

Abstract
Glucokinase (GK) activators represent a class of type 2 diabetes therapeutics actively pursued due to the central role that GK plays in regulating glucose homeostasis. Herein we report a novel C5-alkyl-2-methylurea-substituted pyridine series of GK activators derived from our previously reported thiazolylamino pyridine series. Our efforts in optimizing potency, enzyme kinetic properties, and metabolic stability led to the identification of compound 26 (AM-9514). This analogue showed a favorable combination of in vitro potency, enzyme kinetic properties, acceptable pharmacokinetic profiles in preclinical species, and robust efficacy in a rodent PD model.
Keywords: Type 2 diabetes, glucokinase activator, GKA, methyl urea-substituted pyridines
Type 2 diabetes is a chronic metabolic disease affecting over 300 million people worldwide.1 It is characterized by elevated fasting plasma and hepatic glucose production and insulin resistance. It is a major risk factor for both microvascular and macrovascular diseases.2−4 Despite the availability of multiple antihyperglycemic agents, many patients still remain unable to safely achieve and maintain tight glycemic control, underscoring the need for more effective therapies.
Glucokinase (GK) is a member of the hexokinase family of intracellular enzymes. It is responsible for the conversion of glucose to glucose-6-phosphate, the first step in glucose utilization.5 GK is present in both pancreatic β-cells and liver hepatic cells. In the pancreas, it serves as a glucose sensor to control glucose-stimulated insulin secretion, while hepatic GK plays an important role in regulating net glucose utilization and production.6
It has been demonstrated that glucokinase activators (GKAs) can influence the enzyme’s kinetic profile by modulating both Km for glucose (also known as S0.5) and Vmax (maximal velocity of the glucose phosphorylation reaction catalyzed by GK).7 As a result, GKAs control blood glucose concentrations by enhancing the ability of pancreatic β-cells to sense glucose and increase insulin secretion in a glucose-dependent manner. In addition, GKAs act in the liver to improve glucose disposal by increasing glycogen storage and decreasing hepatic glucose output. The possible synergistic effect of this dual mechanism may provide an advantage in the treatment of type 2 diabetes.8 To date, a number of GK activators have advanced into clinical trials for the treatment of type 2 diabetes.9−9c
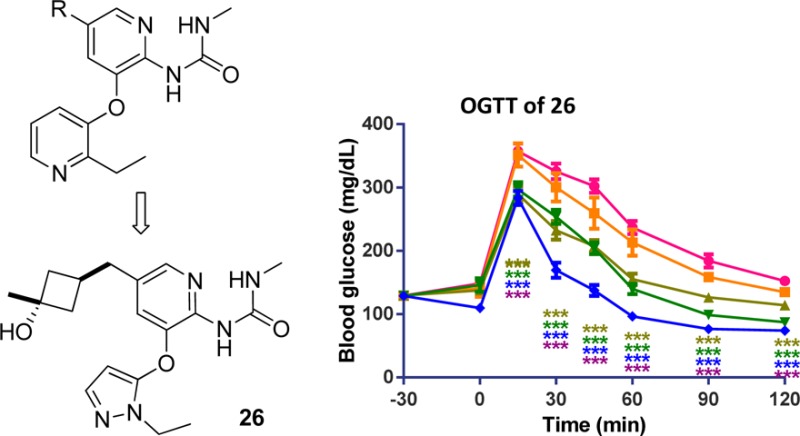
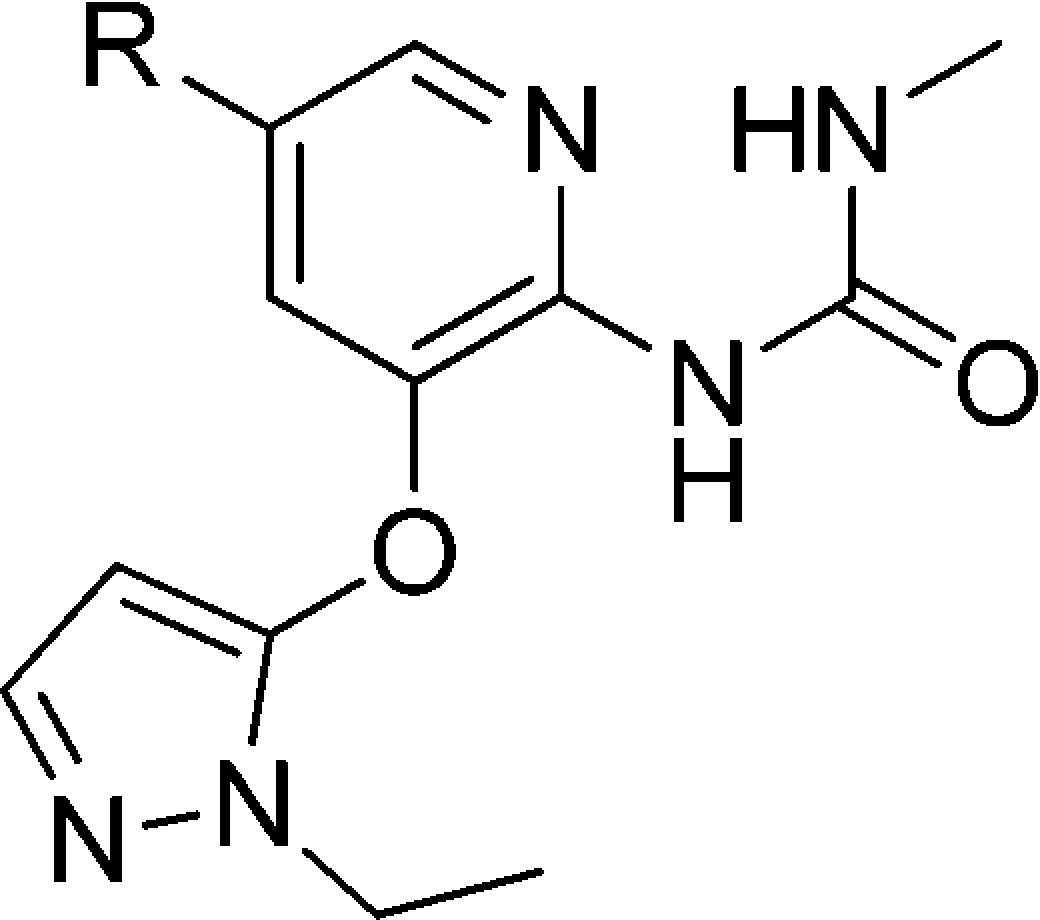
We have previously disclosed a series of thiadiazolylamino pyridine GKAs represented by compound A.10,11 To identify GKAs that are structurally distinct from that, we pursued a novel methyl urea-substituted pyridine series, where the critical bidentate hydrogen bond to Arg63 was maintained by the methyl urea moiety.12 Compound A had thioether substitutions at C5 of the middle pyridine ring. In pursuing more structure diversity, we would like to replace the thioether in compound A with a carbon-based linker in compound B as shown in Figure 1.
Figure 1.

From thioether-substituted thiazolylamino pyridines (A) to methyl urea-substituted pyridines (B).
Hypoglycemia is a key dose limiting adverse effect of glucokinase activators.13−15 In an attempt to mitigate this risk, we targeted GKAs that would not lower the S0.5 below a certain limit nor raise Vmax above a certain limit, preferably with S0.5 > 0.6 mM and Vmax between 0.8 and 1.3 of unactivated GK. A similar strategy has been proposed by others.9b,9c,16 Our threshold values were chosen in part based on data with Piragliatin (S0.5 of 0.28 mM and Vmax of 1.7) and MK-0941 (S0.5 of 0.25 mM and Vmax of 1.3),17 developed by Roche and Merck, respectively. Both compounds were associated with increased risk of hypoglycemia in clinical trials.14,18 In addition, compounds with Vmax lower than 0.8 could result in the phosphorylation rate of the enzyme-activator complex to be lower than the native enzyme at glucose concentrations obtainable in the fed state.19 Therefore, we targeted GKAs with Vmax between 0.8 and 1.3.
A number of C5-alkyl substituted analogues with 3-O-ethylpyridine substitution are summarized in Table 1. The simple isobutyl substituted analogue 1 was potent (EC50 = 0.12 μM) but metabolically unstable, indicated by the relatively high turnover rate in both rat and human liver microsomes. The microsomal stability of this series was improved by the incorporation of a variety of polar functional groups such as alcohols and ethers into the structure (compounds 2 and 4–7). For the ethers, potency improved when elongating the linker from two carbon to three and four (2, 3, and 4). However, the Vmax values for these compounds were lower than the target range. For the alcohols, although they were less potent than their ether analogues (2 vs 5; 4 vs 6), one of the primary alcohols, 6, had acceptable potency, kinetic parameters, and microsomal stability. However, compound 6 had poor stability in primary human hepatocytes (88% was metabolized).20 To reduce the hepatic clearance, the tertiary alcohol 7 was made. Although this improved the stability in primary human hepatocytes (28% metabolized),20 it also reduced the Vmax to an unacceptable level (0.73).
Table 1. Exploration of C5 Substitution of GKAs with 3-O-Ethylpyridine.
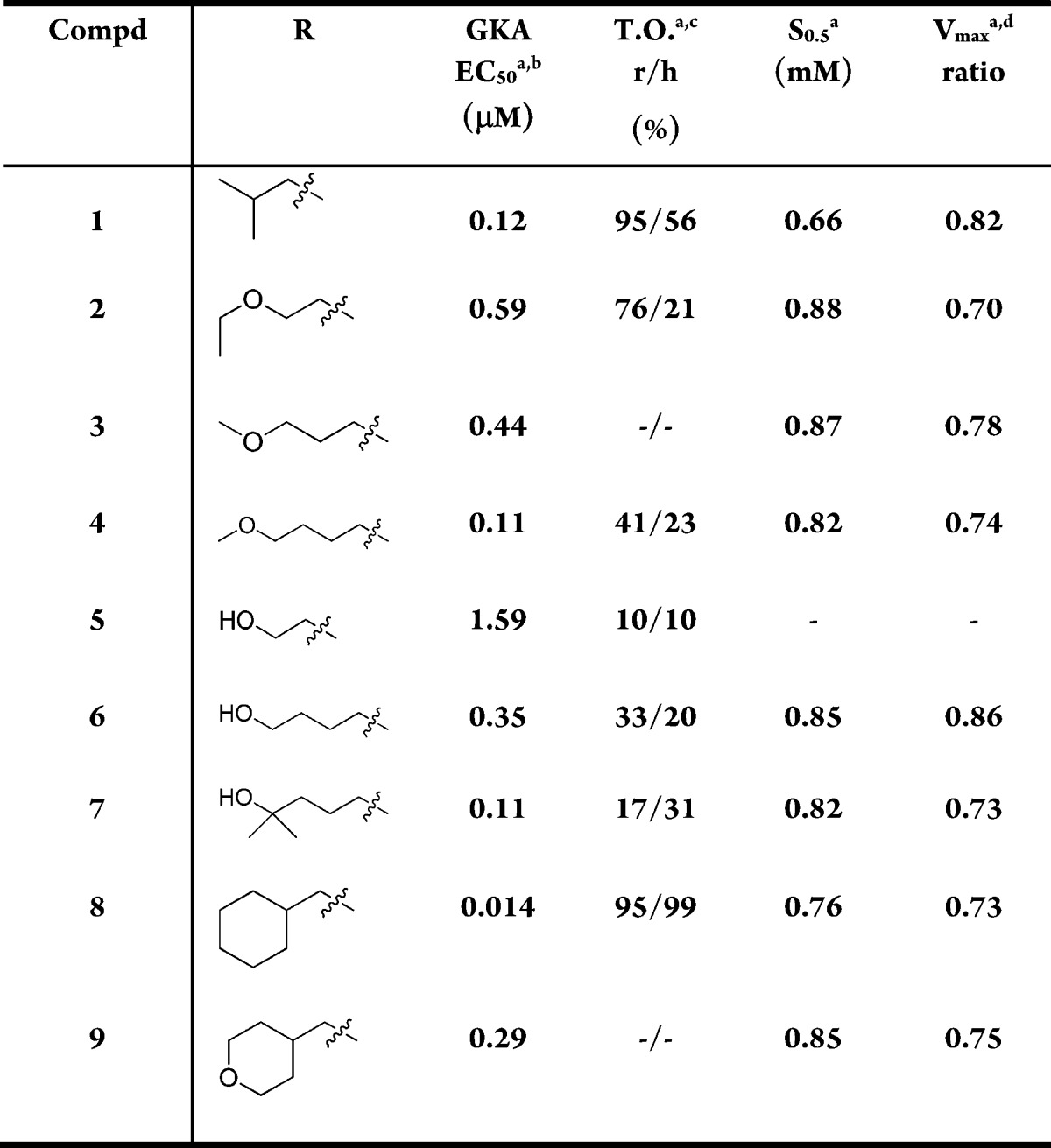
Mean standard deviations for the EC50, S0.5, and Vmax are ±30%, 17%, and 4%, respectively (n = 2). Also, human recombinant GK was used in all the assays.
For the EC50 assay, the glucose concentration was fixed at 5 mM.
Percent turnover: percentage of parent compound being metabolized after incubating in 0.25 mg/mL of rat or human liver microsomes for 30 min at 37 °C with an initial parent concentration of 1 μM.
Maximal velocity of the glucose phosphorylation reaction catalyzed by activated GK/unactivated GK.
We then explored cyclic moieties with one carbon linker such as cyclohexyl methyl or THP methyl substitution. The Vmax values for these compounds (8 and 9) were also low.
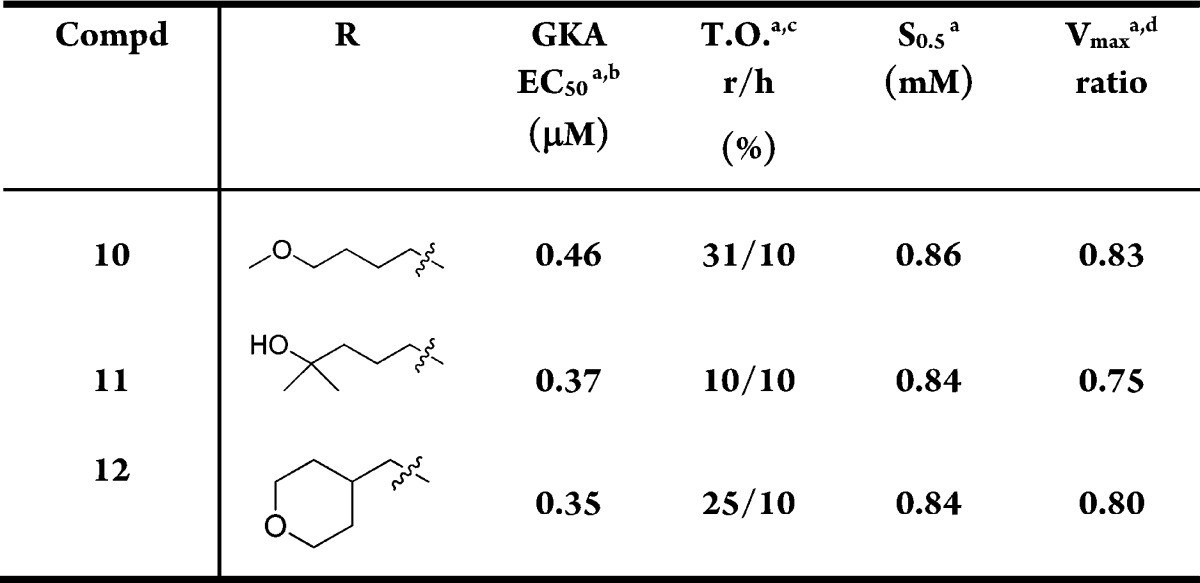
Because of the suboptimal Vmax associated with 3-O-ethylpyridine substitution, we changed the substitution to 3-O-ethylpyrazole. Table 2 shows that the analogues with 3-O-ethylpyrazole had improved Vmax values compared to the 3-O-ethylpyridines, although the potency was reduced somewhat (10 vs 4; 11 vs 7; 12 vs 9). These compounds generally had good microsomal stability.
Table 2. Exploration of C5 Substitution of GKAs with 3-O-Pyrazole.
Mean standard deviations for the EC50, S0.5, and Vmax are ±30%, 17%, and 4%, respectively (n = 2). Also, human recombinant GK was used in all the assays.
For the EC50 assay, the glucose concentration was fixed at 5 mM.
Percent turnover: percentage of parent compound being metabolized after incubating in 0.25 mg/mL of rat or human liver microsomes for 30 min at 37 °C with an initial parent concentration of 1 μM.
Maximal velocity of the glucose phosphorylation reaction catalyzed by activated GK/unactivated GK.
Compound 12 had acceptable potency, kinetic parameters, and good stability in microsomes and primary human hepatocytes (15% metabolized).20 Furthermore, it has a low molecular weight (359) and very good solubility (718 μg/mL at pH 7.4, PBS buffer). Table 3 shows the pharmacokinetic properties of 12 in mice and rats. Although it had modest clearance in both rat and mouse, it had excellent bioavailability in mouse (81%). In addition, we studied the interspecies differences in the kinetic properties of compound 12–GK complex. Compound 12 had a Vmax of 1.12 and a S0.5 of 0.82 in mouse. Although the difference in the Vmax between human and mouse was significant, it was within our target range. Compound 12 (AM-0822) was chosen to proceed into in vivo studies to verify its efficacy.
Table 3. Pharmacokinetics Properties of 12.
| IVa |
POa |
|||||
|---|---|---|---|---|---|---|
| compd | species | CL (L/h/kg) | Vss (L/kg) | T1/2 (h) | F (%) | AUC (μM·h) |
| 12 | rat | 1.5 | 0.8 | 0.41 | 19 | 3.51 |
| 12 | mouse | 1.7 | 1.5 | 0.72 | 81 | 13.3 |
Rat and mouse: IV dose 1 mg/kg, n = 3; PO dose 10 mg/kg, n = 3.
Compound 12 (AM-0822) was tested in a dose–response oral glucose tolerance test (OGTT) using C57Bl/6 mice21 (Figure 2) and showed efficacy at 3 mg/kg, the lowest dose administered. The area-under-the-curve (AUC) for the blood glucose was decreased by 15% at 3 mg/kg and 32% at 10 mg/kg. The 10 mg/kg dose was equally efficacious as the 30 mg/kg dose. The plasma glucose level stabilized at 77 mg/dL between 1.5 and 2 h for the 30 mg/kg dose, well above the hypoglycemic range (<50 mg/dL), thus confirming our target range for the kinetic parameters on GK induced by our GKA.
Figure 2.
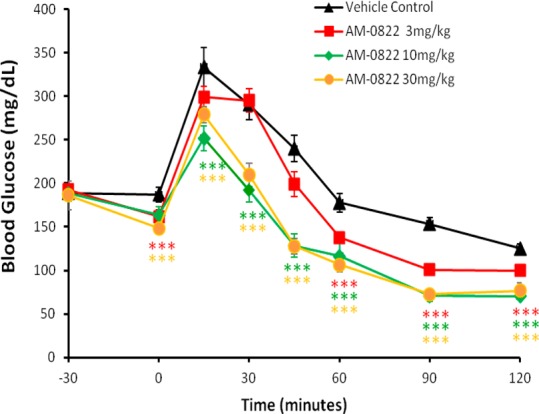
OGTT data for 12 (AM-0822) in C57Bl/6 mice. Statistical significance compared to vehicle treatment is denoted by *(p < 0.05), **(p < 0.01), ***(p < 0.001), and ****(p < 0.0001), as determined by two-way ANOVA, and is color-coded to the treatment in the figure legends.
However, compound 12 had significant brain penetration in rat. The unbound concentrations in the brain were 29% of unbound concentrations in plasma in rat. Because of the fact that GK is also expressed in brain, there will be risk of interfering with counter regulation once GKA penetrates the brain and lowers the brain blood glucose level. Therefore, this was considered to be an undesirable attribute in a potential GKA therapeutic. We then continued to seek GKAs with less brain penetration (less than 10% is desired) in addition to a similar or better overall profile than 12.
Our subsequent efforts were centered on modifications of 12, and Table 4 shows some of the analogues explored. Hydroxyl group substitution at the benzylic position (13), morpholino replacement of THP (14), and alpha substitution to the oxygen of THP (15) all led to compounds with decreased potency. The only compound with similar potency (16) displayed a Vmax less than 0.8.
Table 4. Modifications of the THP Group at C5.
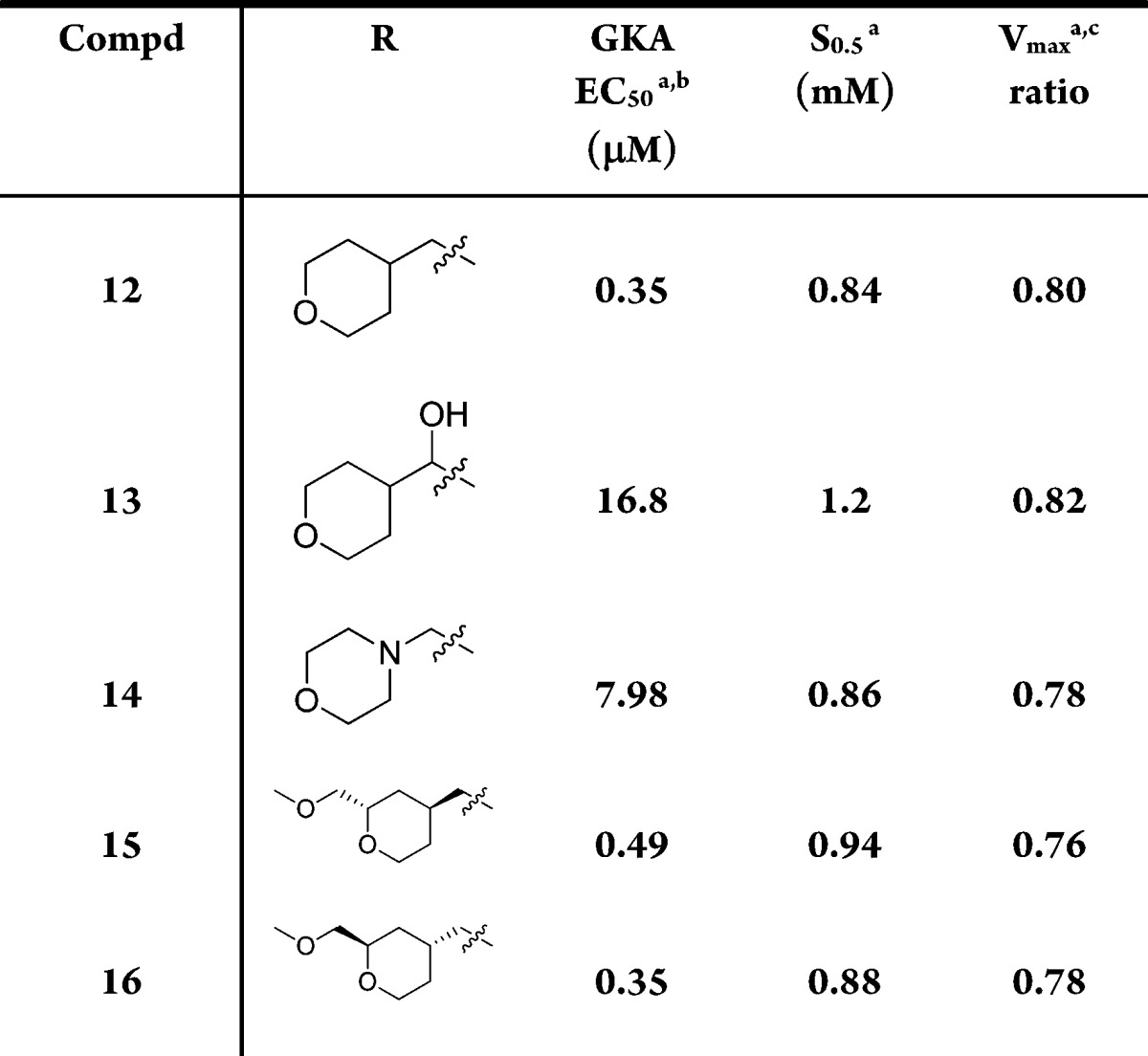
Mean standard deviations for the EC50, S0.5, and Vmax are ±30%, 17%, and 4%, respectively (n = 2). Also, human recombinant GK was used in all the assays.
For the EC50 assay, the glucose concentration was fixed at 5 mM.
Maximal velocity of the glucose phosphorylation reaction catalyzed by activated GK/unactivated GK.
Table 5 shows modifications on the pyrazole ring. These analogues (17 and 18) are also less potent than 12.
Table 5. Modifications on the 3-O-Pyrazole.
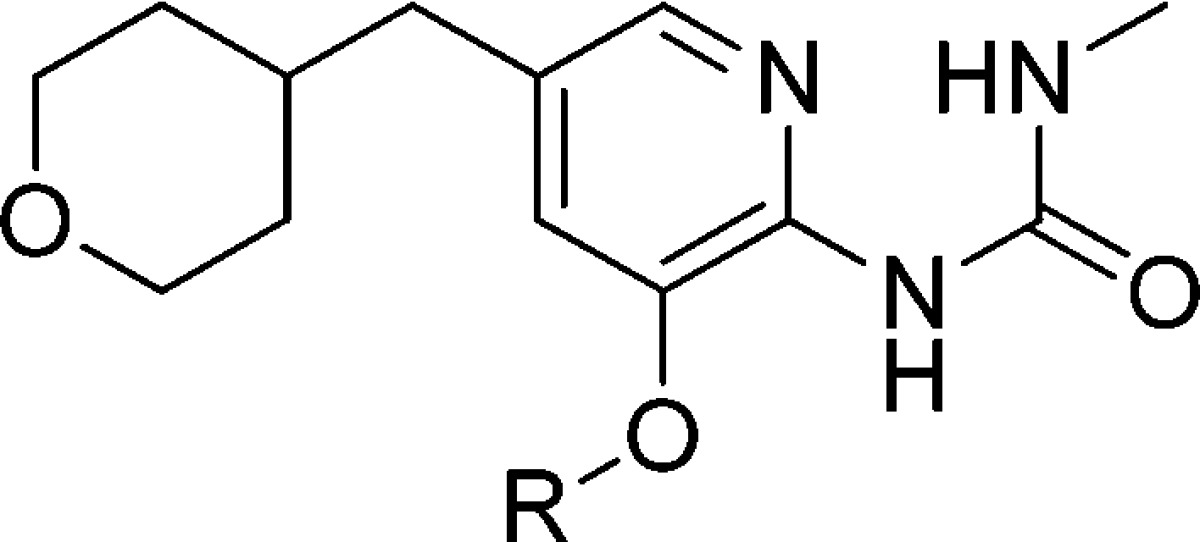
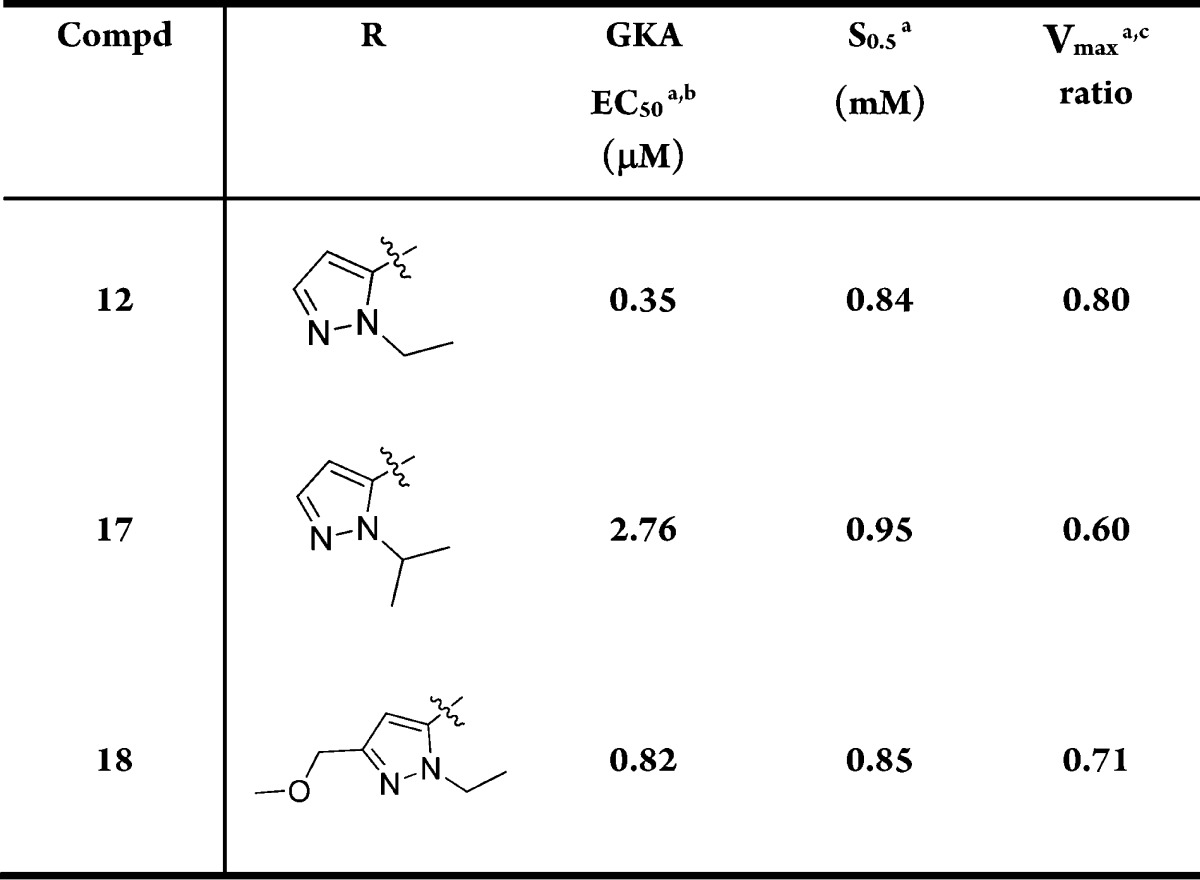
Mean standard deviations for the EC50, S0.5, and Vmax are ±30%, 17%, and 4%, respectively (n = 2). Also, human recombinant GK was used in all the assays.
For the EC50 assay, the glucose concentration was fixed at 5 mM.
Maximal velocity of the glucose phosphorylation reaction catalyzed by activated GK/unactivated GK.
We then investigated cyclic alcohols at C5 (Table 6). Secondary or tertiary hydroxyl would prevent secondary metabolism as shown in 7. Cyclic compounds would also reduce the degrees of freedom in the molecule, which might improve pharmacokinetic properties.22 Another advantage of alcohols is that they generally have increased polarity and polar surface area than ethers, which might lower the brain penetration level.23 The cyclohexanols (19–22) were more potent than 12, but most of them had lower Vmax with the exception of compound 22; however, for 22, the S0.5 was out of the desired range.
Table 6. Cyclic Alcohols at C5.
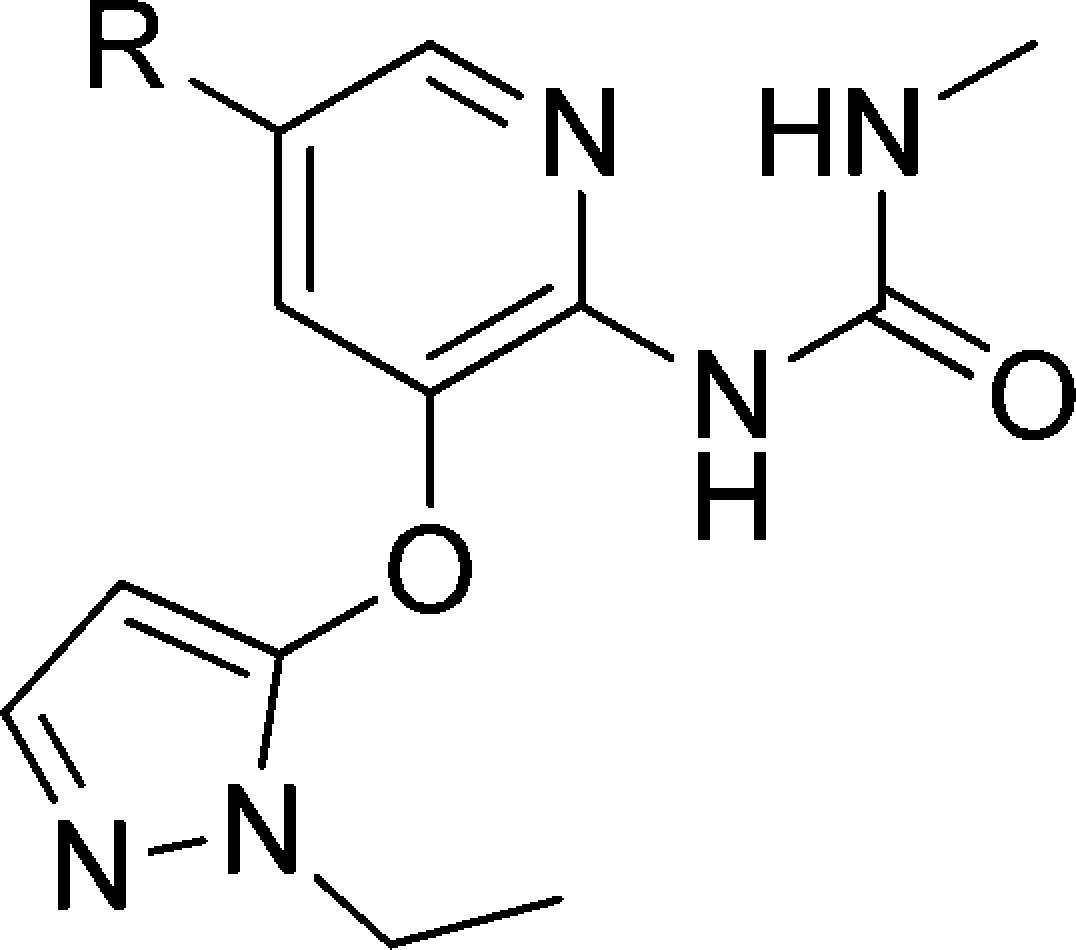
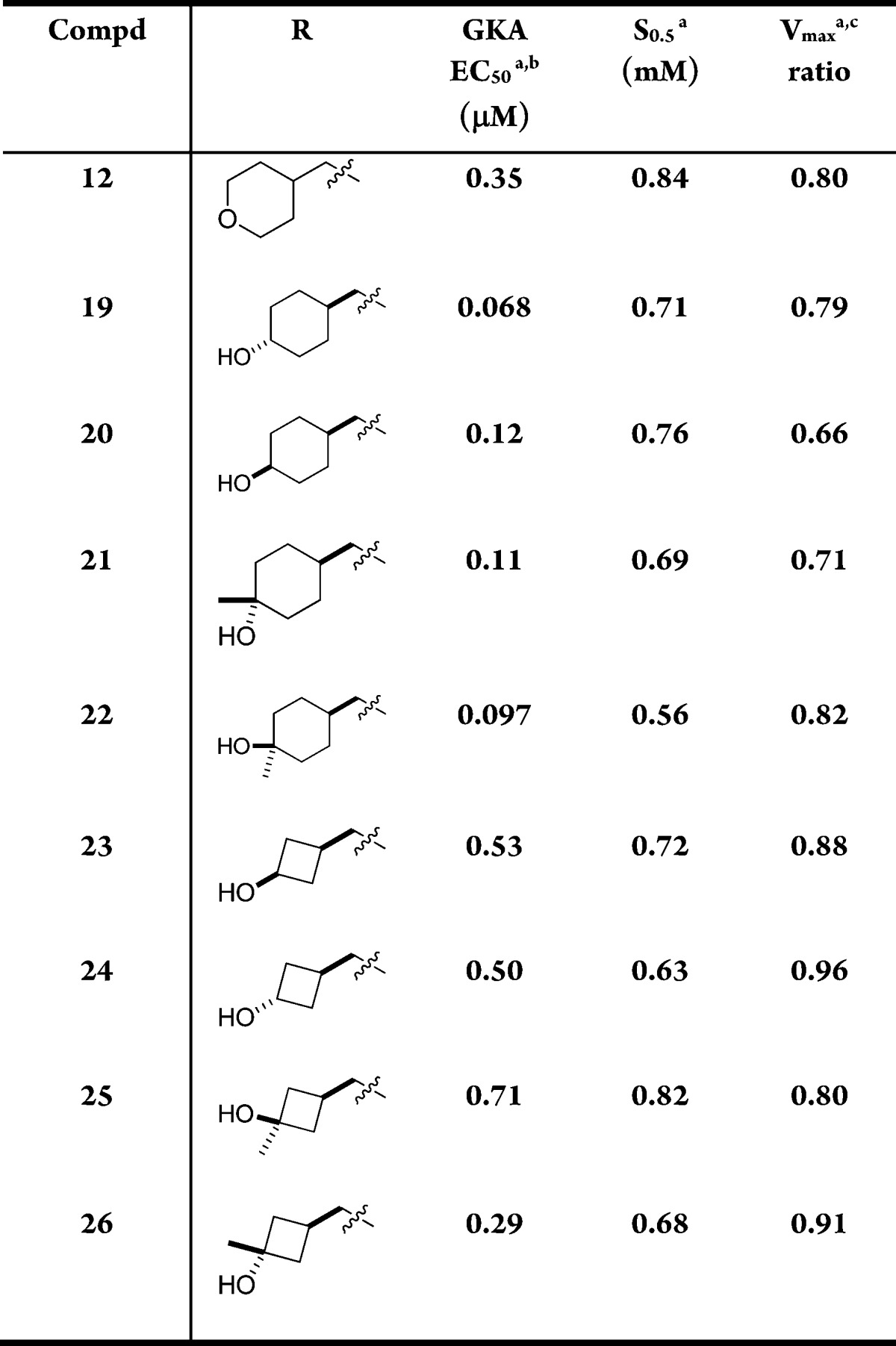
Mean standard deviations for the EC50, S0.5, and Vmax are ±30%, 17%, and 4%, respectively (n = 2). Also, human recombinant GK was used in all the assays.
For the EC50 assay, the glucose concentration was fixed at 5 mM.
Maximal velocity of the glucose phosphorylation reaction catalyzed by activated GK/unactivated GK.
The Vmax parameters in the cyclobutanols were more encouraging. Both cis and trans isomers (23 and 24) had higher Vmax values than 12, with one of the isomers, 24, having a Vmax of 0.96. To improve their potency to a more desirable range, a methyl group was added to form the tertiary cyclobutyl alcohols. One of the isomers, 26, had a Vmax of 0.91 and an EC50 of 0.29 μM, both improved over 12. The S0.5 decreased to 0.68 mM but was still within our target range.
Compound 26 (AM-9514) had excellent solubility, permeability, in vitro clearances and free fractions in plasma protein binding (Figure 3). It had desirable pharmacokinetics in mouse and dog with a clearance of 0.91 L/h/kg in mouse and 0.64 L/h/kg in dog (Table 7). It also had excellent oral bioavailability in dog. It was scaled up for further in vivo studies. The synthesis is shown in Scheme 1.
Figure 3.

In vitro parameters of 26: 4% HSA EC50 (the EC50 measured in the presence of 4% human serum albumin (HSA)).
Table 7. Pharmacokinetic Properties of 26.
| IV |
PO |
|||||
|---|---|---|---|---|---|---|
| compd | species | CL (L/h/kg) | Vss (L/kg) | T1/2 (h) | F (%) | AUC (μM·h) |
| 26 | rata | 1.6 | 0.8 | 2.0 | 9 | 0.39 |
| 26 | mouseb | 0.91 | 1.2 | 1.2 | 44 | 6.84 |
| 26 | doga | 0.64 | 2.2 | 5.5 | 72 | 6.4 |
Rat and dog: IV dose 0.5 mg/kg, n = 3; PO dose 2.0 mg/kg, n = 3.
Mouse: IV dose 1.0 mg/kg, n = 3; PO dose 5.0 mg/kg, n = 3.
Scheme 1. Synthesis of 26.

Reagents and conditions: (a) BnBr, K2CO3, CH3CN, 93%; (b) Tebbe reagent, THF, 58%; (c) H2O2, CCl3CN, DCM, separate the trans/cis isomers, 48%; (d) LiAlH4, THF, 90%; (e) Ac2O, pyridine, DCM, 94%; (f) TBSOTf, DIEPA, DCM, 87%; (g) K2CO3, MeOH, 94%; (h) iodobenzene diacetate, TEMPO, DCM, 85%; (i) 1-(5-bromo-3-((1-ethyl-1H-pyrazol-5-yl)oxy)pyridin-2-yl)-3-methylurea, MeLi, BuLi, aldehyde, THF; then MeSO3H, MeOH, 57% overall yield; (j) MeSO3H, 10% Pd/C, H2, MeOH, 59%.
The synthesis of 26 started from 3-oxocyclobutanecarboxylic acid. It was protected with a benzyl group and then converted to benzyl 3-methylenecyclobutanecarboxylate 26.2 through the Tebbe olefination. Epoxidation of the double bond in 26.2 and isolation of the less polar isomer led to 26.3, which was reduced to diol 26.4 with lithium aluminum hydride. The primary hydroxyl group in 26.4 was selectively protected with an acetate group (26.5), and the tertiary hydroxyl group was protected with a TBS group (26.6). Deprotection of the acetate group in 26.6 freed the primary hydroxyl group for further functional group transformations. Compound 26.7 was oxidized to aldehyde 26.8 under mild conditions. Lithiation of 1-(5-bromo-3-((1-ethyl-1H-pyrazol-5-yl)oxy)pyridin-2-yl)-3-methylurea24 and addition of the organolithium generated to aldehyde 26.8 followed by subsequent deprotection of the TBS protection group resulted in diol 26.9. The benzylic hydroxyl group in 26.9 was removed through hydrogenation under strong acidic conditions to provide compound 26. The cis relationship of the methyl group and the benzylic methyl group relative to the cyclobutane ring in 26 was confirmed by 2D-NMR experiments.
Before further in vivo study, we measured the kinetic properties of 26 on mouse GK (Vmax = 1.14 and S0.5 = 0.69) and found them within our target range and also closer to human parameters compared to compound 12. The antidiabetic efficacy of 26 was tested in an OGTT at doses of 0.3, 1, 3, and 10 mg/kg in fasted ob/ob mice (Figure 4). It showed dose-proportional lowering of blood glucose, with a 43% AUC decrease at 10 mg/kg.
Figure 4.
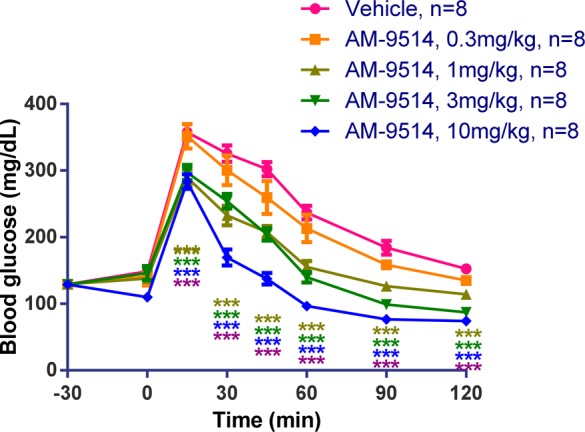
OGTT data of 26 (AM-9514) in ob/ob mice. Compound 26 lowered blood glucose in a dose-dependent manner. Statistical significance compared to vehicle treatment is denoted by *(p < 0.05), **(p < 0.01), ***(p < 0.001), and ****(p < 0.0001), as determined by two-way ANOVA, and is color-coded to the treatment in the figure legends.
Compound 26 was further tested in a normoglycemic glucose lowering test to evaluate its hypoglycemic risk. Fed male C57Bl/6 mice were orally dosed with vehicle or 26 without glucose challenge and then food was removed for 4 h. Blood glucose levels were measured at various time points (Figure 5). The elevation in glucose seen with the vehicle is presumably the result of handling stress on animals. Blood glucose levels were maintained above 85 mg/dL at all time points for both 10 and 30 mg/kg doses, well above its efficacious dose of 1 mg/kg. Therefore, 26 showed no hypoglycemia at the doses tested. Similar results of normoglycemic glucose lowering test were also obtained in rat of compound 26. In addition, compound 26 showed low brain penetration in rat. The unbound concentrations in the brain were 4% of unbound plasma concentrations in rat.
Figure 5.
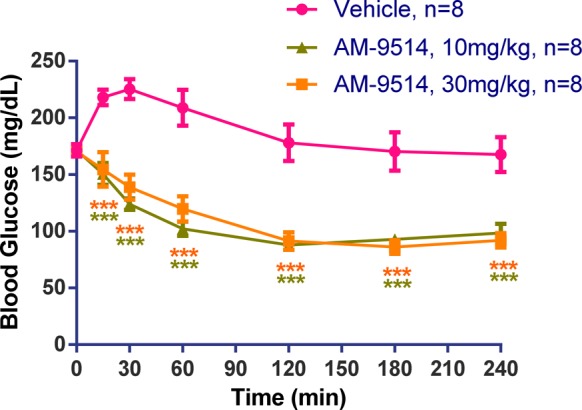
Normoglycemic glucose lowering test of 26 (AM-9514) in C57Bl/6 mice. Compound 26 showed no hypoglycemia during the test. Statistical significance compared to vehicle treatment is denoted by *(p < 0.05), **(p < 0.01), ***(p < 0.001), and ****(p < 0.0001), as determined by two-way ANOVA, and is color-coded to the treatment in the figure legends.
In conclusion, a new class of C5-alkyl-2-methylurea-substituted pyridines as glucokinase activators was developed based on our previous thiazolylamino pyridine GKAs. We optimized the potency and kinetic parameters of this class and identified compound 26 (AM-9514) with good potency, desirable GK kinetic parameters, and favorable in vitro and in vivo profiles including pharmacokinetic properties.
Supporting Information Available
Assay, in vivo procedures, experimental procedures, and data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Danaei G.; Finucane M. M.; Lu Y.; Singh G. M.; Cowan M. J.; Paciorek C. J.; Lin J. K.; Farzdfar F.; Khang Y.-H.; Stevens G. A.; Rao M.; Ali M. K.; Riley L. M.; Robinson C. A.; Ezzati M. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systemic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet 2011, 378, 31–40. [DOI] [PubMed] [Google Scholar]
- Nazimek-Siewniak B.; Moczulski D.; Grzeszczak W. Risk of macrovascular and microvascular complications in Type 2 diabetes: results of longitudinal study design. J. Diabetes Complications 2002, 16, 271–276. [DOI] [PubMed] [Google Scholar]
- Kles K. A.; Vinik A. I. Pathophysiology and treatment of diabetic peripheral neuropathy: the case for diabetic neurovascular function as an essential component. Curr. Diabetes Rev. 2006, 2, 131–145. [DOI] [PubMed] [Google Scholar]
- Rahman S.; Rahman T.; Ismail A. A.; Rashid A. R. Diabetes-associated macrovasculopathy: pathophysiology and pathogenesis. Diabetes Obes. Metab. 2007, 9, 767–780. [DOI] [PubMed] [Google Scholar]
- Iynedjian P. B. Molecular physiology of mammalian glucokinase. Cell. Mol. Life Sci. 2008, 66, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schaftingen E. A protein from rat liver confers to glucokinase the property of being antagonistically regulated by fructose 6-phosphate and fructose 1-phosphate. Eur. J. Biochem. 1989, 179, 179–184. [DOI] [PubMed] [Google Scholar]
- Grimsby J.; Sarabu R.; Corbett W. L.; Haynes N. E.; Bizzarro F. T.; Coffey J. W.; Guertin K. R.; Hilliard D. W.; Kester R. F.; Mahaney P. E.; Marcus L.; Qi L.; Spence C. L.; Tengi J.; Magnuson M. A.; Chu C. A.; Dvorozniak M. T.; Matschinsky F. M.; Grippo J. F. Allosteric activators of glucokinase: potential role in diabetes therapy. Science 2003, 310, 370–373. [DOI] [PubMed] [Google Scholar]
- Matschinsky F. Assessing the potential of glucokinase activators in diabetes therapy. Nat. Rev. Drug Discovery 2009, 8, 399–416. [DOI] [PubMed] [Google Scholar]
- Sarabu R.; Berthel S. J.; Kester R. F.; Tilley J. W. Novel glucokinase activators: a patent review (2008–2010). Expert Opin. Ther. Patents 2011, 21, 13–31. [DOI] [PubMed] [Google Scholar]
- Castro A. Kinase activators as a novel class of antidiabetic agents. Drug Discovery Today 2012, 17, 528–529. [Google Scholar]
- Pfefferkorn J. A. Strategies for the design of hepatoselective glucokinase activators to treat type 2 diabetes. Expert Opin. Drug Discovery 2013, 8, 319–330. [DOI] [PubMed] [Google Scholar]
- Hinklin R. J.; Boyd S. A.; Chicarelli M. J.; Condroski K. R.; Dewolf W. E. Jr.; Lee P. A.; Lee W.; Singh A.; Thomas L.; Voegtli W. C.; Williams L.; Aicher T. D. Identification of a new class of glucokinase activators through structure-based design. J. Med. Chem. 2013, 56, 7669–7678. [DOI] [PubMed] [Google Scholar]
- Lu M.; Li P.; Bandyopadhyay G.; Lagakos W.; Dewolf W. E. Jr.; Alford T.; Chicarelli M. J.; Williams L.; Anderson D. A.; Baer B. R.; McVean M.; Conn M.; Veniant M. M.; Coward P. Characterization of a novel glucokinase activator in rat and mouse models. PloS One 2012, 9, e88431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For detailed transition from the thiazolylamino pyridine series to methyl urea-substituted pyridine series, please see:Hinklin R. J.; Aicher T. D.; Anderson D. A.; Baer B. R.; Boyd S. A.; Condroski K. R.; DeWolf W. E. Jr.; Kraser C. F.; McVean M.; Rhodes S. P.; Sturgis H. L.; Williams L.; Houze J. B. Discovery of 2-pyridylureas as glucokinase activators. J. Med. Chem. 2014, 57, 8180–8186. [DOI] [PubMed] [Google Scholar]
- Bonadonna R. C.; Heise T.; Arbet-Engels C.; Kapitza C.; Avogaro A.; Grimsby J.; Zhi J.; Grippo J. F.; Balena R. Piragliatin (RO4389620), a novel glucokinase activator, lowers plasma glucose both in the postabsorptive state and after a glucose challenge in patients with type 2 diabetes mellitus: a mechanistic study. J. Clin. Endocrinol. Metab. 2010, 95, 5028–5036. [DOI] [PubMed] [Google Scholar]
- Meininger G. E.; Scott R.; Alba M.; Shentu Y.; Luo E.; Amin H.; Davies M. J.; Kaufman K. D.; Goldstein B. J. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care 2011, 34, 2560–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bue-Valleskey J. M.; Schneck K. B.; Sinha V. P.; Wonddmagegnehu E. T.; Kapitza C.; Miller J. WLY2599506, a novel glucokinase activator (GKA), improves fasting and postprandial glucose in patients with type 2 diabetes mellitus (T2DM). Presented at the 71st American Diabetes Association Meeting, San Diego, CA, 2011. [Google Scholar]
- Pfefferkorn J. A.; Guzman-Perez A.; Oates P. J.; Litchfield J.; Aspnes G.; Basak A.; Benbow J.; Bian J.; Choi C.; Freeman-Cook K.; Corbett J. W.; Didiuk M.; et al. Designing glucokinase activators with reduced hypoglycemia risk: discovery of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as a clinical candidate for the treatment of type 2 diabetes mellitus. Med. Chem. Commun. 2011, 2, 828–839. [Google Scholar]
- The S0.5 and Vmax data of Piragliatin and MK-0941 were generated in house.
- Filipski K. J.; Stevens B. D.; Pfefferkorn J. A.. New Therapeutic Strategies for Type 2 Diabetes; RSC Publishing: London, U.K., 2012; pp 88–108. [Google Scholar]
- Grimsby J.; Berthel S. J.; Sarabu R. Glucokinase activators fo rthe potential treatment of type 2 diabetes. Curr. Top. Med. Chem. 2008, 8, 1524–1532. [DOI] [PubMed] [Google Scholar]
- Hepatic stability is the ratio of the observed clearance expressed in mL/min/kg divided by the literature value for the hepatic blood flow values in the species studied (mL/min/kg).
- To save some cost, we used normal C57Bl/6 mice in the OGTT test of compound 12 instead of ob/ob mice.
- Veber D. F.; Johnson S. R.; Cheng Y.-H.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
- Pajouhesh H.; Lenz G. R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Please see Supporting Information for detailed experimental procedure to make 1-(5-bromo-3-((1-ethyl-1H-pyrazol-5-yl)oxy)pyridin-2-yl)-3-methylurea (intermediate 11.3, page 22 in Supporting Information).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.