

Summary
Objective
FOXG1-related disorders are associated with severe intellectual disability, absent speech with autistic features, and epilepsy. Children with deletions or intragenic mutations of FOXG1 also have postnatal microcephaly, morphologic abnormalities of the corpus callosum, and choreiform movements. Duplications of 14q12 often present with infantile spasms, and have subsequent intellectual disability with autistic features. Long term epilepsy outcome and response to treatment has not been studied systematically in a well-described cohort of subjects with FOXG1-related disorders. We report on the epilepsy features and developmental outcome of 23 new subjects with deletions or intragenic mutations of FOXG1, and 7 subjects with duplications.
Methods
Subjects had either chromosomal microarray or FOXG1 gene sequencing performed as part of routine clinical care. Development and epilepsy follow-up data were collected from medical records from treating neurologists and through telephone parental interviews using standardized questionnaires.
Results
Epilepsy was diagnosed in 87% of the subjects with FOXG1-related disorders. The mean age of epilepsy diagnosis in FOXG1 duplications was significantly younger than those with deletions/intragenic mutations (p=0.0002). All of the duplication FOXG1 children with infantile spasms responded to hormonal therapy and only one required long-term anti-epileptic therapy. In contrast, more children with deletions/intragenic mutations required anti-epileptic drugs on follow-up (p<0.0005). All subjects with FOXG1-related disorders had neurodevelopmental disabilities after 3 years of age, regardless of the epilepsy type or intractability of seizures. All had impaired verbal language and social contact, and three duplication subjects were formally diagnosed with autism. Subjects with deletion/intragenic mutations however had significantly worse ambulation (p=0.04) and functional hand use (p<0.0005).
Significance
Epilepsy and developmental outcome characteristics allow clinicians to distinguish among the FOXG1-related disorders. Further genotype-phenotype studies of FOXG1 may help to elucidate why children develop different forms of developmental epilepsy.
Keywords: FOXG1, 14q12, infantile spasms, epilepsy, developmental outcome
Introduction
Abnormalities of the FOXG1 gene cause a developmental brain disorder characterized by severe intellectual disability, absent speech with autistic behavior, movement disorder, and epilepsy1. While initially described as “congenital Rett syndrome”2,3, children with FOXG1-related disorders have clinical features that distinguish from Rett syndrome. Patients with either deletions of 14q12 surrounding FOXG1 or intragenic mutations typically have postnatal microcephaly, simplified gyral pattern in the frontal lobes, morphologic abnormalities of the corpus callosum, severe intellectual disability with absent language, and choreiform movements1,4. Simultaneously, a phenotype was associated with duplications of 14q12 including FOXG1 consisting of normal head size and corpus callosum morphology, but autistic features including absent language and severe intellectual disability5,6. These differential features are illustrated in Figure 1.
Figure 1.
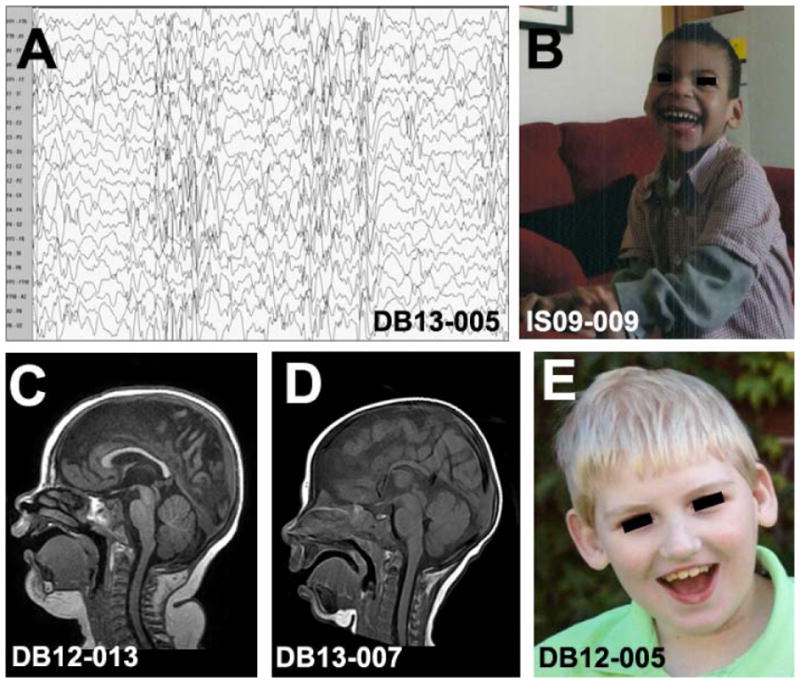
Differential characteristics of subjects with duplications or deletions/intragenic mutations of FOXG1 on 14q12. Infantile spasms with hypsarrhythmia (A) has been commonly described with duplications. Children with smaller 14q12 duplications are non-dysmorphic, and have autistic features (B). MRI in duplications of 14q12 show a normal gyral pattern and intact corpus callosum (C), while children with deletions/intragenic mutations of FOXG1 have hypoplasia of the corpus callosum (D), foreshortened frontal lobes with gyral simplification, postnatal microcephaly, and multiple neurodevelopmental disabilities (E).
Early reports indicated that epilepsy may be an important feature differentiating deletions/truncating mutations from duplications of FOXG1, as infantile spasms were frequently reported in children with duplications6,7. There was evidence that infant-onset epilepsy in children with FOXG1 duplications responded to traditional hormonal therapy8, but long term epilepsy outcome and response to treatment have not been studied systematically in a well-described cohort of subjects with FOXG1-related disorders.
Similarly, most of our knowledge of the epilepsy and developmental characteristics in individuals with deletions and intragenic mutations of FOXG1 comes from relatively small cohorts1,3,4,9,10. Relatively little is known about the correlations between epilepsy types, age of onset, and long-term developmental outcome. While epilepsy is a known phenotypic feature of FOXG1-related disorders11 no study has yet compared the epilepsy subtypes found in duplications FOXG1 patients to deletion/intragenic mutation patients.
We report on the epilepsy features and developmental outcome of 23 new subjects with deletions or intragenic mutations of FOXG1 at 14q12, and 7 subjects with duplications. Two of our subjects with duplications were published previously6,7, and we now provide longitudinal follow up data. We demonstrate that genotype is associated with variation in the epilepsy syndrome, age of onset, response to therapy, and intractability of seizures among the FOXG1-related disorders. FOXG1-related disorders can be distinguished from Rett syndrome based in part on the epilepsy phenotype. These conditions represent an opportunity to study the effects of copy number of a single gene on brain development and epilepsy pathogenesis.
Methods
Subject ascertainment
All subjects were consented through the Infantile Spasms Registry & Genetic Studies or the Genetic Studies of Developmental Brain Disorders. The protocols were approved by the Research Subjects Review Board of the University of Rochester Medical Center.
Genetic studies
All subjects had either chromosomal microarray or FOXG1 gene sequencing performed as part of routine clinical care. Subjects with copy number variants identified by chromosomal microarray had the de novo status of these variants confirmed by fluorescent in situ hybridization of parental samples according to routine clinical practice.
Epilepsy and follow-up developmental data
Development and epilepsy follow-up data was collected retrospectively from medical records obtained from the treating neurologists and through telephone parental interviews using standardized questionnaires. Detailed developmental data were collected from subjects older than 3 years of age. Primary EEG studies were reviewed in 5 subjects, and reports of EEGs were reviewed in the remainder.
Brain imaging
Routine clinical brain MRI scans were reviewed in 6 of the duplication FOXG1 subjects and 11 of the deletion/intragenic mutation subjects.
Statistical analysis
All statistics (t-test) were performed using R v.3.0.1 (http://cran.us.r-project.org/). Duplication subjects were compared to deletion/intragenic mutation subjects, and no multiple comparisons were performed.
Results
Subject characteristics
The mean age of all subjects with FOXG1-related disorders in our cohort was 6.8 years old, although subjects with duplications of FOXG1 were slightly younger (mean age 3.2 years old) than subjects with deletions (mean age 4.4 years old) and both were younger than subjects with intragenic mutations (mean age 9.4 years old) at the time of study. Our cohort included 17 males and 13 females. Five of our seven duplication subjects were male.
Genetic studies
Our cohort included four subjects with de novo deletions of 14q12 ranging in size 0.25–6.4 Mb including FOXG1 who have not been reported previously. Nineteen subjects had intragenic mutations of FOXG1, with fifteen nonsense, frameshift, or intragenic insertions or deletions predicted to cause premature protein truncation, and two missense mutations. We report two sibling pairs (DB12-017a1, DB12-017a2 and DB13-029a1, DB13-029a2) with recurrence of the same FOXG1 intragenic mutation, suggesting parental gonadal mosaicism and the first such cases reported to our knowledge. Additionally, we report the first pair of identical twins (DB13-052a1 and DB13-052a2) with FOXG1 mutations. Two of our duplication 14q12 subjects were reported previously6,7, and we add five novel subjects with FOXG1 duplication. One of these, DB13-005, had a 33.9 Mb duplication of 14q12, the largest such chromosomal duplication reported for this disorder. The duplicated 14q12 material in DB12-018 was associated with a supernumerary marker chromosome derived from a translocation between the proximal long arm of chromosome 14 and the distal long arm of chromosome 6. The SMC (plus one normal chromosome 14) was inherited from the mother, who carried a balanced 6;14 translocation. Chromosomal microarray in DB12-018 demonstrated two de novo genomic imbalances, a 15.7 Mb gain from 14q11.2 to 14q13.2 and a 4.5 Mb gain in 6q27. Subject DB13-016 also had additional de novo gain of ~5.5 Mb from 14q21.1 to 14q21.2.
Different epilepsy diagnoses found in deletions/mutations FOXG1 vs duplications
Epilepsy was diagnosed in 87% of the subjects in our cohort with FOXG1-related disorders. Subjects with deletions and intragenic mutations of FOXG1 had a variety of epilepsy types described, including complex partial seizures, myoclonic seizures, and generalized tonic seizures. There was no classic epilepsy syndrome diagnosis that could be applied to this group, and there was no correlation between seizure type and deletion vs. intragenic mutations. This variability was also illustrated by the discordance of epilepsy in our two sibling pairs affected with the same FOXG1 mutations. Subject DB12-017a1 with a p.Gly172_Met192del mutation had onset of complex partial seizures at 18 months, and remained on one AED. However, sibling DB12-017a2 with the same mutation had never had a seizure. While both siblings DB13-029a1 and DB13-029a2 had onset of epilepsy at two years of age, seizures in DB13-029a2 continued, and required two current AEDs.
In contrast, six of our seven subjects with duplications of FOXG1 had infantile spasms, while this specific epilepsy syndrome was not found in children with deletions/intragenic mutations and to our knowledge that association has not been reported in the literature. All six FOXG1 duplication subjects with infantile spasms received adrenocorticotropin (ACTH), and all six responded with cessation of clinical spasms and resolution of hypsarrhythmia on EEG. Subject IS09-009 had later occurrence of a few tonic seizures, but had been off all AEDs for three years at the time of study. Subject IS14-001 had initial cessation of infantile spasms after 24 hours of ACTH therapy, had normalization of hypsarrhythmia on EEG, but then developed myoclonic seizures 1 year later and is currently on topiramate. The characteristics of epilepsy are summarized in Table 2.
Table 2.
Epilepsy and outcome in 30 subjects with FOXG1-related disorders.
| Subject | FOXG1 abnormality | Age EPI dx | Hosp. in last 12 months | Current number of AEDs | Type of seizures | Walks unassisted | Verbal language | Functional hand use | Makes social contact |
|---|---|---|---|---|---|---|---|---|---|
| DB12-001 | c.651C>G/p.Tyr217X | 1.5 y | 0 | 2 | Tonic seizures, myoclonic seizures | − | − | − | − |
| DB12-002 | p.Gln86X | 15 m | 1 | 3 | Generalized tonic-clonic seizures | − | − | − | +/− |
| DB12-003 | 6.4 Mb Del 26908842-33280997 | N/A* | N/A* | N/A* | None | N/A** | N/A** | N/A** | N/A** |
| DB12-004 | 1.1 Mb Del 27622465-28725069 | 3 y 2 m | 0 | 4 | Complex partial seizures | − | − | − | − |
| DB12-005 | c.430G>T/p.Glu144X | 1 y 6 m | 1 | 3 | Generalized seizures | − | − | − | + |
| DB12-006 | c.460dupG/p.Glu154GlyfsX301 | 19 m | 1 | 3 | Generalized seizures, tonic seizures | − | − | − | + |
| DB12-008 | 0.25 Mb Del 28116191-28370178 | 9 m | 3 | 3 | Tonic seizures | N/A** | N/A** | N/A** | N/A** |
| DB12-009 | c.460dupG/p.Glu154GlyfsX301 | 2 y | 0 | 2 | Generalized tonic-clonic seizures | − | − | − | − |
| DB12-015 | c.133_469del377insACCCACCGCCCC | 1 y | 0 | 0 | Myoclonic seizures | − | − | − | − |
| DB12-016 | c.577G>A/p.Ala193Thr | 8 m | 0 | 3 | Tonic seizures | − | − | + | + |
| DB12-017a1 | c.515_577del63/p.Gly172_Met192del | 18 m | 0 | 1 | Complex partial seizures | − | − | − | + |
| DB12-017a2 | c.515_577del63/p.Gly172_Met192del | N/A* | N/A* | N/A* | None | − | − | − | +/− |
| DB13-006 | c.506delG/p.Gly169AlafsX23 | 11 m | 5 | 3 | Myoclonic seizures, tonic seizures, drop seizures | − | − | − | +/− |
| DB13-007 | c.586C>T/p.Gln196X | 1 y | 0 | 1 | Myoclonic seizures | − | − | − | +/− |
| DB13-012 | c.755G>T/p.Gly252Val | N/A* | N/A* | N/A* | None | − | − | − | +/− |
| DB13-019 | 4.1 Mb Del 26658715-30780974 | 8 m | 0 | 2 | Tonic seizures at age 4 y | − | − | − | + |
| DB13-029a1 | c.460dupG/p.Glu154GlyfsX301 | 2 y | 0 | 0 | Generalized tonic-clonic, status epilepticus prior to age 12 y | − | − | + | +/− |
| DB13-029a2 | c.460dupG/p.Glu154GlyfsX301 | 2 y | 0 | 3 | Generalized tonic-clonic prior to age 11 y | − | − | + | +/− |
| DB13-028 | c.735delC/p.Tyr246ThrfsX80 | 13 m | 0 | 1 | Tonic seizures and generalized tonic-clonic seizures | − | − | − | +/− |
| DB13-033 | c.762G>C/p.Tyr254X | 1 yr | 3 | 2 | Complex partial seizures | N/A** | N/A** | N/A** | N/A** |
| DB13-041 | c.222_223dupGC/p.Pro75ArgfsX118 | 3 y 6 m | 0 | 1 | Tonic seizures | − | − | − | +/− |
| DB13-052a1 | c.460dupG/p.Glu154GlyfsX301 | 4 y | 0 | 1 | Complex partial seizures | − | +/− | − | +/− |
| DB13-052a2 | c.460dupG/p.Glu154GlyfsX301 | 4 y | 0 | 1 | Complex partial seizures | − | +/− | − | +/− |
| IS09-009 | 3.3 Mb Dup 26908812-30254928 | 6 m | 0 | 0 | Infantile spasms | + | − | + | +/− |
| IS10-028 | 3.0 Mb Dup 27165797-30192375 | 10 m | 0 | 0 | Infantile spasms | + | + | + | + |
| DB12-013 | 4.1 Mb Dup 26169335-30237575 | 6 m | 0 | 0 | Infantile spasms | N/A** | N/A** | N/A** | N/A** |
| DB12-018 | 15.6 Mb Dup 19528022-35193884 | 9 m | 0 | 0 | Infantile spasms | N/A** | N/A** | N/A** | N/A** |
| DB13-005 | 33.9 Mb Dup 21927637-55833232 | 8 m | 2 | 0 | Infantile spasms | N/A** | N/A** | N/A** | N/A** |
| DB13-016 | 13.3 Mb Dup 20555119-33885364 | N/A* | N/A* | N/A* | Febrile seizures x 3 | +/− | − | + | − |
| IS14-001 | 4.4 Mb Dup 26558014-30980441 | 6 m | 1 | 1 | Infantile spasms Myoclonic seizures |
N/A** | N/A** | N/A** | N/A** |
N/A* = no diagnosis of epilepsy; N/A** = subject <3 years of age; EPI = epilepsy; AEDs = anti-epileptic drugs
Children with duplications of FOXG1 had earlier diagnosis of epilepsy
Given the common occurrence of infantile spasms in children with FOXG1 duplications, the mean age of epilepsy diagnosis in this group was significantly younger (p=0.0004) at 7.4 months of age, compared to 22.3 months of age children with deletions and intragenic mutations (Figure 2A). However, taken together the mean age of epilepsy diagnosis for both groups was still under 2 years of age.
Figure 2.

Significant differences were found between subjects with FOXG1 deletions/intragenic mutations compared to those with duplications in the areas of mean age of epilepsy diagnosis (A; *p=0.0002), current numbers of anti-epileptic drugs (AED) used (B; **p<0.0005), impaired ambulation (C; ***p=0.04), and impaired functional hand use (D; ****p<0.0005).
Children with deletions/mutations of FOXG1 required significantly more AEDs and had intractable seizures
Subjects with deletions/intragenic mutations of FOXG1 had later onset of epilepsy, and most continued to have active seizures through the time of the study. On average, these subjects were taking 1.7 anti-epileptic drugs (AEDs) at the time of study, and this was significantly different (p<0.0005) than children with duplications only one of whom was on one AED after resolution of infantile spasms (Figure 2B). While there was no significant difference in the number of hospitalizations for seizures in the 12 months preceding the study, we do note that the FOXG1 duplication group was younger, and the hospitalization for ACTH initiation for infantile spasms was included in two of the subjects.
Common c.460dupG mutation has similar epilepsy severity and developmental outcome compared to deletions of FOXG1 and other mutations
Our cohort contained 6 subjects with the common c.460dupG mutation, and we were interested to know if there were any significant differences in epilepsy onset, severity, or developmental outcome compared to other subjects with intragenic mutations and deletions and all FOXG1-related disorders. The mean age of epilepsy diagnosis was 31.2 months in children with the c.460dupG mutation, compared to 17.8 months in all others with deletions or mutations in FOXG1, (p=0.06). There was also no significant difference compared to others with mutations or deletions of FOXG1 in the number of anti-epileptic medications used or number of hospitalizations in the past year, nor were there significant differences in ambulation, language, functional hand use, or social interactions.
Both deletions/mutations and duplications of FOXG1 had neurodevelopmental disability at 3 years regardless of epilepsy
Overall subjects with FOXG1-related disorders had serious neurodevelopmental disabilities when studied after 3 years of age. This was consistent, regardless of the epilepsy type or intractability of seizures. Both deletion/intragenic mutation and duplication FOXG1 groups had impaired verbal language and social contact. Of note, all three FOXG1 duplication subjects that were older than 3 years of age had formal diagnoses of autism. None of the deletion/intragenic mutation subjects had a formal diagnosis of autism, although it is likely this diagnosis would be confounded by concomitant severe cognitive impairment. Subjects with deletion/intragenic mutations however had significantly (p=0.04) worse ability to walk independently (Figure 2C). In fact, none of our subjects older than 3 years of age with deletions or intragenic mutations were independently ambulatory, and this was true whether they had epilepsy or not. We noted similar findings in functional hand use, which was significantly more impaired (p<0.0005) in subjects with deletions or intragenic mutations (Figure 2D). The developmental outcome data are summarized in Table 3.
Table 3.
Key characteristics of epilepsy and development after 3 years of age in subjects with FOXG1-related disorders. Subjects with deletions/intragenic mutations show significant differences compared to those with duplications in mean age of epilepsy diagnosis, current number of anti-epileptic drugs, ambulation, and functional hand use. Verbal language and social contact were impaired equally in both groups.
| Characteristic | Deletions & intragenic mutations | Duplications | P-value |
|---|---|---|---|
| Mean age epilepsy diagnosis (months) | 22.3 | 7.4 | 0.0002 |
| Number of overnight hospitalizations | 0.4 | 0.5 | 1.00 |
| Number of AEDs | 1.7 | 0.16 | <0.0005 |
| Nonambulatory | 1.0 | 0.2 | 0.04 |
| Absent verbal language | 0.95 | 0.7 | 0.48 |
| Poor functional hand use | 0.86 | 0.0 | <0.0005 |
| Impaired social contact | 0.48 | 0.5 | 0.94 |
Brain imaging was consistent with previously published reports
We confirmed that the brain MRI scans from our deletion/intragenic mutation FOXG1 subjects were consistent with well-described findings of others,1,2,3,4 with common foreshortening of the frontal lobes with simplification of the gyral pattern and variable corpus callosum abnormalities ranging from hypogenesis of the genu of the corpus callosum (in intragenic FOXG1 mutations) to complete agenesis (in large 14q12 deletions). Brain imaging of duplication FOXG1 subjects was overall normal.
Discussion
This is the first study to consider FOXG1-related disorders together, comparing the epilepsy and developmental findings in 23 new subjects with deletions or intragenic mutations to the findings in 7 subjects with duplications. Our data make important observations regarding the role of copy number of a single gene on epilepsy pathogenesis and developmental outcome. A detailed study of FOXG1 affords an opportunity to understand why children develop specific forms of developmental epilepsy, and may ultimately lead to different therapeutic approaches.
FOXG1 is a transcriptional repressor that regulates dorsal-ventral brain patterning12, is important in the development of Cajal-Retzius cells, and is known to inhibit gliogenesis and promote neurogenesis13. Forebrain overexpression of FOXG1 leads to thickening of the neuroepithelium, which may be due to a decrease in neuroepithelial apoptosis14. However, the mechanism(s) by which FOXG1 abnormalities lead to epilepsy are not known. Presumably, as a transcription factor important in dorsal-ventral differentiation, FOXG1 abnormalities result in abnormal neuronal and/or glial populations during brain development. As several early infantile epilepsies are caused by genes critical for GABAergic interneuron identity, such as ARX15, or in the case of SCN1A lead to impaired sodium currents in GABAergic interneurons16, we hypothesize that duplications of FOXG1 may lead to impaired ventral GABAergic interneuron development or function. The lack of infantile spasms with deletion or intragenic mutation of FOXG1 and the observation that epilepsy is later in onset in these subjects suggests a different mechanism for epilepsy pathogenesis in loss of function mutations and deletions.
We describe several novel FOXG1 genetic findings. Our cohort contained two sibling pairs with the same intragenic mutations, although in both families the epilepsy severity was discordant. These cases present challenges for genetic counseling, as FOXG1-related disorders are usually mediated by de novo genomic events. Recurrence in these families suggests parental gonadal mosaicism. Six of our subjects had the previously reported c.460dupG mutation4, which appears to be a common FOXG1 abnormality. This mutation is in a known stretch of repeated guanines, confirming that this area is a mutational hotspot. We also report recurrence of this mutation in a sibling pair (DB13-029a1 and DB13-029a2), as well as our pair of identical twins (DB13-052a1 and DB13-052a2), providing an opportunity to follow long-term outcome regarding this particular genotype. Overall, the 6 subjects with the c.460dupG mutation did not have significant differences in severity of epilepsy or in developmental outcome compared to other subjects with mutations or deletions of FOXG1. We also present two novel missense mutations (p.Ala193Thr and p.Gly252Val), and in both note varying severity of seizures in those subjects. We add additional cases of 14q12 duplications, including a supernumerary marker chromosome as well as the largest known duplication (33.9 Mb). In both cases, despite the presence of dysmorphic features and copy gain of large areas of chromosome 14 material, both had infantile spasms that responded uneventfully to ACTH. This suggests that FOXG1 status is a significant, if not the primary, driver of epilepsy phenotype in cases of 14q12 duplication.
Our data confirm the common occurrence of infantile spasms among children with duplications of FOXG15–8,17, and additionally detail their responsiveness in general to ACTH. Given their normal brain MRI findings and nondysmorphic features seen in those with smaller duplications, these children were initially labeled “cryptogenic” at the time of diagnosis of infantile spasms. Even recent literature18–20 would have predicted a favorable outcome. However, we found that despite their initial resemblance to so-called “cryptogenic” cases and response to hormonal therapy, the developmental outcome of subjects with duplications of FOXG1 was nevertheless significantly impacted. This highlights the importance of chromosomal microarray in infants presenting with infantile spasms. Microarray will detect duplications and deletions of 14q12 including FOXG1, but specific gene sequencing, either alone or in a commercially available epilepsy panel, is necessary to identify intragenic mutations. Currently, the yield of testing for exonic deletions/duplications of FOXG1 using multiplex ligation-dependent probe amplification (MLPA) is not known. Several recent reviews are now available to assist clinicians in the genetic evaluation of children presenting with early onset epilepsy21–23.
The epilepsy seen with deletions and intragenic mutations of FOXG1 was more variable, was later in onset, and required ongoing anti-epileptic medication. Overnight hospitalization for seizures in the past 12 months, a measure of epilepsy severity, was not significantly different between the deletion/intragenic mutation and duplication groups. However, in one of the duplication patients the initial hospitalization for ACTH therapy was included in this assessment. It is therefore possible that in a larger cohort with older duplication FOXG1 subjects, this measure of epilepsy severity would be significant as well. Subjects in the deletion/intragenic mutation cohort had a range of severity of seizures, although some had no seizures at all. Particularly noteworthy were the discrepancies in seizure phenotype among siblings with the same mutation. This variability has been described in other neurodevelopmental disorders such as Rett syndrome24,25 and MEF2C haploinsufficiency26, and we hypothesize variability in epilepsy severity may be a common feature of conditions where multiple cell types in the developing forebrain are affected.
The retrospective nature of our study had several disadvantages, particularly in regard to the review of EEG data in a rare syndrome. As not all EEGs were available for our review, important aspects of background and interictal activity may not have been captured. For example, we had insufficient data to conclude whether any of the FOXG1 deletion/intragenic mutation subjects had Lennox-Gastaut syndrome (LGS). Some subjects may have met criteria for LGS, however, EEG data from 5–7 years of age when slow spike wave is most prevalent was not available. LGS is suspected in some subjects due to intractable seizures with multiple seizure types, including tonic seizures. Therefore we argue that follow-up study in this age group is justified.
The brain imaging findings were similar to those previously reported for FOXG1-related disorders. We found consistent frontal lobe and corpus callosum abnormalities in deletion/intragenic mutation FOXG1 subjects. There are not yet data relating the extent of these imaging abnormalities to background and interictal EEG findings such as frontal slowing or low amplitude frontal rhythms. The relationship of both frontal lobe and callosal abnormalities to developmental disability in this cohort will be important for future studies.
This is the largest cohort of subjects with FOXG1-related disorders reported to date, and the first to report the combined epilepsy features and developmental outcome of deletions, intragenic mutations, and duplications. Both deletions/mutations and duplications of FOXG1 had neurodevelopmental disability after three years of age regardless of epilepsy, and our data confirm common clinical findings of impairment of verbal language and social contact in FOXG1-related disorder irrespective of whether the gene is deleted, truncated, or duplicated. When FOXG1 is deleted, additional findings include postnatal microcephaly, hypoplasia of the corpus callosum, impaired ambulation, poor functional hand use. The epilepsy in patients with deletions/intragenic mutations is variable, nonspecific, can be intractable, and is later in onset. In FOXG1 duplications, the findings generally include normocephaly, normal brain imaging, presentation with infantile spasms that responds to therapy, but lack of subsequent epilepsy. All of these factors make FOXG1-related disorders easily distinguishable from Rett syndrome, and given the variability of outcomes and complications specific to Rett syndrome, it is prudent to understand these differences when counseling affected families.
Supplementary Material
Table 1.
Demographic and genomic features of 23 subjects with FOXG1 deletions or mutations and 7 subjects with duplications. Chromosome 14 coordinates are hg18 with the exception of IS14-001 which is reported in hg19.
| Subject | Sex | Age at time of study | Genomic abnormality (chr14) or FOXG1 mutation |
|---|---|---|---|
| DB12-001 | M | 4 y 8 m | c.651C>G/p.Tyr217X |
| DB12-002 | F | 4 y 11 m | p.Gln86X |
| DB12-003 | F | 2 y 6 m | 6.4 Mb Del 26908842-33280997 |
| DB12-004 | F | 3 y 7 m | 1.1 Mb Del 27622465-28725069 |
| DB12-005 | M | 9 y 6 m | c.430G>T/p.Glu144X |
| DB12-006 | M | 6 y 4 m | c.460dupG/p.Glu154GlyfsX301 |
| DB12-008 | F | 1 y 7 m | 0.25 Mb Del 28116191-28370178 |
| DB12-009 | F | 16 y 3 m | c.460dupG/p.Glu154GlyfsX301 |
| DB12-015 | M | 6 y 11 m | c.133_469del377insACCCACCGCCCC |
| DB12-016 | M | 13 y 9 m | c.577G>A/p.Ala193Thr |
| DB12-017a1 | F | 9 y 8 m | c.515_577del63/p.Gly172_Met192del |
| DB12-017a2 | F | 6 y 10 m | c.515_577del63/p.Gly172_Met192del |
| DB13-006 | M | 3 y 11 m | c.506delG/p.Gly169AlafsX23 |
| DB13-007 | M | 3 y 7 m | c.586C>T/p.Gln196X |
| DB13-012 | F | 6 y 6 m | c.755G>T/p.Gly252Val |
| DB13-019 | M | 10 y | 4.1 Mb Del 26658715-30780974 |
| DB13-029a1 | F | 25 y 10 m | c.460dupG/p.Glu154GlyfsX301 |
| DB13-029a2 | M | 22 y | c.460dupG/p.Glu154GlyfsX301 |
| DB13-028 | M | 5 y 6 m | c.735delC/p.Tyr246ThrfsX80 |
| DB13-033 | M | 1 y 9 m | c.762G>C/p.Tyr254X |
| DB13-041 | M | 8 y 1 m | c.222_223dupGC/p.Pro75ArgfsX118 |
| DB13-052a1 | F | 5 y | c.460dupG/p.Glu154GlyfsX301 |
| DB13-052a2 | F | 5 y | c.460dupG/p.Glu154GlyfsX301 |
| IS09-009 | M | 9 y | 3.3 Mb Dup 26908812-30254928 |
| IS10-028 | F | 4 y | 3.0 Mb Dup 27165797-30192375 |
| DB12-013 | M | 1 y 5 m | 4.1 Mb Dup 26169335-30237575 |
| DB12-018 | M | 1 y 11 m | 15.6 Mb Dup 19528022-35193884 |
| DB13-005 | M | 1 y 2 m | 33.9 Mb Dup 21927637-55833232 |
| DB13-016 | M | 3 y | 13.3 Mb Dup 20555119-33885364 |
| IS14-001 | F | 2 y | 4.4 Mb Dup 26558014-30980441 |
Acknowledgments
We wish to acknowledge the FOXG1 Foundation for referring subjects to this study. Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institutes of Health (NIH) under award numbers K12NS066098 (to L.E.S.) R01NS058721 (to W.B.D.), the Child Neurology Foundation Logan Infantile Spasms Award (to A.R.P), and K08NS078054 (to A.R.P.).
Footnotes
Disclosure of Conflicts of Interest
The authors have no conflicts of interest to declare.
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Ariani F, Hayek G, Rondinella D, et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am J Hum Genet. 2008;83:89–93. doi: 10.1016/j.ajhg.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mencarelli MA, Spanhol-Rosseto A, Artuso R, et al. Novel FOXG1 mutations associated with the congenital variant of Rett syndrome. J Med Genet. 2010;47:49–53. doi: 10.1136/jmg.2009.067884. [DOI] [PubMed] [Google Scholar]
- 3.Bahi-Buisson N, Nectoux J, Girard B, et al. Revisiting the phenotype associated with FOXG1 mutations: two novel cases of congenital Rett variant. Neurogenetics. 2010;11:241–9. doi: 10.1007/s10048-009-0220-2. [DOI] [PubMed] [Google Scholar]
- 4.Kortüm F, Das S, Flindt M, et al. The core FOXG1 syndrome phenotype consists of postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and corpus callosum hypogenesis. J Med Genet. 2011;48:396–406. doi: 10.1136/jmg.2010.087528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yeung A, Bruno D, Scheffer IE, et al. 4.5 Mb microduplication in chromosome band 14q12 including FOXG1 in a girl with refractory epilepsy and intellectual impairment. Eur J Med Genet. 2009;52:440–2. doi: 10.1016/j.ejmg.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Brunetti-Pierri N, Paciorkowski AR, Ciccone R, et al. Duplications of FOXG1 in 14q12 are associated with developmental epilepsy, mental retardation, and severe speech impairment. Eur J Hum Genet. 2011;19:102–7. doi: 10.1038/ejhg.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paciorkowski AR, Thio LL, Rosenfeld JA, et al. Copy number variants and infantile spasms: evidence for abnormalities in ventral forebrain development and pathways of synaptic function. European Journal of Human Genetics: EJHG [Internet] 2011 Jun 22; doi: 10.1038/ejhg.2011.121. [cited 2011 Sep 28]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/21694734. [DOI] [PMC free article] [PubMed]
- 8.Bertossi C, Cassina M, De Palma L, et al. 14q12 duplication including FOXG1: Is there a common age-dependent epileptic phenotype? Brain Dev. 2013 Jul 6; doi: 10.1016/j.braindev.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 9.Ellaway CJ, Ho G, Bettella E, et al. 14q12 microdeletions excluding FOXG1 give rise to a congenital variant Rett syndrome-like phenotype. Eur J Hum Genet. 2013;21:522–7. doi: 10.1038/ejhg.2012.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumakura A, Takahashi S, Okajima K, et al. A haploinsufficiency of FOXG1 identified in a boy with congenital variant of Rett syndrome. Brain Dev. 2013 Oct 16; doi: 10.1016/j.braindev.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Guerrini R, Parrini E. Epilepsy in Rett syndrome, and CDKL5- and FOXG1-gene-related encephalopathies. Epilepsia. 2012;53:2067–78. doi: 10.1111/j.1528-1167.2012.03656.x. [DOI] [PubMed] [Google Scholar]
- 12.Hébert JM, Fishell G. The genetics of early telencephalon patterning: some assembly required. Nat Rev Neurosci. 2008;9:678–85. doi: 10.1038/nrn2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brancaccio M, Pivetta C, Granzotto M, et al. Emx2 and Foxg1 inhibit gliogenesis and promote neuronogenesis. Stem Cells. 2010;28:1206–18. doi: 10.1002/stem.443. [DOI] [PubMed] [Google Scholar]
- 14.Ahlgren S, Vogt P, Bronner-Fraser M. Excess FoxG1 causes overgrowth of the neural tube. J Neurobiol. 2003;57:337–49. doi: 10.1002/neu.10287. [DOI] [PubMed] [Google Scholar]
- 15.Colombo E, Collombat P, Colasante G, et al. Inactivation of Arx, the murine ortholog of the X-linked lissencephaly with ambiguous genitalia gene, leads to severe disorganization of the ventral telencephalon with impaired neuronal migration and differentiation. J Neurosci. 2007;25;27:4786–98. doi: 10.1523/JNEUROSCI.0417-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu FH, Mantegazza M, Westenbroek RE, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–9. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- 17.Striano P, Paravidino R, Sicca F, et al. West syndrome associated with 14q12 duplications harboring FOXG1. Neurology. 2011;3;76:1600–2. doi: 10.1212/WNL.0b013e3182194bbf. [DOI] [PubMed] [Google Scholar]
- 18.Karvelas G, Lortie A, Scantlebury MH, et al. A retrospective study on aetiology based outcome of infantile spasms. Seizure. 2009;18:197–201. doi: 10.1016/j.seizure.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 19.Partikian A, Mitchell WG. Neurodevelopmental and epilepsy outcomes in a North American cohort of patients with infantile spasms. J Child Neurol. 2010;25:423–8. doi: 10.1177/0883073809341664. [DOI] [PubMed] [Google Scholar]
- 20.Riikonen RS. Favourable prognostic factors with infantile spasms. Eur J Paediatr Neurol. 2010;14:13–8. doi: 10.1016/j.ejpn.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 21.Paciorkowski AR, Thio LL, Dobyns WB. Genetic and biologic classification of infantile spasms. Pediatr Neurol. 2011;45:355–67. doi: 10.1016/j.pediatrneurol.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kamien BA, Cardamone M, Lawson JA, et al. A genetic diagnostic approach to infantile epileptic encephalopathies. J Clin Neurosci. 2012;19:934–41. doi: 10.1016/j.jocn.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 23.Mastrangelo M, Leuzzi V. Genes of early-onset epileptic encephalopathies: from genotype to phenotype. Pediatr Neurol. 2012;46:24–31. doi: 10.1016/j.pediatrneurol.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 24.Cardoza B, Clarke A, Wilcox J, et al. Epilepsy in Rett syndrome: association between phenotype and genotype, and implications for practice. Seizure. 2011;20:646–9. doi: 10.1016/j.seizure.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 25.Halbach NSJ, Smeets EEJ, Van den Braak N, et al. Genotype-phenotype relationships as prognosticators in Rett syndrome should be handled with care in clinical practice. Am J Med Genet A. 2012;158A:340–50. doi: 10.1002/ajmg.a.34418. [DOI] [PubMed] [Google Scholar]
- 26.Paciorkowski AR, Traylor RN, Rosenfeld JA, et al. MEF2C Haploinsufficiency features consistent hyperkinesis, variable epilepsy, and has a role in dorsal and ventral neuronal developmental pathways. Neurogenetics. 2013;14:99–111. doi: 10.1007/s10048-013-0356-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.