

Abstract
AIM: To investigate the function of Pea3 in colorectal carcinoma (CRC) invasion and metastatic potential.
METHODS: The expression of Pea3 during clinical progression of human CRC was investigated using Oncomine Research Edition. To assay Pea3 expression in established CRC cell lines, we performed western blotting of cell lysates. We employed shRNA-mediated knockdown of Pea3 in HCT116 (HCT) and LS174T CRC cells which was confirmed by real-time quantitative PCR (qPCR) and western blotting. Transwell invasion assays, MTS proliferation assays, anoikis assays, and fluorometric matrix metalloprotease (MMP) assays were performed to determine the effects of Pea3 knockdown on invasion, proliferation, anoikis and MMP activity in CRC cells in vitro. Alterations in epithelial-mesenchymal transition (EMT) and matrix metalloprotease (MMP) mRNA levels were determined by qPCR. CRC cells were injected into the flanks of nude mice to generate xenografts and tumor growth monitored with serial calliper measurements. To assay metastatic potential, CRC cells were injected into the spleen of nude mice, and histological analysis performed on the livers 21 d later.
RESULTS: We demonstrated that reduction of Pea3 expression in CRC cells significantly impaired their invasive capacity (HCT.shPea3, 0.28 ± 0.04 fold, P < 0.01; LS.shPea3, 0.15 ± 0.04 fold; SW.shPea3, 0.23 ± 0.03, P < 0.01), reduced anoikis resistance (HCT.shPea3 75.4% ± 1.9% viable cells vs HCT.shCtrl 88.6% ± 0.6% viable cells, P < 0.01; LS.shPea3 71.7% ± 0.5% viable cells vs LS.Ctrl 89.6% ± 0.3% viable cells, P < 0.005, but had no effect on proliferation (HCT.shCtrl AUC 5098 ± 123 vs HCT.shPea3 5689 ± 151, P < 0.05; LS.shCtrl AUC 5600 ± 324.1 vs LS.shPea3 6423 ± 400, P < 0.05). In vivo, HCT.shPea3 and HCT.shCtrl tumour xenografts grew at a similar rate (HCT.shPea3 2.64 ± 0.82 fold vs HCT.shCtrl 2.88 ± 0.80 fold, P > 0.05). In keeping with a pro-metastatic function for Pea3 in CRC, several EMT markers and MMPs were downregulated in shPea3-expressing cells, suggesting that Pea3 may exert its effects through these processes. A reduction in overall MMP activity was observed in HCT.shPea3 cells compared to their control counterparts (HCT.shPea3 0.61 ± 0.04 fold, P < 0.005). This translated in vivo to the complete absence of metastases in the livers of mice that were grafted with CRC cells lacking Pea3. Conversely, CRC cells expressing Pea3 formed liver metastases in all mice.
CONCLUSION: Our study implicates Pea3 as a mediator of metastases, and provides a biological rationale for the adverse prognosis associated with elevated Pea3 expression in human CRC.
Keywords: Colorectal cancer, Pea3, Epithelial-mesenchymal transition, Liver metastasis
Core tip: Colorectal carcinoma (CRC) is one of the leading causes of cancer mortality. Pea3 is a transcription factor that has been implicated in the pathogenesis of CRC. We demonstrate that Pea3 directly influences metastatic potential in CRC using a model of liver metastases. We provide supporting findings that CRC invasiveness, anoikis and matrix metalloproteinase activity are influenced by Pea3, and that this may collectively contribute to altered metastatic potential. Our research provides an important biological rationale to explain previously reported clinical findings that elevated Pea3 expression in human tumors is correlated with increased metastatic potential and decreased overall survival.
INTRODUCTION
Colorectal adenocarcinoma describes cancers of glandular origin that arise in the colon, rectum and anus. As normal glandular cells accumulate neoplastic changes, dysplastic cells transition through the classical, step-wise normal-adenoma-adenocarcinoma sequence. Some of the most common genetic abnormalities in colorectal carcinoma (CRC) occur in genes whose products are involved in vital signalling cascades, such as the WNT/β-catenin pathway (APC gene), the MAPK and PI3K pathways (KRAS gene), and the TGF-β signalling pathway (SMAD4), while other gene products are involved in DNA damage pathways (TP53 gene, MLH1/2 genes)[1]. Importantly, our evolving understanding of these mechanisms, as well as of other intersecting pathways directs advances in the development of prognostic markers and targeted therapy for CRC[2]. Thus, identification of novel molecules involved in CRC carcinogenesis is a crucial endeavour.
One such molecular marker is Pea3 (E1AF/ETV4), which is a transcription factor of the ETS family[3]. Pea3 is clustered with ETV1 and ETV5 in the Pea3 subfamily (reviewed in[4]). Molecular cloning of the human Pea3 gene (as E1AF) was first described in 1993[5], and various reports have been published on the physiological roles of Pea3 in development, such as in organogenesis of the kidney[6], mammary gland[7], and limb buds[8]. Importantly, an association of Pea3 with oncogenesis has also been described by various groups, in that the expression of Pea3 may correlate with HER2/Neu overexpression, increased tumour grade and adverse prognosis in breast cancer[9-11]. Similarly, Pea3 and the other two subfamily members have been studied in the context of other tumours such as prostate, esophageal, gastric and lung cancer[12-14] (and reviewed in[4]). Perhaps unsurprisingly, the correlation of Pea3 overexpression in CRC prognosis was also published; overall and disease-free survival periods were shorter in patients with Pea3-positive tumours[15]. Various matrix metalloproteases (MMPs) and cyclooxygenase-2 (COX-2) have been suggested as possible transcriptional targets of Pea3[15-19]. Pea3 has been demonstrated to function as a mediator in different malignancies including breast cancer[20,21], non-small cell lung cancer[22], prostate cancer[23], and fibrosarcoma[24].
While research to date establishes Pea3 as a promising prognostic marker in CRC[15,17,25], to our knowledge, there have been no studies directly investigating the biological function and associated mechanisms of Pea3 in CRC tumorigenicity and metastases. Here, we report our findings using shRNA-mediated interference of Pea3 expression in human CRC cell lines. We show that reduced levels of Pea3 decrease the ability of CRC cells to invade through basement membrane matrix, as well as to survive in anchorage-independent conditions. Strikingly, CRC xenografts with reduced Pea3 expression suffer a significant disadvantage in their ability to form liver metastases. To our knowledge, this is the first report to utilize shRNA interference and a metastatic xenograft model to study the role of Pea3 in CRC. We believe that our findings provide a rationale for the adverse prognosis associated with elevated Pea3 expression in human CRC.
MATERIALS AND METHODS
Cell lines and cell culture
Human colorectal adenocarcinoma (LS174T, LS180, HCT116, SW480, SW1222, SW620, COLO 205, 293T) cell lines were purchased from American Type Culture Collection (ATCC; VA, United States). Early passage cell lines were cultured in Dulbecco’s modified Eagle medium (DMEM) containing 4.5 g/L glucose (Invitrogen, Ontario, Canada) supplemented with 10% fetal bovine serum (FBS) (Invitrogen, Ontario, Canada) and penicillin-streptomycin (Invitrogen, Ontario, Canada); the culture medium is hereafter referred to as 10% DMEM. Cells were maintained in a 37 °C incubator with 5% CO2. Cell lines were passaged before they reached 75% confluency and were regularly tested with MycoAlert (Lonza, Ontario, Canada) to ensure the absence of mycoplasma contamination. Cell morphology was regularly checked.
Transcriptome analysis of Pea3 expression in normal-adenocarcinoma sequence
Oncomine™ (Compendia Bioscience, Ann Arbor, MI) was used for analysis and visualization.
Generation of shPea3 cells
Human Pea3-specific and scramble control shRNA cassettes in retroviral vectors were purchased from Origene (Rockville, MD, United States). For the generation of transfectants, 3 × 105 cells were seeded into 6-well plates, then 16 h later, vectors encoding shPea3 or the control shRNA were transiently transfected into SW480 cells using Lipofectamine 2000 (Invitrogen, Ontario, Canada) as per manufacturer’s recommendations, and 24 h later, the transfectants were used for cell lysates, proliferation assays, invasion assays, and/or anoikis assays (described below). Transductants were generated using 293T cell supernatant. Briefly, retroviral transductants were generated via triple-transfection of 293T cells using the Lipofectamine 2000 reagent (Invitrogen, Ontario, Canada) with a retroviral vector encoding the Pea3-shRNA, or control shRNA, as well as the VSV-G gag/pol and env vectors. Supernatants were collected at 48 h. 2 × 105 HCT116 were spin-infected in 6-well plates using the retroviral supernatants, 20% DMEM, and 8 μg/mL of polybrene at 1000 g for 1.5 h. Transductants were selected and maintained using puromycin (50 μg/mL). For the comparison of shPea3 with the variant shRNA, we transiently transfected HCT116 or SW480 cells with the constructs encoding the respective Pea3 shRNA or control shRNA constructs using the protocol described above.
Real-time quantitative PCR
Total RNA was extracted using the RNeasy Mini kit (Qiagen, Ontario, Canada) and cDNA synthesized using Omniscript RT kit (Qiagen, Ontario, Canada) as per manufacturer’s instructions. Expression levels of genes of interest were quantified through quantitative real-time PCR using the SYBR Select (Life Technologies, Ontario, Canada) on the StepOnePlus Real-time PCR system. Expression levels were calculated using the comparative Ct method via StepOne Software (Life Technologies, Ontario, Canada), and relative expression levels normalized to GAPDH. Primer sequences are listed in Table 1.
Table 1.
Primer sequences used in quantitative polymerase chain reaction analysis
| Target gene | Forward | Reverse |
| GAPDH | ACCCACTCCTCCACCTTTGA | CATACCAGGAAATGAGCTTGACAA |
| PEA3 | AGGAGACGTGGCTCGCTGA | AACCTAGCTTTCCACAGCCCC |
| FSP1 | TGATCCTGACTGCTGTCATGG | TACTCTTGGAAGTCCACCTCG |
| TWIST | GACCTCGGGGCCATCCACACC | CGCCGCCGCCGCCACAGC |
| VIMENTIN | GCGTGACGTACGTCAGCAATATGA | GTTCCAGGGACTCATTGGTTCCTT |
| SLUG | ACGCCTCCAAAAAGCCAAACTACA | CTTCAGGGCGCCCAGGCTCACATA |
| SNAIL | AGGCCCTGGCTGCTACAAG | TGCCCTCCCTCCACAGAAAT |
| ZEB-1 | TTGCCGCTGTTGCTGATGTG | GTGCCCTGCCTCTGGTCCTC |
| E-CADHERIN | CTTCACCGACTTACCTACT | GTGCCATACACTTAATTCTC |
| MMP1 | ATTCTACTGATATCGGGGCTTTGA | ATGTCCTTGGGGTATCCGTGTAG |
| MMP2 | TATTTGATGGCATCGCTCAG | GCCTCGTATACCGCATCAAT |
| MMP3 | ATGCCCACTTTGATGATGATGAAC | CCACGCCTGAAGGAAGAGATG |
| MMP7 | TGCTCACTTCGATGAGGATG | TGGGGATCTCCATTTCCATA |
| MMP9 | TTGACAGCGACAAGAAGTGG | CCCTCAGTGAAGCGGTACAT |
| MMP10 | GAGTGGGGCAGCAAAAGAGGAG | TGGGCCATCAAAAGAGTAAAAGTC |
| MMP13 | CTTCGGCTTAGAGGTGACTGG | TTTTGCCGGTGTAGGTGTAGATAG |
| MMP14 | AGGCCGACATCATGATCTTCTTTG | GGTGGGGTTTTTGGGTTTATCAGG |
| TIMP1 | ACCCCCGCCATGGAGAGTGTC | CCGGAAGAAAGATGGGAGTG |
| TIMP2 | CGCAGCAAACACATCCGTAGAAGG | CCGAGGAGGGGGCCGTGTAGATAA |
| TIMP3 | CCCTTCGGCACGCTGGTCTACA | TCAGCGGGAAGGGAGGGAAGTG |
| TIMP4 | CGTCGGGGCGGGATTGG | ACTGCTTCTGGCTGTTGGCTTCTA |
Sequences were designed with Primer 3 software (http://primer3.sourceforge.net/) or with the DNASTAR Lasergene Suite (Madison, WI, United States). Sequences were checked against the human transcript database via PrimerBlast (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) for cross-amplification of unintended transcripts. MMP: Matrix metalloproteinase; TIMP: Tissue inhibitor of metalloproteinase.
Western blotting
Cells were lysed in ice-cold radioimmunoassay precipitation assay (RIPA) lysis buffer [50 mmol/L Tris pH7.5, 150 mmol/L NaCl, 2 mmol/L EDTA Ph 8.0, 0.5% (v/v) Triton X-100, and Complete protease inhibitor cocktail (Roche, Quebec, Canada)]. Cell debris and insoluble material were removed by centrifugation at 12000 g at 4 °C for 20 min. Following protein quantitation using the Bradford protein assay (Bio-Rad, Ontario, Canada), 25 g of lysate was loaded per lane and proteins resolved by 4%-20% gradient SDS-PAGE gel, wet-transferred to polyvinylidene fluoride membranes (EMD Millipore, MA, United States), and the membranes were incubated in 5% nonfat dry milk in Tris-buffered saline Tween-20 (TBST) (10 mmol/L Tris-Base, 150 mmol/L NaCl, 0.05% Tween-20; pH 7.4) for 1 h at room temperature to block nonspecific antibody binding, followed by incubation with primary antibody in 5% milk in TBST overnight at 4 °C with gentle agitation. The membranes were washed three times for 10 min each in TBST, then incubated in TBST at room temperature for 1 h, followed by three 10-min washes with TBST. Protein-antibody binding on the membranes was detected with the use of enhanced chemiluminescence (ECL) Plus solution (GE Healthcare Life Sciences, Quebec, Canada) followed by exposure of the membranes to X-ray film (FujiFilm, Ontario, Canada). Anti-human-β-actin antibodies were purchased from Cell Signalling Technology (Danvers, MA, United States) and anti-human-Pea3 antibodies were purchased from Santa Cruz Biotechnology (Paso Robles, CA, United States); secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA, United States).
Matrigel transwell invasion assay
Cells were serum starved cells overnight (0.1% DMEM), then 2 × 105 cells were seeded on top of 8 μm transwell inserts (BD Biosciences, Ontario, Canada) with 0.1% DMEM and pre-coated with Matrigel basement membrane matrix (Becton, Dickinson and Company, Ontario, Canada) under 1% O2; 10% DMEM was used as a chemoattractant. After 24 h cells that had invaded through the Matrigel coated transwell inserts were fixed, stained by Kwik-Diff Stain (Thermo Fisher Scientific, Ontario, Canada) and number of invading cells counted under 10x objective power of a Leica DM LB2 microscope (Leica Microsystems, Ontario, Canada).
In vitro cellular proliferation assay
Cells were plated in increasing numbers in 96-well plates in 10% DMEM. After 72 h of incubation, the number of viable cells was quantified by the MTS assay (CellTitre 96® AQueous Cell Proliferation Assay, Promega Corporation, WI, United States) according to the manufacturer’s instructions, and absorbance at 495 nm was measured using the Benchmark Plus multi-well plate reader (Bio Rad Laboratories Inc., Toronto, Canada).
Anoikis assay
Cells were plated on 6-well ultra-low attachment plates (Corning Incorporated, Tewksbury, MA, United States) for 2-5 d under usual tissue culture conditions. Cell death was analyzed by dye exclusion using a cell counter or FACS analysis.
MMP activity assay
The fluorometric generic MMP kit was purchased from AnaSpec (Fremont, CA, United States). For cell lysates, 106 cells were plated in 100-mm petri dishes for 24 h, and cell lysis procedure was carried out in a 0.1% Triton (v/v) solution according to manufacturer’s recommendations. Lysates were incubated for 3 h with 1 mmol/L APMA, and cleavage of the fluorometric MMP reagent was assessed using a standard plate reader.
Mice
All experiments involving mice were performed according to University of Toronto and Sunnybrook Research Institute guidelines, using a peer-reviewed animal protocol. Balb/c athymic nude mice were purchased from Harlan (Ontario, Canada).
In vivo tumour growth assay
Five million (HCT-scr, HCT-shPea3, LS-scr, LS-shPea3) cells were resuspended in 50 μL of FBS-free culture medium and mixed with 50 μL Matrigel (Becton, Dickinson and Company, Ontario, Canada) and injected subcutaneously into the right flanks of 6- to 7-wk-old female athymic nude mice (Harlan, Ontario, Canada). Caliper measurements were made and tumour growth was plotted as fold increase in size compared to starting.
In vivo liver metastasis assay
Briefly, 1 million cells (HCT116-scr or HCT116-shPea3) were resuspended in 20 μL of FBS-free culture medium and injected into the spleens of 6- to 7-wk-old female athymic nude mice (Harlan, Ontario, Canada). Three weeks following injection, mice were euthanized by cervical dislocation, and their livers were collected for H&E staining (see below).
Immunohistochemistry
Five micron tumour sections were stained with haematoxylin and eosin, as previously described[26].
Statistical analysis
All statistical tests were two-sided, and the statistical analysis was performed using the GraphPad Prism version 5.0 program (GraphPad Software, CA, United States). Statistical significance was defined as P ≤ 0.05. The Student t-test was used to compare the mean values between two groups. Data are presented as mean values with standard deviations.
RESULTS
Pea3 transcript expression is increased through the adenoma-carcinoma sequence
In order to investigate the expression of Pea3 during the clinical progression of benign colon to malignant CRC in humans, we performed data mining using Oncomine (Compendia Bioscience, Ann Arbor, MI), a repository of gene expression profiles of normal and cancer tissues. Interestingly, Pea3 expression increased through the normal-adenoma-carcinoma sequence (Figure 1A), suggesting a role for Pea3 in the carcinogenesis of CRC.
Figure 1.
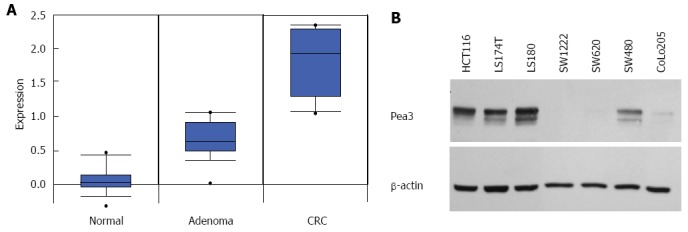
Pea3 expression in normal-adenoma-carcinoma (colon) and human colorectal carcinoma cell lines. A: Box plot of Pea3 transcript expression in normal colon tissue, adenoma, and adenocarcinoma samples. Oncomine™ compendium was utilized for analysis (Skrzypczak 2 colon sample set). Expression is shown as log2 transformation of median-centered intensity. Black line represents the median; blue box, 25/75 percentile; bars, 10/90 percentile; dots, minimum and maximum values; B: Western blot analysis of Pea3 expression in human colorectal cancer cell lines. Shown are blots for Pea3 and β-actin loading control. Representative of three independent experiments.
Pea3 expression in CRC cell lines and Pea3 shRNA
We then assayed a panel of human CRC cell lines for Pea3 protein expression by western blotting. Our results showed detectable Pea3 expression in a number of established CRC cell lines (Figure 1B). From this panel, we chose the HCT116, LS174T, and SW480 cell lines, all of which express Pea3, to study Pea3 function in CRC. We utilised Pea3-specific shRNA to interfere with Pea3 expression in the HCT116 and SW480 cell lines (Figure 2A). Using quantitative PCR (qPCR), we observed a significant reduction in Pea3 expression in the Pea3-shRNA cell lines (Figure 2B; HCT.shPea3, 0.15 ± 0.01 fold; LS.shPea3, 0.51 ± 0.02 fold; SW.shPea3 0.22 ± 0.02 fold, P < 0.005) relative to their control-shRNA counterparts (HCT.shCtrl, LS.shCtrl, SW.shCtrl), thereby validating our RNA interference strategy. Similarly, western blot analysis showed reduced Pea3 protein levels in the Pea3-shRNA cells relative to control-shRNA cells (Figure 2C). We also tested a variant Pea3-shRNA with a different nucleotide sequence [shPea3(v2)] in its ability to result in a reduction of Pea3 expression. In comparison to the original Pea3-shRNA construct we used, transfection of shPea3(v2) construct into the HCT116 cells resulted in a modest but reproducible reduction of Pea3 protein (Figure 2D). Thus, the Pea3-shRNA was successful in reducing Pea3 transcript and protein levels in these cell lines.
Figure 2.
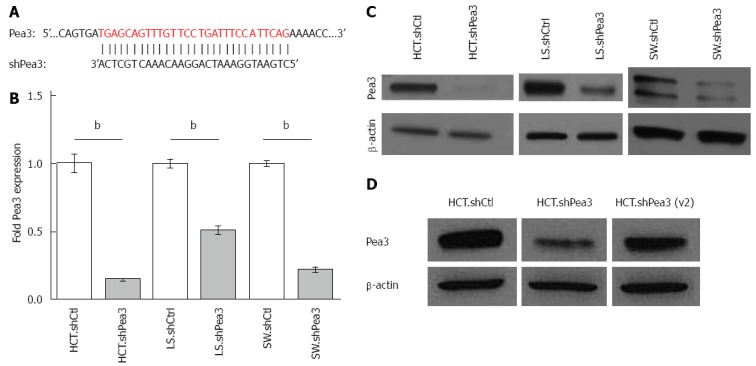
Generation and characterization of short hairpin RNA (shRNA)-mediated interference with Pea3 expression. A: Nucleotide alignment of Pea3 sequence with shRNA. The shown Pea3 sequence is identical in four published Pea3 splice variants (accession numbers NM_001986.2, NM_001079675.1, NM_001261437.1 and NM_001261438.1). Vertical lines indicate complementary bases; B: Quantitative polymerase chain reaction (qPCR) analysis of Pea3 expression in HCT116, LS174T, and SW480 cells expressing Pea3-specific shRNA (HCT.shPea3, LS.shPea3, and SW.shPea3) or control shRNA (HCT.shCtrl, LS.shCtrl, and SW.shCtrl). Obtained ∆Ct values are normalized to Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) values, and expression is shown as fold change in Pea3 expression relative to cells expressing control shCtrl, bP < 0.01 vs control shRNA; C: Western blot analysis of Pea3 expression in HCT116, LS174T and SW480 cells expressing Pea3-specific shRNA (HCT.shPea3, LS.shPea3, and SW.shPea3) or control shRNA (HCT.shCtrl, LS.shCtrl, and SW.shCtrl). Shown are blots for Pea3 and β-actin loading control. Representative of three independent experiments; D: Western blot analysis of Pea3 expression in HCT116 cells expressing two different Pea3-specific shRNA [HCT.shPea3 and HCT.shPea3(v2)] or control shRNA (HCT.shCtrl). Shown are blots for Pea3 and β-actin loading control. Representative of three independent experiments.
Functional consequences of reduction of Pea3 expression in vitro
To assess the functional consequences of shRNA-mediated silencing of Pea3, we carried out a number of in vitro functional assays. As the ability of cancer cells to invade from their primary site and then into tissues at the site of metastasis is a crucial step in the formation of metastases[27,28], we tested the invasive capacity of Pea3-shRNA cell lines using a transwell invasion assay. Briefly, serum-starved cells were seeded on transwell inserts coated with basement membrane matrix, serum-supplemented culture medium was used as a chemoattractant, and 24 h later, cells that had invaded through the inserts were counted[29]. In keeping with a pro-metastatic role for Pea3, the number of cells invading through the inserts was significantly reduced in shPea3 cells in comparison to the control counterparts (Figure 3A; HCT.shPea3, 0.28 ± 0.04 fold; LS.shPea3, 0.15 ± 0.04 fold; SW.shPea3, 0.23 ± 0.03, P < 0.01). To exclude a phenomenological effect of the particular shRNA construct we used in these assays, we also tested ability of the shPea3(v2) construct to affect in vitro invasion. In keeping with our observations with the original shPea3 construct, HCT116 and SW480 cells transfected with the shPea3(v2) construct also displayed a significant reduction in their ability to invade (Figure 3A; HCT.shPea3(v2), 0.71 ± 0.01 fold; SW.shPea3(v2), 0.46 ± 0.02 fold P < 0.005). Thus, Pea3 expression confers a distinct advantage in the ability of CRC cells to invade in vitro. Another phenotype necessary in the generation of metastases from the primary tumour is the ability to overcome anchorage-dependent cell death, or anoikis[30]. We thus carried out anoikis studies, where we assessed the ability of these CRC cells to survive in non-adherent culture conditions. Similar to the invasion assay, fewer viable cells were observed in HCT.shPea3 and LS.shPea3 cells compared to control cells (Figure 3B; HCT.shPea3 75.4% ± 1.9% vs HCT.shCtrl 88.6% ± 0.6%, P < 0.01; LS.shPea3 71.7% ± 0.5% vs LS.Ctrl 89.6% ± 0.3%, P < 0.005). Of note, we were unable to induce anoikis in SW cells in our culture conditions (greater than 95% cell viability), and thus, we were unable to conclude whether Pea3 influences anoikis resistance in these cells. Therefore, Pea3 is also involved in providing CRC cells with the ability to survive in an anchorage-independent manner. Lastly, we tested the impact of Pea3 expression on cell proliferation using a MTS assay. In this context, however, HCT.shCtrl cells did not possess a proliferative advantage over the shPea3 cells in vitro (Figure 3C left panel, HCT.shCtrl AUC 5098 ± 123 vs HCT.shPEA3 5689 ± 151, P < 0.05). Similarly, the proliferative capacities of LS.shCtrl (Figure 3C right panel, LS.shCtrl AUC 5600 ± 324.1 vs LS.shPEA3 6423 ±400, P < 0.05) and SW.shCtrl cells (data not shown) did not statistically differ from their shPea3 counterparts. To address the influence of Pea3 expression on CRC proliferation in vivo, we generated subcutaneous xenografts in the flanks of Balb/c athymic nude mice, and monitored tumour growth. The HCT.shPea3 and HCT.shCtrl tumours grew at a similar rate (Figure 3D, HCT.shPea3 2.64 ± 0.82 fold vs HCT.shCtrl 2.88 ± 0.80 fold, P > 0.05). Interestingly, LS.shPea3 tumours grew faster than their shCtrl counterparts (Figure 3D, LS.shPea3 4.62 ± 1.37 fold vs LS.shCtrl 2.80 ± 0.69 fold, P < 0.05). Collectively, our in vitro and in vivo results indicate that the loss of Pea3 expression is not associated with a proliferative disadvantage. However, Pea3 expression contributes to CRC cell invasiveness and increased anoikis resistance, which support a pro-metastatic role for Pea3 in CRC.
Figure 3.
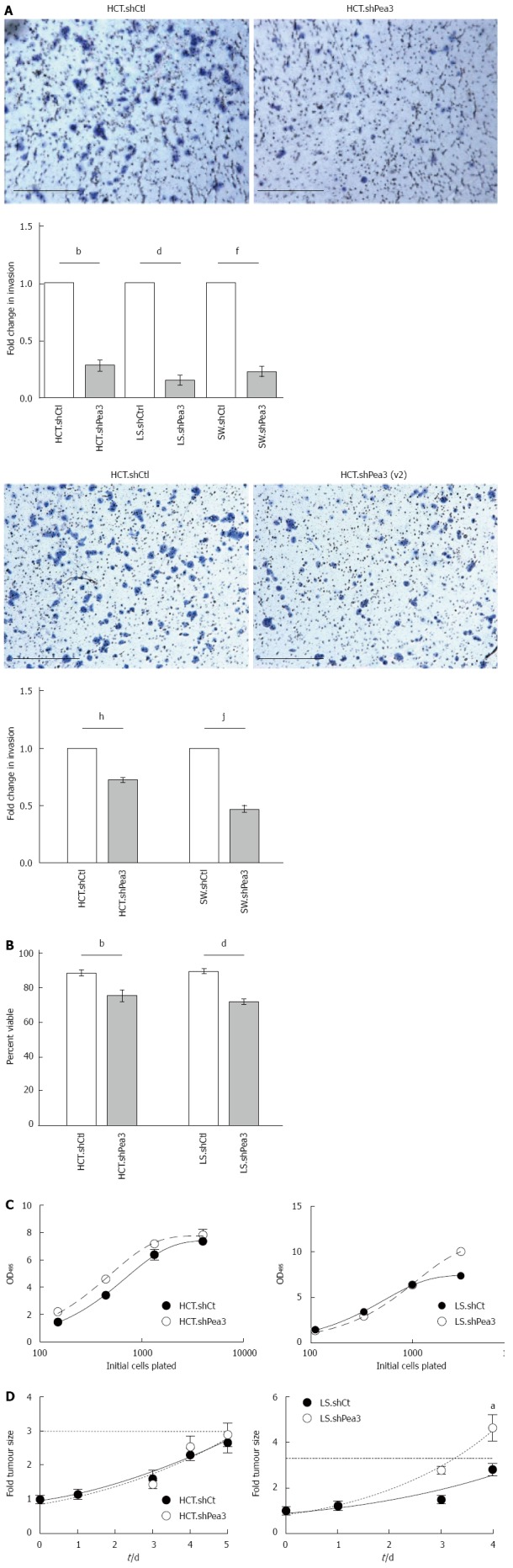
Characterization of invasiveness, anoikis resistance, and proliferative capacity of colorectal carcinoma cells expressing the short hairpin RNA Pea3 (shPea3) construct. A: Representative images of the 24-h transwell invasion assay using HCT.shPea3, HCT.shPea3(v2) and HCT.shCtrl cells with quantification of the transwell invasion assays. Data are shown as fold change in invasion of shPea3 or shPea3(v2) expressing cells relative to their shCtrl counterparts. bP < 0.01, HCT.shCtrl vs HCT.shPea3; dP < 0.01, LS.shCtrl vs LS.shPea3; fP < 0.01, SW.shCtrl vs SW.shPea3; hP < 0.01, LS.shCtrl vs LS.shPea3; B: Anoikis assay using the stable HCT116 and LS174T transductants. Cell death was assessed using dye exclusion, and the fraction of surviving cells after 2 d (HCT116) or 5 d (LS174T) was plotted. bP < 0.01, HCT.shCtrl vs HCT.shPea3; dP < 0.01, LS.shCtrl vs LS.shPea3; C: In vitro proliferation assay of HCT.shPea3 and HCT.shCtrl cells (left panel), as well as LS.shPea3 and LS.shCtrl cells (right panel). Cells were plated in 96-wells in 4-fold dilution series, followed by OD reading at 495 nm for quantification (shown). Area under curve analysis was performed for statistical significance; D: In vivo tumour xenograft growth assay of HCT.shPea3 and LS.shPea3 cells. 5 × 106 cells were injected s.c. into the flanks of mice (8 per group) and calliper measurements of the tumours were taken. Shown are fold change in tumour sizes compared to day zero. Dotted horizontal line indicates 3-fold starting tumour size. aP < 0.05 vs final tumour measurements. Statistical analysis was performed using Student’s t-test for the final tumour measurements.
In vivo metastatic potential of Pea3-reduced CRC cells
Given our in vitro findings, we hypothesized that Pea3-expressing CRC tumours may possess a metastatic advantage. Since liver is the primary site of CRC metastasis in patients, we utilized an in vivo model of liver metastasis. We injected HCT cells into the spleens of Balb/c athymic nude mice and enumerated liver metastases[31,32]. Histological analysis of the livers at 21 d following splenic injection revealed that mice injected with HCT.shPea3 cells formed no metastases in the livers, while all mice injected with HCT.shCtrl cells showed metastases (Figure 4A, P < 0.05). Our observations indicated that reduction of Pea3 expression greatly impairs metastatic potential in the HCT cell line, confirming a prominent role for Pea3 in promoting liver metastases in CRC.
Figure 4.
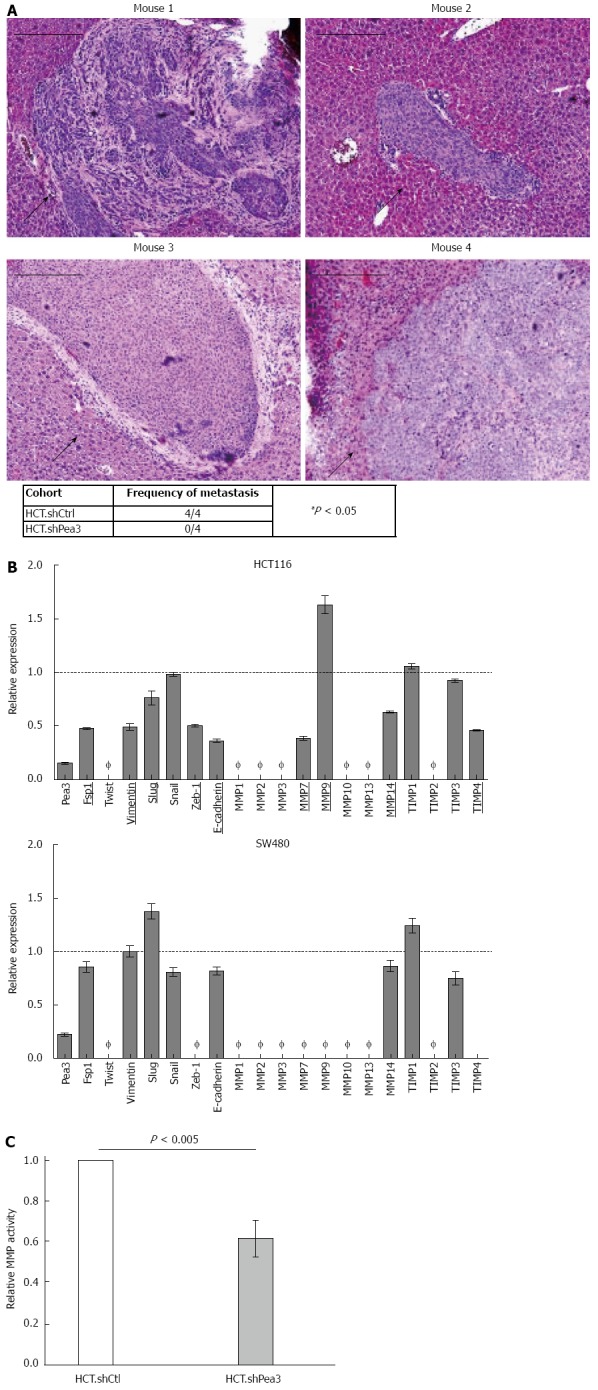
In vivo xenograft assay for the formation of liver metastases by HCT.shPea3 cells, and differential expression of epithelial-mesenchymal transition markers and matrix metalloproteases. A: Ten million HCT cells were injected into spleens of athymic nude mice (4 per group), and livers were analyzed by immunohistochemistry (haematoxylin and eosin) at 21-d post injection. Representative images of metastases from the HCT.shCtrl cohort. Black arrows indicate the metastatic mass. Scale bar indicates 200 μm. Summary of the metastases observed in the shPea3 vs shCtrl cohort. Result of the χ2 test is shown; B: qPCR analysis of select epithelial-mesenchymal transition markers and a panel of MMP transcripts in HCT116 (top panel) or SW480 cells (bottom panel). Obtained ∆Ct values are normalized to GAPDH values, and expression is shown as fold change in Pea3 expression relative to cells expressing control shCtrl. Ø indicates expression is below detection threshold (Ct > 32 cycles). Targets that display differential expression between shPea3 and shCtrl consistently of at least 20% are underlined and bolded. Dashed horizontal line indicates the expression of the shCtrl cohort. Representative of three independent experiments; C: Results of the generic MMP activity assay, given as relative light units normalized to the amount of total protein in the sample. Data shown as fold change in MMP activity of HCT.shPea3 expressing cells relative to HCT.shCtrl. Representative of four independent experiments. MMP: Matrix metalloproteases.
Potential downstream targets of Pea3
Since our data indicated that Pea3 expression increased the ability of CRC cells to invade (in vitro) and to establish liver metastases (in vivo), we explored the potential mechanisms of Pea3 action. The two pathways we investigated were epithelial to mesenchymal transition (EMT), and MMP expression, which are two important processes implicated in CRC metastasis[33,34]. We performed qPCR analysis of mRNA expression of common EMT markers and MMPs in HCT.shPea3 cells relative to HCT.shCtrl cells (Figure 4B). In keeping with a pro-metastatic role for Pea3, several EMT markers and MMPs were downregulated in shPea3-expressing cells, suggesting that Pea3 may exert its effects through these processes (Figure 4B). In contrast to HCT116, the SW480 cell line expressed fewer of these markers, and the levels of reduction were much more modest, possibly as a result of transient expression of the shPea3 construct (Figure 4B). We carried out fluorometric MMP activity assays and identified a reduction in overall MMP activity in HCT.shPea3 cells compared to their control counterparts (Figure 4C; HCT.shPea3 0.61 ± 0.04 fold, P < 0.005). Thus, Pea3 may potentially modulate EMT and MMPs to confer a pro-metastatic phenotype in CRC.
DISCUSSION
Colorectal cancer is one of the most prevalent cancers in the world, and a leading cause of cancer death, largely due to its propensity for early metastatic spread. In line with its significance, a large body of research has been devoted to studying various aspects of CRC biology, including identification of novel prognostic markers and drug targets, as well as their respective mechanisms of action. Pea3 is one such marker, which is being explored in the context of human CRC as well as in other cancers (reviewed in[4]). Previous reports on the association of increased Pea3 expression in tumours with reduced patient survival constitutes an important clinical observation and establishes Pea3 as a compelling molecular target to study in cancer[15,35]. Previous research directed at elucidating the mechanism(s) of action for Pea3 in CRC implicated MMP-1 and MMP-7 as downstream effectors[15,17,19], while others suggest that signalling pathways such as osteopontin are involved[35,36].
To date, no study has yet directly investigated the in vivo function of Pea3 in CRC using a metastatic model. As far as we are aware this is the first report to directly study the effects of Pea3 expression by using shRNA-mediated knockdown technology in CRC both in vitro and in vivo. Our results demonstrate that a reduction in Pea3 levels results in a significant decline in the cells’ ability to invade in vitro. Interestingly, this deficiency is also paralleled by a reduction in the transcript levels of two MMPs (MMP-7 and MMP-14) and a decline in total MMP activity. Thus, our results corroborate previous studies that reported a positive regulatory role for Pea3 in MMP expression[15,17,37,38]. Nonetheless, we cannot formally rule out the possibility that Pea3 may promote MMP action via additional mechanisms such as transcriptional repression of MMP inhibitors or stimulation of MMP activators that have yet to be identified.
We demonstrate for the first time that interference with Pea3 expression significantly impairs the ability of CRC cells to form liver metastases. This provides important experimental evidence to explain the previously reported clinical findings regarding the association of Pea3 with overall survival and distant metastases in CRC[15]. Thus, this study provides a meaningful contribution to our understanding of the role of Pea3 in CRC. We employed female mice for our studies, and although to our knowledge, CRC cells are not estrogen or androgen-dependent for their growth, future studies could utilize both male and female mice to rule out any gender-specific differences for Pea3 in CRC tumorigenesis and metastasis.
A potential involvement of Fsp1 (S100A4) constitutes yet another interesting observation in our study. Previously, Fsp1 has been suggested to positively regulate the expression of MMPs in certain cancers, such as MMP-9 in thyroid cancer and osteocarcinoma[39,40], and MMP-13 in breast cancer[41]. Therefore, it is possible that PEA3 may regulate MMP expression through the regulation of Fsp1. Moreover, Fsp1 has been suggested to promote metastasis (reviewed in[42]) by increasing angiogenesis, tissue invasion, and cell motility. Thus, if Fsp1 is indeed regulated by Pea3, this may provide the cancer cells with multiple advantages in forming metastases. While not formally tested, it is interesting to speculate that the decrease in anoikis resistance observed in Pea3-shRNA cells may also stem from a reduction in Fsp1 expression.
Currently, we perceive a number of avenues regarding future research on Pea3 in cancer. The potential utility of Pea3 as a prognostic marker has already been considered in CRC[15,36], as well as in additional cancers[4]. Future research will be needed to identify other cancers in which Pea3 may play a pro-metastatic role in attempt to establish a more complete framework of the importance of Pea3 in cancer. From a clinical standpoint, the frequency of Pea3 overexpression in these cancers and its impact on patient outcomes will have to be established. However, research on Pea3 in cancer does not necessarily have to be limited to prognostics. While targeting of intracellular molecules in cancer therapeutics is challenging, the potential utility of RNA interference is already being evaluated in solid tumours[43]. Furthermore, identification of downstream targets may also yield potential drug targets. For this purpose, future research should aim to identify transcriptional targets of Pea3, and given our findings on MMP activity and anoikis resistance, molecules known to be involved in these pathways,such as Fsp1 and MMP-14, would be promising targets.
COMMENTS
Background
Colorectal carcinoma (CRC) is a leading cause of cancer mortality largely due to its propensity for early metastatic spread. There is a need to understand the biological mechanisms underlying the process of CRC metastasis to assist in the future development of targeted therapies.
Research frontiers
Overexpression of the Pea3 transcription factor in CRC patients has been correlated with distant metastasis, and reduced overall survival, making Pea3 a compelling target to study in CRC. Engagement of epithelial-mesenchymal transition (EMT) and matrix-metalloproteases (MMPs) downstream of Pea3 have been previously implicated as processes involved in metastatic spread.
Innovations and breakthroughs
The authors have demonstrated that interference with Pea3 expression significantly impairs the ability of CRC cells to form liver metastases. Additionally, Pea3 is required for efficient invasiveness, induction of MMP activity, and expression of certain EMT markers. Together, this provides important experimental evidence to explain the previously reported clinical findings regarding the correlation of Pea3 with overall survival and distant metastases in CRC.
Applications
Pea3 is required for efficient metastatic spread in CRC and thus implicates it as a promising therapeutic target.
Terminology
Epithelial-mesenchymal transition is the process through which epithelial cells lose cell polarity and adhesion, and gain migratory and invasive properties which can promote metastasis. Matrix metalloproteases are involved in degrading the components of the extracellular matrix which may promote cancer cell migration, invasion and survival.
Peer review
In this manuscript, the author investigated the biological function of Pea3 in CRC and explored some possible mechanisms of Pea3 in CRC. This has not been reported in the literature previously and warrants publication, and this study is original and has some clinical significance.
Footnotes
Supported by Early investigator award to Liu SK from the Ontario Institute for Cancer Research; and the Canada Foundation for Innovation - MEDI ORF
P- Reviewer: El-Tawil AM, Wang G S- Editor: Ma YJ L- Editor: A E- Editor: Ma S
References
- 1.Arends MJ. Pathways of colorectal carcinogenesis. Appl Immunohistochem Mol Morphol. 2013;21:97–102. doi: 10.1097/PAI.0b013e31827ea79e. [DOI] [PubMed] [Google Scholar]
- 2.Heinemann V, Douillard JY, Ducreux M, Peeters M. Targeted therapy in metastatic colorectal cancer -- an example of personalised medicine in action. Cancer Treat Rev. 2013;39:592–601. doi: 10.1016/j.ctrv.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 3.Shindoh M, Higashino F, Kohgo T. E1AF, an ets-oncogene family transcription factor. Cancer Lett. 2004;216:1–8. doi: 10.1016/j.canlet.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 4.Oh S, Shin S, Janknecht R. ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochim Biophys Acta. 2012;1826:1–12. doi: 10.1016/j.bbcan.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Higashino F, Yoshida K, Fujinaga Y, Kamio K, Fujinaga K. Isolation of a cDNA encoding the adenovirus E1A enhancer binding protein: a new human member of the ets oncogene family. Nucleic Acids Res. 1993;21:547–553. doi: 10.1093/nar/21.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chotteau-Lelièvre A, Desbiens X, Pelczar H, Defossez PA, de Launoit Y. Differential expression patterns of the PEA3 group transcription factors through murine embryonic development. Oncogene. 1997;15:937–952. doi: 10.1038/sj.onc.1201261. [DOI] [PubMed] [Google Scholar]
- 7.Chotteau-Lelievre A, Montesano R, Soriano J, Soulie P, Desbiens X, de Launoit Y. PEA3 transcription factors are expressed in tissues undergoing branching morphogenesis and promote formation of duct-like structures by mammary epithelial cells in vitro. Dev Biol. 2003;259:241–257. doi: 10.1016/s0012-1606(03)00182-9. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Z, Verheyden JM, Hassell JA, Sun X. FGF-regulated Etv genes are essential for repressing Shh expression in mouse limb buds. Dev Cell. 2009;16:607–613. doi: 10.1016/j.devcel.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benz CC, O’Hagan RC, Richter B, Scott GK, Chang CH, Xiong X, Chew K, Ljung BM, Edgerton S, Thor A, et al. HER2/Neu and the Ets transcription activator PEA3 are coordinately upregulated in human breast cancer. Oncogene. 1997;15:1513–1525. doi: 10.1038/sj.onc.1201331. [DOI] [PubMed] [Google Scholar]
- 10.Fleming FJ, Myers E, Kelly G, Crotty TB, McDermott EW, O’Higgins NJ, Hill AD, Young LS. Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer; a predictive role for SRC-1. J Clin Pathol. 2004;57:1069–1074. doi: 10.1136/jcp.2004.016733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Myers E, Hill AD, Kelly G, McDermott EW, O’Higgins NJ, Young LS. A positive role for PEA3 in HER2-mediated breast tumour progression. Br J Cancer. 2006;95:1404–1409. doi: 10.1038/sj.bjc.6603427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keld R, Guo B, Downey P, Cummins R, Gulmann C, Ang YS, Sharrocks AD. PEA3/ETV4-related transcription factors coupled with active ERK signalling are associated with poor prognosis in gastric adenocarcinoma. Br J Cancer. 2011;105:124–130. doi: 10.1038/bjc.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keld R, Guo B, Downey P, Gulmann C, Ang YS, Sharrocks AD. The ERK MAP kinase-PEA3/ETV4-MMP-1 axis is operative in oesophageal adenocarcinoma. Mol Cancer. 2010;9:313. doi: 10.1186/1476-4598-9-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sloan KA, Marquez HA, Li J, Cao Y, Hinds A, O’Hara CJ, Kathuria S, Ramirez MI, Williams MC, Kathuria H. Increased PEA3/E1AF and decreased Net/Elk-3, both ETS proteins, characterize human NSCLC progression and regulate caveolin-1 transcription in Calu-1 and NCI-H23 NSCLC cell lines. Carcinogenesis. 2009;30:1433–1442. doi: 10.1093/carcin/bgp129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horiuchi S, Yamamoto H, Min Y, Adachi Y, Itoh F, Imai K. Association of ets-related transcriptional factor E1AF expression with tumour progression and overexpression of MMP-1 and matrilysin in human colorectal cancer. J Pathol. 2003;200:568–576. doi: 10.1002/path.1387. [DOI] [PubMed] [Google Scholar]
- 16.Asting AG, Carén H, Andersson M, Lönnroth C, Lagerstedt K, Lundholm K. COX-2 gene expression in colon cancer tissue related to regulating factors and promoter methylation status. BMC Cancer. 2011;11:238. doi: 10.1186/1471-2407-11-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boedefeld WM, Soong R, Weiss H, Diasio RB, Urist MM, Bland KI, Heslin MJ. E1A-F is overexpressed early in human colorectal neoplasia and associated with cyclooxygenase-2 and matrix metalloproteinase-7. Mol Carcinog. 2005;43:13–17. doi: 10.1002/mc.20093. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Borchert GL, Phang JM. Polyoma enhancer activator 3, an ets transcription factor, mediates the induction of cyclooxygenase-2 by nitric oxide in colorectal cancer cells. J Biol Chem. 2004;279:18694–18700. doi: 10.1074/jbc.M308136200. [DOI] [PubMed] [Google Scholar]
- 19.Lynch CC, Crawford HC, Matrisian LM, McDonnell S. Epidermal growth factor upregulates matrix metalloproteinase-7 expression through activation of PEA3 transcription factors. Int J Oncol. 2004;24:1565–1572. [PubMed] [Google Scholar]
- 20.Yuen HF, Chan YK, Grills C, McCrudden CM, Gunasekharan V, Shi Z, Wong AS, Lappin TR, Chan KW, Fennell DA, Khoo US, Johnston PG, El-Tanani M. Polyomavirus enhancer activator 3 protein promotes breast cancer metastatic progression through Snail-induced epithelial-mesenchymal transition. J Pathol. 2011;224:78–89. doi: 10.1002/path.2859. [DOI] [PubMed] [Google Scholar]
- 21.de Launoit Y, Chotteau-Lelievre A, Beaudoin C, Coutte L, Netzer S, Brenner C, Huvent I, Baert JL. The PEA3 group of ETS-related transcription factors. Role in breast cancer metastasis. Adv Exp Med Biol. 2000;480:107–116. doi: 10.1007/0-306-46832-8_13. [DOI] [PubMed] [Google Scholar]
- 22.Hakuma N, Kinoshita I, Shimizu Y, Yamazaki K, Yoshida K, Nishimura M, Dosaka-Akita H. E1AF/PEA3 activates the Rho/Rho-associated kinase pathway to increase the malignancy potential of non-small-cell lung cancer cells. Cancer Res. 2005;65:10776–10782. doi: 10.1158/0008-5472.CAN-05-0060. [DOI] [PubMed] [Google Scholar]
- 23.Aytes A, Mitrofanova A, Kinkade CW, Lefebvre C, Lei M, Phelan V, LeKaye HC, Koutcher JA, Cardiff RD, Califano A, et al. ETV4 promotes metastasis in response to activation of PI3-kinase and Ras signaling in a mouse model of advanced prostate cancer. Proc Natl Acad Sci USA. 2013;110:E3506–E3515. doi: 10.1073/pnas.1303558110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Habelhah H, Okada F, Kobayashi M, Nakai K, Choi S, Hamada J, Moriuchi T, Kaya M, Yoshida K, Fujinaga K, et al. Increased E1AF expression in mouse fibrosarcoma promotes metastasis through induction of MT1-MMP expression. Oncogene. 1999;18:1771–1776. doi: 10.1038/sj.onc.1202465. [DOI] [PubMed] [Google Scholar]
- 25.Nosho K, Yoshida M, Yamamoto H, Taniguchi H, Adachi Y, Mikami M, Hinoda Y, Imai K. Association of Ets-related transcriptional factor E1AF expression with overexpression of matrix metalloproteinases, COX-2 and iNOS in the early stage of colorectal carcinogenesis. Carcinogenesis. 2005;26:892–899. doi: 10.1093/carcin/bgi029. [DOI] [PubMed] [Google Scholar]
- 26.Liu SK, Bham SA, Fokas E, Beech J, Im J, Cho S, Harris AL, Muschel RJ. Delta-like ligand 4-notch blockade and tumor radiation response. J Natl Cancer Inst. 2011;103:1778–1798. doi: 10.1093/jnci/djr419. [DOI] [PubMed] [Google Scholar]
- 27.Kassis J, Lauffenburger DA, Turner T, Wells A. Tumor invasion as dysregulated cell motility. Semin Cancer Biol. 2001;11:105–117. doi: 10.1006/scbi.2000.0362. [DOI] [PubMed] [Google Scholar]
- 28.Wells A. Tumor invasion: role of growth factor-induced cell motility. Adv Cancer Res. 2000;78:31–101. doi: 10.1016/s0065-230x(08)61023-4. [DOI] [PubMed] [Google Scholar]
- 29.Kleinman HK, Jacob K. Invasion assays. Curr Protoc Cell Biol. 2001;Chapter 12:Unit 12.2. doi: 10.1002/0471143030.cb1202s00. [DOI] [PubMed] [Google Scholar]
- 30.Taddei ML, Giannoni E, Fiaschi T, Chiarugi P. Anoikis: an emerging hallmark in health and diseases. J Pathol. 2012;226:380–393. doi: 10.1002/path.3000. [DOI] [PubMed] [Google Scholar]
- 31.Bresalier RS, Raper SE, Hujanen ES, Kim YS. A new animal model for human colon cancer metastasis. Int J Cancer. 1987;39:625–630. doi: 10.1002/ijc.2910390514. [DOI] [PubMed] [Google Scholar]
- 32.Ishizu K, Sunose N, Yamazaki K, Tsuruo T, Sadahiro S, Makuuchi H, Yamori T. Development and characterization of a model of liver metastasis using human colon cancer HCT-116 cells. Biol Pharm Bull. 2007;30:1779–1783. doi: 10.1248/bpb.30.1779. [DOI] [PubMed] [Google Scholar]
- 33.Zucker S, Vacirca J. Role of matrix metalloproteinases (MMPs) in colorectal cancer. Cancer Metastasis Rev. 2004;23:101–117. doi: 10.1023/a:1025867130437. [DOI] [PubMed] [Google Scholar]
- 34.Bhangu A, Wood G, Mirnezami A, Darzi A, Tekkis P, Goldin R. Epithelial mesenchymal transition in colorectal cancer: Seminal role in promoting disease progression and resistance to neoadjuvant therapy. Surg Oncol. 2012;21:316–323. doi: 10.1016/j.suronc.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 35.Martinez C, Churchman M, Freeman T, Ilyas M. Osteopontin provides early proliferative drive and may be dependent upon aberrant c-myc signalling in murine intestinal tumours. Exp Mol Pathol. 2010;88:272–277. doi: 10.1016/j.yexmp.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 36.Mole DJ, O’Neill C, Hamilton P, Olabi B, Robinson V, Williams L, Diamond T, El-Tanani M, Campbell FC. Expression of osteopontin coregulators in primary colorectal cancer and associated liver metastases. Br J Cancer. 2011;104:1007–1012. doi: 10.1038/bjc.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bièche I, Tozlu S, Girault I, Onody P, Driouch K, Vidaud M, Lidereau R. Expression of PEA3/E1AF/ETV4, an Ets-related transcription factor, in breast tumors: positive links to MMP2, NRG1 and CGB expression. Carcinogenesis. 2004;25:405–411. doi: 10.1093/carcin/bgh024. [DOI] [PubMed] [Google Scholar]
- 38.Gum R, Lengyel E, Juarez J, Chen JH, Sato H, Seiki M, Boyd D. Stimulation of 92-kDa gelatinase B promoter activity by ras is mitogen-activated protein kinase kinase 1-independent and requires multiple transcription factor binding sites including closely spaced PEA3/ets and AP-1 sequences. J Biol Chem. 1996;271:10672–10680. doi: 10.1074/jbc.271.18.10672. [DOI] [PubMed] [Google Scholar]
- 39.Jia W, Gao XJ, Zhang ZD, Yang ZX, Zhang G. S100A4 silencing suppresses proliferation, angiogenesis and invasion of thyroid cancer cells through downregulation of MMP-9 and VEGF. Eur Rev Med Pharmacol Sci. 2013;17:1495–1508. [PubMed] [Google Scholar]
- 40.Zhang G, Li M, Jin J, Bai Y, Yang C. Knockdown of S100A4 decreases tumorigenesis and metastasis in osteosarcoma cells by repression of matrix metalloproteinase-9. Asian Pac J Cancer Prev. 2011;12:2075–2080. [PubMed] [Google Scholar]
- 41.Wang L, Wang X, Liang Y, Diao X, Chen Q. S100A4 promotes invasion and angiogenesis in breast cancer MDA-MB-231 cells by upregulating matrix metalloproteinase-13. Acta Biochim Pol. 2012;59:593–598. [PubMed] [Google Scholar]
- 42.Boye K, Maelandsmo GM. S100A4 and metastasis: a small actor playing many roles. Am J Pathol. 2010;176:528–535. doi: 10.2353/ajpath.2010.090526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burnett JC, Rossi JJ, Tiemann K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol J. 2011;6:1130–1146. doi: 10.1002/biot.201100054. [DOI] [PMC free article] [PubMed] [Google Scholar]