

Abstract
Many common causes of blindness involve the death of retinal photoreceptors, followed by progressive inner retinal cell remodeling. For an inducible model of retinal degeneration to be useful, it must recapitulate these changes. Intravitreal administration of adenosine triphosphate (ATP) has recently been found to induce acute photoreceptor death. The aim of this study was to characterize the chronic effects of ATP on retinal integrity. Five-week-old, dark agouti rats were administered 50 mM ATP into the vitreous of one eye and saline into the other. Vision was assessed using the electroretinogram and optokinetic response and retinal morphology investigated via histology. ATP caused significant loss of visual function within 1 day and loss of 50% of the photoreceptors within 1 week. At 3 months, 80% of photoreceptor nuclei were lost, and total photoreceptor loss occurred by 6 months. The degeneration and remodeling were similar to those found in heritable retinal dystrophies and age-related macular degeneration and included inner retinal neuronal loss, migration, and formation of new synapses; Müller cell gliosis, migration, and scarring; blood vessel loss; and retinal pigment epithelium migration. In addition, extreme degeneration and remodeling events, such as neuronal and glial migration outside the neural retina and proliferative changes in glial cells, were observed. These extreme changes were also observed in the 2-year-old P23H rhodopsin transgenic rat model of retinitis pigmentosa. This ATP-induced model of retinal degeneration may provide a valuable tool for developing pharmaceutical therapies or for testing electronic implants aimed at restoring vision. J. Comp. Neurol. 522:2928–2950, 2014. © 2014 Wiley Periodicals, Inc.
Keywords: retinitis pigmentosa (RP), age-related macular degeneration (AMD or ARMD), neural degeneration, retinal remodeling, rodent
Retinal degeneration from photoreceptor death, as occurs in age-related macular degeneration (AMD) and in inherited disorders such as retinitis pigmentosa, accounts for 50% of blindness in the Western world (Taylor et al., 2005). These diseases are characterized by loss of photoreceptors, followed by inner retinal neuronal cell death and progressive remodeling (Fletcher and Kalloniatis, ,; Gargini et al., 2007; Jones et al., ,; Marc and Jones, 2003; Marc et al., ,; Pignatelli et al., 2004; Strettoi et al., 2002). There are few treatment strategies for these diseases, but those that exist involve therapies to slow photoreceptor death using pharmacological agents and, more recently, electronic implants (O'Brien et al., 2012; Stingl et al., 2013a,b; Trifunović et al., 2012). In particular, investigation into the efficacy of retinal implants has fuelled the need for the development of inducible animal models of retinal degeneration that mimic the human retinal disease phenotype and are experimentally tractable. Thus, although there are many transgenic or natural genetic mutations in small animals, including genetic models of retinitis pigmentosa (Fletcher et al., 2011), few inducible models of retinal degeneration are available for which the timing can be regulated.
Among the available inducible models, acute light damage has recently become popular and has been extensively researched (Hunter et al., 2012; Marc et al., 2008; Wenzel et al., 2005). This model is effective for inducing photoreceptor death and recapitulates many aspects of retinal degeneration. However, this method is limited in application, because, although it is very effective in albino animals, pigmented animals are relatively resistant to light-induced damage (LaVail and Gorrin, 1987). Also, other aspects of the genetic background, even between albino strains, can moderate the effectiveness of this method (Danciger et al., 2007; LaVail et al., 1987a,b). Pharmacological treatments to induce retinal degeneration have also been developed, but many of these, like light damage, have their drawbacks. Systemic administration of N-methyl-N-nitrosourea (MNU) has been found to induce tumor formation in addition to photoreceptor death (Tsubura et al., 2011), which makes it a poor choice for long-term studies of retinal implants. Similarly, methanol administration has detrimental systemic effects on the central nervous system and affects both inner and outer retinal neurons to induce blindness in rodents and humans (Eells et al., 1996; Murray et al., 1991). Systemic administration of iodoacetate has also been used as a model of retinal degeneration in rodents but induces bilateral vision loss (Noell, 1951; Scott et al., 2011; Wang et al., 2011). Other novel inducible models of retinal degeneration that can be administered to one eye only without systemic side effects are desirable.
Recently, intravitreal administration of adenosine triphosphate (ATP) has been found to be effective for inducing photoreceptor death and loss of retinal function in the injected eye, suggesting it as a potential candidate for an inducible model of retinal degeneration (Notomi et al., ,; Puthussery and Fletcher, 2009). ATP is a naturally occurring molecule traditionally associated with cellular energy metabolism and with purinergic neurotransmission outside the cell (Burnstock, 2012). Extracellular ATP mediates fast excitatory neurotransmission through activation of two classes of purine receptor: the ionotropic, ligand-gated P2X receptors and the metabotropic, G protein-coupled P2Y receptors. In the rat eye, injection of ATP (45 mM) into the vitreous was found to induce photoreceptor-specific death within 18 hours in the rat via a P2X-receptor mediated mechanism (Puthussery and Fletcher, 2009). The photoreceptors were the only neurons affected; the RPE and inner retinal neurons were spared. Among the purine receptor classes expressed in the eye, the P2X7 receptor (P2X7-R) has been implicated in the mechanisms underlying photoreceptor death in response to high concentrations of ATP (Notomi et al., ,; Puthussery and Fletcher, 2009). Previous work from our laboratory has demonstrated the expression of P2X7-Rs on the synaptic terminals of rod and cone photoreceptors in the rat, whereas many of the other P2 subtypes investigated were not (for review see Ward et al., 2010; P2X7: Puthussery and Fletcher, 2004; P2X2: Puthussery and Fletcher, 2006; P2X3: Puthussery and Fletcher, 2007; P2X7: Puthussery et al., 2006; P2Y1: Ward and Fletcher [2009]). In addition, although the P2X7-R has been shown to mediate a physiological function as a neurotransmitter receptor in the rodent retina (Vessey and Fletcher, 2012), recent work shows that, if ATP concentrations exceed what can be adequately degraded by ectonucleotidases, excessive stimulation of P2X7-Rs on photoreceptors causes apoptotic cell death (Notomi et al., ,; Puthussery and Fletcher, 2009). This appears to proceed via a P2X7-R specific mechanism, because high doses of a P2X7-R ligand, BzATP, largely fail to induce photoreceptor apoptosis when injected into the eye of P2X7-R knockout mice (Notomi et al., 2011). Overall this work suggests that the mechanism of ATP-induced photoreceptor death is specifically via a P2X7-R mediated neurotoxic response akin to NMDA-mediated neurotoxicity in the inner retina (Olney et al., 1987).
Development of inducible models of retinal degeneration is important for trialing vision-restorative therapies. The aim of this study was to characterize the effects of ATP on the integrity of retinal neurons and glia in the rat over 6 months and determine whether this may be a useful model system. In addition, we compared some unusual and “extreme” aspects of remodeling characterized in the ATP model with a heritable model of retinal degeneration, the P23H rhodopsin transgenic rat. Our findings indicate that ATP-induced photoreceptor death recapitulates many of the anatomical changes that occur in retinal degenerations, providing a viable model for the investigation of the effects of photoreceptor death on retinal function and second-order neuronal remodeling.
MATERIALS AND METHODS
Animals
Five-week-old, dark agouti rats (n = 45) were obtained from the Animal Resource Center (ARC), Australia. Heterozygous Pro23His-line 3 transgenic rats (P23H rats) were obtained by crossing homozygous Pro23His rats with Sprague-Dawley rats. The Pro23His rats were originally produced by Xerogen Biosciences (formerly Chrysalis DNX Transgenic Sciences, Princeton, NJ) and developed and supplied with support of the National Eye Institute by Prof. Matthew LaVail, Beckman Vision Centre, University of California San Francisco School of Medicine. The breeding pairs for the work described in this study were kindly donated by Dr. Krisztina Valter-Kocsi and Prof. Jonathan Stone at The Australian National University, Canberra, and bred at the University of Melbourne. Rats were housed under <30 lux cage illumination at the University of Melbourne animal facility on a 12-hour light–dark cycle. Food and water were available ad libitum. All experiments adhered to the Association for Research in Vision and Ophthalmology (ARVO, Rockville, MD) standards for the ethical treatment of animals and complied with the requirements of the Animal Ethics Committee of the University of Melbourne (ethics number 1011840).
Intraocular injections of ATP in dark agouti rats
At 5 weeks of age, dark agouti rats were anesthetized by an intramuscular administration of a mixture of ketamine (60 mg/kg; Provet, Victoria, Australia) and xylazine (5 mg/kg; Provet). In addition, the corneal reflex was anesthetized with topical administration of Alcaine (0.5%; Alcon Laboratories, Victoria, Australia). Using a 30-G needle, a single injection of 2 μl of 1 M ATP prepared in sterile saline vehicle (0.9%) was injected into the vitreous of one eye using a Hamilton syringe. The contralateral control eye received 2 μl saline vehicle (Dureau et al., 2001). Based on an estimated vitreal volume of 40 μl, the concentration of ATP at the retina was 50 mM.
Visual acuity
At 1, 3, and 6 days after administration of ATP, awake, behaving rats (n = 6) were placed in an OptoMotry system (CerebralMechanics Inc., Lethbridge, Alberta, Canada [Douglas et al., 2005; Prusky et al., 2004]) and tested for visual responsiveness under photopic conditions using a virtual optokinetic drum. Spatial frequency thresholds were assessed by monitoring the head-tracking response to a range of spatially modulated, sinusoidal gratings (contrast 100%, mean photopic luminance 100 cd/m2), presented using a simple staircase protocol rotating at a constant velocity (12°/second). Motion detected in the clockwise direction represents the left eye response and motion detected in the anti-clockwise direction represents the right eye response, so separate spatial frequency thresholds could be determined for ATP- and saline-injected eyes independently (Douglas et al., 2005). All measurements were made by an observer who was blinded to the stimulus condition. The spatial frequency threshold (cycles/degree) was defined as the highest spatial frequency that elicited a reliable head-tracking response.
Electroretinogram
To assess retinal function after ATP administration, electroretinograms (ERGs) were recorded from saline- and ATP-treated eyes of n = 9 rats at 1 and 6 days after ATP, as previously described (Vessey et al., 2011). Animals were dark adapted overnight. On the next day, the rats were anesthetized using a mixture of ketamine (60 mg/kg) and xylazine (5 mg/kg). The cornea was anesthetized (Alcaine; proparacaine hydrochloride 0.5%; Alcon), the pupil was dilated (Midriacyl; tropicamide 0.5%; Alcon), and to maintain body temperature rats were placed on a heating pad. Using an Ag/AgCl electrode that was placed gently on the center of the cornea and a reference electrode placed in the mouth, ERGs were recorded. Responses were amplified (gain ×5,000, −3 dB at 1 Hz and 1 kHz; ADInstruments, Castle Hill, New South Wales, Australia) and digitized at 10 kHz. To elicit the ERG response, a full-field flash of 2.1 log cd · second/m2 was generated by a Nikon photography flash (Nikon SB900) and delivered via a custom-made Ganzfeld. Two consecutive flashes (0.8-sec interstimulus interval) were used to assess the rod and cone responses independently (Jobling et al., 2013; Lyubarsky and Pugh, 1996; Lyubarsky et al., 1996). The first flash elicited responses from both the rod and cone pathways (mixed response). The second flash elicited responses only from the cones. The rod responses were isolated by digital subtraction of the cone response from the mixed response.
ERG component analysis
With the paucity of response from the ATP-treated eyes, it was not possible to model the ERG responses. Instead, the rod photoreceptor response of the ERG, the a-wave, was determined by measuring the amplitude (μV) from the prestimulus baseline to the trough of the waveform. The amplitude of the rod post-photoreceptor response, the b-wave, was measured from the a-wave trough to the peak of the waveform or, if no a-wave was present, from the prestimulus baseline. The implicit times of the a- and b-waves were measured from the time of flash presentation to the trough of the a-wave or the peak of the b-wave, respectively. The cone post-photoreceptor response, b-wave, was analyzed by measuring the amplitude, from the prestimulus baseline to the peak of the waveform, and the implicit time, the time until the maximum response was reached.
Gross histology
The gross retinal morphology of ATP- and saline-injected eyes was ascertained using hematoxylin- and eosin-stained paraffin sections and toluidine blue-stained resin sections (Ho et al., 2012). Rats were anesthetized with intramuscular administration of a mixture of ketamine and xylazine (60:5 mg/kg; Provet) and killed using sodium pentobarbital (120 mg/kg; Provet). The eyes were isolated, and the anterior portions of the eye and lens were removed by dissection. For paraffin sectioning, whole eyes were fixed overnight in 4% paraformaldehyde containing 3% sucrose, 5% acetic acid, and 60% ethanol. Eyes were then dehydrated in graded alcohols before being embedded in paraffin wax. Retinae were then sectioned at 4 μm, placed on poly-L-lysine-coated slides, and incubated overnight at 37°C. Sections were deparaffinized, stained with Mayer’s hematoxylin and eosin, and coverslipped. For resin sections, eyecups were fixed overnight in 1% paraformaldehyde, 2.5% glutaraldehyde, 3% sucrose, and 0.01% calcium chloride in 0.1 M phosphate buffer, pH 7.4 (PB). Eyecups were washed in PB and then dehydrated in a graded series of methanol (75%, 85%, 95%, and 100%) and acetone (100%). Tissues were embedded in an epon resin (ProSciTech, Queensland, Australia) and polymerized overnight at 60°C. Retinae were sectioned (1 μm) on an ultramicrotome (Reichert-Jung Ultracut S; Reichert, Depew, NY) and stained with 1% toluidine blue.
A microscope (Axioplan; Carl Zeiss, Göttingen, Germany) was used to view retinal sections with a ×40 oil objective, and images were captured using a digital camera and associated computer software? (Spot version 3.5.2; Diagnostic Instruments). Images were converted to gray scale and adjusted for white levels, brightness, and contrast in Adobe Photoshop CS4 (Adobe Systems, San Jose, CA). Retinal cellular layers (multiple sections, n > 2) in saline- and ATP-treated eyes were imaged within 150 μm of the optic nerve at 1 week (n = 7, paraffin), 3 months (n = 6, resin), and 6 months (n = 6, resin). The numbers of rows of photoreceptor and inner nuclear layer cells were quantified manually at five regions per image in a cell counting application (ImageJ 1.43; National Institutes of Health, Bethesda, MD). The number of ganglion cells across the retina was quantified and expressed per millimeter of retinal length. In addition, to determine whether there was an eccentricity-dependent effect of ATP on photoreceptor number, full-tile scans of toluidine blue-stained retinae from optic nerve to ciliary body were collected using an LSM confocal microscope (Carl Zeiss). These images were used to generate “spider plot” analyses (Mittag et al., 1999), in which retinae were divided into 10 locations from optic nerve (ON) to the ciliary body, and the number of rows of photoreceptors was counted and averaged. An Abercrombie correction was used to normalize cell counts and account for differences in section thickness between paraffin and resin sections (Abercrombie and Johnson, 1946).
Fluorescence immunohistochemistry
Fluorescence immunohistochemistry was used to assess morphology of retinal cell classes using previously described techniques (Vessey and Fletcher, 2012). The posterior eye cups of saline- and ATP-treated eyes at 3 months (n = 6) and 6 months (n = 6) postinjection were fixed for 30 minutes in 4% paraformaldehyde in PB, washed three times in PB, and cryoprotected in a series of graded sucrose solutions (10%, 20%, and 30% in PB). Saline- and ATP-treated eyes were then embedded together in optimal cutting temperature compound (Tissue-Tek O.C.T.; Sakura, Torrance, CA), frozen, and sectioned transversely at 14 μm on a cryostat at −20°C (Microm, Walldorf, Germany). Sections were collected on poly-L-lysine-coated slides (Menzel-Gläser, Braunschweig, Germany) and stored at −20°C.
For immunofluorescent labeling, slides were defrosted and washed in PB, and sections were incubated in a blocking solution (10% normal goat serum [NGS], 1% bovine serum albumin [BSA], 0.5% Triton-X in PB) for 1 hour before incubation overnight, at room temperature, in primary antibody (Table1) diluted in antibody buffer (3% NGS, 1% BSA, 0.5% Triton-X in PB). After being washed in PB, sections were incubated with appropriate secondary antibody: goat anti-guinea pig, goat anti-mouse, or goat anti-rabbit conjugated to fluorescent dyes (AlexaFluor 488, AlexaFluor 594, or AlexaFluor 643; diluted 1:500; Life Sciences, Victoria, Australia) and a nuclear dye, 4′,6-diamidino-2-phenylindole (DAPI; diluted 1:300; Life Sciences) for 90 minutes. The sections were washed in PB, coated in a glycerol/Mowiol-based mounting media, and covered with a glass coverslip. All antibodies listed were used as tissue markers and were found to label cellular morphology and distribution, as carefully demonstrated in previous publications (Table1).
Table 1.
Primary Antibodies Used for Immunohistochemical Analysis
| Cell type/target | Antibody/lectin | Dilution | Source | Immunogen |
|---|---|---|---|---|
| Cones | Fluorescein-labeled peanut agglutinin (PNA) from Arachis hypogaea (peanuts) | 1:250 | Catalog No. FL-1071; Vector Laboratories, Burlingame, CA | NA |
| Photoreceptor and bipolar cell terminals | Guinea pig antivesicular glutamate transporter (VGLUT1), polyclonal | 1:1,000 | Catalog No. AB 5905; Millipore, Merck, VIC, Australia | C-terminal peptide of rat VGLUT1, GATHSTVQPPRPPPPVRDY |
| Horizontal cells | Mouse anticalbindin D28k, monoclonal | 1:4,000 | Catalog No. AB 300; Swant, Bellinzona, Switzerland | Purified bovine kidney calbindin-D28K |
| Rod bipolar cells | Mouse antiprotein kinase Cα (PKC), monoclonal (MC5) | 1:4,000 | Catalog No. P5704; Sigma-Aldrich, NSW, Australia | Peptide amino acids 296–317 of PKC |
| Amacrine and ganglion cells | Mouse anticalretinin, monoclonal | 1:1,000 | Catalog No. 63B; Swant, Bellinzona, Switzerland | Recombinant human calretinin-22k |
| Blood vessels | Lectin from Bandeiraea simplicifolia BS-I isolectin B4 FITC | 1:75 | Catalog No. L2895; Sigma,-Aldrich, NSW, Australia | N/A |
| Microglia | Rabbit anti-ionized calcium-binding adaptor molecule 1 (IbA1), polyclonal | 1:1,500 | Catalog No. 019–19741; Wako Pure Chemical Industries, Richmond, VA, USA | Synthetic peptide corresponding to the C-terminus of IbA1: PTGPPAKKAISELP |
| Müller cells | Mouse antiglutamine synthetase (GS), monoclonal (GS-6) | 1:1,000 | Catalog No. MAB302; Millipore, Merck, VIC, Australia | Glutamine synthetase purified from sheep brain |
| Astrocytes and gliotic Müller cells | Rabbit antiglial fibrillary acid protein (GFAP), polyclonal | 1:20,000 | Catalog No. Z0334;Dako,Carpinteria, CA, USA | Bovine spinal cord GFAP |
| Cells in G1–S transition | Rabbit anticyclin-D1, monoclonal (SP4) | 1:2 | Catalog No. AB21699; Abcam, Sapphire Bioscience, NSW, Australia | A synthetic peptide from the C-terminus of human cyclin D1 (amino acids 250–295, Uniprot seq: P24385) |
| All cycling cells, those in G1, S, G2, and M | Rabbit anti-1,098-bp Ki67 motif-containing cDNA fragment, monoclonal (SP6) | 1:1,000 | Catalog No. AB16667; Abcam, Saphire Bioscience, NSW, Australia | A synthetic peptide from the C-terminus of human Ki-67 (amino acids 2319–2323, FKELF, within amino acids 2300–2400, Uniprot seq: P46013) |
All antibodies listed were used as tissue markers and were found to label cellular morphology and distribution as demonstrated in previous publications.
Fluorescein-labeled peanut agglutinin (PNA) was used to visualize cone photoreceptors. PNA binds preferentially to a commonly occurring structure, galactosyl(b-1,3)N-acetylgalactosamine, and has previously been found to label cone photoreceptors of rodents (Vessey et al., 2011) and many other species (Hageman and Johnson, 1986).
Guinea pig anti-Vesicular Glutamate Transporter (VGLUT1) was used to label photoreceptor and bipolar cell terminals as has been shown previously for rodent retinae (Johnson et al., 2003; Vessey et al., 2012). It is affinity purified using the immunogenic peptide and recognizes one band of 60 kDa on immunoblot of synaptic membrane fraction from rat cerebral cortex (Melone et al., 2005).
Mouse anti-Calbindin D-28k was used to label horizontal cells as has been previously shown in rodent retinae (Ho et al., 2012; Vessey and Fletcher, 2012). It recognizes one band of 28 kDa on immunoblot of synaptic membrane fraction from rat cerebral cortex and combinant calbindin D-28k (manufacturer’s product sheet).
Mouse anti-Protein Kinase Cα (PKC) was used to label rod bipolar cells as has been previously shown in rodent retinae (Greferath et al., 1990; Ho et al., 2012; Vessey et al., 2012). The antibody reacts with the 80-kD polypeptide of PKC on immunoblot of bovine brain PKC, extracts of rat glioma and murine NIH 3T3 cell lines, and rat brain (manufacturer’s product sheet).
Mouse anti-Calretinin was used to label amacrine and ganglion cells as has been previously shown in rodent retinae (Ho et al., 2012; Vessey et al., 2012). In the rodent retina, calretinin is present in choline acetyltrasferase (ChAT) amacrine cells as well as other GABAergic widefield amacrine cells and ganglion cells (Araki and Hamassaki-Britto, 2000; Gábriel and Witkovsky, 1998). It recognizes a band of 29 kDa on immunoblot of synaptic membrane fraction from rat cerebral cortex. In mouse cerebral cortex, it labels a subpopulation of neurons but does not label cerebral cortex of calretinin knockout mice (manufacturer’s product sheet).
Lectin from Bandeiraea simplicifolia BS-I isolectin B4 FITC was used to label blood vessels. This marker has been previously established to label the retinal microvessels via histochemistry (Tyler and Burns, 1991) and has been used in a number of studies as an identifier for rodent retinal blood vessels (van Wijngaarden et al., 2007; Vessey et al., 2011).
Rabbit polyclonal anti-ionized calcium-binding adaptor molecule 1 (IbA1) was used to label microglia (Vessey et al., 2011). The specificity of this antiserum has been previously demonstrated by testing in immunoblots of rodent cortex proteins, in which it was shown to react with a unique band of the expected molecular size, 17 kDa (Ito et al., 1998).
Mouse anti-Glutamine synthetase (GS) was used to label Müller cells as has been previously shown for rodent retinae (Vessey et al., ,). The glutamine synthetase antibody produces a single expected band of 45 kDa on immunoblots from rat brain (see manufacturer’s data sheet) and in mouse retina (Chen and Weber, 2002; Nasonkin et al., 2011).
Rabbit polyclonal anti-glial fibrillary acid protein (GFAP) was used to label astrocytes and gliotic Müller cells (Vessey et al., 2011). The specificity of this antiserum has been previously demonstrated by testing in immunoblots of rodent retinal proteins, in which it was shown to react with a unique band of the expected molecular size, 51 kDa (Chen and Weber, 2002).
Rabbit monoclonal anti-Cyclin-D1 was used to label cells in G1-S transition as has been shown previously in retinae (Albarracin and Valter, 2012; Bienvenu et al., 2010). The anti-Cyclin-D1 antibody produces an expected band of about 33 kDa on immunoblots from mouse testes (see manufacturer’s data sheet and McIver et al., 2012) and regenerating mouse skeletal muscle (Galatioto et al., 2010).
Rabbit monoclonal anti-1098 bp Ki-67 motif-containing cDNA fragment (Ki-67) was used to label all cycling cells, those in G1, S, G2 and M, as has been shown previously in retinae (Glaschke et al., 2011) and in the mouse small intestine (Bergner et al., 2014). The gene encodes 15 exons with a large exon 13 containing 16 homologous, highly conserved 22-amino-acid-sequence elements called the “Ki67 motif.” Nine of the Ki67 motif regions include a highly immunogenic amino acid sequence (amino acids 2319–2323; FKELF; Kubbutat et al., 1994) that forms the epitope for many Ki67 monoclonal antibodies, including the SP6 clone (Pathmanathan and Balleine, 2013). According to the manufacturer, the SP6 clone recognizes a band of 356 kDa on Western blots of SKBR3 cell lysates, matching reports using other Ki-67 clones (Key et al., 1993).
Images were taken witjh an LSM 5 Meta confocal laser scanning microscope (Zeiss) using a ×20 air or ×40/1.3 oil immersion objective at a resolution of 1,024 × 1,024 pixels. Gain settings were at the same level when taking images for saline- and ATP-treated tissue sections. Scale bars were digitally added to the images in Zeiss LSM Image Browser software (v4.2.0.121; Zeiss). Images were adjusted for black levels, contrast, and brightness in Adobe Photoshop CSE version 4 using the same settings for consistency among samples. In all cases, two separate representative images of ATP-treated retinae are presented in order to show variation in the degenerative process across the retina and in particular to highlight differences between regions where photoreceptor nuclei remained or were absent.
Statistical analysis
Results are expressed as the mean ± standard error of the mean (SEM). ERG responses and spatial frequency thresholds across time (1, 3, and 6 days) and treatment (saline vs. ATP) were analysed by two-way analysis of variance (ANOVA). Similarly, a two-way ANOVA was used to determine changes in cell counts over time (1 week, 3 months, and 6 months) and treatment (saline vs. ATP). A Bonferroni post hoc test was used to analyze the effect of ATP treatment at individual ages (Graphpad Prism v4; GraphPad Software, San Diego, CA). In all figures, statistical significance is expressed as *P <0.05.
RESULTS
Intravitreal administration of ATP induces loss of photoreceptors
The time course of the effects of 50 mM ATP on the structure and function of the retina was assessed in rats over 1 week, 3 months, and 6 months. Retinal function was assessed using the electroretinogram at 1 and 6 days after ATP administration (Fig. 1). A twin-flash paradigm was used to isolate rod and cone pathway function separately, and representative waveforms are shown (Fig. 1A,D, rod and cone, respectively). Rod photoreceptor (a-wave) function and post-photoreceptor (b-wave) function were significantly reduced but still detectable 1 day after ATP treatment (Fig. 1B,C). By 6 days after ATP injection, the rod pathway response was indistinguishable from baseline recording noise (for both a- and b-wave; two-way ANOVA for time P < 0.001 and treatment P < 0.0001; Bonferroni post hoc for saline vs ATP, P < 0.05 at 1 and 6 days). Cone pathway function was negligible within 1 day and was still undetectable at 6 days after ATP (Fig. 1E; cone b-wave; two-way ANOVA for time P = 0.25 and treatment P < 0.0001, Bonferroni post hoc for saline vs ATP, P < 0.05 at 1 and 6 days). Similarly, photopic visual function, as measured by optokinetic spatial frequency threshold, was negligible and significantly reduced compared with the saline-treated fellow eye at 1, 3, and 6 days after ATP injection (Fig. 1F; two-way ANOVA for time P = 0.98, treatment P < 0.0001; Bonferroni post hoc for saline vs. ATP, P < 0.05 at 1, 3, and 6 days). In the saline-treated fellow eye, spatial frequency threshold was ∼0.53 ± 0.5 cycles/°, and, although slightly lower, perhaps due to contributions from ipsilateral (ATP-treated) visual centers, this response was not significantly different from the response of the uninjected rats (0.58 ± 0.01, P > 0.05). This suggests that ATP induces loss of cone-derived retinal responses and loss of functional photopic vision within 24 hours and complete loss of both rod and cone derived retinal function within 1 week.
Figure 1.
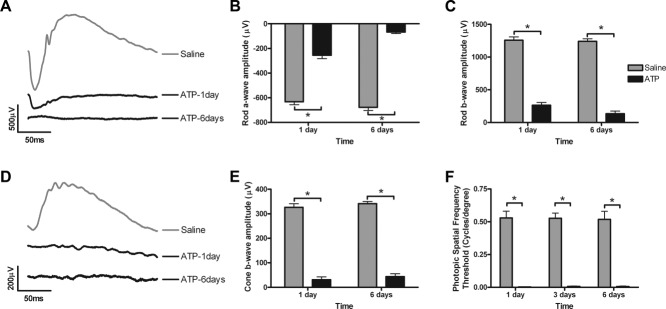
Time course of the effects of 50 mM ATP on retinal function at 1 and 6 days. Twin, full-field 2.1 log cd · second/m2 intensity flashes were used to isolate the rod and cone pathway electroretinogram responses of saline- and ATP-treated eyes. A: Representative rod pathway waveforms from saline-treated (gray) and ATP-treated (black) eyes. Rod a-wave amplitude (B) and rod b-wave amplitude (C) were assessed for saline-treated (gray bar, n = 9) and ATP-treated (black, n = 9) eyes at 1 and 6 days after intravitreal injection. D: Representative cone pathway waveforms from saline-treated (gray) and ATP-treated (black) eyes. E: Cone b-wave amplitudes were assessed for saline-treated (gray bar, n = 9) and ATP-treated (black, n = 9) eyes at 1 and 6 days after intravitreal injection. F: Spatial frequency threshold in saline- and ATP-treated eyes was assessed under photopic conditions using the optokinetic response at 1, 3, and 6 days after injection. Significance of P < 0.05 between saline- and ATP-treated eyes indicated by an asterisk.
In line with this loss of visual function, the structure of the retina after intravitreal administration of ATP was found to be aberrant (Fig. 2). In all cases, two separate, representative images of ATP-treated retinae are presented to show variation in the degenerative process across the retina. One week after ATP treatment, there was specific loss of the photoreceptors across most regions of the retina, but the inner retina was relatively preserved (Fig. 2B,C). At 3 months after ATP treatment, the photoreceptor layer was further reduced, with only a few rows of nuclei and inner–outer segments detected in some regions (Fig. 2E), and in other regions photoreceptors were completely absent (Fig. 2F). At 6 months, the photoreceptor layer was completely degenerated (Fig. 2H), and in some regions the retinal pigment epithelium (RPE) had migrated into the retina (Fig. 2H, arrowhead).The inner retinal neurons also appeared affected (Fig. 2I). In contrast, the morphology of saline-treated eyes (Fig. 2A,D,G) was similar to uninjected control rat eyes at each age (data not shown).
Figure 2.
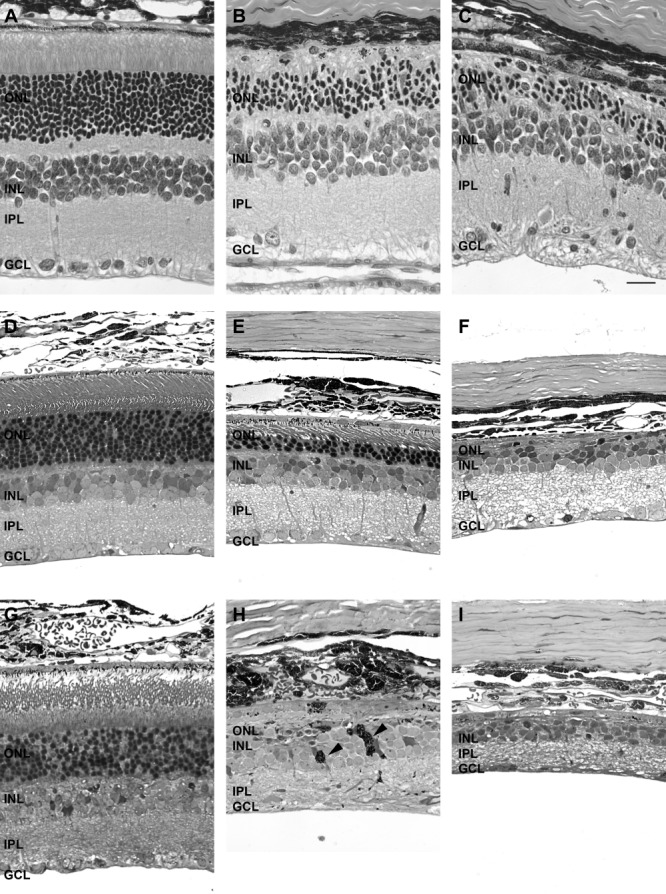
Time course of the effects of 50 mM ATP on the gross structure of the retina at 1 week, 3 months, and 6 months. Gross retinal structure was assessed in saline-treated and ATP-treated regions of retina at 1 week (A: saline; B,C: ATP, paraffin), 3 months (D: saline; E,F: ATP, resin), and 6 months (G: saline; H,I: ATP, resin). For each time point, two example regions of ATP-treated retinae are presented. Arrowheads in H indicate aberrant pigmented (RPE) cells within the neural retina. ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bar = 20 μm.
To quantify the gross retinal changes induced by ATP, the numbers of rows of photoreceptor nuclei and inner nuclear layer nuclei were determined, and the numbers of nuclei across the ganglion cell layer were also counted (Fig. 3). ATP treatment induced a specific and significant loss of photoreceptor nuclei: approximately 50% at 1 week and ∼80% at 3 months, and by 6 months there was, on average, less than one layer of cells (Fig. 3A; two-way ANOVA, time P = 0.0003, treatment P < 0.0001; Bonferroni post hoc for saline vs ATP, P < 0.05 at 1 week, 3 months, and 6 months). When quantified across the retina as a function of distance from the optic nerve, ATP was found to alter photoreceptor row number differentially across eccentricity. At 3 months, on average, there were no photoreceptors centrally and some residual photoreceptors in the far periphery (Fig. 3B). At 6 months, the number of photoreceptors in the periphery was reduced to less than one layer (Fig. 3C). In contrast, although the number of rows of nuclei in the inner nuclear layer decreased with age in ATP- and saline-treated eyes, there was no significant effect of the treatment at any time (Fig. 3D; two-way ANOVA, time P < 0.0001, treatment P = 0.15). ATP treatment had no effect on the number of cells in the ganglion cell layer at 1 week or 3 months after the initial insult; however, after 6 months, cell numbers were significantly reduced in the ATP-treated eye (Fig. 3E; two-way ANOVA, time P = 0.0016, treatment P = 0.0352; Bonferroni post hoc for saline vs ATP, P < 0.05 at 6 months only). This suggests that 50 mM ATP provides a single insult that induces photoreceptor dysfunction and death within 1 week. Furthermore, over time, almost all photoreceptors and also some amacrine and/or ganglion cells in the ganglion cell layer are lost as part of an ongoing degenerative process.
Figure 3.
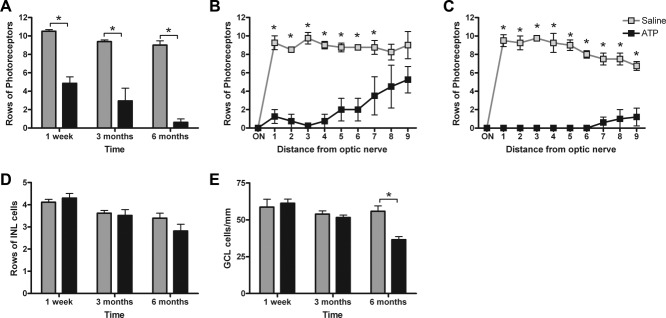
Quantification of the effects of 50 mM ATP on the gross structure of the retina at 1 week, 3 months, and 6 months. A: The average number of rows of photoreceptor nuclei was quantified for saline-treated (gray bars) and ATP-treated (black bars) eyes at 1 week, 3 months, and 6 months. B,C: To determine whether there was an effect across eccentricity, rows of photoreceptor nuclei were also quantified at 10 regions across the retina from the optic nerve (ON) to the peripheral ciliary body at 3 months (B) and 6 months (C). D: The average number of rows of inner nuclear layer cells quantified in saline-treated (gray bars) and ATP-treated (black bars) eyes at 1 week, 3 months, and 6 months. E: The average number of cells across the ganglion cell layer were also quantified. Significance of P < 0.05 between saline-treated (n = 6) and ATP-treated (n = 6) eyes indicated by an asterisk.
ATP induces photoreceptor degeneration and remodeling of inner retinal neurons
To investigate further the time course of changes following ATP treatment, various retinal cell markers were used to probe the neurodegenerative process. Retinae from saline- and ATP-treated eyes were labeled for cone photoreceptors (PNA, green), photoreceptor and bipolar cell terminals (VGLUT1, red), and cell nuclei (DAPI, blue) at 3 months (Fig. 4A–C) and 6 months (Fig. 4D-E) post-injection. In the saline-treated eyes, PNA-labeled cone photoreceptor inner/outer segments and terminals were apparent across the outer retina and outer plexiform layers at 3 months (Fig. 4A) and 6 months (Fig. 4D), respectively. Cone terminals were found to colocalize with VGLUT1-labeled synapses in the outer plexiform layer in a manner consistent with healthy retinal tissue (Sherry et al., 2003). In ATP-treated eyes, although there were still regions of photoreceptor nuclei at 3 months (Fig. 4B), PNA-labeled cone inner/outer segments were absent, and cone photoreceptor terminals were displaced or absent from the outer plexiform layer. In addition, VGLUT1-labeled photoreceptor terminals were displaced into the outer nuclear layer (Fig. 4B, arrowhead). In other regions where photoreceptor nuclei were absent, a few, sparse VGLUT1-positive photoreceptor terminals remained (Fig. 4C). By 6 months after ATP administration, evidence of PNA-labeled cones and VGLUT1-positive photoreceptor terminals was negligible (Fig. 4E,F). Also, in some regions, the inner retina had begun to remodel significantly, with large holes representing pigmented cells from the RPE and/or fluid-filled vacuoles located in the inner retina (Fig. 4E, asterisk), as has been described previously (Marc and Jones, 2003), and loss of VGLUT1-positive bipolar cell terminals in the inner plexiform layer (Fig. 4F).
Figure 4.

Cone and rod photoreceptors degenerate following ATP treatment. Sections of retinae from saline- and ATP-treated eyes were labeled for cone photoreceptors (PNA, green), photoreceptor and bipolar cell terminals (VGLUT1, red), and cell nuclei (DAPI, blue) at 3 months (A: saline; B,C: ATP) and 6 months (D: saline; E,F: ATP) post-injection. For each time point, two example regions of ATP-treated retinae are presented. Arrowhead in B indicates an abnormal clump of VGLUT1-positive photoreceptor terminals displaced in the ONL. Asterisks in E indicate RPE cell migration and/or vacuole structures in the retina. ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bars = 20 μm.
To investigate the response of inner retinal neurons, retinae were labeled for rod bipolar cells with an antibody against PKCα (Fig. 5). At 3 months post-ATP injection, rod bipolar cell morphology was relatively well maintained in most regions of the retina (Fig. 5B) and, in general, was similar to saline-injected (Fig. 5A) or healthy retinae (Uesugi et al., 1992). In regions where photoreceptors remained, rod bipolar cell nuclei were located in the outer regions of the inner nuclear layer, their dendrites contacted remnant VGLUT1-positive photoreceptor terminals in the outer plexiform layer, and PKCα/VGLUT1-positive terminals were laminated in the inner most region of the inner nuclear layer (Fig. 5B). However, in regions devoid of photoreceptors, rod bipolar cell morphology was more irregular (Fig. 5C). By 6 months after ATP injection, PKCα-positive rod bipolar cells were still present, although they appeared reduced in number, their overall morphology was abnormal, and they laminated irregularly in the inner plexiform layer (Fig. 5E,F). In addition, there were instances of extreme remodeling events with rod bipolar cell dendrites exiting the neural retina and forming VGLUT1-positive contacts in the choroid (Fig. 5F, arrowhead). These instances of neural retina emigration into the choroid were also observed at the light microscopic level (Fig. 5G, arrowheads). Given the unusual nature of this neuronal remodeling, it was of interest to determine whether this change was representative of heritable retinal degenerations. Two-year-old P23H rat retinae were compared (Fig. 5H). The P23H rat had no outer nuclear layer, regions of reduced and remodeled inner nuclear layer, and an abnormal distribution of VGLUT1-positive synapses. In particular, there were distinct regions of PKCα-positive/VGLUT1-positive synapses in the choroid (Fig. 5H, arrow), similar to those seen in the ATP-treated retina at 6 months.
Figure 5.

Rod bipolar cell morphology following ATP-induced photoreceptor degeneration and in P23H rat retinae. Sections of retinae from saline- and ATP-treated rat eyes and a 2-year-old P23H rat retina for comparison were labeled for rod bipolar cells (PKCα, green), photoreceptor and bipolar cell terminals (VGLUT1, red), and cell nuclei (DAPI, blue). A–C: ATP-treated rat retinae at 3 months (A: saline; B,C: ATP). D–F: ATP-treated rat retinae at six months (D: saline; E,F: ATP). For each time point for ATP-treated retinae, two example regions are presented. G: Light microscopic image of ATP-treated rat retina at 6 months. H: Two-year-old P23H rat retina. Arrowheads in F,H indicate extreme remodeling event of VGLUT1/PKCα-positive synapses displaced into the choroid in 6 month ATP retina (F) and 2-year-old P23H rat retina (H). ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bars = 20 μm.
Next, horizontal cells were investigated in the ATP-treated eyes. Horizontal cells, labeled with an antibody against calbindin, were evident at both 3 and 6 months (Fig. 6). At 3 months after ATP injection, calbindin-positive cell bodies were present in the inner nuclear layer adjacent to the outer plexiform layer (Fig.6B,C), much like those observed in the saline-treated eye (Fig. 6A). However, their processes were altered compared with those of saline-treated eyes and had remodeled to contact the few remaining VGLUT1-positive photoreceptor terminals (Fig. 6B,C). By 6 months after ATP administration, calbindin-positive cells had begun to migrate within the inner nuclear layer (Fig. 6E) and were found to extend processes into the inner plexform layer to form contacts with VGLUT1-positive bipolar cell terminals (Fig. 6E). In other areas, as was seen with the PKC positive rod bipolar cells, horizontal cell processes exited the neural retina and formed connections with VGLUT1-positive synapses in the choroid (Fig. 6F, arrowheads).
Figure 6.

Horizontal cell morphology following ATP-induced photoreceptor degeneration. Sections of retinae from saline- and ATP-treated eyes were labeled for horiztontal cells (calbindin D28k, green), photoreceptor and bipolar cell terminals (VGLUT1, red), and cell nuclei (DAPI, blue) at 3 months (A: saline; B,C: ATP) and 6 months (D: saline; E,F: ATP) post-injection. For each time point, two example regions of ATP-treated retinae are presented. Arrowheads in F indicate extreme remodeling event of horizontal cell processes associating with VGLUT1-positive synapses displaced into the choroid. ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bars = 20 μm.
Amacrine and ganglion cells were identified with an antibody against calretinin (Fig. 7). At 3 months following ATP, calretinin-positive amacrine cells in the inner nuclear layer and ganglion cells/amacrine cells in the ganglion cell layer were generally well preserved (Fig. 7B,C) and of similar morphology to saline-injected retinae (Fig. 7A). Furthermore, in regions of ATP-injected eyes with remnant photoreceptors, the processes of these cells were found to laminate in three layers within the inner plexiform layer (Fig. 7B), similar to control retinae (Fig. 7A) and consistent with healthy rodent retinae (Sherry et al., 2003). In regions where photoreceptor nuclei were absent, calretinin-positive processes appeared irregular yet still formed three distinct laminae (Fig. 7C). By 6 months after ATP injection, calretinin-positive cells were less regular in number and spacing, and their processes were frequently disorganized (Fig. 7E). In some regions, severe remodeling had occurred, with processes found to extend into the choroid (Fig. 7F, arrowheads). Overall, the effects of ATP treatment on neuronal remodeling correlate well with events common to retinal degenerations (Marc et al., 2003). However, there were instances of neurons and process connections outside the neural retinae, an extreme and unusual remodeling event that is not commonly described for retinal degenerations but was also found to be apparent in the 2-year-old P23H rat retina.
Figure 7.

Amacrine and ganglion cell morphology following ATP-induced photoreceptor degeneration. Sections of retinae from saline- and ATP-treated eyes were labelled for a subset of amacrine and ganglion cells (calretinin, green) and cell nuclei (DAPI, blue) at 3 months (A: saline; B,C: ATP) and 6 months (D: saline; E,F: ATP) post-injection. For each time point, two example regions of ATP-treated retinae are presented. Arrowheads in F indicate extreme remodeling events of amacrine cell processes displaced into the choroid. ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bars = 20 μm.
ATP induces loss and remodeling of intraretinal blood vessel profiles
To determine whether ATP-induced retinal degeneration had effects on blood vessels, sections were labeled with IB4 lectin (Fig. 8). In saline-treated control retinae, blood vessel profiles labeled with IB4 lectin (green) were apparent in the ganglion cell layer/nerve fiber layer (superficial plexus), at the border of the inner plexiform and inner nuclear layer (inner plexus), and also in the outer plexiform layer (deep plexus; Fig. 8A,D). This lamination of the intraretinal blood vessels was consistent with healthy rodent retinae (Vessey et al., 2011). Three months after ATP administration, in regions where photoreceptors were still present, blood vessel profiles were similar to those seen in saline-treated eyes (Fig. 8B). In regions where photoreceptors were absent, the vasculature appeared abnormal, and the number of blood vessel profiles appeared to be reduced (Fig. 8C). At 6 months after ATP, the number of blood vessel profiles was reduced, and morphology was abnormal, although vessels in the superficial plexus were still apparent in all regions (Fig. 8E,F).
Figure 8.

Blood vessel morphology following ATP-induced photoreceptor degeneration. Sections of retinae from saline- and ATP-treated eyes were labeled for blood vessels (isolectin B4, green) and cell nuclei (DAPI, blue) at 3 months (A: saline; B,C: ATP) and 6 months (D: saline; E,F: ATP) post-injection. For each time point, two example regions of ATP-treated retinae are presented. ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bars = 20 μm.
ATP induces Müller cell gliosis, scarring, and extraretinal migration
The main macroglia of the retina, Müller cells, were labeled with an antibody against glutamine synthetase (GS; green), and the astrocytes and gliotic Müller cells were labeled with an antibody against GFAP (red; Fig. 9). In the saline-treated eyes, GS-positive Müller cell bodies were present in the middle of the inner nuclear layer, and their processes extended across the transverse retina to form the inner and outer limiting membrane (Fig. 9A,D). In these control eyes, GFAP-labeled astrocytes were found in the nerve fiber layer at the base of the ganglion cell layer. No GFAP immunoreactivity was present in the Müller cell processes, which indicates that the saline-injected rat retinae were healthy. Three months after ATP injection, in regions where photoreceptor nuclei were still present in a defined outer nuclear layer, the structure of the GS-labeled Müller glia was consistent with that seen in saline-injected eyes; however, all Müller cells were also positive for GFAP, i.e., gliotic (Fig. 9B). In regions devoid of photoreceptors, the Müller glia not only were GFAP-positive but also were remodeled, with nuclei at both the top and the bottom of the inner nuclear layer and processes that ran diagonally across the retina (Fig. 9C). There was also evidence of Müller cell hypertrophy and glial scar formation at the margins of the retina (Fig. 9C). By 6 months after ATP, there appeared to be fewer GS-positive Müller cells, and the remaining cells were significantly remodeled. In addition, large holes, likely fluid-filled vacuoles or pigmented displaced RPE cells, encapsulated by the Müller glia, were apparent in the inner nuclear layer (Fig. 9E, asterisk); in other regions, GFAP/GS-positive Müller cells were found to leave the neural retina and enter the choroid (Fig. 9F, arrowhead).
Figure 9.

Glial cell morphology following ATP-induced photoreceptor degeneration. Sections of retinae from saline- and ATP-treated eyes were labeled for Müller glial cells (GS, green), astrocytes and Müller cell gliosis (GFAP, red), and cell nuclei (DAPI, blue) at 3 months (A: saline; B,C: ATP) and 6 months (D: saline; E,F: ATP) post-injection. For each time point, two example regions of ATP-treated retinae are presented. Asterisk in E indicates holes representing fluid-filled vacuoles and/or RPE cell migration into the retina. Arrowhead in F indicate extreme remodeling events of GFAP/GS-positive Müller cell processes displaced into the choroid. ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bars = 20 μm.
ATP induces Müller cell expression of cell cycling markers
Another aspect of the late-stage retinal degenerative process recently highlighted in the literature is that Müller glial cells begin to express markers of cell cycling and proliferation (Albarracin and Valter, 2012; Wan et al., 2008). However, this has not yet been described for heritable retinal degenerations. To investigate this, retinae from 2-year-old P23H rats and 3 and 6 month ATP-treated rats were labeled for Müller cells (GS, green) and the cell cycling marker cyclin D1, a marker of G1/S phase (cyclin D1, red; Fig. 10). In P23H rats, there were regions of retina where Müller cell expression of GS was reduced, suggesting a change in glutamate–glutamine recycling in these cells. In these regions, also, the Müller cells labelled for cyclin D1 (Fig. 10A). This response was apparent in the ATP-treated eyes at 3 months (Fig.10C–F) and 6 months (Fig.10G–J) but never in saline-treated, contralateral control eyes (Fig. 10B). In particular, at 6 months after ATP, there was evidence of RPE cell expression of cyclin D1 (Fig. 10G, arrowhead) and strong labeling of Müller cells. In addition, cyclin D1 expression was apparent in Müller cells that had migrated outside the neural retina into the choroid (Fig. 10I, arrow).
Figure 10.

Müller cells express the cell cycling marker cyclin D1 in P23H rat retinae and ATP-induced photoreceptor degeneration. Sections of retinae from 2-year-old P23H rat model of retinitis pigmentosa (A), saline-treated rat eyes at 6 months (B), ATP-treated rat eyes at 3 months (C: tile scan; D–F: magnification of three areas from C), and ATP-treated rat eyes at 6 months (G: tile scan; H–J: magnification of three areas from G) post-injection were labeled for Müller glial cells (GS, green), cell cycling (cyclin D1, red), and cell nuclei (DAPI, blue). Arrowhead in G indicates RPE cyclin D1 expression. Arrow in I indicates extreme remodeling events of cyclin D1-positive Müller cells displaced into the choroid. RPE, retinal pigment epithelium; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bars = 100 μm.
To probe further whether Müller cells were indeed expressing markers of re-entry into the cell cycle, 3 and 6 month ATP-treated retinae were labelled with an antibody against Ki-67, a protein that is present during all active phases of the cell cycle, G1, S, G2, and mitosis (Abdouh and Bernier, 2006). Ki67 was not present in saline-treated control eyes (Fig. 1A,D) or in regions of 3 month, ATP-treated retina in which photoreceptor nuclei were still present (Fig. 1B). Ki67 labeling was apparent in GS-positive Müller cells and also other cells in regions of ATP-treated retina in which the photoreceptor layer was absent at both 3 and 6 months (Fig. 1C,E,F). Taken together, these results suggest that ATP treatment induces retinal degeneration and neuronal and Müller glial remodeling consistent with that seen in retinal degenerations. Furthermore, this model recapitulates the more extreme instances of remodeling, neuronal migration into the choroid, and Müller cell expression of cell cycle markers seen in the aged P23H rat retina.
Figure 11.
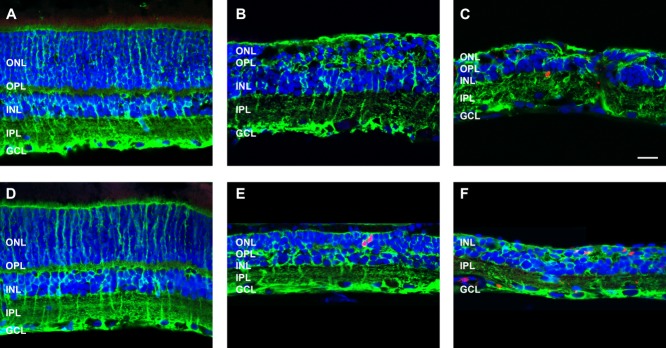
Müller cells express the cell cycling marker Ki67 following ATP-induced photoreceptor degeneration. Sections of retinae from saline- and ATP-treated eyes were labeled for Müller glial cells (GS, green), cell cycling (Ki67, red), and cell nuclei (DAPI, blue) at 3 months (A: saline; B,C: ATP) and 6 months (D: saline; E,F: ATP) post-injection. ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bar = 20 μm.
DISCUSSION
This study characterizes the effects of a single, intravitreal injection of 50 mM ATP on the integrity of rat retinal neurons and glia over 6 months. ATP caused complete loss of visual acuity within 1 day, loss of rod and cone pathway retinal function and 50% of the photoreceptors within 1 week, and ongoing retinal degeneration. At 3 months, 80% of the photoreceptor nuclei were lost, and inner retinal cells and the general integrity of the inner plexiform layer and ganglion cell layer remained intact. At 6 months, photoreceptors were absent, and extreme degenerative and remodeling events, such as neuronal and glial migration outside the neural retina and phenotypic changes in glial cell markers, were observed. These extreme changes were very similar to those observed in the 2-year-old P23H rat model of retinitis pigmentosa. Overall, the use of intravitreal ATP administration to induce photoreceptor death and retinal remodeling recapitulates many of the changes that occur in retinal degeneration. This model may provide a valuable tool for the development of pharmaceutical therapies aimed at slowing vision loss or for the testing of electronic implants aimed at restoring vision in retinal degeneration.
ATP induces photoreceptor death, visual dysfunction, and retinal remodeling consistent with previous reports of retinal degenerations
Many common causes of blindness involve the death of retinal photoreceptors. This is followed by progressive inner retinal cell remodeling (Fletcher and Kalloniatis, ,; Gargini et al., 2007; Jones et al., ,; Marc and Jones, 2003; Marc et al., ,; Pignatelli et al., 2004; Strettoi et al., 2002; Zhu et al., 2013). For an inducible model to be useful, it must recapitulate these changes. In the present study, ATP was found to induce complete loss of photopic visual responsiveness within 1 day, as measured by the cone electroretinogram and the optokinetic response. By 6 days after ATP administration, both rod and cone pathway electroretinogram responses were negligible. In contrast, vision in the saline-injected, contralateral control eye was normal, suggesting no systemic side effects. This finding is in line with work by Puthussery and Fletcher (2009), which showed that the function of the retina, as measured by the ERG, was absent 5 days after ATP administration. In addition to loss of functional vision, ATP induced photoreceptor specific death and an ongoing degenerative process. These changes occurred despite the likelihood that the ATP would be broken down by endogenous ectonucleoside triphosphate diphosphohydrolases (E-NTPDases) in the eye within the first day after injection (Nedeljkovic et al., 2005; Ricatti et al., 2009). Previous work has shown that ATP induces retinal degeneration via a direct neurotoxic action on the photoreceptors rather than the RPE or microglia or the inner retinal neurons, likely via a P2X7-R-mediated action (Notomi et al., ,; Puthussery and Fletcher, 2009). Our data support this and furthermore show that ATP induces an initial insult that causes photoreceptor failure and subsequent loss, followed by ongoing retinal degeneration and remodeling.
In retinal degenerations, both heritable and acquired, the remodeling process has been suggested to occur in three stages, documented and reviewed thoroughly by Marc et al. (2003). Broadly, phase 1 involves a primary insult to the photoreceptors and photoreceptor death, phase 2 is characterized by extensive photoreceptor death and changes in the inner retinal neurons and glia, and phase 3 occurs in response to the loss of the last of the photoreceptors and involves global remodeling of the remnant retina. With regard to ATP-induced photoreceptor death, phase 1 was found to occur within the first week after injection, during which the initial ATP insult induced rapid photoreceptor dysfunction and death, affecting both rods and cones. At 3 months after ATP administration, phase 2 degeneration was found to occur in regions where the photoreceptors remained, and early phase 3 was found to occur in regions devoid of photoreceptor nuclei and terminals. By 6 months after ATP administration, evidence of cones and photoreceptor terminals was negligible, and many examples of phase 3 remodeling (early, middle, and late) were apparent (Marc et al., 2003).
It has been suggested that, as long as photoreceptors remain, retinal remodeling is subtle and abstruse; however, when all photoreceptors are lost, significant remodeling occurs, and it is similar in most retinal degenerations (Jones et al., ,). In general, our findings relating to neuronal, glial, and blood vessel remodeling at 3 months after ATP induced photoreceptor death seem concordant with this hypothesis. At 3 months, evidence of phase 2 degeneration and remodeling (Marc et al., 2003) in regions where photoreceptor nuclei remained included displaced photoreceptor terminals into the remaining outer nuclear layer; rod bipolar and horizontal cell dendrite retraction and extension to contact remnant photoreceptor terminals; relative preservation of the inner retina, specifically bipolar cell terminals and amacrine/ganglion cells and their processes; and Müller glia being structurally sound but gliotic; blood vessel profiles were similar to those seen in saline-treated eyes. Evidence of early phase 3 remodeling in regions devoid of photoreceptors included Müller glia hypertrophy; scar/seal formation and remodeling, with Müller cell nuclei at both the top and bottom of the inner nuclear layer and aberrant processes; and abnormal vasculature, with the number of blood vessel profiles reduced. Taken together, the retinal changes in neurons, glia, and blood vessels observed at 3 months after ATP-induced photoreceptor death best represent phase 2 and early phase 3 stages of retinal degeneration and remodeling (Marc et al., 2003).
By 6 months after ATP administration, phase 3 remodeling was apparent (Marc et al., 2003). Evidence of phase 3 remodeling events affecting neurons included a reduction in number of amacrine and/or ganglion cells in the ganglion cell layer, migration of horizontal and bipolar cell toward the inner plexiform layer, migration of amacrine and ganglion cells toward the glial seal and development of multicellular fascicles and microneuromas; for example, horizontal cells had begun to migrate within the inner nuclear layer and were found to extend processes into the inner plexform layer to make contact with bipolar cell terminals. Evidence of phase 3 remodeling events affecting Müller glia included hypertrophy and gliosis, consolidation of the glial seal at the margins of the transverse retina, and Müller cell processes surrounding invading RPE cells and new neurites. In addition, there was evidence of late phase 3 remodeling events affecting the blood vessel and RPE, including abnormal morphology, vessel and RPE cell loss, and invasion of RPE into the neural retina, consistent with changes seen in genetic rat models of retinal degeneration with age (Villegas-Pérez et al., 1998; Wang et al., 2003). Ten major types of restructuring of the retina have been suggested to occur in phase 3 that are consistent across heritable retinal degenerations in humans and mice (Jones et al., 2003); we see many examples of these changes in the ATP-treated eyes at 3 months and all of these changes at 6 months (Table2).
Table 2.
In the ATP-Treated Eyes There Are Remodeling Events Consistent With the 10 Major Types of Restructuring of the Retina Seen in Retinal Degenerations (Jones et al., 2003)
| Remodeling event | 3 Months | 6 Months | |
|---|---|---|---|
| Neurons | Cell death including ganglion cell loss | − | + |
| Relocation of all types of surviving neurons | − | + | |
| Fragmentation of the inner plexiform layer lamination | − | + | |
| Evolution of new neurites in complex fascicles surrounded by Müller cell processes | − | + | |
| Formation of new synapses throughout the retina | + | + | |
| Müller glia | Migration | + | + |
| Hypertrophic: column formation | + | + | |
| Glial seal at the distal margin of the retina | + | + | |
| Blood vessel | Atrophy and remodelling | + | + |
| RPE | Invasion of RPE into the retina | + | + |
Evidence of the remodeling event is indicated by a plus sign, absence by a minus sign.
ATP induces extreme neuronal and glial remodeling consistent with the P23H rat model of retinitis pigmentosa
In addition to the retinal remodeling changes detailed above, two types of extreme remodeling event were also observed: 1) glial and neural cell exit from the neural retina into the choroid and formation of new extraretinal neuronal connections and 2) alterations in Müller glia phenotype, including loss of glutamine synthetase and expression of cell cycling markers. These changes were not specific to ATP-induced retinal degeneration but were also observed in the P23H rat model of retinitis pigmentosa at 2 years. This suggests that these extreme remodeling events are a late-stage consequence of photoreceptor death and the degenerative process.
With regard to glial and neural cell exit from the neural retina into the choroid, this type of remodeling event has been described in the light-damage-induced model of retinal degeneration in the rat, in which emigration of Müller cells and neurons of all classes from the retina into the choroid was observed (Marc et al., 2008). Marc et al. (2008) suggest, in an unpublished observation, that this event has been observed in human retinitis pigmentosa. In addition, it may occur in human AMD as well (Pow and Sullivan, 2007; Sullivan et al., 2007). We suggest that neuronal and glial emigration and new neuronal synapse formation in the choroid may be an extreme remodeling event that is consistent across retinal degenerations in both retinitis pigmentosa and AMD in humans and animal models. We hypothesize that this occurs in response to failure and loss of the RPE as part of an ongoing degenerative process. The RPE and Bruch’s membrane, a layered structure consisting mainly of collagen, provide a structural barrier between the choroid and the retina (Guymer et al., 1999). In regions where the RPE dies or remodels and invades the retina, the barrier between the retina and choroid would be weakened. This would allow migrating glia and neurons to exit the neural retina and enter the choroid; however, more research is required to confirm whether failure of RPE integrity precedes neuronal/glial migration into the choroid.
In addition, we have shown that in both the P23H rat and also the ATP-induced model, Müller glia undergo extreme changes in phenotype, including loss of GS and concomitant increase in expression of cell cycling markers cyclin D1 and Ki67. GS is a key enzyme expressed by Müller cells that is important in the degradation of glutamate released in neurotransmission to glutamine for recycling (Bringmann et al., 2009). The loss of GS suggests that in some regions Müller glia are no longer able to perform one of their key neuroregulatory functions. A loss of GS and/or increase in glutamate levels in Müller glia has been reported previously for retinal degenerations such as light damage (Albarracin and Valter, 2012; Marc et al., 2008), retinal detachment (Lewis et al., 1994; Marc et al., 1998), and retinitis pigmentosa (Fletcher and Kalloniatis, 1997). However, in addition to this, we see Müller cell expression of cell cycling markers in these regions of depleted GS. Cyclin D1 in Müller cells has been reported following light damage (Albarracin and Valter, 2012) and in NMU models of retinal degeneration (Wan et al., 2008). Albarracin and Valter suggest that Müller cells are undergoing proliferative changes to restore retinal structure, similar to the role of fibroblasts in skin injuries and scarring. However, an alternative hypothesis is that Müller glia are de-differentiating to a progenitor-like state as occurs in fish (Moshiri et al., 2004; Raymond et al., 2006) and birds (Fischer, 2005; Fischer and Omar, 2005). In these species, Müller cells are able to de-differentiate and re-enter the mitotic cell cycle to regenerate retinal neurons, restoring some level of functional vision. For mammals, it is believed that this process is halted by lack of appropriate differentiation factor expression. However, it has been reported that in rodents these proliferating glia are able, with the correct stimulus, to form inner retinal neurons such as bipolar and amacrine cells (Karl et al., 2008) as well as rhodopsin-expressing photoreceptors (Wan et al., 2008). The present model of ATP-induced retinal degeneration may prove useful for testing which factors support glial de-differentiation.
Other applications and limitations of ATP-induced retinal degeneration
The heterogeneity of retinal degenerations and the variability in the progression of these diseases means that investigating therapeutic strategies to ameliorate these disorders can be a problem. The advantage of an inducible model of retinal degeneration is that the start and process of the disease can be controlled, and in this case the agent, ATP, can be administered to one eye only, so systemic side effects are negligible, and viable vision remains in the other eye. In these respects, the ATP-induced retinal degeneration model has advantages over many of the other models, such as NMU, which affects both eyes and induces cancer with time (Tsubura et al., 2011); iodoacetate, which affects both eyes (Noell, 1951; Scott et al., 2011; Wang et al., 2011); and light damage, which has limited applicability in pigmented animals (Hunter et al., 2012; Marc et al., 2008).
The ATP-induced retinal degeneration model does have potential limitations. One of these is that both the rod and the cone photoreceptors appear to undergo simultaneous degeneration. This is unlike many heritable retinal degenerations such as retinitis pigmentosa, in which rods degenerate before cones (Fletcher et al., 2011), but is similar to AMD, in which both rods and cones are affected (Curcio, 2001). In addition, as purinergic receptors are located throughout the rodent retina (Ward et al., 2010), there is the potential for ATP to induce significant damage to neurons other than photoreceptors (Hu et al., 2010). Inner retinal neurons, such as calretinin-positive amacrine and ganglion cells, express P2X7-Rs (Vessey and Fletcher, 2012). Thus, although these inner retinal neurons remain morphologically intact up to 3 months after the ATP-induced insult, their function may be altered. Despite these potential limitations, our preliminary evidence suggests that ATP-induced photoreceptor degeneration is a viable model in larger vertebrates, such as the cat, providing a useful model for testing of electronic devices aimed at restoring vision in humans (unpublished observation). Our data for the rat suggest that the optimal time to test such devices would be 3 months after ATP administration, because this is a time when most photoreceptors are lost but retinal remodeling is not rampant and ganglion cell numbers have not been significantly reduced. However, testing of retinal implants at later stages may also be beneficial for comparison with late-stage retinal degenerations in humans.
CONCLUSIONS
The present study shows that intravitreal ATP administration induces photoreceptor death and retinal remodeling. We find that it replicates many of the changes that occur in retinal degenerations and that it induces some extreme remodeling events that are also common to heritable retinal degenerations and AMD. This model provides a valuable tool for investigating the degenerative process, glial cell de-differentiation and proliferation, and therapies aimed at slowing vision loss or electronically restoring vision.
Acknowledgments
The authors thank Dr. Krisztina Valter-Kocsi and Professor Jonathan Stone (Australian National University) for providing breeding pairs of Pro23His rats. In addition, we are indebted to the support of Ms. Andrea Rassell and Ms. Lidia Trogrlic for technical assistance with this project. The Bionics Institute acknowledges the support it receives from the Victorian Government through its Operational Infrastructure Support Program.
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest.
ROLE OF AUTHORS
All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: ELF, KAV. Acquisition of data: KAV, ELF, FPA, UG. Analysis and interpretation of data: KAV, ELF, FPA, UG. Drafting of the manuscript: KAV, UG, FPA, AIJ, JAP, TH, RUdI, ELF. Critical revision of the manuscript for important intellectual content: KAV, UG, FPA, AIJ, JAP, TH, RUdI, ELF. Statistical analysis: KAV, FPA. Obtained funding: ELF. Administrative, technical, and material support: ELF. Study supervision: KAV, ELF.
LITERATURE CITED
- Abdouh M, Bernier G. In vivo reactivation of a quiescent cell population located in the ocular ciliary body of adult mammals. Exp Eye Res. 2006;83:153–164. doi: 10.1016/j.exer.2005.11.016. [DOI] [PubMed] [Google Scholar]
- Abercrombie M, Johnson ML. Quantitative histology of Wallerian degeneration: I. Nuclear population in rabbit sciatic nerve. J Anat. 1946;80:37–50. [PubMed] [Google Scholar]
- Albarracin R, Valter K. 670 nm red light preconditioning supports Muller cell function: evidence from the white light-induced damage model in the rat retina. Photochem Photobiol. 2012;88:1418–1427. doi: 10.1111/j.1751-1097.2012.01130.x. [DOI] [PubMed] [Google Scholar]
- Araki CM, Hamassaki-Britto DE. Calretinin co-localizes with the NMDA receptor subunit NR1 in cholinergic amacrine cells of the rat retina. Brain Res. 2000;869:220–224. doi: 10.1016/s0006-8993(00)02364-7. [DOI] [PubMed] [Google Scholar]
- Bergner AJ, Stamp LA, Gonsalvez DG, Allison MB, Olson DP, Myers MG, Jr, Anderson CR, Young HM. Birthdating of myenteric neuron subtypes in the small intestine of the mouse. J Comp Neurol. 2014;522:514–527. doi: 10.1002/cne.23423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienvenu F, Jirawatnotai S, Elias JE, Meyer CA, Mizeracka K, Marson A, Frampton GM, Cole MF, Odom DT, Odajima J, Geng Y, Zagozdzon A, Jecrois M, Young RA, Liu XS, Cepko CL, Gygi SP, Sicinski P. Transcriptional role of cyclin D1 in development revealed by a genetic-proteomic screen. Nature. 2010;463:374–378. doi: 10.1038/nature08684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Biedermann B, Francke M, Iandiev I, Grosche J, Wiedemann P, Albrecht J, Reichenbach A. Role of retinal glial cells in neurotransmitter uptake and metabolism. Neurochem Int. 2009;54:143–160. doi: 10.1016/j.neuint.2008.10.014. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Purinergic signalling: Its unpopular beginning, its acceptance and its exciting future. Bioessays. 2012;34:218–225. doi: 10.1002/bies.201100130. [DOI] [PubMed] [Google Scholar]
- Chen H, Weber AJ. Expression of glial fibrillary acidic protein and glutamine synthetase by Muller cells after optic nerve damage and intravitreal application of brain-derived neurotrophic factor. Glia. 2002;38:115–125. doi: 10.1002/glia.10061. [DOI] [PubMed] [Google Scholar]
- Curcio CA. Photoreceptor topography in ageing and age-related maculopathy. Eye. 2001;15:376–383. doi: 10.1038/eye.2001.140. [DOI] [PubMed] [Google Scholar]
- Danciger M, Yang H, Ralston R, Liu Y, Matthes MT, Peirce J, Lavail MM. Quantitative genetics of age-related retinal degeneration: a second F1 intercross between the A/J and C57BL/6 strains. Mol Vis. 2007;13:79–85. [PMC free article] [PubMed] [Google Scholar]
- Douglas RM, Alam NM, Silver BD, McGill TJ, Tschetter WW, Prusky GT. Independent visual threshold measurements in the two eyes of freely moving rats and mice using a virtual-reality optokinetic system. Vis Neurosci. 2005;22:677–684. doi: 10.1017/S0952523805225166. [DOI] [PubMed] [Google Scholar]
- Dureau P, Bonnel S, Menasche M, Dufier JL, Abitbol M. Quantitative analysis of intravitreal injections in the rat. Curr Eye Res. 2001;22:74–77. doi: 10.1076/ceyr.22.1.74.6974. [DOI] [PubMed] [Google Scholar]
- Eells JT, Salzman MM, Lewandowski MF, Murray TG. Formate-induced alterations in retinal function in methanol-intoxicated rats. Toxicol Appl Pharmacol. 1996;140:58–69. doi: 10.1006/taap.1996.0197. [DOI] [PubMed] [Google Scholar]
- Fischer AJ. Neural regeneration in the chick retina. Prog Ret Eye Res. 2005;24:161–182. doi: 10.1016/j.preteyeres.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Omar G. Transitin, a nestin-related intermediate filament, is expressed by neural progenitors and can be induced in Muller glia in the chicken retina. J Comp Neurol. 2005;484:1–14. doi: 10.1002/cne.20406. [DOI] [PubMed] [Google Scholar]
- Fletcher EL, Kalloniatis M. Neurochemical architecture of the normal and degenerating rat retina. J Comp Neurol. 1996;376:343–360. doi: 10.1002/(SICI)1096-9861(19961216)376:3<343::AID-CNE1>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Fletcher EL, Kalloniatis M. Neurochemical development of the degenerating rat retina. J Comp Neurol. 1997;388:1–22. [PubMed] [Google Scholar]
- Fletcher EL, Jobling AI, Vessey KA, Luu C, Guymer RH, Baird PN. Animal models of retinal disease. Prog Mol Biol Transl Sci. 2011;100:211–286. doi: 10.1016/B978-0-12-384878-9.00006-6. [DOI] [PubMed] [Google Scholar]
- Gábriel R, Witkovsky P. Cholinergic, but not the rod pathway-related glycinergic (All), amacrine cells contain calretinin in the rat retina. Neurosci Lett. 1998;247:179–182. doi: 10.1016/s0304-3940(98)00323-1. [DOI] [PubMed] [Google Scholar]
- Galatioto J, Mascareno E, Siddiqui MA. CLP-1 associates with MyoD and HDAC to restore skeletal muscle cell regeneration. J Cell Sci. 2010;123:3789–3795. doi: 10.1242/jcs.073387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargini C, Terzibasi E, Mazzoni F, Strettoi E. Retinal organization in the retinal degeneration 10 (rd10) mutant mouse: a morphological and ERG study. J Comp Neurol. 2007;500:222–238. doi: 10.1002/cne.21144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaschke A, Weiland J, Del Turco D, Steiner M, Peichl L, Glösmann M. Thyroid hormone controls cone opsin expression in the retina of adult rodents. J Neurosci. 2011;31:4844–4851. doi: 10.1523/JNEUROSCI.6181-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greferath U, Grünert U, Wässle H. Rod bipolar cells in the mammalian retina show protein kinase C-like immunoreactivity. J Comp Neurol. 1990;301:433–442. doi: 10.1002/cne.903010308. [DOI] [PubMed] [Google Scholar]
- Guymer R, Luthert P, Bird A. Changes in Bruch’s membrane and related structures with age. Prog Ret Eye Res. 1999;18:59–90. doi: 10.1016/s1350-9462(98)00012-3. [DOI] [PubMed] [Google Scholar]
- Hageman GS, Johnson LV. Biochemical characterization of the major peanut-agglutinin-binding glycoproteins in vertebrate retinae. J Comp Neurol. 1986;249:499–510, 482–493. doi: 10.1002/cne.902490406. [DOI] [PubMed] [Google Scholar]
- Ho T, Vessey KA, Cappai R, Dinet V, Mascarelli F, Ciccotosto GD, Fletcher EL. Amyloid precursor protein is required for normal function of the rod and cone pathways in the mouse retina. PLoS One. 2012;7:e29892. doi: 10.1371/journal.pone.0029892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Lu W, Zhang M, Zhang X, Argall AJ, Patel S, Lee GE, Kim YC, Jacobson KA, Laties AM, Mitchell CH. Stimulation of the P2X7 receptor kills rat retinal ganglion cells in vivo. Exp Eye Res. 2010;91:425–432. doi: 10.1016/j.exer.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JJ, Morgan JI, Merigan WH, Sliney DH, Sparrow JR, Williams DR. The susceptibility of the retina to photochemical damage from visible light. Prog Ret Eye Res. 2012;31:28–42. doi: 10.1016/j.preteyeres.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res. 1998;57:1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- Jobling AI, Vessey KA, Waugh M, Mills SA, Fletcher EL. A naturally occurring mouse model of achromatopsia: characterization of the mutation in cone transducin and subsequent retinal phenotype. Invest Ophthalmol Vis Sci. 2013;54:3350–3359. doi: 10.1167/iovs.13-11831. [DOI] [PubMed] [Google Scholar]
- Johnson J, Tian N, Caywood MS, Reimer RJ, Edwards RH, Copenhagen DR. Vesicular neurotransmitter transporter expression in developing postnatal rodent retina: GABA and glycine precede glutamate. J Neurosci. 2003;23:518–529. doi: 10.1523/JNEUROSCI.23-02-00518.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BW, Watt CB, Frederick JM, Baehr W, Chen CK, Levine EM, Milam AH, Lavail MM, Marc RE. Retinal remodeling triggered by photoreceptor degenerations. J Comp Neurol. 2003;464:1–16. doi: 10.1002/cne.10703. [DOI] [PubMed] [Google Scholar]
- Jones BW, Kondo M, Terasaki H, Lin Y, McCall M, Marc RE. Retinal remodeling. Jpn J Ophthalmol. 2012;56:289–306. doi: 10.1007/s10384-012-0147-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karl MO, Hayes S, Nelson BR, Tan K, Buckingham B, Reh TA. Stimulation of neural regeneration in the mouse retina. Proc Natl Acad Sci U S A. 2008;105:19508–19513. doi: 10.1073/pnas.0807453105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Key G, Petersen JL, Becker MH, Duchrow M, Schluter C, Askaa J, Gerdes J. New antiserum against Ki-67 antigen suitable for double immunostaining of paraffin wax sections. J Clin Pathol. 1993;46:1080–1084. doi: 10.1136/jcp.46.12.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Key G, Duchrow M, Schluter C, Flad HD, Gerdes J. Epitope analysis of antibodies recognising the cell proliferation associated nuclear antigen previously defined by the antibody Ki-67 (Ki-67 protein) J Clin Pathol. 1994;47:524–528. doi: 10.1136/jcp.47.6.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVail MM, Gorrin GM. Protection from light damage by ocular pigmentation: analysis using experimental chimeras and translocation mice. Exp Eye Res. 1987;44:877–889. doi: 10.1016/s0014-4835(87)80050-7. [DOI] [PubMed] [Google Scholar]
- LaVail MM, Gorrin GM, Repaci MA. Strain differences in sensitivity to light-induced photoreceptor degeneration in albino mice. Curr Eye Res. 1987a;6:825–834. doi: 10.3109/02713688709034850. [DOI] [PubMed] [Google Scholar]
- LaVail MM, Gorrin GM, Repaci MA, Thomas LA, Ginsberg HM. Genetic regulation of light damage to photoreceptors. Invest Ophthalmol Vis Sci. 1987b;28:1043–1048. [PubMed] [Google Scholar]
- Lewis GP, Guerin CJ, Anderson DH, Matsumoto B, Fisher SK. Rapid changes in the expression of glial cell proteins caused by experimental retinal detachment. Am J Ophthalmol. 1994;118:368–376. doi: 10.1016/s0002-9394(14)72962-9. [DOI] [PubMed] [Google Scholar]
- Lyubarsky A, Nikonov S, Pugh EN., Jr The kinetics of inactivation of the rod phototransduction cascade with constant Ca2+i. J Gen Physiol. 1996;107:19–34. doi: 10.1085/jgp.107.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyubarsky AL, Pugh EN., Jr Recovery phase of the murine rod photoresponse reconstructed from electroretinographic recordings. J Neurosci. 1996;16:563–571. doi: 10.1523/JNEUROSCI.16-02-00563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marc RE, Jones BW. Retinal remodeling in inherited photoreceptor degenerations. Mol Neurobiol. 2003;28:139–147. doi: 10.1385/MN:28:2:139. [DOI] [PubMed] [Google Scholar]
- Marc RE, Murry RF, Fisher SK, Linberg KA, Lewis GP. Amino acid signatures in the detached cat retina. Invest Ophthalmol Vis Sci. 1998;39:1694–1702. [PubMed] [Google Scholar]
- Marc RE, Jones BW, Watt CB, Strettoi E. Neural remodeling in retinal degeneration. Prog Ret Eye Res. 2003;22:607–655. doi: 10.1016/s1350-9462(03)00039-9. [DOI] [PubMed] [Google Scholar]
- Marc RE, Jones BW, Watt CB, Vazquez-Chona F, Vaughan DK, Organisciak DT. Extreme retinal remodeling triggered by light damage: implications for age related macular degeneration. Mol Vis. 2008;14:782–806. [PMC free article] [PubMed] [Google Scholar]
- McIver SC, Stanger SJ, Santarelli DM, Roman SD, Nixon B, McLaughlin EA. A unique combination of male germ cell miRNAs coordinates gonocyte differentiation. PLoS One. 2012;7:e35553. doi: 10.1371/journal.pone.0035553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melone M, Burette A, Weinberg RJ. Light microscopic identification and immunocytochemical characterization of glutamatergic synapses in brain sections. J Comp Neurol. 2005;492:495–509. doi: 10.1002/cne.20743. [DOI] [PubMed] [Google Scholar]
- Mittag TW, Bayer AU. Light-induced retinal damage in mice carrying a mutated SOD I gene. Exp Eye Res. 1999;69:677–683. doi: 10.1006/exer.1999.0748. [DOI] [PubMed] [Google Scholar]
- Moshiri A, Close J, Reh TA. Retinal stem cells and regeneration. Int J Dev Biol. 2004;48:1003–1014. doi: 10.1387/ijdb.041870am. [DOI] [PubMed] [Google Scholar]
- Murray TG, Burton TC, Rajani C, Lewandowski MF, Burke JM, Eells JT. Methanol poisoning. A rodent model with structural and functional evidence for retinal involvement. Arch Ophthalmol. 1991;109:1012–1016. doi: 10.1001/archopht.1991.01080070124049. [DOI] [PubMed] [Google Scholar]
- Nasonkin IO, Lazo K, Hambright D, Brooks M, Fariss R, Swaroop A. Distinct nuclear localization patterns of DNA methyltransferases in developing and mature mammalian retina. J Comp Neurol. 2011;519:1914–1930. doi: 10.1002/cne.22613. [DOI] [PubMed] [Google Scholar]
- Nedeljkovic N, Banjac A, Horvat A, Stojiljkovic M, Nikezic G. Developmental profile of NTPDase activity in synaptic plasma membranes isolated from rat cerebral cortex. Int J Dev Neurosci. 2005;23:45–51. doi: 10.1016/j.ijdevneu.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Noell WK. The effect of iodoacetate on the vertebrate retina. J Cell Physiol. 1951;37:283–307. doi: 10.1002/jcp.1030370209. [DOI] [PubMed] [Google Scholar]
- Notomi S, Hisatomi T, Kanemaru T, Takeda A, Ikeda Y, Enaida H, Kroemer G, Ishibashi T. Critical involvement of extracellular ATP acting on P2RX7 purinergic receptors in photoreceptor cell death. Am J Pathol. 2011;179:2798–2809. doi: 10.1016/j.ajpath.2011.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notomi S, Hisatomi T, Murakami Y, Terasaki H, Sonoda S, Asato R, Takeda A, Ikeda Y, Enaida H, Sakamoto T, Ishibashi T. Dynamic increase in extracellular ATP accelerates photoreceptor cell apoptosis via ligation of P2RX7 in subretinal hemorrhage. PLoS One. 2013;8:e53338. doi: 10.1371/journal.pone.0053338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien EE, Greferath U, Vessey KA, Jobling AI, Fletcher EL. Electronic restoration of vision in those with photoreceptor degenerations. Clin Exp Optom. 2012;95:473–483. doi: 10.1111/j.1444-0938.2012.00783.x. [DOI] [PubMed] [Google Scholar]
- Olney J, Price M, Salles KS, Labruyere J, Frierdich G. MK-801 powerfully protects against N-methyl aspartate neurotoxicity. Eur J Pharmacol. 1987;141:357–361. doi: 10.1016/0014-2999(87)90552-8. [DOI] [PubMed] [Google Scholar]
- Pathmanathan N, Balleine RL. Ki67 and proliferation in breast cancer. J Clin Pathol. 2013;66:512–516. doi: 10.1136/jclinpath-2012-201085. [DOI] [PubMed] [Google Scholar]
- Pignatelli V, Cepko CL, Strettoi E. Inner retinal abnormalities in a mouse model of Leber’s congenital amaurosis. J Comp Neurol. 2004;469:351–359. doi: 10.1002/cne.11019. [DOI] [PubMed] [Google Scholar]
- Pow DV, Sullivan RK. Nuclear kinesis, neurite sprouting and abnormal axonal projections of cone photoreceptors in the aged and AMD-afflicted human retina. Exp Eye Res. 2007;84:850–857. doi: 10.1016/j.exer.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Prusky GT, Alam NM, Beekman S, Douglas RM. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest Ophthalmol Vis Sci. 2004;45:4611–4616. doi: 10.1167/iovs.04-0541. [DOI] [PubMed] [Google Scholar]
- Puthussery T, Fletcher E. Extracellular ATP induces retinal photoreceptor apoptosis through activation of purinoceptors in rodents. J Comp Neurol. 2009;513:430–440. doi: 10.1002/cne.21964. [DOI] [PubMed] [Google Scholar]
- Puthussery T, Fletcher EL. Synaptic localization of P2X7 receptors in the rat retina. J Comp Neurol. 2004;472:13–23. doi: 10.1002/cne.20045. [DOI] [PubMed] [Google Scholar]
- Puthussery T, Fletcher EL. P2X2 receptors on ganglion and amacrine cells in cone pathways of the rat retina. J Comp Neurol. 2006;496:595–609. doi: 10.1002/cne.20889. [DOI] [PubMed] [Google Scholar]
- Puthussery T, Fletcher EL. Neuronal expression of P2X3 purinoceptors in the rat retina. Neuroscience. 2007;146:403–414. doi: 10.1016/j.neuroscience.2007.01.055. [DOI] [PubMed] [Google Scholar]
- Puthussery T, Yee P, Vingrys AJ, Fletcher EL. Evidence for the involvement of purinergic P2X receptors in outer retinal processing. Eur J Neurosci. 2006;24:7–19. doi: 10.1111/j.1460-9568.2006.04895.x. [DOI] [PubMed] [Google Scholar]
- Raymond PA, Barthel LK, Bernardos RL, Perkowski JJ. Molecular characterization of retinal stem cells and their niches in adult zebrafish. BMC Dev Biol. 2006;6:36. doi: 10.1186/1471-213X-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricatti MJ, Alfie LD, Lavoie EG, Sevigny J, Schwarzbaum PJ, Faillace MP. Immunocytochemical localization of NTPDases1 and 2 in the neural retina of mouse and zebrafish. Synapse. 2009;63:291–307. doi: 10.1002/syn.20605. [DOI] [PubMed] [Google Scholar]
- Scott PA, Kaplan HJ, Sandell JH. Anatomical evidence of photoreceptor degeneration induced by iodoacetic acid in the porcine eye. Exp Eye Res. 2011;93:513–527. doi: 10.1016/j.exer.2011.06.017. [DOI] [PubMed] [Google Scholar]
- Sherry DM, Wang MM, Bates J, Frishman LJ. Expression of vesicular glutamate transporter 1 in the mouse retina reveals temporal ordering in development of rod vs. cone and ON vs. OFF circuits. J Comp Neurol. 2003;465:480–498. doi: 10.1002/cne.10838. [DOI] [PubMed] [Google Scholar]
- Stingl K, Bach M, Bartz-Schmidt KU, Braun A, Bruckmann A, Gekeler F, Greppmaier U, Hörtdörfer G, Kusnyerik A, Peters T, Wilhelm B, Wilke R, Zrenner E. Safety and efficacy of subretinal visual implants in humans: methodological aspects. Clin Exp Optom. 2013a;96:4–13. doi: 10.1111/j.1444-0938.2012.00816.x. [DOI] [PubMed] [Google Scholar]
- Stingl K, Bartz-Schmidt KU, Besch D, Braun A, Bruckmann A, Gekeler F, Greppmaier U, Hipp S, Hörtdörfer G, Kernstock C, Koitschev A, Kusnyerik A, Sachs H, Schatz A, Stingl KT, Peters T, Wilhelm B, Zrenner E. Artificial vision with wirelessly powered subretinal electronic implant alpha-IMS. Proc Biol Sci. 2013b;280:20130077. doi: 10.1098/rspb.2013.0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strettoi E, Porciatti V, Falsini B, Pignatelli V, Rossi C. Morphological and functional abnormalities in the inner retina of the rd/rd mouse. J Neurosci. 2002;22:5492–5504. doi: 10.1523/JNEUROSCI.22-13-05492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan RK, Woldemussie E, Pow DV. Dendritic and synaptic plasticity of neurons in the human age-related macular degeneration retina. Invest Ophthalmol Vis Sci. 2007;48:2782–2791. doi: 10.1167/iovs.06-1283. [DOI] [PubMed] [Google Scholar]
- Taylor HR, Keeffe JE, Vu HT, Wang JJ, Rochtchina E, Pezzullo ML, Mitchell P. Vision loss in Australia. Med J Aust. 2005;182:565–568. doi: 10.5694/j.1326-5377.2005.tb06815.x. [DOI] [PubMed] [Google Scholar]
- Trifunović D, Sahaboglu A, Kaur J, Mencl S, Zrenner E, Ueffing M, Arango-Gonzalez B, Paquet-Durand F. Neuroprotective strategies for the treatment of inherited photoreceptor degeneration. Curr Mol Med. 2012;12:598–612. doi: 10.2174/156652412800620048. [DOI] [PubMed] [Google Scholar]
- Tsubura A, Lai YC, Miki H, Sasaki T, Uehara N, Yuri T, Yoshizawa K. Review: animal models of N-methyl-N-nitrosourea-induced mammary cancer and retinal degeneration with special emphasis on therapeutic trials. In Vivo. 2011;25:11–22. [PubMed] [Google Scholar]
- Tyler NK, Burns MS. Comparison of lectin reactivity in the vessel beds of the rat eye. Curr Eye Res. 1991;10:801–810. doi: 10.3109/02713689109013875. [DOI] [PubMed] [Google Scholar]
- Uesugi R, Yamada M, Mizuguchi M, Baimbridge KG, Kim SU. Calbindin D-28k and parvalbumin immunohistochemistry in developing rat retina. Exp Eye Res. 1992;54:491–499. doi: 10.1016/0014-4835(92)90127-e. [DOI] [PubMed] [Google Scholar]
- van Wijngaarden P, Brereton HM, Coster DJ, Williams KA. Genetic influences on susceptibility to oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2007;48:1761–1766. doi: 10.1167/iovs.06-0531. [DOI] [PubMed] [Google Scholar]
- Vessey KA, Fletcher EL. Rod and cone pathway signalling is altered in the P2X7 receptor knock out mouse. PLoS One. 2012;7:e29990. doi: 10.1371/journal.pone.0029990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessey KA, Wilkinson-Berka JL, Fletcher EL. Characterization of retinal function and glial cell response in a mouse model of oxygen-induced retinopathy. J Comp Neurol. 2011;519:506–527. doi: 10.1002/cne.22530. [DOI] [PubMed] [Google Scholar]
- Vessey KA, Greferath U, Jobling AI, Phipps JA, Ho T, Waugh M, Fletcher EL. Ccl2/Cx3cr1 knockout mice have inner retinal dysfunction but are not an accelerated model of AMD. Invest Ophthalmol Vis Sci. 2012;53:7833–7846. doi: 10.1167/iovs.12-10650. [DOI] [PubMed] [Google Scholar]
- Villegas-Pérez MP, Lawrence JM, Vidal-Sanz M, Lavail MM, Lund RD. Ganglion cell loss in RCS rat retina: a result of compression of axons by contracting intraretinal vessels linked to the pigment epithelium. J Comp Neurol. 1998;392:58–77. [PubMed] [Google Scholar]
- Wan J, Zheng H, Chen ZL, Xiao HL, Shen ZJ, Zhou GM. Preferential regeneration of photoreceptor from Muller glia after retinal degeneration in adult rat. Vis Res. 2008;48:223–234. doi: 10.1016/j.visres.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Wang S, Villegas-Pérez MP, Holmes T, Lawrence JM, Vidal-Sanz M, Hurtado-Montalbán N, Lund RD. Evolving neurovascular relationships in the RCS rat with age. Curr Eye Res. 2003;27:183–196. doi: 10.1076/ceyr.27.3.183.16053. [DOI] [PubMed] [Google Scholar]
- Wang W, Fernandez de Castro J, Vukmanic E, Zhou L, Emery D, Demarco PJ, Kaplan HJ, Dean DC. Selective rod degeneration and partial cone inactivation characterize an iodoacetic acid model of swine retinal degeneration. Invest Ophthalmol Vis Sci. 2011;52:7917–7923. doi: 10.1167/iovs.11-7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward MM, Fletcher EL. Subsets of retinal neurons and glia express P2Y1 receptors. Neuroscience. 2009;160:555–566. doi: 10.1016/j.neuroscience.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Ward MM, Puthussery T, Vessey KA, Fletcher EL. The role of purinergic receptors in retinal function and disease. Adv Exp Med Biol. 2010;664:385–391. doi: 10.1007/978-1-4419-1399-9_44. [DOI] [PubMed] [Google Scholar]
- Wenzel A, Grimm C, Samardzija M, Remé CE. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Ret Eye Res. 2005;24:275–306. doi: 10.1016/j.preteyeres.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Mistra S, Nivison-Smith L, Acosta ML, Fletcher EL, Kalloniatis M. Mapping cation entry in photoreceptors and inner retinal neurons during early degeneration in the P23H-3 rat retina. Vis Neurosci. 2013:1–11. doi: 10.1017/S0952523813000047. [DOI] [PubMed] [Google Scholar]