

Abstract
Expression of recombinant proteins in bacterial or eukaryotic systems often results in aggregation rendering them unavailable for biochemical or structural studies. Protein aggregation is a costly problem for biomedical research. It forces research laboratories and the biomedical industry to search for alternative, more soluble, non-human proteins and limits the number of potential “druggable” targets. In this study we present a highly reproducible protocol that introduces the systematic use of an extensive number of detergents to solubilize aggregated proteins expressed in bacterial and eukaryotic systems. We validate the usefulness of this protocol by solubilizing traditionally difficult human protein targets to milligram quantities and confirm their biological activity. We use this method to solubilize monomeric or multimeric components of multi-protein complexes and demonstrate its efficacy to reconstitute large cellular machines. This protocol works equally well on cytosolic, nuclear and membrane proteins and can be easily adapted to a high throughput format.
Keywords: Protein purification, Detergents, Multiprotein complexes, Membrane receptors
Introduction
The study of protein–protein interactions has contributed critically to development of most biological sciences fields. Yet, one of the most frequently encountered problems in protein chemistry is protein aggregation. To tackle this problem, efforts have been concentrated on development and optimization of protein expression systems [1–8] that have successfully produced folded (recombinantly-expressed) proteins. However, in spite of these advances, and the use of higher eukaryote expression systems, a large number of proteins continue to aggregate inside host cells or upon cell lysis, rendering them unavailable for biochemical and structural studies. Moreover, insoluble proteins are usually of mammalian origin and are frequently critical targets for drug discovery. Protein aggregation has hampered biochemical and drug-discovery studies and has forced structural biologists to opt for shorter versions of full-length proteins or for the more soluble “homologous versions” of the protein found in other species [9–12], particularly thermostable proteins from Archea species.
Several factors can trigger protein aggregation [13,14] some of these include: (1) monomer to oligomer transitions produced by electrostatic or hydrophobic interactions on proteins with complimentary surfaces; or covalent associations due to disulfide bond formation; (2) aggregation initiated by the presence of hydrophobic or highly charged electrostatic patches of partially unfolded intermediates; (3) aggregation of chemically modified products, such as proteolysis fragments and oxidized proteins. Small molecules capable of counteracting aggregating factors, could potentially improve protein solubility. Amphiphilic compounds, such as detergents, containing both hydrophilic “head” and hydrophobic “tail” groups, are great candidates to achieve solubility conditions in non-ideal environments such as protein lysates.
Detergents are classified according to their head group charge as ionic, if they have positive (cationic), negative (anionic), or both, positive and negative (zwitterionic) charges; and non-ionic if they lack head group charges. Hydrocarbon tails can be saturated al-kanes (with different chain lengths), branched non-saturated al-kenes or aromatic. With over a hundred detergents commercially available, a particular combination of hydrophilic head group and hydrocarbon tail length can interact favorably with charged surface residues and shield hydrophobic patches on subunits of multi-protein complexes or partially unfolded intermediates. Detergents have played critical roles in solubilization of membrane proteins; however their use as solubilizing agents for protoplasmic proteins has never been explored methodically and has been limited to the empiric use of few ionic and non-ionic detergents.
In this study we present a general method that features the systematic use of detergents to solubilize and purify – biomedically-relevant – human proteins to homogeneity, and apply this strategy to solubilize monomeric and multimeric components of multi-protein complexes (MPCs)3 towards their reconstitution.
Methods and results
Extraction of target proteins from insoluble pellets
Our approach towards protein solubilization involves the following steps (Fig. 1).
Fig. 1.
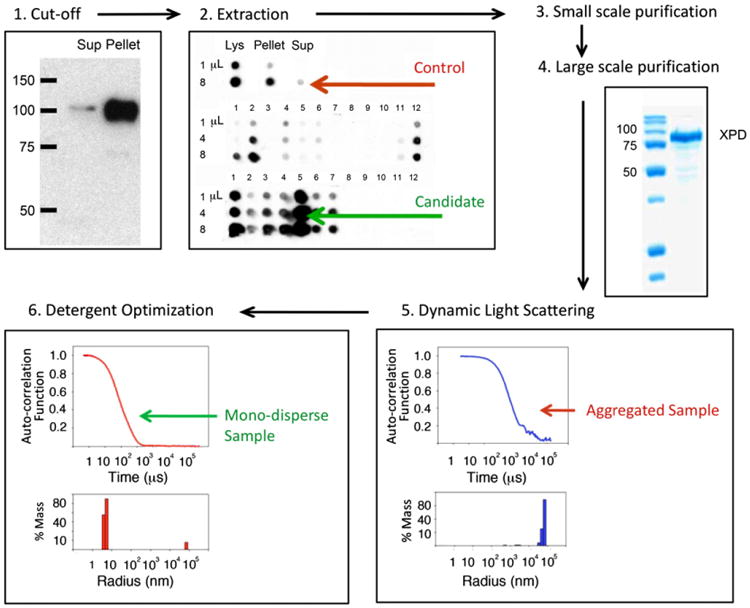
Overall strategy. See text for details. (1) Cut-off: Proteins whose soluble fraction is less than 30% are subject to a detergent screen as illustrated for the human nucleotide excision repair protein (NER) XPD expressed in a baculovirus-infected Sf9 system. (2) Detergent extraction candidates: recombinant protein is extracted from insoluble pellets using a panel of detergents and the efficiency of each detergent to solubilize sample is quantified using immuno dot blots. (3) Small scale purification allows us to fine-tune salt and detergent concentration for sample binding to affinity beads and tag removal. (4) Large-scale purification with best detergent candidate. (5) Dynamic Light Scattering (DLS) and Multi Angle Light Scattering (MALS) experiments to test monodispersity. Aggregated samples (defined here as proteins whose measured radius (by DLS) is three times larger that it's predicted one) will be subject to additional detergent screening. (6) Detergent optimization: purified samples with large particle size are rescreened and analyzed in batch form using DLS. Detergents that can successfully produce monodisperse samples (measured radius <3× predicted radius) are selected.
Selecting a target for detergent extraction
To decide whether a protein will be subject to a solubilization protocol, we first perform immunoblot experiments of the supernatant and pellet after cell lysis to identify the fraction of insoluble protein. Visual inspection of western blots allows determination of the soluble and insoluble fractions (Fig. 1, panel: cut-off). We routinely perform detergent extraction and solubilization protocols for those samples whose soluble fraction is less than 30–50% of the total lysate. This is based on the observation that proteins with borderline or low solubility are not generally monodisperse during dynamic light scattering experiments.
Detergent extraction
The following is a general approach that we have taken for a large number of aggregated yeast and human proteins purified from Escherichia coli (E. coli), Saccharomyces cerevisiae or baculovirus-infected Spodoptera frugiperda (Sf9) cells. Protein samples with soluble fractions below 30–50% are subject to detergent extraction. Since it is hard to predict which detergent will have a positive effect on protein solubilization a wide range of surfactants including ionic and non-ionic and zwitterionic species are used. There are several commercially available detergent kits that provide a good starting point for the screen; among them is a 96-well block format screen from Hampton Research (detergent screen HT catalog number HR2-406). Approximately 0.5 g of cells (E. coli or Sf9) expressing a target protein are re-suspended in 9.5 mL of buffer containing 200 mM NaCl, 50 mM Hepes pH 7.5, and 2 mM β-mercaptoethanol (BME) (buffer A). In our hands, this initial solution conditions work well for most targets. However, salt type and concentration as well as buffer type and pH might be adapted for other protein targets. After brief sonication, cell lysates are aliquoted in 100 fractions containing 100 μL of cell lysate each, and spun down for 30 min. After centrifugation, the supernatant is removed and each pellet containing the insoluble target protein is re-suspended in 75 μL of buffer A plus 25 μL from each detergent in the 96-well format HR screen, leaving four control samples for re-suspension with buffer only. Since most detergents in the screen are 10× CMC (for each detergent), aliquots will be at 2.5× CMC. The CMC of a detergent is defined as the minimal detergent concentration that will allow formation of micelles in solution. Samples are incubated for 20 min at 4 °C. Protein extraction may be enhanced by room-temperature or longer incubation times, however careful monitoring is required to avoid proteolysis of degradation-prone proteins. After the detergent incubation period, samples are centrifuged at 17,000 RCF again for 30 min and the supernatants loaded on a dot blot apparatus for quantification using a specific antibody (against the target protein or an engineered affinity tag). Three different volumes of sample are loaded 1, 4 and 8 μL. However, the sample volume loaded in the dot blot apparatus will depend of the quality of the antibody used to detect the target protein and might need to be determined empirically. Analysis of dot blot experiments (Fig. 1, panel: extraction, and Supplementary Fig. 1) allows selection of the best extracting detergents candidates compared to the control, “buffer only” aliquot.
Small scale purification
After selection of the best extracting detergents (from dot-blot experiments), we carry out small-scale purifications using the best 6–12 detergents. The reason to perform a small-scale purification step is twofold: first, to test the effect of extracting detergents on affinity beads, since some detergents could interfere with sample binding, and second – when purifying protein complexes – to test whether the presence of a detergent could disrupt subunit interactions.
We perform this screen by sonicating 0.5 g of E. coli or Sf9 cells expressing the desired protein in 10 mL of buffer A and aliquot in 6–12 equal fractions. Each extracting detergent (at 2.5× CMC) is added to an aliquot and incubated for 20 min at 4 °C. Samples are centrifuged for 30 min at 17,000 RCF to separate pellet and supernatant fractions. Supernatants are mixed with 6–12 aliquots of 50–100 μL of affinity beads (previously equilibrated with buffer A plus 2.5× CMC of the corresponding extracting detergent) and incubated over an hour. We have used the following affinity beads with high success: (1) nickel beads for poly-His tagged proteins (Sigma); (2) antibody beads (IgG, GE healthcare) for protein-A-tagged (PA-tag) proteins; (3) amylose resin (New England Biolabs) for proteins fused to maltose binding protein tag and, (4) chitin beads for proteins purified using the IMPACT-twin purification system (New England Biolabs). Following incubation, beads are thoroughly washed several times with buffer A plus 2.5× CMC of the corresponding detergent, and eluted (or boiled); individual fractions are subject to SDS–PAGE to quantify protein extraction and quality of purification. Alternatively, for protein expressing at low levels, we perform western blots to quantify purification efficiency. It is important to note that detergents can inhibit the ability of certain enzymes to remove affinity tags [15]. An SDS–PAGE of a representative small-scale experiment during purification of the human TFIIH subunits XPB and p52 is illustrated in Fig. 2. The integrity of the XPB–p52–PA complex is preserved in most detergent solubilized conditions and clear enrichment of the complex is observed with the use of Fos-Choline-12 (lane 4) and the two-detergent mixture Brij-58 and Fos-Choline-12 (lane 3). Similarly, small-scale purification of the XPB–p62–p44–p34 complex (performed over Ni2+ beads) shows clear enrichment of detergent-solubilized versus buffer-only samples (see Supplementary Fig. 2). A small fraction of p62–p44–p34 may be recovered to a certain degree in the absence of detergent, however, the presence of a surfactant was necessary to co-purify the subunits with XPB.
Fig. 2.
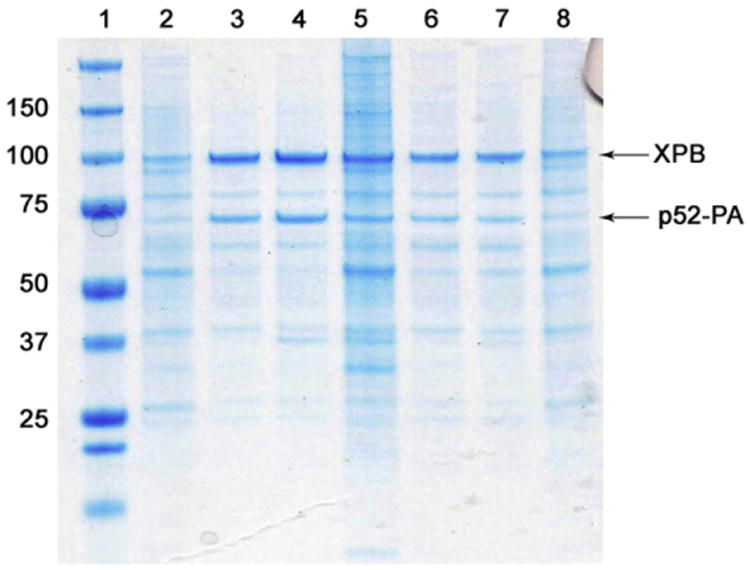
Sf9 cells (0.5 g) co-infected with two viruses expressing 10His–XPB and p52–PA were lysed in 4.5 mL of buffer A and aliquoted in seven equal samples. Buffer only (lane 2), and six extracting detergents (at 2.5× CMC) including a combination of two detergents Fos-Choline 12 and Brij-58 (lane 3), Fos-Choline-12 alone (lane 4), ZW 3–12 (lane 5), Brij-58 (lane 6), C12E8 (lane 7) and Anapoe-80 (lane 8) were added to the lysates and used throughout the experiment. Samples were incubated with Ni2+ beads (50 μL) for 4 h and washed extensively with buffer A and buffer A + 40 mM imidazole. After removal of wash buffer, beads were boiled and ran on a SDS–PAGE for analysis. The following conclusions can be drawn from this figure: (1) samples purified in the presence of buffer-A only (lane 2) or Anapoe-80 (lane 8) shows no enrichment of 10His XPB–p52–PA heterodimer. (2) Integrity of XPB–p52–PA complex is preserved in most detergent solubilized conditions (lanes 3–7) demonstrating that samples are correctly folded. (3) A clear enrichment of the XPB–p52–PA heterodimer is observed with Fos-Choline-12, in addition the presence of detergent helped to eliminate most samples impurities.
Large scale purification
Most favorable conditions found during small scale experiments can be extrapolated to large scale purifications but we recommend using the following guidelines for optimal results: (a) Extracting detergent is added before cell lysis. High protein concentrations have been recorded in the cytosol of most organisms; for example, it is estimated that the concentration of protein in the E. coli cell environment is 200–320 mg/mL [16]. It is possible that weak, non-specific protein–protein interactions in a crowded cellular environment might help solubilize a target protein. However, this “solubilizing” environment disappears as soon as cells are lyzed and proteins are diluted in buffer. In our experience, the presence of a detergent during cell disruption enhances solubility of the target protein. (b) Non-ionic detergents with μM CMCs may require mM concentrations for optimal extraction. (c) Detergents can sometimes interfere with binding of affinity tags to beads, therefore we perform overnight batch binding experiments or load the lysate containing the protein of interest at very low flow rates, e.g. 0.2 mL/min. (d) Extracting detergent is present at all times during purification at 2.5× CMC, however the concentration may vary depending on the protein and could be decreased to 1.5× CMC, this concentration must be determined empirically using light scattering experiments.
Testing sample monodispersity using dynamic light scattering experiments (DLS)
The presence of an extracting detergent does not ensure that samples will be monodisperse. Therefore it is essential to determine the aggregation state of purified proteins in solution (Fig. 1, panel: Dynamic light scattering). Since the intensity of the light scattered by a particle is proportional to the cube of the particle radius, light scattering techniques are particularly effective for detecting the presence of large particles versus monomers/oligomers in solution [17–19]. DLS data analysis provides the distribution of the total protein mass contributed by ensembles of particles with different radii [20,21]. Using DLS techniques, we have established a protocol to identify high throughput solubility conditions for protein samples. If a target protein is aggregated (defined here as proteins whose calculated molecular weight is three times larger that its predicted one) it is subjected to further screening to determine if an additional/new detergent can yield a monodisperse sample with a molecular weight close to the predicted one (samples with these characteristics will be simply referred as monodisperse). For detergent optimization, 96 aliquots of 15 μL of purified protein are mixed with 5 μL of individual detergents from the HR detergent screen giving a final detergent concentration of 2.5× CMC in 20 μL. After a short incubation period of approximately 10–30 min, all samples are centrifuged for 20 min at 17,000 RCF, loaded into a 384-well Corning plate and analyzed in batch form using the Wyatt DynaPro Plate Reader Plus. This instrument is equipped with a sensitive and fast solid state detector and a fast multi channel auto-correlator and it can operate in batch mode measuring up to 384 samples (these characteristics make the instrument ideal for screening a large numbers of solubility conditions). Moreover this instrument can accurately measure the size of protein standards in the range of 10–500 kDa (Supplementary Fig. 3A). DLS data is collected using a small number of short acquisitions (e.g. 20 acquisitions, lasting 5 s each) to identify detergent conditions that disrupt protein aggregates. For these experiments, a highly concentrated sample is not required, however protein homogeneity is critical. Samples with protein concentrations above 0.2 mg/mL can be easily tested.
During purification of the human XPD helicase, DLS data showed that XPD had a large particle radius in the presence of the initial extracting detergent (Fos-Choline-12), see Fig. 1, panel: Large scale purification. An additional detergent screen identified a second detergent (polyoxyethylene(8)dodecyl ether C12E8) that produces a monodisperse and functional XPD (Fig. 4d).
Fig. 4.
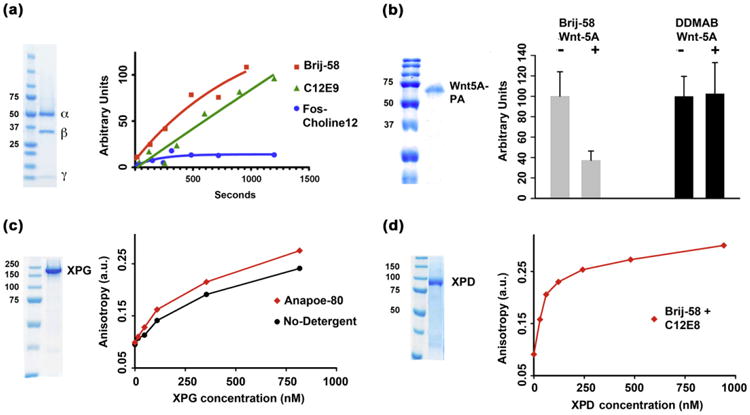
Biological activity of detergent-solubilized samples. (a) GαSβγ GTPγS binding activity comparison in select detergents. GTPγS binding of 800 nM GαSβγ was measured in the presence of 0.5 mM Brij-58 alone (red trace), 0.25 mM C12E9 (green trace) and 4 mM Fos-Choline-12 (blue trace) over 20 min. Each sample was passed through nitrocellulose membranes and GTPγS binding was quantified by measuring [35S]GTPγS-GαSβγ scintillation. (b) HepG2 cells cultured in the presence of Wnt-5A solubilized in Brij-58 but not DDMAB for 48 h led to a noteworthy decrease in TOPFLASH reporter activity. (c) Binding of human XPG to the Y shape DNA substrate. Increasing amounts of human XPG was added to 25 nM fluorescently labeled DNA substrate. The equilibrium dissociation constant, Kd, was estimated as the amount of XPG that was necessary to get to 50% maximum binding. (d) Binding of human XPD to a single strand 39mer DNA substrate. Increasing amounts of human XPD were added to 25 nM fluorescently-labeled DNA substrate. The equilibrium dissociation constant, Kd, was estimated as 58 nM using non-linear fit of the anisotropy to the XPD concentration.
Optimizing solubility conditions using DLS
After performing the screen to identify detergents that can disrupt protein aggregates, we investigate the effect of protein concentration on particle radius as samples are concentrated using centrifugal concentrators (Millipore and GE). Samples are taken at different intervals during concentration, loaded on a plate (corning, 384-well) and analyzed by DLS using the plate reader in batch mode. For this analysis, DLS parameters should be adjusted to enhance scattering signals from small particles by increasing the number of acquisitions to 50–100 and the acquisition time to 10–20 s. The purpose of this step is to identify detergents that can yield monodisperse samples at higher protein concentrations (see Supplementary Fig. 3B); and determine the upper limit of target solubility in terms of protein concentration. In most cases the radii and molecular weight (MW) of (concentrated) detergent-solubilized samples measured by DLS are very close to the expected ones (Fig. 3 and Supplementary Fig. 3B). This last suggests that nuclear or cytosolic proteins solubilized by detergents (at 1-2× CMC) are generally not embedded within a micelle but have small number of detergent molecules shielding hydrophobic patches on their surface. In some circumstances, as protein is concentrated, an increase in its MW results from the formation of oligomers (dimers, trimers or tetramers). However, one must be careful since it is possible that during protein concentration using ultrafiltration devices, the molarity of a detergent might increase well above the CMC-observed mostly when concentrating large sample volumes solubilized using detergents with high molecular weights or very low CMCs (see Supplementary materials, note on the use of detergents).
Fig. 3.
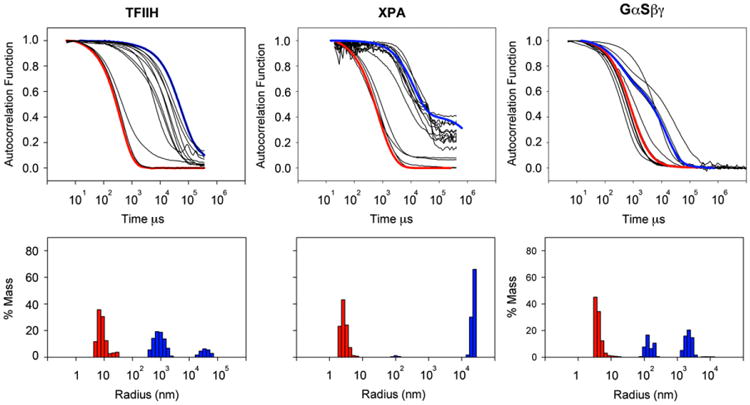
Effect of detergent solubilization on particle radius for S. cerevisiae core TFIIH complex (cTFIIH, comprises Rad3, Tfb1, Tfb2, Ssl1 and Tfb4 [25]), human XPA, and human GαSβγ. The predicted radii for cTFIIH is 9.1 nm, human XPA is 2.5 nm and GαSβγ is 6.6 nm. Proteins whose purifications were initially successful but had large particle sizes were subject to DLS experiments in (batch mode) to find conditions that lead to monodisperse samples. Correlograms measure the exponential time decay of the autocorrelation function of the scattered light of particles in solution diffusing with Brownian motion and they contain information on the diffusion speed of different particle size groups in solution. After standard data analysis, from correlograms, shown in black in the top panel it is possible to estimate the radii of diffusing particles in solution. Larger decay times correspond to larger particle radii. Control correlograms are shown in top panels. Two examples of particle radius distributions are shown in lower panels. Indicated in blue are examples of detergent conditions with still large aggregates in solution. Examples of detergent conditions that had a dramatic effect on the particle radius are indicated in red.
If the detergent used to extract a protein sample from the cell pellet is different from that producing a monodisperse sample, we utilize the former during cell lysis, extraction, column loading and washing steps. We then switch to the latter gradually – while the sample is still on the column – by running a 10–20 column-volumes gradient. After the gradient, the sample is washed extensively and eluted in the presence of the “monodispersing” detergent. Each protein is tested for function to insure that detergents do not interfere with protein activity (see below).
Functional studies to confirm protein folding and the presence of a biologically active sample
Proteins could unfold or become inactive during detergent extraction or solubilization, it is therefore critical to test the biological activity of the protein in the presence of detergent. To this end we routinely perform functional studies on several solubilized protein samples. For GαSβγ (Fig. 4a), three detergents, n-DodecylphosphoCholine (Fos-Choline-12), polyethylene glycol (20) monohexadecyl ether (Brij-58) and C12E9 (among few others) were capable of producing monodisperse folded samples – as evidenced by the integrity of the heterotrimer – however GTP exchange was impaired in the sample solubilized with Fos-Choline-12 (Fig. 4a). As another example, Wnt-5A purified and solbilized in the presence of DDMAB did not inhibit β-catenin-TCE/LEF transactivation using a reporter system in cultured hepatocellular cancer cells that harbor a constitutively active β-catenin owing to a deletion in CTNNB1. However, Brij-58-solubilized Wnt-5A successfully inhibited β-catenin activation [22] (Fig. 4b). Biological activity of detergent solubilized proteins might be compromised in the presence of the solubilizing surfactant, in such cases it will be necessary to test for activity in the presence of other detergents. Lastly, DNA binding to appropriate DNA substrates for the human XPG and XPD NER proteins were confirmed using fluorescence anisotropy experiments after solubilizing XPG in Brij-58 (Fig. 4c) and XPD in C12E8 (Fig. 4d). Purified monodisperse human XPG (expressed and purified from baculovirus infected Sf9 cells) bound to a Y shaped DNA with equilibrium dissociation constant, Kd, of approximately 250 nM (see Supplementary protocols 2). Previous gel mobility shift experiments indicated that human XPG bound to a similar Y substrate in the nM range at low ionic strength [23]. Similarly highly purified, monodisperse XPD was expressed and purified from insect cells was found to bind to a ssDNA (39mer)with an equilibrium dissociation constant of Kd = 58 nM. For comparison, a previous equilibrium dissociation constant, of 270 nM was determined experimentally by fluorescence anisotropy for Sulfolobus acidocaldarius XPD binding to a 15 nt ssDNA [24].
In addition to affecting protein activity, detergents can disrupt protein–protein interactions. In our experience this does not happen frequently. Affinity purification of yeast TFIIH (using a TAP tag on Tfb4) yields a five-component core TFIIH [25] (cTFIIH) comprising Rad3, Tfb1, Tfb2, Ssl1 and Tfb4. However, the use of two detergents, β-d-Fructopyranosyl-α-d-glucopyranoside monodode-canoate (sucrose monolaureate) and C12E8, during purification of cTFIIH dislodged Rad3, Tfb1 and Tfb2 from cTFIIH (Supplementary Fig. 4). This finding provided a critical clue for co-expression experiments of the human homologs (p44 and p34) in insect cells (see below).
The best 8
It is possible to propose a list of surfactants (Table 1) that can be used (as first attempt) to solubilize protein targets from cell pellets. Moreover, if a detergent with good solubilizing capabilities is found, additional members of the same surfactant family with different alkyl chain-lengths should be explored. It is also likely that detergents belonging to the same detergent group provide similar solubilization capabilities, however the lower CMCs associated with longer carbon chain lengths could provide some benefits in further purifications steps or have a significantly lower (or none) inhibitory effect on biological activity (Fig. 4a). For example, solubilization of GαSβγ was accomplished using both Fos-Choline-12 and n-tetradecylphosphocholine (Fos-Choline-14), however GTP-exchange was inhibited by the former (CMC = 1.5 mM) but not by the latter (CMC = 0.15 mM). Testing the extracting capabilities of these eight detergents can be performed following the protocols discussed in detail under small-scale purifications.
Table 1.
Detergents, to be used as a first line approach for protein extraction, and proteins solubilized by each of them.
| Detergent type | Protein | CMC (mM) |
|---|---|---|
| Sarkosyl: Sodium dodecanoyl sarcosine (ionic) | XPG, PTHR1, CD3δ, CD3ζ, LRP5 | 14 |
| Anapoe-58: Polyethylene glycol(20)monohexadecyl ether (Brij-58) (non-ionic) | XPC, XPB, XPD, Wnt-5A P44–P52–P62, GαSβγ | 0.004 |
| Anapoe-80: Polyoxyethylene(80)sorbitan monolaurate (non-ionic) | XPG, XPF–ERCC1, XPC | 0.012 |
| Anapoe-C13E8: Polyoxyethylene(8)dodecyl ether (non-ionic) | XPA, CD3-δ, P62–P42–P34 | 0.1 |
| Anapoe-C12E8: Polyoxyethylene(8)dodecyl ether (non-ionic) | GαSβγ, XPD, PTHR1, CD3δ, CD3ζ | 0.1 |
| Zwittergent-3-10: n-decyl-N,N-dimethyl-3-ammonio-1-propanesulfonate (zwittergent) | TFIIF, TFG2, TF2K, FCP1 | 40 |
| Fos-Choline-12: n-dodecylphosphocholine (zwittergent) | XPD, Fzd4, PTHR1, DRA1-DRB1, LRP5, CD3δ | 1.5 |
| DDMAB: n-dodecyl-N,N-(dimethylammonio)butyrate (zwittergent) | Wnt5A, XPC, FCP1 | 4.3 |
Difficult targets
Some proteins are particularly hard to solubilize with a single detergent, in such cases we have successfully employed two surfactants mixtures, ionic–non-ionic or non-ionic–non-ionic for their solubilization [26–28]. For example, the ionic–non-ionic combination is particularly useful for extraction of proteins from E. coli pellets. The combination of N-methyl-N-(1-oxododecyl)-glycine, (Sarkosyl) (another detergent identified by dot-blot analysis to be a good candidate for extraction) and polyoxyethylene(80)sorbitan monolaurate (Anapoe-80) could extract most XPG (Fig. 4d) from the insoluble fraction. Removal of Sarkosyl from the samples was accomplished successfully – with samples still bound to the affinity column – by gradually decreasing the concentration of Sarkosyl (using a 10–20 column volumes gradient) followed by overnight wash. Other successful combinations include C12E8/Brij-58, Ana-poe-80/Brij-58 and Fos-Choline-14/Brij-58.
Solubilization of membrane proteins and membrane associated proteins
We have applied detergent solubilization protocols to several membrane proteins including: (1) single-pass transmembrane receptors such as CD3δ CD3ζ expressed in E. coli (Fig. 5a and b and Supplementary Fig. 1); (2) G-protein coupled receptors (GPCRs), among them frizzled-4 (Fzd4) (Fig. 5c and Supplementary Fig. 5) and the parathormone receptor-1 (PTHR1) expressed in E. coli (Fig. 5d); (3) monotopic membrane associated proteins such as GαSβγ, and lipid modified secreted protein such as Wnt-5A, both expressed in insect cells. The protocols employed to extract and solubilize these proteins were no different to those used to purify non-membrane proteins; however most single-pass trans-membrane receptors, as well as E. coli expressed proteins required the presence of two detergents for extraction.
Fig. 5.
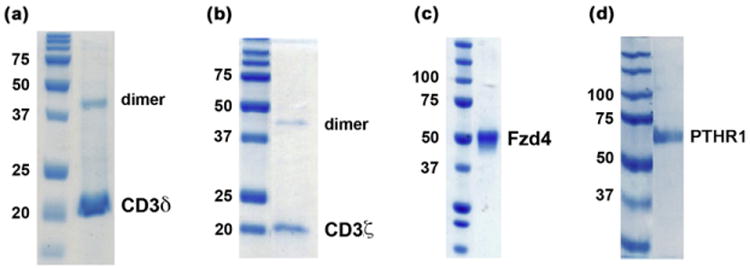
Solubilization of membrane proteins. (a and b) SDS–PAGE of E. coli-expressed human receptors CD3δ and CD3ζ. (c) SDS–PAGE of human Fzd-4 expressed in Sf9 cells and purified to homogeneity in the presence of Fos-Choline-12. (d) SDS–PAGE of human PTHR1 expressed in E. coli and purified to homogeneity in the presence of Fos-Choline-14.
Solubilization and reconstitution of multi-protein complex (MPCs)
Multi-protein complexes are specially challenging systems since components might interact transiently and with low affinity, making recovery of full (stoichiometric) complexes almost impossible; therefore, biochemical reconstitution of MPCs often requires expression and purification of individual monomeric and/or multimeric components. Using methods outlined above for single proteins, we have successfully reconstituted several MPCs including yeast transcriptional complexes and the human TFIIH complex.
Reconstitution of yeast transcriptional complexes
We aimed to reconstitute the minimal set of proteins required for promoter-dependent transcription [29,30] (mPIC) in complex with double-stranded DNA (Supplementary Fig. 6a). The mPIC includes RNA polymerase II (Pol II) and the general transcription factors TFIIF (comprising three subunits, TFG1, TFG2, and TFG3, MW = 105, 40 and 30 kDa respectively), TFIIB, and the TATA-binding protein (TBP, the 30 kDa subunit of the TFIID complex). In this case the rate limiting step was purification of the TFIIF heterotrimer which had consistently shown low protein yields due to aggregation [30,31] We performed a detergent screen and successfully purified TAP-tagged [31,32] TFIIF (protein-A (PA) tag plus calmodulin-binding-peptide (CBP-tag)) in the presence of 0.1% (w/v) ZW3–10 (Supplementary Fig. 7). DLS experiments revealed a particle of 6 nm, equivalent to an average radius of MW 250 kDa. The steps involved in mPIC assembly are illustrated in Fig. 6a (I–IV). Using similar protocols, we have reconstituted other yeast tran-scriptional complexes including (among several others) a Pol II– TFIIF–coreTFIIH–TFIIB (Fig. 6b and Supplementary Fig. 6b).
Fig. 6.
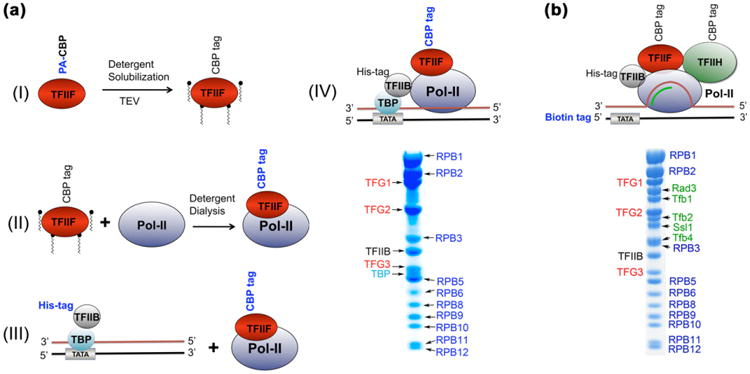
Reconstitution of yeast transcriptional complexes. (a) (I) TFIIF was purified over IgG resin in the presence of 20 mM ZW3-10 and eluted using TEV protease (Note: affinity tag used in a given purification stage is shown in blue). Further purification was accomplished using a Heparin column (GE Healthcare). Protein was eluted using an ammonium sulfate gradient (250–600 mM) in the presence of ZW3–10. It is important to note that the concentration of ZW3–10 to maintain a monodisperse TFIIF was later found to be below the CMC of the detergent (0.1× CMC). (II) Pol II was purified individually as described before [37] and the two proteins were mixed together (Pol II:TFIIF molar ratio = 1.5, to insure a stoichiometric complex) under ZW3–10 (10 mM) and high salt (250 mM ammonium sulfate). Overnight dialysis into a buffer containing no detergent and low salt (120 mM KCl) allowed purification of a transcriptionally active Pol II–TFIIF complex (using calmodulin binding peptide (CBP) tag on TFG2) over calmodulin resin (Supplementary Fig. 7). (III) The TFIIB-TBP complex with a 53-mer double-stranded DNA (dsDNA) was purified as described before [38] and mixed with Pol II–TFIIF complex (see Supplementary protocols 1). (IV) SDS–PAGE of the resulting complex purified over calmodulin resin (GE-Healthcare) using the calmodulin binding peptide (CBP) tag on TFG2. (b) SDS-PAGE of a S. cerevisiae transcribing complex comprising Pol II, TFIIF, coreTFIIH37, TFIIB, a 51-mer transcribing bubble (Supplementary Fig. 6b).
Solubilization of individual subunit of MPCs
We next evaluated the efficacy of this method to isolate individual (monomeric) as well as dimeric and multimeric subunit of MPCs. To this end, we expressed all core subunits of the human TFIIH complex as monomers, dimers, and other multimers. The TFIIH complex includes [33]: (1) a 7-subunit core (cTFIIH) composed of p62, p52, p44, p34, p8, and the helicases XPB and XPD; and (2) a 3-subunit CDK-activating kinase module comprising cyclin-H, MAT1 and Cdk7. We expressed most components of cTFIIH individually in Sf9 cells (except for p8, expressed in E. coli). Four subunits, XPB, XPD, p52 and p44 had good expression levels, but co-fractionated with the insoluble pellet. Individual detergent screens were performed, and all four proteins were purified successfully in the presence of detergents (Fig. 7a (I–IV). DLS measurements confirmed that all samples were monodisperse and had particle sizes within the expected molecular weight (Supplementary Fig. 8a). Expression levels for p34 and p62 were low when expressed individually, and therefore had to be co-expressed with another core member; however, we did not exhaust all available expression optimization protocols. We co-express successfully p34 in complex with p44 [34] (Fig. 7b and Supplementary Fig. 8b (left panel)); p62 was expressed in complex with p44 and p34 (Fig. 7c (I) and Supplementary Fig. 8b (right panel)). Other TFIIH sub-assemblies expressed and purified in the presence of detergents include: (1) XPB, p62 p44 and p34 (Fig. 7c (II)); (2) XPB– p52 (Fig. 2); (3) XPB–p52–p8–GST (Fig. 7d (I) and Supplementary Fig. 8c); and (4) p62–p44–p34–p8–GST (Fig. 7d (II)). Moreover, reconstitution of a stoichiometric 6-subunit TFIIH complex was accomplished by mixing an XPB–p62–p44–p34 sub-complex (Fig. 7c (II)) with detergent solubilized p52 (Fig. 7a (III) and p8– GST (Fig. 7a (V)), both expressed individually. Isolation of a fully monodisperse complex was achieved using size exclusion chromatography (Fig. 7e and Supplementary Fig. 8d). This strategy has allowed us to uncover new interactions among TFIIH components such as those taking place between XPB with p62–p44–p34 (Fig. 7c (II)) and p8 with p62–p44–p34 (Fig. 7d (II)).
Fig. 7.
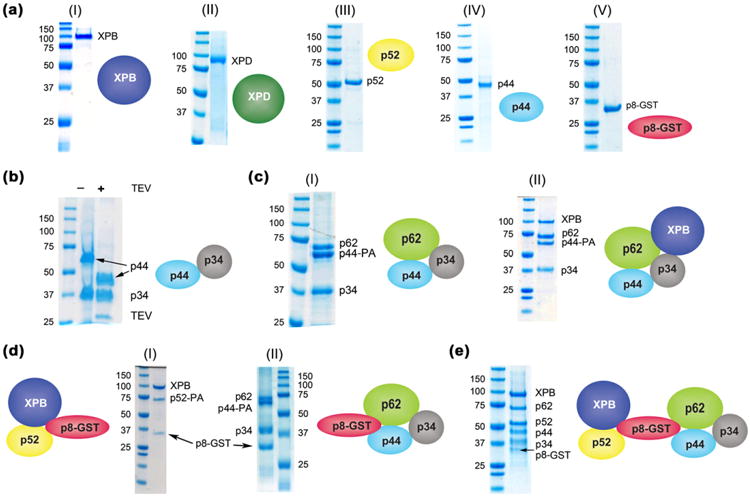
Solubilization of core-TFIIH: from individual subunits to sub-assemblies. (a) SDS–PAGE of core-TFIIH subunits solubilized as monomers in the presence of detergent (I–IV), GST–p8 (V) did not require detergent for purification. See Supplementary protocols 1 for detailed purification methods. (b) SDS–PAGE of p44-p34 complex. The heterodimer was purified using two affinity tags. A deca-histidine tag at the C-terminus of p34 was used first (lane 2). In a second step, protein was loaded on IgG beads and complex was eluted by clipping the PA-tag on the C-terminus of p44 (lane 2) using TEV protease. (c) SDS–PAGE of a p62–p44–PA–p34 (I) complex and of an XPB–p62–p44–p34 (II) both purified in the presence of 0.5 mM Brij-58 and 0.2 mM C13E8 over Ni2+ beads. Note: The PA-tag at the C-terminus of p44 did not interfere with complex assembly. (d) (I) SDS–PAGE of XPB–p52–GST–p8 complex. XPB–p52 heterodimer was purified using a deca-histidine tag on XPB in the presence of Brij-58. The heterodimer was supplemented with 1.5× molar excess of GST-p8. The complex was purified over nickel beads after extensive washing with 40 mM imidazole. (II) SDS–PAGE of a p62–p44–p34–GST-P8 complex. Detergent-solubilized heterotrimer p62 –p44 –PA –p34 (c (I)) was supplemented with 1.5× molar excess GST-p8 and loaded on a nickel column washed extensively with 40 mM imidazole to remove excess, unbound GST-p8 and eluted with 500 mM imidazole. Note: all buffers were supplemented with 0.5 mM Brij-58. (e) SDS–PAGE of a reconstituted XPB–p62–p52–p44–p34–GST-p8 complex. Brij-58-solubilized XPB–p62–p44–p34 component (d (II)) was mixed with 1.5 M excess of Brij-58-solubilized p52 and with GST-p8. Sample was applied to a superdex-200 size exclusion column (GE Healthcare) for separation.
Discussion
We have developed an efficient protocol for large-scale solubilization of biologically active, membrane and protoplasmic proteins by exploring systematically the use of detergents. In most instances, these proteins were previously unavailable or not in sufficient quantities to perform functional and structural studies [35,36]. This strategy has worked for all expression systems tested, bacteria, yeast, insect and mammalian cells.
Solubilization of individual components of MPCs (as demonstrated for human TFIIH) could provide a powerful approach to investigate protein–protein interactions within the complex, and therefore establish its topology and identify subunits responsible for interactions with known – and possibly new – binding partners. In addition, individual subunits of MPCs can be used to supplement sub-stoichiometric components (often observed during co-expression experiments) for functional or structural studies.
There is always the possibility that the use of detergents to solubilize samples could hamper their biological activity, however, an advantage of performing broad screens for sample solubilization is that the biological activity for a particular sample can be tested using several detergents. In most cases one can find a surfactant with minimal effect on protein function.
Protein purification is an essential tool to many areas of study in the biomedical field. Successful implementation of these protocols should have significant contributions to: (1) biochemical and cell biological experiments to test protein–protein interactions; (2) large scale production of human proteins and receptors for structural and biophysical studies (such as isothermal titration calorimetry or surface plasmon resonance); (3) biochemical reconstitution of MPCs for structural, biochemical and biophysical studies; (4) generation of protein samples for antibody production; and (5) finding novel network interactions using quantitative proteomics experiments.
Supplementary Material
Acknowledgments
University of Pittsburgh Medical School startup funds to G.C. supported most of this work. Additional support was contributed by the National Institutes of Health Grant R01ES019566 (B.V.H., J.G.S. and G.C.) and the Ri.MED Foundation, Italy (F.P., A.C., G.C. and S.P.S.M.). We would like to thank Angela Gronenborn PhD for helpful suggestions.
Footnotes
Abbreviations used: MPCs, multi-protein complexes; BME, b-mercaptoethanol; cTFIIH, core TFIIH; GPCRs, G-protein coupled receptors; Fzd4, frizzled-4; PTHR1, parathormone receptor-1; CBP, calmodulin binding peptide; dsDNA, double-stranded DNA.
Appendix A. Supplementary material: Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.pep.2012.10.007.
References
- 1.Menzella HG. Comparison of two codon optimization strategies to enhance recombinant protein production in Escherichia coli. Microb Cell Factor. 2011;10:15. doi: 10.1186/1475-2859-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Midgett CR, Madden DR. Breaking the bottleneck: eukaryotic membrane protein expression for high-resolution structural studies. J Struct Biol. 2007;160:265–274. doi: 10.1016/j.jsb.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Miller LK. Baculoviruses for foreign gene expression in insect cells. Biotechnology. 1988;10:457–465. doi: 10.1016/b978-0-409-90042-2.50029-5. [DOI] [PubMed] [Google Scholar]
- 4.Bishop DH. Gene expression using insect cells and viruses. Curr Opin Biotechnol. 1990;1:62–67. doi: 10.1016/0958-1669(90)90011-9. [DOI] [PubMed] [Google Scholar]
- 5.Noirclerc-Savoye M, Gallet B, Bernaudat F, Vernet T. Large scale purification of linear plasmid DNA for efficient high throughput cloning. Biotechnol J. 2010;5:978–985. doi: 10.1002/biot.201000132. [DOI] [PubMed] [Google Scholar]
- 6.Dortay H, Akula UM, Westphal C, Sittig M, Mueller-Roeber B. High-throughput protein expression using a combination of ligation-independent cloning (LIC) and infrared fluorescent protein (IFP) detection. PLoS One. 2011;6:e18900. doi: 10.1371/journal.pone.0018900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sitaraman K, Chatterjee DK. High-throughput protein expression using cell-free system. Methods Mol Biol. 2009;498:229–244. doi: 10.1007/978-1-59745-196-3_15. [DOI] [PubMed] [Google Scholar]
- 8.Chambers SP, Swalley SE. Designing experiments for high-throughput protein expression. Methods Mol Biol. 2009;498:19–29. doi: 10.1007/978-1-59745-196-3_2. [DOI] [PubMed] [Google Scholar]
- 9.Kitamoto N, Yamagata H, Kato T, Tsukagoshi N, Udaka S. Cloning and sequencing of the gene encoding thermophilic beta-amylase of Clostridium thermosulfurogenes. J Bacteriol. 1988;170:5848–5854. doi: 10.1128/jb.170.12.5848-5854.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsukagoshi N, Ihara H, Yamagata H, Udaka S. Cloning and expression of a thermophilic alpha-amylase gene from Bacillus stearothermophilus in Escherichia coli. Mol Gen Genet. 1984;193:58–63. doi: 10.1007/BF00327414. [DOI] [PubMed] [Google Scholar]
- 11.Yonath A. The search and its outcome: high-resolution structures of ribosomal particles from mesophilic, thermophilic, and halophilic bacteria at various functional states. Annu Rev Biophys Biomol Struct. 2002;31:257–273. doi: 10.1146/annurev.biophys.31.082901.134439. [DOI] [PubMed] [Google Scholar]
- 12.Cava F, Hidalgo A, Berenguer J. Thermus thermophilus as biological model. Extremophiles. 2009;13:213–231. doi: 10.1007/s00792-009-0226-6. [DOI] [PubMed] [Google Scholar]
- 13.Philo JS, Arakawa T. Mechanisms of protein aggregation. Curr Pharm Biotechnol. 2009;10:348–351. doi: 10.2174/138920109788488932. [DOI] [PubMed] [Google Scholar]
- 14.Wang W, Nema S, Teagarden D. Protein aggregation – pathways and influencing factors. Int J Pharm. 2010;390:89–99. doi: 10.1016/j.ijpharm.2010.02.025. [DOI] [PubMed] [Google Scholar]
- 15.Vergis JM, Wiener MC. The variable detergent sensitivity of proteases that are utilized for recombinant protein affinity tag removal. Protein Expr Purif. 2011;78:139–142. doi: 10.1016/j.pep.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elowitz MB, Surette MG, Wolf PE, Stock JB, Leibler S. Protein mobility in the cytoplasm of Escherichia coli. J Bacteriol. 1999;181:197–203. doi: 10.1128/jb.181.1.197-203.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berne BJ, Pecora R. Laser light-scattering from liquids. Annu Rev Phys Chem. 1974;25:233–253. [Google Scholar]
- 18.Hole P, Montes-Burgos I, Walczyk D, Smith J, Lynch I, Dawson K. Characterisation of nanoparticle size and state prior to nanotoxicological studies. J Nanopart Res. 2010;12:47–53. [Google Scholar]
- 19.Pullara F, Emanuele A. Early stages of beta2-microglobulin aggregation and the inhibiting action of alphaB-crystallin. Proteins. 2008;73:1037–1046. doi: 10.1002/prot.22122. [DOI] [PubMed] [Google Scholar]
- 20.Pecora R. Quasi-elastic light-scattering from macromolecules. Annu Rev Biophys Bioeng. 1972;1:257–276. doi: 10.1146/annurev.bb.01.060172.001353. [DOI] [PubMed] [Google Scholar]
- 21.Aragon SR, Pecora R. Theory of dynamic light-scattering from polydisperse systems. J Chem Phys. 1976;64:2395–2404. [Google Scholar]
- 22.Yuzugullu H, Benhaj K, Ozturk N, Senturk S, Celik E, Toylu A, Tasdemir N, Yilmaz M, Erdal E, Akcali KC, Atabey N, Ozturk M. Canonical Wnt signaling is antagonized by noncanonical Wnt5a in hepatocellular carcinoma cells. Mol Cancer. 2009;8:90. doi: 10.1186/1476-4598-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hohl M, Thorel F, Clarkson SG, Scharer OD. Structural determinants for substrate binding and catalysis by the structure-specific endonuclease XPG. J Biol Chem. 2003;278:19500–19508. doi: 10.1074/jbc.M213155200. [DOI] [PubMed] [Google Scholar]
- 24.Liu H, Rudolf J, Johnson KA, McMahon SA, Oke M, Carter L, McRobbie AM, Brown SE, Naismith JH, White MF. Structure of the DNA repair helicase XPD. Cell. 2008;133:801–812. doi: 10.1016/j.cell.2008.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takagi Y, Komori H, Chang WH, Hudmon A, Erdjument-Bromage H, Tempst P, Kornberg RD. Revised subunit structure of yeast transcription factor IIH (TFIIH) and reconciliation with human TFIIH. J Biol Chem. 2003;278:43897–43900. doi: 10.1074/jbc.C300417200. [DOI] [PubMed] [Google Scholar]
- 26.Beck A, Tsamaloukas AD, Jurcevic P, Heerklotz H. Additive action of two or more solutes on lipid membranes. Langmuir. 2008;24:8833–8840. doi: 10.1021/la800682q. [DOI] [PubMed] [Google Scholar]
- 27.Nagaranjan . Micellization of binary surfectant mixtures. In: Holland, Rubingh, editors. Mixed Surfectant Systems. Chapter 4 ACS; Washington, DC: 1992. [Google Scholar]
- 28.Ogino, Abe . Physicochemical properties of mixed surfectant systems. In: Holland, Rubingh, editors. Mixed Surfectant Systems. Chapter 8 ACS; Washington, DC: 1992. [Google Scholar]
- 29.Tyree CM, George CP, Lira-DeVito LM, Wampler SL, Dahmus ME, Zawel L, Kadonaga JT. Identification of a minimal set of proteins that is sufficient for accurate initiation of transcription by RNA polymerase II. Genes Develop. 1993;7:1254–1265. doi: 10.1101/gad.7.7a.1254. [DOI] [PubMed] [Google Scholar]
- 30.Parvin JD, Sharp PA. DNA topology and a minimal set of basal factors for transcription by RNA polymerase II. Cell. 1993;73:533–540. doi: 10.1016/0092-8674(93)90140-l. [DOI] [PubMed] [Google Scholar]
- 31.Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- 32.Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- 33.Egly JM, Coin F. A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair. 2011;10:714–721. doi: 10.1016/j.dnarep.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 34.Jawhari A, Uhring M, Crucifix C, Fribourg S, Schultz P, Poterszman A, Egly JM, Moras D. Expression of FLAG fusion proteins in insect cells: application to the multi-subunit transcription/DNA repair factor TFIIH. Protein Expr Purif. 2002;24:513–523. doi: 10.1006/prep.2001.1597. [DOI] [PubMed] [Google Scholar]
- 35.Aboussekhra A, Biggerstaff M, Shivji MK, Vilpo JA, Moncollin V, Podust VN, Protic M, Hubscher U, Egly JM, Wood RD. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell. 1995;80:859–868. doi: 10.1016/0092-8674(95)90289-9. [DOI] [PubMed] [Google Scholar]
- 36.Araujo SJ, Tirode F, Coin F, Pospiech H, Syvaoja JE, Stucki M, Hubscher U, Egly JM, Wood RD. Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes Develop. 2000;14:349–359. [PMC free article] [PubMed] [Google Scholar]
- 37.Cramer P, Bushnell DA, Fu J, Gnatt AL, Maier-Davis B, Thompson NE, Burgess RR, Edwards AM, David PR, Kornberg RD. Architecture of RNA polymerase II and implications for the transcription mechanism. Science. 2000;288:640–649. doi: 10.1126/science.288.5466.640. [DOI] [PubMed] [Google Scholar]
- 38.Kosa PF, Ghosh G, DeDecker BS, Sigler PB. The 2.1-A crystal structure of an archaeal preinitiation complex: TATA-box-binding protein/transcription factor (II)B core/TATA-box. Proc Natl Acad Sci USA. 1997;94:6042–6047. doi: 10.1073/pnas.94.12.6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.