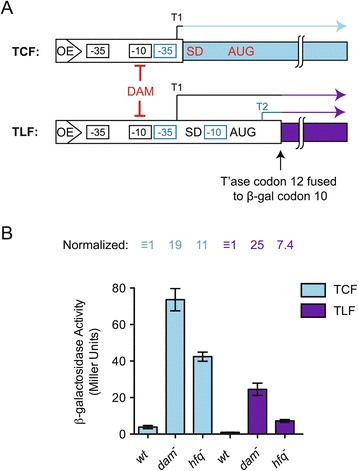
Transposase- lacZ translational and transcriptional fusion reporter assays in wt , dam − and hfq − strains. (A) Schematic of the IS50-lacZ transcriptional fusion (TCF; upper) and translational fusion (TLF; lower) reporters. The TCF reporter encodes the first 80 bp of IS50-Right (white rectangle) fused to lacZ (light blue rectangle). This fusion encodes only the first 15 nucleotides of the transposase (T1) transcript, which is expressed from the native promoter; the -35/-10 elements are shown in black. The inhibitor transcript is not expressed as the promoter for the inhibitor is missing its -10 region. The TLF encodes the first 128 bp of IS50-Right. This includes up to the 12th codon of T1, which is fused in-frame to the 10th codon of lacZ (purple rectangle). T1 and T2 and their respective promoter elements (-35/-10 sequences) are color-coded. Note that the start codon for the inhibitor protein has been mutated so that only transposase expression will give rise to β-galactosidase activity. Also note that the transposase promoter in both the TCF and the TLF is sensitive to Dam methylation. (B) β-galactosidase activity (given in Miller Units) for isogenic strains (wt, dam − or hfq −) harboring either the TCF or TLF in single-copy in the chromosome of E. coli. For each fusion, the activity was normalized to that of the wt strain. The data sets shown for the TCF and TLF were compiled from two and three independent experiments, respectively, with each experiment including at least three replicates. Mean and standard error values are shown.
To further test this possibility, we constructed a TLF reporter (pDH908) wherein the IS50 transposase promoter was replaced by a heterologous promoter (from the lpp gene) whose regulation is not sensitive to hfq status [10]. An isogenic plasmid (pDH795) in which the TLF contained the IS50 transposase promoter was also constructed. Cells (wt or hfq−) were transformed with plasmids containing these constructs and reporter expression was measured as above. The results presented in Figure 4 show that transposase expression increased approximately 9-fold for the construct containing the IS50 promoter and less than 2-fold for the construct containing the lpp promoter. These results support our contention that Hfq-directed regulation of IS50 transposase expression occurs at the transcriptional level because the absence of the IS50 promoter and not the presence of the IS50 5′ UTR was the dominant factor in observing strong up-regulation of reporter expression under conditions of hfq deficiency.
Figure 4.
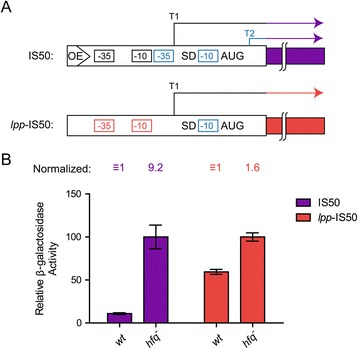
Heterologous promoter-transposase- lacZ translational fusion reporter assays in wt and hfq − strains. (A) Schematic of the IS50-lacZ translational fusion with IS50 transposase and lpp promoters. The IS50 translational fusion (TLF) is as described in Figure 3A. The lpp-IS50 TLF contains the lpp promoter (-35 and -10 elements) fused to the IS50 transposase gene such that only IS50 sequences at, and downstream of the T1 transcriptional start, are present. (B) β-galactosidase activity for isogenic strains (wt or hfq −) harboring the indicated TLF on a multicopy plasmid. For each fusion, the activity was normalized to that of the wt strain. The data sets shown were compiled from two independent experiments, respectively, with each experiment including at least three replicates. Mean and standard error values are shown.
Hfq impacts steady-state levels of full-length IS50 transposase mRNA
To further assess the impact of hfq deficiency on transposase gene expression, we looked at both the steady-state level and the stability of the transposase transcript (T1) in hfq+ and hfq− cells. For the steady-state analysis, total RNA was isolated from various strains (wt or hfq−) (DBH33 background) containing a multi-copy plasmid encoding the full-length transposase gene under the control of its native promoter. In addition to the wt version of this plasmid (pDH533), we also analyzed a mutant form containing mutations in the overlapping dam methylation sites in the transposase promoter (pDH752) (see Figure 1A); these mutations make this construct DAM insensitive. Primer extension was used to detect both T1 and T2 transcripts, as well as the lpp transcript (loading control). As expected for a dam-sensitive promoter, levels of T1 increased substantially (approximately 8-fold) in wt cells containing the plasmid with the dam-insensitive promoter versus wt cells containing the wt promoter (compare lanes 3 to 7 with lanes 8 to 12 in Figure 5A and bar graph in Figure 5B). In contrast, there was no significant change in T2 levels in the above samples. In hfq− (wt promoter) there was also a substantial increase in T1 levels (11-fold) versus the wt strain (compare lanes 3 to 7 with lanes 14 to 18) and no significant change in T2 levels. Thus in an hfq− background there was an increase in the steady-state level of transposase transcript and this increase was slightly greater than that observed when methylation of the transposase promoter was blocked.
Figure 5.
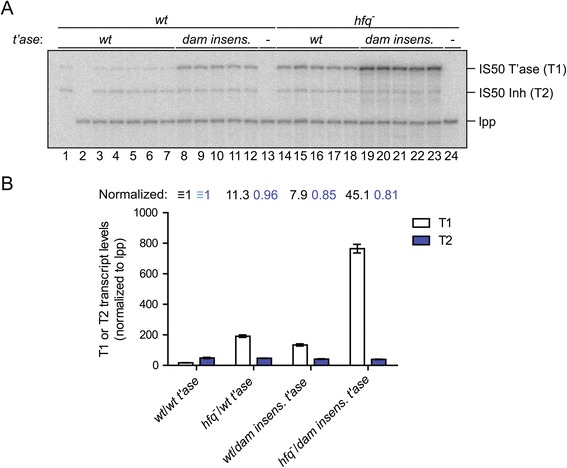
Steady-state levels of IS50 transposase mRNA in wt and hfq − cells. (A) Plasmids encoding wt or DAM-Insensitive IS50 transposase genes were transformed into wt (DBH33) or hfq − (DBH16) E. coli strains. Total RNA was isolated from five different clones grown to mid-log phase for each of the indicated strains. Primer extension reactions were multiplexed using 32P-labeled primers complimentary to IS50 transposase (primer oDH230) and lpp (primer oDH390) RNAs. The corresponding cDNAs were analyzed on a 10% sequencing gel. T1 and T2 are defined in Figure 1. Note that transcription of lpp is known to be insensitive to hfq status [10]. (B) Summary of data in (A).
We also looked at the combined impact of knocking out Hfq and blocking DAM methylation on T1 levels (lanes 19 to 23 in Figure 5A). In comparison to wt, the ‘double mutant’ situation resulted in a 45-fold increase in T1 levels. Based on the observed synergy, we think it unlikely that the observed impact of deleting hfq is linked to the regulation of dam expression.
To directly test if a component of Hfq-directed repression of IS50 transposase expression is post-transcriptional, we compared the stability of the IS50 transposase mRNA (T1) in isogenic wt and hfq− strains. Total RNA was isolated from a pair of rifampicin-sensitive strains (TM338 and TM618) containing a plasmid encoding IS50 transposase (pDH533) before and after rifampicin treatment as shown in Figure 6. Transposase mRNA was detected by primer extension. In the hfq− strain the half-life of the T1 transcript increased by approximately 1.7-fold, revealing that hfq status does impact on transposase mRNA stability.
Figure 6.
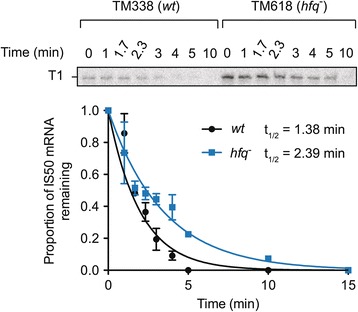
IS50 transposase mRNA half-life analysis. Strains TM338 (wt) and TM618 (hfq −) were transformed with IS50 transposase encoding plasmid pDH533 and total RNA was isolated either before or after the addition of rifampicin (at the indicated time points). Transposase RNA was detected as described in Figure 5. The bands were quantified (ImageQuant) and T1 normalized to un-extended primer before plotting the proportion of RNA remaining after rifampicin addition (time zero = 1.0). The data was fit to a one-phase exponential decay curve by non-linear regression (Prism) to determine the half-life (t1/2). The data shown is a compilation from two independent experiments.
Taken together, the results from Figures 3, 4, 5 and 6 show that IS50 transposase expression is substantially reduced in an hfq+ relative to an hfq− strain and that hfq status primarily affects transposase transcription.
Regulation of Tn5 transposase expression by global transcriptional regulators
As Hfq does not typically function directly in transcription, we set out to define a transcription factor that down-regulates IS50 transposase transcription and is itself regulated by Hfq. Toward this end, we asked if disrupting genes for two global transcription factors, Crp and Lrp [18], had an impact on IS50 transposase expression. Note that we had to construct new TCF reporter strains for this work because the available crp and lrp disruption strains we used to transduce DBH107 to either crp− or lrp− were marked with the same antibiotic resistance gene used to select for a lysogen with a chromosomal copy of the TCF. We show in Figure 7A that crp− but not lrp− had a substantial impact on transposase expression. For example, in cells grown in exponential phase in Luria broth (LB), there was up-regulation of transposase expression (approximately 4-fold) in both crp− (DBH307) and hfq− (DBH306) strains but not in the lrp− strain (DBH315). We also performed semi-quantitative RT-PCR and show that transposase-lacZ transcript levels increased similarly in crp− and hfq− strains (Figure 7B). These results are consistent with Crp being a negative regulator of IS50 transposase transcription.
Figure 7.
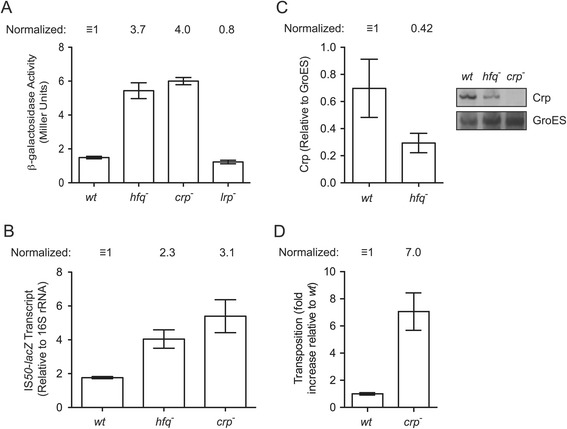
Gene expression and Tn5 transposition assays in strains harboring disruptions of global transcriptional regulators. (A) β-galactosidase activity for isogenic strains (wt, hfq −, crp − and lrp −) harboring the TCF in single-copy in the chromosome (DBH303 and derivatives). Cells were grown to mid-log phase in Luria broth (LB). Mean and standard error values of duplicate experiments, each of which included at least three replicates, are shown. (B) IS50-lacZ transcript levels. Total RNA was extracted from cells described in panel (A), and subjected to RT-PCR. (C) Western blot analysis of Crp levels in cellular extracts from wt and hfq − cells grown in LB. As a negative control, crp − cells were also analyzed. A representative image is shown in the inset. Crp levels were normalized to GroES, which is known to be insensitive to hfq status [19]. (D) Tn5 transposition from the chromosome of DBH179 (wt) and DBH345 (crp−) was measured by the conjugal ‘mating out’ assay as described in Methods. The data is from a single experiment wherein five independent clones of each strain were tested. Mean and standard error values are shown. The average transposition frequency was 1.70 × 10−4 events per mL of mating mix for the wt strain and for purposes of comparison this value was set at 1 and the ‘crp’ value was normalized to this. In two other independent experiments the fold increase in Tn5 transposition for crp − versus wt did not differ by more than 20% compared to the experiment shown (data not shown). For experiments in (A-C), mean and standard error values from at least three independent isolates are shown.
We next asked if Crp expression was regulated by Hfq. Notably, work done in Yersinia pestis has shown that Hfq positively regulates Crp expression at the post-transcriptional level [20]. Towards this end we performed Western blot analysis with a Crp antibody on E. coli cell extracts from wt (DBH303), hfq− (DBH306) and crp− (DBH307) strains (Figure 7C). The results show that lower levels of Crp are present in the hfq− strain, which is consistent with Hfq also being a positive regulator of Crp expression in E. coli.
Finally, we assessed the impact of knocking out crp on Tn5 transposition frequency using the ‘mating out’ assay (Figure 7D). In the absence of crp, Tn5 transposition increased 7-fold, which is consistent with results from the transposase expression experiments.
IS50 transposase expression and Tn5 transposition are up-regulated by over-expression of the sRNA SgrS
Over-expression of sRNAs can alter Hfq-regulated networks by limiting the availability of Hfq [21,22]. Given our findings that Tn5 transposition and transposase gene expression are affected by hfq status, we asked if IS50 transposase expression might be sensitive to Hfq-titration. Towards this end, we measured transposase expression from the TLF under conditions where a single sRNA was over-expressed from an IPTG inducible promoter (pLlacO) in DBH33, which is lacIq. Our initial screen included four different Hfq-dependent sRNAs, including RybB, RyeB, MicC and SgrS, all of which are expected to tightly bind Hfq in vivo; apparent equilibrium dissociation constants of approximately 3.3 nM and < 20 nM have been measured for MicC and SgrS, respectively [23-25]. Cells were grown in M9 glucose and sRNA expression was induced for 4 hours in exponential phase. We show in Figure 8A that only one of the sRNAs tested, SgrS, had a significant impact on transposase expression. Induction of SgrS increased transposase expression just over three-fold. Given the comparable Hfq binding affinities of the sRNAs tested, it seemed unlikely that SgrS expression was increasing transposase expression through an Hfq-titration mechanism.
Figure 8.
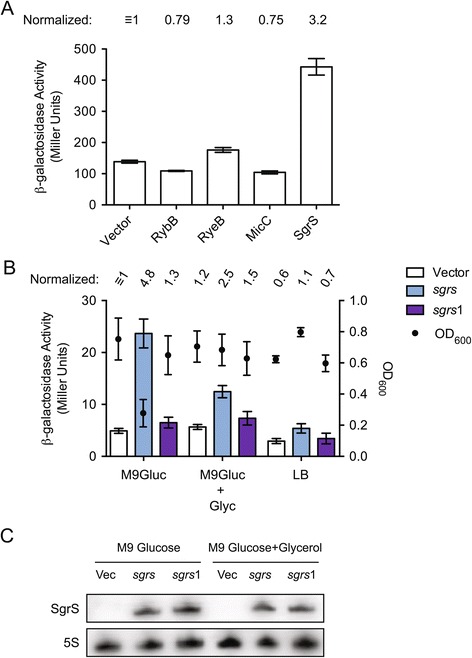
Transposase- lacZ expression assays in cells over-expressing sRNAs. (A) Transposase expression from an IS50 translational fusion (TLF) (see Figure 3A) present on a low-copy plasmid (pDH798) was measured in the presence of a compatible plasmid expressing one of the indicated sRNAs from the inducible pLlacO promoter in DBH33. Cells were grown in M9 glucose and 0.1 mM IPTG was added to subcultures to induce sRNA expression. Transposase expression was measured 4 hours after IPTG addition. Expression levels were normalized to the strain with the vector only control. (B) The impact of different growth media on SgrS-induced up-regulation of transposase expression was evaluated using a single-copy TCF fusion (see Figure 3A) present in the chromosome of DBH265. Note that the sgrS1 allele of SgrS contains a two-nucleotide mutation that inhibits its ability to down-regulate expression of the glucose transporter encoded by ptsG. Subcultures were grown in either M9 glucose, M9 glucose + glycerol, or Luria broth (LB), as indicated. β-galactosidase activity was measured approximately 4 to 6 hours after subcultures were started. In (A) and (B) mean and standard error values of duplicate experiments, each of which included at least three replicates, are shown. (C) Northern blot of RNA isolated from cells in (B). RNA was extracted from cells immediately before starting the Miller assay and visualized by Northern blotting with 32P-labeled RNA probes complementary to either SgrS or the 5S rRNA (internal control).
SgrS down-regulates the expression of several known targets, including the primary glucose transporter encoded by the ptsG gene, a mannose transporter encoded by manXYZ and it up-regulates the expression of yigL, a phosphatase involved in phospho-sugar detoxification [26]. As we observed up-regulation of IS50 transposase expression in cells over-expressing SgrS in M9 glucose media, we considered the possibility that this effect was a response to glucose limitation. In fact, we show in Additional file 2 that induction of SgrS in M9 glucose resulted in a substantial slowing of bacterial growth, as would be expected if nutrients had become growth-rate limiting. To further test the glucose limitation hypothesis, we performed a similar experiment in rich media (LB) and in M9 glucose supplemented with glycerol, a carbon source whose import is not dependent on glucose transporters [27]. We also tested the response of the reporter to over-expression of an SgrS mutant, sgrS1, that is incapable of down-regulating glucose import [28]. In these experiments we used a Tn5 TCF as a reporter in the DBH107 strain background; DBH107 has a complete deletion of the lac operon and consequently the plasmid-encoded sRNA genes are constitutively expressed. To avoid problems in growing these cells, cultures were initially propagated in either LB or M9 glucose/glycerol and then where indicated, switched to other media.
We show in Figure 8B that after approximately 4 hours of SgrS over-expression in M9 glucose, reporter expression increased close to 5-fold relative to a ‘vector’ control. In contrast, over-expression of SgrS1 was incapable of up-regulating reporter expression under these same conditions, suggesting that SgrS must be able to down-regulate glucose import and or retention in order to increase transposase transcription. When cells were grown in M9 glucose supplemented with glycerol, expression of SgrS as above caused only an approximately 2-fold increase in transposase expression. Importantly, the reduced effects of SgrS on transposase expression under ‘glycerol’ conditions cannot be explained by differential expression of the respective sRNAs, as levels of SgrS and SgrS1 were similar in M9 glucose with or without glycerol (Figure 8C). Also, we failed to see significant transposase induction when SgrS was over-expressed in LB media where there are multiple carbon sources. Finally, consistent with the glucose limitation hypothesis, we also show in Figure 8B that increased transposase expression resulting from SgrS expression in M9 glucose was the only condition that inhibited cell growth.
Given that transposition frequency is expected to be roughly proportional to transposase expression, we also asked if glucose limitation had an impact on Tn5 transposition. Cells encoding a chromosomal copy of Tn5 were transformed with an SgrS-expressing plasmid (or vector only control) and the frequency of Tn5 transposition was measured using the ‘mating out’ assay. Note that cells were grown in M9 glucose media and SgrS expression was induced only when donor strains were subcultured on the day of mating. We show in Figure 9 that induction specifically of SgrS resulted in a 5-fold increase in Tn5 transposition relative to the vector only control. Notably, when cells were grown in M9 supplemented with glucose and glycerol, induction of SgrS did not result in a significant increase in Tn5 transposition. Also, we observed a reduced growth rate only in cultures where SgrS was induced in M9 glucose media (data not shown). The results of the ‘mating out’ analysis are thus entirely consistent with the gene expression experiments presented in Figure 8.
Figure 9.
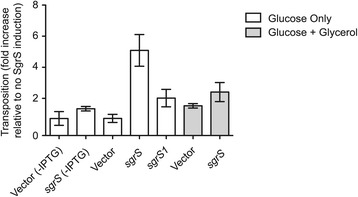
Impact of SgrS expression on Tn5 transposition. Transposition of a chromosomal copy of Tn5 was measured in DBH179 using the ‘mating out’ assay. DBH179 containing one of the indicated plasmids was grown overnight in M9 glucose and then subcultured in either M9 glucose or M9 glucose plus glycerol before mating with the recipient strain and plating on selective media as described in Figure 2. IPTG was added to the subculture (to 0.1 mM) to induce SgrS expression, except where indicated (-IPTG). The average transposition frequency for the ‘no SgrS’ control was 5.52 × 10−5 events per mL of mating mix. All other transposition frequencies were normalized to this value. Mean and standard error values of duplicate experiments, each of which included at least five replicates for each experimental group, are shown.
Discussion
Hfq is a global regulator of gene expression in bacteria. However, until recently, Hfq had not been linked to the control of transposable elements. Work in the Tn10/IS10 system provided the first example of Hfq inhibiting a transposon [7]. In the current work, we asked if the transposition of a related element, Tn5/IS50, is also regulated by Hfq. We show that Tn5 transposition and IS50 transposase expression are repressed by Hfq; however, the mechanism of repression is atypical for Hfq, involving predominantly a block in IS50 transposase transcription. Preliminary evidence is presented that is consistent with Hfq modulating IS50 transposase transcription through regulation of Crp. We also show that transposase transcription and Tn5 transposition are activated by over-expression of the sRNA SgrS and provide evidence that this is a transcriptional response to glucose limitation.
Hfq negatively regulates Tn5 transposition
The results of ‘mating out’ experiments were consistent with Hfq acting as a strong negative regulator of Tn5 transposition. Tn5 transposition increased close to 75-fold in one hfq− strain (DBH179 background). The magnitude of this increase was somewhat surprising given that up-regulation of Tn10 in hfq−, under essentially antisense-minus conditions, was about 7-fold [7]. However, in a different hfq− strain (DBH261 background) Tn5 transposition increased only 9-fold. At this point it is unclear why there was such a large discrepancy in the ‘mating out’ values for the two strains. One possibility is that colony counts in the DBH179 ‘mating out’ (hfq−) included clones that had ‘jack-pot’ events. That is, colonies were counted that did not derive from independent transposition events. This could explain the high standard error associated with the transposition frequency in the hfq− column in Figure 2A. If, for example, we removed the 3 most prominent outliers from the (DBH179) hfq− data set, the fold increase in transposition dropped to 15-fold, which is more in line with what we observed in the DBH261 strain background and for Tn10 in single copy [7].
A trans-complementation (Figure 2A) experiment provided definitive proof that the increase in Tn5 transposition detected in one of our hfq− ‘mating out’ strains (DBH179 background) was in fact due to hfq deficiency. In addition, the failure of two Hfq RNA-binding face mutants to provide complementation was consistent with Hfq-directed inhibition of Tn5 transposition relying on functions of Hfq required in canonical Hfq-directed regulatory pathways [17]. That is, Hfq must retain the ability to bind both mRNAs and sRNAs to influence Tn5 transposition.
Hfq, Crp and IS50 transposase gene expression
Evidence that hfq status influences IS50 transposase expression came from two types of experiments. First, the expression of transposase-lacZ reporter genes in both transcriptional and translational fusion constructs increased significantly under conditions of hfq deficiency. Second, the steady-state level of the native transposase transcript also increased significantly in hfq−. Importantly, the large increase in steady-state transcript level (11-fold) coincided with a less substantial increase in transposase mRNA stability (less than 2-fold increase in half-life). In addition, up-regulation of reporter expression in hfq− for a TLF was almost completely abrogated when the IS50 transposase promoter was replaced by a heterologous promoter. Taken together, these results are consistent with Hfq (or a factor regulated by Hfq) suppressing IS50 transposase expression predominantly at the level of transcription. Notably the suppressive effect of Hfq on IS50 transposase transcription was remarkably specific, as the level of a second transcript (T2) encoded by IS50 was not affected by hfq status.
As Hfq does not typically act directly in gene transcription, we think it likely that Hfq acts indirectly on the IS50 transposase promoter. In addition to DAM, only one other transcription factor, LexA, has been implicated as a regulator of transposase transcription. There is a weak LexA-binding site in the transposase promoter (Figure 1A); however, lexA deficiency was shown to increase transposase transcription only two to three-fold in a TCF [16]. As we have seen increases in transposase expression of up to 11-fold for a TCF in hfq−, it seems unlikely that Hfq would be working through LexA. In contrast, transposase expression increased in dam− to a level more in line with that observed in hfq− (less than two-fold difference in the TCF). However, the observed synergy between hfq− and mutations that rendered the IS50 transposase promoter DAM-insensitive led us to conclude that Hfq does not regulate IS50 transcription by impacting DAM levels (and, therefore, promoter methylation). These results provided motivation to search for other targets of Hfq that impinge on IS50 transposase transcription. This search identified Crp as an additional negative regulator of IS50 transposase transcription. Notably, transposase expression increased to approximately the same level in crp− and hfq− in the experiment in Figure 7. The similar magnitude of up-regulation of transposase expression in hfq− and crp− could be indicative of Hfq acting upstream of Crp to inhibit transposase expression. We did in fact find evidence of Hfq positively regulating Crp protein levels (Figure 7C). This observation is consistent with work recently published in the Y. pestis system where it was found that Crp protein levels decreased approximately five-fold in an hfq disruption strain [20].
Crp is a known activator/repressor of transcription [18] and, therefore, more likely than Hfq to be directly involved in regulating IS50 transposase expression at the transcriptional level. Given our evidence that Hfq positively regulates crp expression, a plausible scenario explaining our expression data is that the observed up-regulation of IS50 transposase transcription in hfq− is a result of decreased Crp protein levels. Crp may act either directly or indirectly on the IS50 transposase promoter to repress transcription. This is currently a working model as we have not yet tested the possibility that Crp binds the IS50 transposase promoter and it may only be coincidental that transposase expression increased to similar levels in hfq− and crp− strains. Notably, we also found that Tn5 transposition increased when the crp gene was disrupted, although the extent of the increase was smaller than that observed in the isogenic hfq disruption strain. This could be indicative of additional factors in the Hfq regulon impinging on Tn5 transposition.
There is precedent for Crp down-regulating the transcription of a transposase gene. In the case of IS2, transposase transcription increased close to 200-fold in crp−. It was also shown through protein-DNA footprinting that Crp binds directly to the IS2 transposase promoter [29]. Interestingly, based on the consensus binding sequence for Crp, the authors of the above study predicted that Crp would bind to the IS50 transposase gene. However, the predicted crp binding site is located downstream of the transposase promoter and is not present in our TCF (where we detected increased transposase expression in crp−). Nevertheless, it would be worthwhile to test for Crp binding to the IS50 transposase promoter as the results of Crp ChIP-chip studies revealed the presence of thousands of weak crp binding sites scattered throughout the E. coli genome [30]. It is also possible that Crp acts indirectly on the IS50 transposase promoter by regulating the expression of another transcription factor.
Tn5 transposition and metabolic stress
We also identified conditions that activate transposase expression and transposition; over-expression of the sRNA SgrS increased transposase expression and transposition approximately five-fold. We favor the possibility that this induction is a consequence of glucose limitation but cannot formally rule out the possibility that SgrS targets an as yet undefined regulatory pathway that impinges on transposase expression. Our reasoning for this is that we observed induction of transposase expression and transposition specifically when cells were grown with glucose as the major carbon source and SgrS is known to prevent expression and function of the major glucose transporter encoded by the ptsG gene [26]. Consistent with this idea, we found that transposase induction levels correlated with a reduced growth rate. Furthermore, we demonstrated that: (i) an allele of SgrS (sgrS1) that is incapable of down-regulating ptsG expression failed to induce transposase expression in M9 glucose; (ii) under conditions where SgrS was expressed in M9 glucose media supplemented with glycerol, we failed to see induction of transposase expression to the same extent as when glycerol was absent; (iii) SgrS expression did not impact transposase expression when cells were grown in rich media (LB) and (iv) over-expression of 3 other sRNAs (RybB, RyeB and MicC) that are not expected to influence glucose transport did not increase transposase expression in M9 glucose [31-33]. Precedent for nutritional stress influencing transposition comes from earlier work in the IS903 system where mutations in a gene (aspA) required for fermentative metabolism during anaerobic growth caused transposition to occur at an accelerated rate [34].
At this point it is unclear as to what factors are driving the induction of the transposase gene under SgrS over-expression conditions. With regard to further defining the mechanism of IS50 transposase up-regulation under SgrS over-expression conditions, it would also be advantageous to find alternative experimental conditions for achieving this increased expression. If, for example, simply starving cells by restricting a carbon source during growth achieves the same end as over-expressing SgrS in M9 glucose media, an unbiased screen to search for genetic factors that are necessary for the up-regulation of transposase expression could be performed to reveal the regulatory network impinging on the transposase promoter. As it stands, any factors that influence SgrS expression would interfere with the outcome of such a screen. Alternatively, if it was found that restricting glucose is not sufficient for inducing transposase expression, the possibility that SgrS plays a more direct role in controlling transposase expression would have to be considered.
Conclusions
In this work, we have identified several genes that impact on IS50 transposase expression, including hfq, crp and sgrS. Hfq and Crp proteins are negative regulators and SgrS RNA (under specific growth conditions) is a positive regulator of transposase gene expression. Exactly how these factors impinge on transposase expression remains to be worked out and at this point it is not clear if we are seeing modulation of the same regulatory network in opposite directions when hfq and crp genes are disrupted and SgrS RNA is over-expressed. Tn5/IS50 is the second transposon identified that is affected by disruption of the hfq gene and the first that does not encode an antisense RNA. This raises the possibility that Hfq influences the transposition frequency of many other bacterial transposons.
Methods
Plasmids, bacteriophage and strains
The IS50 translational fusion plasmid (pDH798) is a pWKS30-derivative containing base pairs 1 to 431 of IS50 (nucleotides 1 to 366 of T1) fused to codon 10 [35] of the E. coli lacZ gene. The IS50 transcriptional fusion plasmid (pDH682) is a pUC18-derivative containing base pairs 1 to 80 of IS50 (nucleotides 1 to 15 of T1) fused to nucleotide -16 (relative to the translational start codon) of lacZ. Plasmids encoding sRNAs (pDH764, sgrS; pDH766, rybB; pDH768, micC; pDH772, ryeB) and the corresponding empty vector control (pDH763) were kindly provided by S Gottesman. The plasmid encoding sgrS1 (pDH895) was kindly provided by C Vanderpool. Plasmids encoding Hfq (pDH700, wt) and mutant derivatives (pDH701, K56A; pDH713, Y25A) are described in Ross et al [8]. Details of plasmid constructions are provided in Additional file 3 and a list of oligonucleotides used in this work is provided in Additional file 4.
Lambda phages encoding IS50 transcriptional (λDBH849 and λDBH888) and translational (λDBH812) reporters were generated by cloning IS50 expression cassettes marked with an antibiotic resistance gene (either kanR or cmR) into the his operon of pNK81 and then infecting a strain harboring one of these plasmids with λNK1039, which also contains the his operon. Antibiotic resistant lysogens from the above crosses were selected by replica plating and subsequently phage released from the lysogens were purified, giving rise to λDBH849 (IS50-lacZ-kanR TCF), λDBH888 (IS50-lacZ-CmR TCF) and λDBH812 (IS50-lacZ-KanR TLF).
E. coli strains for the ‘mating out’ assay were constructed by P1 transduction of Tn5 from ER2507 (NEB) into DBH33, DBH344 and DBH259. Strains containing chromosomal IS50-lacZ fusions were generated by lysogenizing DBH107 with λDBH849 (DBH265), λDBH888 (DBH303) or λDBH812 (DBH281). Mutant derivatives of these strains were generated by P1 transduction. A list of all of the strains, plasmids and bacteriophage used in this work is presented in Table 1.
Table 1.
Plasmids, bacteriophage and strains
| Strain or Plasmid | Relevant genotype | Use | Source or reference |
|---|---|---|---|
| E. coli | |||
| DBH13 | HB101 [F− leu − pro −]; StrR | ‘Mating out’ recipient | [36] |
| ER2507 | zjc::Tn5; KanR | Source of zjc::Tn5 | NEB |
| DBH179 | NK5830 [recA − arg −/F’ lacpro +] zjc::Tn5; KanR | ‘Mating out’ donor | This study |
| DBH184 | DBH179 hfq-1::Ωcat; KanRCmR | ‘Mating out’ donor | This study |
| DBH228 | RZ211/pOX38Gen | Source of pOX38Gen | [37] |
| DBH233 | HW-5 [phoA4(Am) his-45 recA1 rpsL99 met-54 F−]; StrR | Parent strain | [38] |
| DBH259 | DBH233/pOX38Gen; StrRGenR | Parent strain | This study |
| DBH261 | DBH259 zjc::Tn5; StrRGenRKanR | ‘Mating out’ donor | This study |
| DBH271 | DBH261 hfq-1::Ωcat; StrRGenRKanRCmR | ‘Mating out’ donor | This study |
| DBH272 | DBH261 dam::Tn9cat; StrRGenRKanRCmR | ‘Mating out’ donor | This study |
| DBH107 | MC4100 [F− Δ(argF-lac)169* rpsL150]; StrR | Parent strain | [39] |
| DBH265 | DBH107/λDBH849; StrRKanR | Miller Assay | This study |
| DBH267 | DBH265 hfq-1::Ωcat; StrRCmRKanR | Miller Assay | This study |
| DBH268 | DBH265 dam::Tn9cat; StrRCmRKanR | Miller Assay | This study |
| DBH281 | DBH107/λDBH812; StrRKanR | Miller Assay | This study |
| DBH283 | DBH281 hfq-1::Ωcat; StrRCmRKanR | Miller Assay | This study |
| DBH285 | DBH281 dam::Tn9cat; StrRCmRKanR | Miller Assay | This study |
| DBH303 | DBH107/λDBH888; StrRCmR | Miller Assay | This study |
| DBH306 | DBH303 Δhfq722::kan; StrRCmRKanR | Miller Assay | This study |
| DBH307 | DBH303 Δcrp765::kan; StrRCmRKanR | Miller Assay | This study |
| DBH315 | DBH303 Δlrp787::kan; StrRCmRKanR | Miller Assay | This study |
| DBH33 | NK5830 [recA − arg −/F’ lacpro +] | Parent strain | [40] |
| DBH16 | DBH33 hfq-1::Ωcat; CmR | Parent strain | [7] |
| DBH241 | DBH33 dam::Tn9cat; CmR | Parent strain | This study |
| DBH238 | DBH33/λDBH849; KanR | Miller Assay | This study |
| DBH239 | DBH238 hfq-1::Ωcat; KanRCmR | Miller Assay | This study |
| DBH240 | DBH238 dam::Tn9cat; KanRCmR | Miller Assay | This study |
| DBH208 | DBH33/λDBH812; KanR | Miller Assay | This study |
| DBH210 | DBH208 hfq-1::Ωcat; KanRCmR | Miller Assay | This study |
| DBH237 | DBH208 dam::Tn9cat; KanRCmR | Miller Assay | This study |
| DBH323 | DBH107 recA −; StrR | Miller Assay | This study |
| DBH326 | DBH107 recA − hfq-1::Ωcat; StrRCmR | Miller Assay | This study |
| DBH242 | DBH33 Δcrp765::kan | Parent strain | This study |
| DBH344 | DBH242 Δcrp765; KanS | Parent strain | This study |
| DBH345 | DBH344 zjc::Tn5; Kan R | ‘Mating out’ donor | This study |
| TM338 | W3110mlc rne-Flag-cat; rifSCmR | RNA half-life measurements | [41] |
| TM618 | W3110mlc rne-Flag-cat Δhfq; rifSCmR | RNA half-life measurements | [42] |
| DH5α | recA − | Plasmid propagation | Invitrogen |
| Plasmids | |||
| pWKS30 | pSC101-derived; low copy-number ori ; ApR | ‘Empty vector’ for Hfq expression | [35] |
| pDH700 | pWKS30-P3-hfq WT; ApR | HfqWT expression | [7] |
| pDH701 | pWKS30-P3-hfq K56A; ApR | HfqK56A expression | [7] |
| pDH713 | pWKS30-P3-hfq Y25A; ApR | HfqY25A expression | [8] |
| pDH533 | pUC18-derivative; Tn5 t’ase M56A; ApRCmR | Source of Tn5 transposase (No Inh.) | [43] |
| pDH752 | pDH533 with t’ase mutated to G53A,C61A; ApRCmR | DAM-insensitive t’ase | This study |
| pDH828 | pDH533 with t’ase mutated to D97A; ApRCmR | Catalytic− t’ase | This study |
| pNK81 | pBR333-derivative; encodes his operon; ApR | Lambda crosses | [44] |
| pDH682 | pUC18-derivative; IS50-lacZ TCF; ApR | Source of TCF | This study |
| pDH838 | pDH682-derivative; TCF ‘marked’ with kanR | Parent of pDH849 | This study |
| pDH883 | pDH682-derivative; TCF ‘marked’ with cmR | Parent of pDH888 | This study |
| pDH849 | TCF-kanR from pDH682 cloned into BclI-cut pNK81; ApRKanR | For crossing TCF onto λ | This study |
| pDH888 | TCF-cmR cloned onto BclI-cut pNK81; ApRCmR | For crossing TCF onto λ | This study |
| pDH658 | pRZ9905-derivative; full-length IS50-lacZ TLF; ApR | Parent of pDH795 | This study |
| pDH795 | pDH658-derivative; ‘deletion’ TLF used in this study; ApR | Parent of pDH804 | This study |
| pDH804 | pDH795-derivative; TLF ‘marked’ with kanR | Parent of pDH812 | This study |
| pDH812 | TLF-kanR cloned into BclI-cut pNK81; ApRKanR | For crossing TLF onto λ | This study |
| pDH753 | pWKS30-derivative; contains IS50-lacZ TLF from pDH658; ApR | Parent of pDH798 | This study |
| pDH798 | pDH753-derivative; ApSKanR | Miller Assay | This study |
| pDH763 | pBR-plac; ApR | Vector for sRNA-induction | [45] |
| pDH764 | pBR-plac-sgrS; ApR | SgrS-induction | [46] |
| pDH895 | pBR-plac-sgrS1; ApR | SgrS1-induction | [47] |
| pDH766 | pBR-plac-rybB; ApR | RybB-induction | [48] |
| pDH768 | pBR-plac-micC; ApR | MicC-induction | [48] |
| pDH772 | pBR-plac-ryeB; ApR | RyeB-induction | [48] |
| pDH908 | pDH795-derivative; Lpp-TLF | Miller Assay | This study |
| Phage | |||
| λNK1039 | Encodes his operon | Parent phage | [49] |
| λDBH812 | IS50-lacZ translational fusion (TLF) from pDH812 marked with kanR | Chromosomal TLF construction | This study |
| λDBH849 | IS50-lacZ transcriptional fusion (TCF) marked with kanR | Chromosomal TCF construction | This study |
| λDBH888 | IS50-lacZ transcriptional fusion (TCF) marked with cmR | Chromosomal TCF construction | This study |
‘Mating out’ assay
Conjugal ‘mating out’ experiments were performed essentially as described for single-copy chromosomal transposons in Ross et al. [7], except that for measuring transposition in hfq− versus wt, donor growth was carried out in M9 glucose media supplemented with kanamycin (25 μg/mL) and amino acids, instead of LB. DBH13 was used as the recipient. Total exconjugants and transposition events with DBH179 and derivatives were scored by plating mating mixes on M9 glucose plates supplemented with leucine, thiamine and streptomycin (150 μg/mL) or streptomycin and kanamycin (25 μg/mL), respectively. Total exconjugants and transposition events with DBH261 and derivatives were scored by plating mating mixes on M9 glucose plates supplemented with leucine, thiamine, streptomycin (150 μg/mL) and gentamicin (12.5 μg/mL) or streptomycin, gentamicin and kanamycin (25 μg/mL), respectively.
β-galactosidase assays
Cells were grown in M9 glucose (with arginine and thiamine) or LB. In situations where strains contained plasmids, plasmids were maintained by including the appropriate antibiotic. Overnight cultures (0.05 mL) were used to seed subcultures (1.5 mL), which typically were grown to mid-log phase before being processed for the Miller assay as previously described [7].
RNA isolation, primer extension and Northern blot analysis
Total RNA was isolated essentially as described in [50]. For steady-state analysis, cells were grown to mid-log phase in LB before RNA isolation. For half-life analysis, rifampicin (dissolved in dimethyl sulfoxide; DMSO) was added to cell cultures (to 200 μg/mL) to arrest transcription and RNA was isolated immediately before and after rifampicin addition at the indicated time intervals. Primer extension analysis was carried out using 32P-labeled primers oDH230 and oDH390, end-labeled with OptiKinase (USB, Cleveland, OH, USA) according to manufacturer’s instructions. Extension reactions used 5 μg of RNA, and Superscript III reverse transcriptase essentially as described in [51], except that annealing was performed at 65°C (with no ice treatment) before extending at 55°C for 45 minutes. Extension products were resolved on 6% and 10% denaturing polyacrylamide gels. For Northern blot analysis, 2 μg of RNA was mixed with an equal volume of denaturing load dye (95% deionized formamide [v/v], 10 mM EDTA, 0.5× TBE, 3% xylene cyanol [w/v]), heated to 95°C for 2 minutes, and resolved on a 6% polyacrylamide gel containing 7 M urea. Separated RNAs were electro-transferred to Hybond N (GE Healthcare, Mississauga, ON, Canada) in 0.5× TBE and fixed with UV. Annealing and washing was performed in ULTRAhyb buffer (Ambion, Burlington, ON, Canada) according to the manufacturer’s instructions, using RNA probes complimentary to SgrS or the 5S rRNA (internal standard). To construct the radiolabeled RNA probes, DNA templates for in vitro transcription were made by PCR with primers oDH232/233 (SgrS) and oDH234/235 (5S rRNA) - note that, for each primer pair, the forward primer includes the T7 core promoter. These templates were transcribed in vitro in the presence of 32P-UTP to generate uniformly labeled RNA probes. In vitro transcription reactions were performed in 25 μL volumes with approximately 1 μg DNA template, 1 × T7 RNA polymerase buffer (NEB, Beverly, MA, USA), 20 units RNasin (Promega, Madison, WI, USA), 4 mM dithiothreitol (DTT), 0.16 mg/mL BSA, 0.4 mM each of GTP, CTP and ATP, 0.01 mM UTP, 50 μCi [α-32P]UTP, and 100 units of T7 RNA polymerase.
Western blot
Cells were centrifuged (2 minutes at 21,000 × g), resuspended in SDS load mix (2% [w/v] SDS, 10% [v/v] glycerol, 50 mM Tris-HCl pH 6.8, 0.25% [w/v] bromophenol blue, 0.8 M β-mercaptoethanol) and heated at 95°C for 5 minutes. To normalize for differences in growth between the various samples, the OD600 of each sample was measured and the volume spun normalized to give an equivalent to OD600 approximately equal to 0.35. The resulting lysates were subjected to SDS-PAGE on a 12% polyacrylamide gel, proteins transferred to PVDF (Roche, Indianapolis, IN, USA) and Crp was detected by Western blot with a polyclonal rabbit anti-Crp antibody (kind gift of H Aiba). The primary antibody was diluted 1:20,000 in TBST; the secondary antibody (anti-rabbit IgG-horseradish peroxidase (HRP) conjugate; Promega, Madison, WI, USA) was used at 1:5,000. Crp was visualized with a Pierce ECL 2 Western blotting substrate (Thermo Scientific, Rockford, IL, USA) and PhosphorImager (GE Healthcare). The membranes were stripped and GroES detected (rabbit anti-GroES antibody from Sigma-Aldrich (St Louis, MO, USA) at 1:10,000) for use as an internal standard; GroES is not sensitive to hfq status [19]. Bands were quantified using ImageQuant software (GE Healthcare) and Crp levels plotted relative to GroES.
Acknowledgements
We thank Michael Ellis for providing comments on the manuscript and for useful discussions, and Claire Young for assistance with P1 transductions. We also thank S Gottesman and C Vanderpool for providing pLlacO-sRNA expression plasmids and the sgrS1 allele, respectively, and H Aiba for providing an anti-Crp antibody. Finally, we thank W Reznikoff for providing IS50 transposase plasmids and T Naas for providing an E. coli strain harboring pOX38-Gen. This work was supported by a grant to DBH (MOP 11281) from Canadian Institutes of Health Research. JR and CM were supported by OGS and NSERC scholarships.
Abbreviations
- BSA
bovine serum albumin
- Crp
cyclic AMP-receptor protein
- DAM
DNA adenine methylase
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- LB
Luria broth
- PAGE
polyacrylamide gel electrophoresis
- PCR
polymerase chain reaction
- RT-PCR
reverse transcription polymerase chain reaction
- sRNA
small RNA
- SDS
sodium dodecyl sulfate
- TBE
Tris borate-EDTA buffer
- TBST
20 mM Tris-HCl (pH 7.5), 150 mM sodium chloride, 0.5% Tween-20
- TCF
transcriptional fusion
- TIR
translation initiation region
- TLF
translational fusion
- UTR
untranslated region
- UV
ultraviolet light
Additional files
Mapping Tn5 transposition events. Southern blot and ST-PCR characterization of Tn5 transposition events in wt and hfq − strains.
Impact of SgrS over-expression on growth rate in M9 glucose. Growth curves of cells in which SgrS RNA was or was not induced by IPTG addition and corresponding Northern blot showing SgrS levels.
Details of plasmids constructed for this work.
List of oligonucleotides used in this work.
Footnotes
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
JR performed ‘mating out’ assays, β-galactosidase assays in Figures 3, 7 and 8, RT-PCR assays, Western blot assays, participated in the design of the study and helped draft the manuscript. RT performed the β-galactosidase assay in Figure 4, steady-state transcript measurements and RNA half-life measurements and prepared figures. MDB helped construct chromosomal reporter strains. CRM made the initial discovery that Tn5 transposition is up-regulated under conditions of hfq deficiency and constructed IS50-reporter plasmids and expression plasmids. DBH participated in the design of the study, helped in constructing strains and plasmids, performed the ‘mating out’ experiment in Figure 7D and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Joseph A Ross, Email: jross46@uwo.ca.
Ryan S Trussler, Email: rtrussl@uwo.ca.
Morgan D Black, Email: mblack46@uwo.ca.
Crystal R McLellan, Email: crystalmclellan5@gmail.com.
David B Haniford, Email: haniford@uwo.ca.
References
- 1.Roberts D, Hoopes BC, McClure WR, Kleckner N. IS10 transposition is regulated by DNA adenine methylation. Cell. 1985;43(1):117–130. doi: 10.1016/0092-8674(85)90017-0. [DOI] [PubMed] [Google Scholar]
- 2.Yin JC, Krebs MP, Reznikoff WS. Effect of dam methylation on Tn5 transposition. J Mol Biol. 1988;199(1):35–45. doi: 10.1016/0022-2836(88)90377-4. [DOI] [PubMed] [Google Scholar]
- 3.Raleigh EA, Kleckner N. Quantitation of insertion sequence IS10 transposase gene expression by a method generally applicable to any rarely expressed gene. Proc Natl Acad Sci U S A. 1986;83(6):1787–1791. doi: 10.1073/pnas.83.6.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krebs MP, Reznikoff WS. Transcriptional and translational initiation sites of IS50. Control of transposase and inhibitor expression. J Mol Biol. 1986;192(4):781–791. doi: 10.1016/0022-2836(86)90028-8. [DOI] [PubMed] [Google Scholar]
- 5.Simons RW, Kleckner N. Translational control of IS10 transposition. Cell. 1983;34(2):683–691. doi: 10.1016/0092-8674(83)90401-4. [DOI] [PubMed] [Google Scholar]
- 6.Ma C, Simons RW. The IS10 antisense RNA blocks ribosome binding at the transposase translation initiation site. EMBO J. 1990;9(4):1267–1274. doi: 10.1002/j.1460-2075.1990.tb08235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ross JA, Wardle SJ, Haniford DB. Tn10/IS10 transposition is downregulated at the level of transposase expression by the RNA-binding protein Hfq. Mol Microbiol. 2010;78(3):607–621. doi: 10.1111/j.1365-2958.2010.07359.x. [DOI] [PubMed] [Google Scholar]
- 8.Ross JA, Ellis MJ, Hossain S, Haniford DB. Hfq restructures RNA-IN and RNA-OUT and facilitates antisense pairing in the Tn10/IS10 system. RNA. 2013;19(5):670–684. doi: 10.1261/rna.037747.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat Rev Microbiol. 2011;9(8):578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le Derout J, Boni IV, Regnier P, Hajnsdorf E. Hfq affects mRNA levels independently of degradation. BMC Mol Biol. 2010;11:17. doi: 10.1186/1471-2199-11-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahillon J, Chandler M. Insertion sequences. Microbiol Mol Biol Rev. 1998;62(3):725–774. doi: 10.1128/mmbr.62.3.725-774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kennedy AK, Guhathakurta A, Kleckner N, Haniford DB. Tn10 transposition via a DNA hairpin intermediate. Cell. 1998;95(1):125–134. doi: 10.1016/S0092-8674(00)81788-2. [DOI] [PubMed] [Google Scholar]
- 13.Bhasin A, Goryshin IY, Reznikoff WS. Hairpin formation in Tn5 transposition. J Biol Chem. 1999;274(52):37021–37029. doi: 10.1074/jbc.274.52.37021. [DOI] [PubMed] [Google Scholar]
- 14.Mahnke Braam LA, Goryshin IY, Reznikoff WS. A mechanism for Tn5 inhibition. carboxyl-terminal dimerization. J Biol Chem. 1999;274(1):86–92. doi: 10.1074/jbc.274.1.86. [DOI] [PubMed] [Google Scholar]
- 15.Reznikoff WS. Transposon Tn5. Annu Rev Genet. 2008;42:269–286. doi: 10.1146/annurev.genet.42.110807.091656. [DOI] [PubMed] [Google Scholar]
- 16.Kuan CT, Tessman I. LexA protein of Escherichia coli represses expression of the Tn5 transposase gene. J Bacteriol. 1991;173(20):6406–6410. doi: 10.1128/jb.173.20.6406-6410.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mikulecky PJ, Kaw MK, Brescia CC, Takach JC, Sledjeski DD, Feig AL. Escherichia coli Hfq has distinct interaction surfaces for DsrA, rpoS and poly(A) RNAs. Nat Struct Mol Biol. 2004;11(12):1206–1214. doi: 10.1038/nsmb858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez-Antonio A, Collado-Vides J. Identifying global regulators in transcriptional regulatory networks in bacteria. Curr Opin Microbiol. 2003;6(5):482–489. doi: 10.1016/j.mib.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Guisbert E, Rhodius VA, Ahuja N, Witkin E, Gross CA. Hfq modulates the sigmaE-mediated envelope stress response and the sigma32-mediated cytoplasmic stress response in Escherichia coli. J Bacteriol. 2007;189(5):1963–1973. doi: 10.1128/JB.01243-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lathem WW, Schroeder JA, Bellows LE, Ritzert JT, Koo JT, Price PA, Caulfield AJ, Goldman WE. Posttranscriptional regulation of the Yersinia pestis cyclic AMP receptor protein Crp and impact on virulence. mBio. 2014;5(1):e01038-01013. doi: 10.1128/mBio.01038-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hussein R, Lim HN. Disruption of small RNA signaling caused by competition for Hfq. Proc Natl Acad Sci U S A. 2011;108(3):1110–1115. doi: 10.1073/pnas.1010082108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moon K, Gottesman S. Competition among Hfq-binding small RNAs in Escherichia coli. Mol Microbiol. 2011;82(6):1545–1562. doi: 10.1111/j.1365-2958.2011.07907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wassarman KM, Repoila F, Rosenow C, Storz G, Gottesman S. Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev. 2001;15(13):1637–1651. doi: 10.1101/gad.901001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishikawa H, Otaka H, Maki K, Morita T, Aiba H. The functional Hfq-binding module of bacterial sRNAs consists of a double or single hairpin preceded by a U-rich sequence and followed by a 3′ poly(U) tail. RNA (New York, NY) 2012;18(5):1062–1074. doi: 10.1261/rna.031575.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fender A, Elf J, Hampel K, Zimmermann B, Wagner EG. RNAs actively cycle on the Sm-like protein Hfq. Genes Dev. 2010;24(23):2621–2626. doi: 10.1101/gad.591310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papenfort K, Vogel J. Small RNA functions in carbon metabolism and virulence of enteric pathogens. Front Cell Infect Microbiol. 2014;4:91. doi: 10.3389/fcimb.2014.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agre P, Bonhivers M, Borgnia MJ. The aquaporins, blueprints for cellular plumbing systems. J Biol Chem. 1998;273(24):14659–14662. doi: 10.1074/jbc.273.24.14659. [DOI] [PubMed] [Google Scholar]
- 28.Rice JB, Vanderpool CK. The small RNA SgrS controls sugar-phosphate accumulation by regulating multiple PTS genes. Nucleic Acids Res. 2011;39(9):3806–3819. doi: 10.1093/nar/gkq1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu ST, Wang HC, Lei GS, Wang SH. Negative regulation of IS2 transposition by the cyclic AMP (cAMP)-cAMP receptor protein complex. J Bacteriol. 1998;180(10):2682–2688. doi: 10.1128/jb.180.10.2682-2688.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grainger DC, Hurd D, Harrison M, Holdstock J, Busby SJ. Studies of the distribution ofEscherichia colicAMP-receptor protein and RNA polymerase along the E. coli chromosome. Proc Natl Acad Sci U S A. 2005;102(49):17693–17698. doi: 10.1073/pnas.0506687102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johansen J, Eriksen M, Kallipolitis B, Valentin-Hansen P. Down-regulation of outer membrane proteins by noncoding RNAs: unraveling the cAMP-CRP- and sigmaE-dependent CyaR-ompX regulatory case. J Mol Biol. 2008;383(1):1–9. doi: 10.1016/j.jmb.2008.06.058. [DOI] [PubMed] [Google Scholar]
- 32.Frohlich KS, Papenfort K, Berger AA, Vogel J. A conserved RpoS-dependent small RNA controls the synthesis of major porin OmpD. Nucleic Acids Res. 2012;40(8):3623–3640. doi: 10.1093/nar/gkr1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen S, Zhang A, Blyn LB, Storz G. MicC, a second small-RNA regulator of Omp protein expression in Escherichia coli. J Bacteriol. 2004;186(20):6689–6697. doi: 10.1128/JB.186.20.6689-6697.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Twiss E, Coros AM, Tavakoli NP, Derbyshire KM. Transposition is modulated by a diverse set of host factors in Escherichia coli and is stimulated by nutritional stress. Mol Microbiol. 2005;57(6):1593–1607. doi: 10.1111/j.1365-2958.2005.04794.x. [DOI] [PubMed] [Google Scholar]
- 35.Wang RF, Kushner SR. Construction of versatile low-copy number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene. 1991;100:195–199. doi: 10.1016/0378-1119(91)90366-J. [DOI] [PubMed] [Google Scholar]
- 36.Bolivar F, Backman K. Plasmids of Escherichia coli as cloning vectors. Methods Enzymol. 1979;68:245–267. doi: 10.1016/0076-6879(79)68018-7. [DOI] [PubMed] [Google Scholar]
- 37.Cuzon G, Naas T, Nordmann P. Functional characterization of Tn4401, a Tn3-based transposon involved in blaKPC gene mobilization. Antimicrob Agents Chemother. 2011;55(11):5370–5373. doi: 10.1128/AAC.05202-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoffman EP, Wilhelm RC. Genetic mapping and dominance of the amber suppressor, Su1 (supD), in Escherichia coli K-12. J Bacteriol. 1970;103(1):32–36. doi: 10.1128/jb.103.1.32-36.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casadaban MJ. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol. 1976;104(3):541–555. doi: 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
- 40.Foster TJ, Davis MA, Roberts DE, Takeshita K, Kleckner N. Genetic organization of transposon Tn10. Cell. 1981;23(1):201–213. doi: 10.1016/0092-8674(81)90285-3. [DOI] [PubMed] [Google Scholar]
- 41.Morita T, Kawamoto H, Mizota T, Inada T, Aiba H. Enolase in the RNA degradosome plays a crucial role in the rapid decay of glucose transporter mRNA in the response to phosphosugar stress in Escherichia coli. Mol Microbiol. 2004;54(4):1063–1075. doi: 10.1111/j.1365-2958.2004.04329.x. [DOI] [PubMed] [Google Scholar]
- 42.Morita T, Maki K, Aiba H. RNase E-based ribonucleoprotein complexes: mechanical basis of mRNA destabilization mediated by bacterial noncoding RNAs. Genes Dev. 2005;19(18):2176–2186. doi: 10.1101/gad.1330405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whitfield CR, Wardle SJ, Haniford DB. The global bacterial regulator H-NS promotes transpososome formation and transposition in the Tn5 system. Nucleic Acids Res. 2009;37(2):309–321. doi: 10.1093/nar/gkn935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Way JC, Davis MA, Morisato D, Roberts DE, Kleckner N. New Tn10 derivatives for transposon mutagenesis and for construction of lacZ operon fusions by transposition. Gene. 1984;32(3):369–379. doi: 10.1016/0378-1119(84)90012-X. [DOI] [PubMed] [Google Scholar]
- 45.Guillier M, Gottesman S. Remodelling of the Escherichia coli outer membrane by two small regulatory RNAs. Mol Microbiol. 2006;59(1):231–247. doi: 10.1111/j.1365-2958.2005.04929.x. [DOI] [PubMed] [Google Scholar]
- 46.Vanderpool CK, Gottesman S. Involvement of a novel transcriptional activator and small RNA in post-transcriptional regulation of the glucose phosphoenolpyruvate phosphotransferase system. Mol Microbiol. 2004;54(4):1076–1089. doi: 10.1111/j.1365-2958.2004.04348.x. [DOI] [PubMed] [Google Scholar]
- 47.Wadler CS, Vanderpool CK. Characterization of homologs of the small RNA SgrS reveals diversity in function. Nucleic Acids Res. 2009;37(16):5477–5485. doi: 10.1093/nar/gkp591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mandin P, Gottesman S. Integrating anaerobic/aerobic sensing and the general stress response through the ArcZ small RNA. EMBO J. 2010;29(18):3094–3107. doi: 10.1038/emboj.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haniford DB, Chelouche AR, Kleckner N. A specific class of IS10 transposase mutants are blocked for target site interactions and promote formation of an excised transposon fragment. Cell. 1989;59(2):385–394. doi: 10.1016/0092-8674(89)90299-7. [DOI] [PubMed] [Google Scholar]
- 50.Garrey SM, Mackie GA. Roles of the 5′-phosphate sensor domain in RNase E. Mol Microbiol. 2011;80(6):1613–1624. doi: 10.1111/j.1365-2958.2011.07670.x. [DOI] [PubMed] [Google Scholar]
- 51.Wilkinson KA, Merino EJ, Weeks KM. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat Protoc. 2006;1(3):1610–1616. doi: 10.1038/nprot.2006.249. [DOI] [PubMed] [Google Scholar]