

Abstract
Background
BAY 94-9027 is a B-domain-deleted recombinant factor VIII (rFVIII) with site-specific attachment of poly(ethylene glycol) that has shown an extended half-life in animal models of hemophilia.
Objectives
To assess the pharmacokinetics and safety of BAY 94-9027 after single and repeated administration in subjects with severe hemophilia A.
Patients/Methods
This 8-week, prospective, multicenter, open-label, phase I trial was conducted in 14 subjects aged 21–58 years with FVIII of < 1%, ≥ 150 days of exposure to FVIII, and no history of FVIII inhibitors. After a ≥ 3-day washout, subjects received a single dose of sucrose-formulated rFVIII (rFVIII-FS) (cohort 1 [n = 7], 25 IU kg−1; cohort 2 [n = 7], 50 IU kg−1) for a 48-h pharmacokinetic (PK) study. After another ≥ 3-day washout, cohort 1 received twice-weekly BAY 94-9027 at 25 IU kg−1 (16 doses), and cohort 2 received once-weekly BAY 94-9027 at 60 IU kg−1 (nine doses). A 168-h PK study was performed after the first and last BAY 94-9027 doses.
Results
BAY 94-9027 showed equivalent recovery and an improved PK profile vs. rFVIII-FS, with a half-life of ∼ 19 h (vs. ∼ 13.0 h for rFVIII-FS). BAY 94-9027 was well tolerated, and no immunogenicity was observed.
Conclusions
This phase I study demonstrates that BAY 94-9027 has an extended half-life in subjects with hemophilia A and, after multiple dosing, was well tolerated with no immunogenicity during the 8-week trial. A phase III study in a larger number of subjects is underway to fully characterize how this prolonged half-life will permit less frequent prophylaxis dosing for patients with hemophilia.
Keywords: clinical trial, Phase 1; factor VIII; hemophilia A; pharmacokinetics; recombinant proteins
Introduction
Observational studies in patients with hemophilia A have suggested that prophylactic factor replacement therapy is more efficacious in preventing joint bleeding episodes and retarding the progression of arthropathy than on-demand treatment 1. Two randomized prospective trials have confirmed the benefit of prophylaxis in preventing joint damage 2,3. Therefore, prophylaxis has become the standard of care for children and adolescents with severe hemophilia 4. Current prophylaxis strategies can be divided into those that aim to maintain factor VIII (FVIII) activity at > 1 IU dL−1 and those guided by the patients' clinical bleeding patterns 5.
Various challenges to prophylaxis exist in everyday practice, including cost 6, difficulties with venipuncture, and complications of venous access devices, particularly in young children 7. du Treil reported that 50% of patients on high-intensity regimens were poorly adherent 8. The inconvenience and time-consuming nature of prophylaxis appear to be major reasons for poor adherence 9. Because joint damage occurs after only a few hemorrhages into affected joints 10, adherence is critical for attaining good long-term outcomes 11. An FVIII replacement therapy with a longer half-life, which would allow for a longer treatment interval for prophylaxis, could potentially reduce the burdens associated with prophylaxis and may in turn improve clinical outcomes.
Several strategies have been successfully employed to extend the half-life of approved therapeutic proteins, including conjugation with poly(ethylene glycol) (PEG) 12. For many proteins, PEGylation prolongs the half-life by decreasing renal clearance of the molecule. This process is not relevant to FVIII because FVIII is so large that renal clearance is minimal 13. Instead, PEGylation-mediated interruption of FVIII interactions with the low-density lipoprotein receptor-related protein (LRP) family of receptors may decrease hepatic clearance of FVIII 14. Previous attempts to PEGylate FVIII by using lysine methylation or terminal amine methylation led to decreased coagulation activity because of impairment of FVIII binding to its multiple partners, including von Willebrand factor (VWF) 15.
Mei et al. 16 described a novel strategy for FVIII PEGylation. Using site-directed mutagenesis, they modified a B-domain-deleted recombinant FVIII (BDD-rFVIII) through introduction of a single cysteine specifically conjugated to a 60-kDa PEG molecule. The site-specific PEGylated BDD-rFVIII (BAY 94-9027; Fig.1) retained coagulant activity in chromogenic and one-stage assays with ellagic acid as the activator. It also had improved pharmacokinetics in rabbits and hemophilic mice and showed efficacy comparable to that of unmodified FVIII in stopping acute bleeds, but had a prolonged duration of action in bleeding models in hemophilic mice 16 and dogs 17. Preclinical studies in mice with hemophilia A and normal rats and rabbits indicated that BAY 94-9027 is less immunogenic than non-PEGylated rFVIII 16,18.
Fig 1.

Schematic of the structure of BAY 94-9027. PEG, poly(ethylene glycol). Adapted from Ivens et al. [19].
We report the first-in-human study of BAY 94-9027. This open-label study demonstrated the extended half-life and other improved pharmacokinetics, clinical safety and lack of immunogenicity of BAY 94-9027 following single and multiple doses in previously treated subjects with severe hemophilia A.
Materials and methods
Study design
This was a non-randomized, prospective, open-label, parallel-group, phase I study (ClinicalTrials.gov, NCT01184820) conducted at four hemophilia centers in the USA. The study was performed in accordance with the US Code of Federal Regulations and the International Conference on Harmonisation Guidelines on Good Clinical Practices. Before the initiation of study procedures, the trial was reviewed and approved by institutional review boards at participating institutions. All subjects gave written informed consent before the initiation of any study-related procedures.
The study objectives were to describe the pharmacokinetics of BAY 94-9027 after single and repeated doses and to assess the safety and immunogenicity of BAY 94-9027 administered over an 8-week period. The trial design is summarized in Fig.2. The study was conducted in two cohorts; each cohort was planned to accrue six to eight subjects to allow for at least six evaluable subjects. VWF antigen levels were measured at baseline in all subjects in both cohorts.
Fig 2.

Study design. PK, pharmacokinetic; rFVIII-FS, sucrose-formulated recombinant factor VIII.
Cohort 1
After a ≥ 3-day washout period, subjects in cohort 1 received a single intravenous 25-IU kg−1 dose of sucrose-formulated rFVIII (rFVIII-FS; Bayer, Tarrytown, NY, USA) over a period of 10 min. A pharmacokinetic (PK) study was performed with measurement of FVIII activity by chromogenic and one-stage coagulation assays at 0.25, 0.5, 1, 3, 6, 8, 24 and 48 h after administration. After another ≥ 3-day washout period, subjects were given the first intravenous dose of 25 IU kg−1 BAY 94-9027 over a period of 10 min. A PK study was repeated as above with additional samples at 72, 96–144 and 168 h after infusion. Subjects were then treated at home with 25 IU kg−1 BAY 94-9027 twice weekly for 8 weeks. A 168-h PK study was repeated with the last dose of BAY 94-9027. During 8 weeks of BAY 94-9027 treatment, subjects received 16 doses.
Cohort 2
The pharmacokinetics of the first four evaluable subjects of cohort 1 were analyzed to determine the dose for cohort 2. A 60-IU kg−1 dose was chosen, to give an expected mean peak FVIII level of ≤ 150% at 0.5 h postinfusion. After a ≥ 3-day washout period, subjects in cohort 2 received a 50-IU kg−1 dose of rFVIII-FS intravenously over a period of 10 min, and a 48-h PK study was performed. After another ≥ 3-day washout period, subjects were infused with the first dose of 60 IU kg−1 BAY 94-9027, and this was followed by a 168-h PK study. Subjects were then treated at home with 60 IU kg−1 BAY 94-9027 once weekly for 8 weeks. A 168-h PK study was repeated with the last dose. During 8 weeks of BAY 94-9027 treatment, subjects received nine doses.
Because the in vivo recovery and half-life of BAY 94-9027 in humans were unknown at study initiation, bleeding events occurring during the study after initiation of BAY 94-9027 treatment were to be treated with standard rFVIII-FS. For acute bleeds, rFVIII-FS was administered at a dose of ≤ 50 IU kg−1, determined by the investigator on the basis of bleeding severity. However, if a bleeding event occurred on the day of a scheduled BAY 94-9027 infusion, the episode was to be treated with the planned dose of study drug. Subjects kept a written log of all infusions.
Subjects
Eligible subjects were men aged 18–65 years with severe hemophilia A, with a documented FVIII activity level of < 1% and ≥ 150 exposure days (EDs) to FVIII. Subjects were immunocompetent, with a CD4 count of > 400 mm−3, and had no history or current evidence of inhibitors (titer of < 0.6 Bethesda units). Subjects had no bleeding disorders other than hemophilia A.
Treatment product
BAY 94-9027 is a BDD-rFVIII variant that is site-specifically conjugated with a 60-kDa branched PEG (Fig.1) 16,19; site-specific conjugation is carried out at an introduced cysteine (K1804C) in the light chain A3 domain of FVIII.
Laboratory methods
FVIII activity levels were measured by the use of chromogenic and one-stage clotting assays with ellagic acid as the activator. For the one-stage assay, FVIII activity was determined in citrated plasma with a turbidimetric assay, with activated partial thromboplastin time assessment calibrated with standard human plasma. FVIII inhibitors were measured at screening, at the first BAY 94-9027 infusion, and 168 h after the last dose of BAY 94-9027, with the Nijmegen-modified Bethesda assay. Antibodies against BAY 94-9027 and PEG were measured with a ligand-binding assay at the first infusion of BAY 94-9027, 1, 2, 4 and 8 weeks after the first infusion, and 168 h after the last infusion. The precision for positive control samples ranged from 11.3% to 27.3% for sample screening, and from 11.2% to 20.3% for confirmatory assays.
Safety assessments included monitoring of adverse events (AEs), wellness assessments, physical examinations, vital signs, complete blood counts, serum chemistry, (including renal and hepatic function) electrocardiograms at screening and at the last visit, and urinalysis.
Pharmacokinetics
PK parameters were calculated with WinNonlin (Pharsight, Mountain View, CA, USA). PK parameters calculated included peak concentration (Cmax), dose-normalized Cmax, area under the plasma concentration–time curve from time zero to infinity (AUC), dose-normalized AUC, half-life, volume of distribution at steady state, and clearance.
The PK profile and half-life for each subject, calculated with non-compartmental methods, was used to extrapolate the time when the concentrations would reach 1%, 3% or 5% for that particular subject.
Statistical analysis
Concentration–time courses of rFVIII-FS and BAY 94-9027 were summarized for each cohort and visit. Arithmetic and geometric means, standard deviations and coefficients of variation were calculated for each sampling point with the sas software package, release 9.1 (SAS Institute, Cary, NC, USA). Data were analyzed by Bayer statisticians. All authors were given access to the clinical trial data.
With analysis of variance of the natural log of PK variables and 90% two-sided confidence intervals for the ratio of means for each comparison, preplanned statistical comparisons of the PK parameters half-life, AUC, and Cmax were analyzed for: (i) a single dose between rFVIII-FS and BAY 94-9027 for each cohort; (ii) multiple doses and a single dose of BAY 94-9027 for each cohort; (iii) a single dose of BAY 94-9027 between cohorts 1 and 2; and (iv) multiple doses of BAY 94-9027 between cohorts 1 and 2.
Results
Subject disposition and demographics
Baseline demographic information is presented in Table1. Fourteen subjects (mean age, 36.1 years [range, 21–58 years]) were enrolled between October 2010 and October 2011. There were seven subjects in each cohort. All subjects completed the study and were evaluable.
Table 1.
Baseline demographics
| Cohort 1 (n = 7) | Cohort 2 (n = 7) | Total (n = 14) | |
|---|---|---|---|
| Race, n (%) | |||
| White | 7 (100.0) | 5 (71.4) | 12 (85.7) |
| Black | 0 | 1 (14.3) | 1 (7.1) |
| Asian | 0 | 1 (14.3) | 1 (7.1) |
| Age (years) | |||
| Mean ± SD | 36.7 ± 16.0 | 35.4 ± 13.3 | 36.1 ± 14.1 |
| Median (range) | 29 (21–56) | 31 (24–58) | 30 (21–58) |
| Weight (kg) | |||
| Mean ± SD | 85.6 ± 16.8 | 80.5 ± 20.3 | 83.0 ± 18.1 |
| Median (range) | 90 (66–114) | 78 (53–107) | 80 (53–114) |
| Height (cm) | |||
| Mean ± SD | 176.8 ± 7.8 | 173.8 ± 6.9 | 175.3 ± 7.2 |
| Median (range) | 177 (166–192) | 170 (168–185) | 174 (166–192) |
SD, standard deviation.
Pharmacokinetics
Mean half-life and AUC values for BAY 94-9027 were similar with the one-stage and the chromogenic methods, with differences ranging from − 10.3% to 4.9% for half-life, and from − 2.0% to 1.7% for AUC, following single and multiple doses (data not shown). However, mean Cmax values were higher for BAY 94-9027 with the one-stage assay (∼ 25% higher on average after single or multiple doses). The data presented here are based on the chromogenic assay because this method is used for BAY 94-9027 potency assignment.
PK parameters are shown in Table2. Infusion of a single dose of BAY 94-9027 at 25 IU kg−1 or 60 IU kg−1 yielded similar Cmax values as similar doses of rFVIII-FS. Dose-proportional increases in plasma concentrations were seen between BAY 94-9027 doses (Fig.3). The Cmax of BAY 94-9027 was not substantially changed after multiple doses. There was a slight increase in concentration following multiple doses of 25 IU kg−1 BAY 94-9027 twice weekly as compared with rFVIII-FS, and there was no further change in concentration following multiple doses of 60 IU kg−1 once weekly. The AUC was greater following infusion of a single dose of BAY 94-9027 than with rFVIII-FS, and the mean half-life of BAY 94-9027 following a single 25-IU kg−1 or 60-IU kg−1 dose was 18.4 h (range, 13.7–28.1 h) vs. 13.0 h (range, 8.4–18.7 h) for rFVIII-FS following a single dose. All subjects with data for the single dose of rFVIII-FS and the last dose of BAY 94-9027 (n = 13) showed an increase in half-life when given BAY 94-9027 as compared with rFVIII-FS (Fig.4). The half-life of BAY 94-9027 was 18.7 h (range, 12.1–30.0 h) when averaged across the two cohorts and across single-dose and multiple-dose data, and was significantly longer than that of rFVIII-FS (P < 0.0001). PK parameters of BAY 94-9027 were similar after single and multiple doses. VWF levels at baseline were similar between groups, but there was a tendency for there to be a longer BAY 94-9027 half-life among subjects with higher VWF levels (Fig.5).
Table 2.
Mean (range) pharmacokinetic parameters following a single dose of sucrose-formulated recombinant factor VIII (rFVIII-FS) and single and multiple doses of BAY 94-9027 (chromogenic assay)
| Dose (IU kg−1) | t½ (h) | Cmax (IU dL−1) | Cmax,norm (kg dL−1) | AUC (IU h−1 dL–1) | AUCnorm (kg h−1 dL–1) | CL (dL h−1 kg–1) | Vss (dL kg−1) | |
|---|---|---|---|---|---|---|---|---|
| Cohort 1 (n = 7) | ||||||||
| rFVIII-FS* | 25 | 12.9 (8.4–18.7) | 69.6 (40.8–91.3) | 2.8 (1.6–3.7) | 1109 (580–2136) | 44.3 (23.2–85.4) | 0.023 (0.012–0.043) | 0.42 (0.31–0.66) |
| BAY 94-9027, single dose | 25 | 18.2 (13.7–28.1) | 63.7 (53.2–80.3) | 2.5 (2.1–3.2) | 1577 (1285–2825) | 61.3 (51.1–113.0) | 0.016 (0.009–0.020) | 0.43 (0.35–0.50) |
| BAY 94-9027, twice weekly (16 doses) | 25 | 18.6 (12.1–30.0) | 79.8 (62.0–92.9) | 3.2 (2.5–3.7) | 2036 (1452–3314) | 81.4 (57.8–132.5) | 0.012 (0.008–0.017) | 0.33 (0.30–0.40) |
| Cohort 2 (n = 7) | ||||||||
| rFVIII-FS | 50 | 13.0 (10.2–15.9) | 228.6 (171–376) | 4.6 (3.4–7.5) | 2502 (1736–4891) | 50.0 (34.7–97.8) | 0.020 (0.010–0.029) | 0.36 (0.23–0.51) |
| BAY 94-9027, single dose | 60 | 18.5 (15.1–23.4) | 172.0 (125–245) | 2.9 (2.1–4.1) | 4329 (3087–8578) | 72.1 (51.4–143.0) | 0.014 (0.007–0.019) | 0.38 (0.25–0.45) |
| BAY 94-9027, once weekly (nine doses) | 60 | 19.5 (15.0–25.6) | 184.7 (139–243) | 3.1 (2.3–4.1) | 4723 (3306–7589) | 78.5 (55.2–126.5) | 0.013 (0.008–0.018) | 0.34 (0.26–0.41) |
AUC, area under the curve from time zero to infinity; AUCnorm, dose-normalized area under the curve from time zero to infinity; CL, clearance; Cmax, maximum concentration; Cmax,norm, dose-normalized maximum concentration; t½, half-life; Vss, volume of distribution at steady state.
n = 6, except for Cmax and Cmax,norm, which are based on n = 7.
Fig 3.

Concentration–time curves of chromogenic factor VIII activity for a single dose of sucrose-formulated recombinant FVIII (rFVIII-FS) given at (A) 25 IU kg−1 and (B) 50 IU kg−1, and after single and multiple doses of BAY 94-9027 given at (A) 25 IU kg−1 and (B) 60 IU kg−1. The data shown are mean ± 90% confidence interval.
Fig 4.
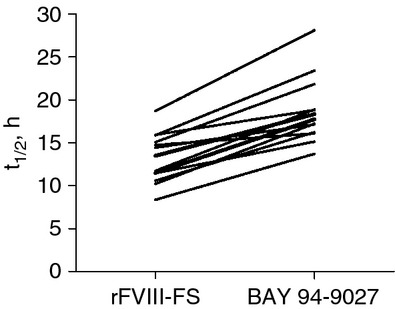
Individual subject half-life (t½) values for sucrose-formulated recombinant factor VIII (rFVIII-FS) and BAY 94-9027 (last dose). Data were derived from all subjects from both cohorts who had data for the single dose of rFVIII-FS and the last dose of BAY 94-9027 (n = 13).
Fig 5.
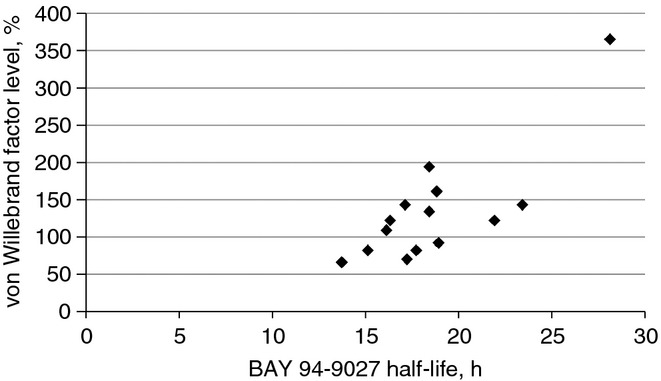
Relationship of baseline von Willebrand factor levels to half-life following the initial BAY 94-9027 dose.
Safety
BAY 94-9027 was well tolerated. No inhibitory or non-inhibitory antibodies were detected against FVIII, BAY 94-9027, or PEG. One serious AE (a bleed into the muscles of the right pelvis requiring hospitalization) occurred in a subject in cohort 1, 5 days after the last 25-IU kg−1 BAY 94-9027 dose during PK sample collection. The event was judged by the investigator to be unrelated to treatment. In this subject, FVIII levels 4 days after BAY 94-9027 dosing (the last sample collected before the serious AE occurred) were below the quantitation limits of 3% for the chromogenic assay and 1.5% for the one-stage assay. Non-serious treatment-emergent AEs were reported in three other subjects: diarrhea, nausea, and vomiting in one subject; root canal pain in one subject; and headache, arthralgia, and pain in the extremities in one subject. None of these AEs was judged to be treatment related. There were no significant changes in any clinical laboratory parameters.
Bleeding
Although determination of the efficacy of BAY 94-9027 in the prevention or treatment of bleeding episodes was not a specific study objective, data on observed bleeding episodes and infusions were recorded during safety monitoring. No data are available on the types of bleeding event (e.g. trauma related or spontaneous).
There were a total of 32 bleeding events reported during the entire study period, 19 of which occurred during the BAY 94-9027 treatment period (n = 4 in the low-dose cohort; n = 15 in the high-dose cohort).
In the low-dose cohort, four subjects had bleeding episodes 5–6 days after a PK dose of BAY 94-9027 at 25 IU kg−1 (one occurred 5 days after the first dose, and three occurred 5–6 days after the last dose); these episodes were treated with rFVIII-FS. As per protocol, the PK analysis samples were collected over a period of 7 days, during which the subject did not receive another dose. All bleeds in this cohort occurred when FVIII levels were predicted to be < 3% (lower limit of quantitation for the chromogenic assay).
In the high-dose cohort, there were 15 bleeds in six subjects (≥ 2 bleeds per subject) during BAY 94-9027 treatment. Eight bleeds occurred when FVIII levels were predicted to be in the range of 5–81 IU dL−1 on the basis of subject pharmacokinetics, and the remaining seven bleeds occurred when FVIII levels were predicted to be < 3%.
Bleeding events treated with rFVIII-FS were resolved as expected. Five bleeding events in the high-dose cohort were treated with BAY 94-9027, either because the bleed occurred on the day of scheduled infusion, or because of inadvertent home use of BAY 94-9027 by the subject; these bleeds were reported by the subject to have resolved after a single 60 IU kg−1 infusion of BAY 94-9027.
Simulations of BAY 94-9027 activity
One of the seven subjects had FVIII levels predicted to be > 1% for 5 days after a single dose of 25 IU kg−1 BAY 94-9027, and six of the seven had FVIII levels predicted to be > 1% for 5 days after a single dose of 60 IU kg−1 (Table3). Furthermore, following multiple doses of 25 IU kg−1 and 60 IU kg−1, respectively, two and six of the seven subjects had FVIII levels predicted to be > 1% for 5 days. After multiple dosing with 60 IU kg−1, two of the seven subjects had FVIII levels predicted to be > 1% for ≥ 7 days. Additionally, one of the seven subjects had FVIII trough levels predicted to be > 3% for 5 days after a single dose of 25 IU kg−1 or 60 IU kg−1. During the multiple-dose period, one of seven (25 IU kg−1) and two of seven (60 IU kg−1) subjects had FVIII levels predicted to be > 3% for 5 days. After multiple dosing with 60 IU kg−1, two of the seven subjects had FVIII levels predicted to be > 3% for 6 days. The predicted numbers of subjects achieving FVIII trough levels of > 1% or > 3% following single doses of rFVIII-FS are also shown in Table3. The doses and dosing schedules used in the ongoing BAY 94-9027 pivotal trial took into consideration several factors, including the simulations of FVIII activity summarized in Table3.
Table 3.
Predicted numbers of subjects achieving factor VIII trough levels of >1% or > 3% following single or multiple doses of BAY 94-9027 or single doses of sucrose-formulated recombinant FVIII (rFVIII-FS)
| Days after dosing | BAY 94-9027 |
rFVIII-FS |
||||
|---|---|---|---|---|---|---|
| 25 IU kg−1, single dose (n = 7) | 25 IU kg−1, multiple doses (n = 7) | 60 IU kg−1, single dose (n = 7) | 60 IU kg−1, multiple doses (n = 7) | 25 IU kg−1, single dose (n = 7) | 50 IU kg−1, single dose (n = 7) | |
| Subjects with FVIII trough levels of > 1%, n | ||||||
| 2 | 7 | 7 | 7 | 7 | 6 | 7 |
| 3 | 7 | 7 | 7 | 7 | 4 | 6 |
| 4 | 5 | 6 | 7 | 7 | 1 | 3 |
| 5 | 1 | 2 | 6 | 6 | 1 | 1 |
| 6 | 1 | 2 | 2 | 3 | 0 | 0 |
| ≥ 7 | 1 | 1 | 1 | 2 | 0 | 0 |
| Subjects with FVIII trough levels of > 3%, n | ||||||
| 2 | 7 | 7 | 7 | 7 | 4 | 7 |
| 3 | 4 | 6 | 7 | 7 | 1 | 4 |
| 4 | 1 | 2 | 6 | 6 | 0 | 1 |
| 5 | 1 | 1 | 1 | 2 | 0 | 0 |
| 6 | 0 | 0 | 1 | 2 | 0 | 0 |
| ≥ 7 | 0 | 0 | 0 | 0 | 0 | 0 |
Discussion
The objectives of this first-in-human study were to evaluate the safety and pharmacokinetics of BAY 94-9027. The results indicate that BAY 94-9027 was well tolerated, with no serious treatment-related AEs, and demonstrate improved pharmacokinetics, including an increased half-life of FVIII activity in the circulation in subjects with severe hemophilia A.
PEGylation has been shown to improve the pharmacokinetics of approved drugs in various therapeutic areas, with good tolerance and safety profiles and no immunogenicity 12,19. No inhibitor formation and no anti-BAY 94-9027 or anti-PEG antibody formation was detected in this population of previously treated subjects who had ≥ 150 previous EDs with FVIII. Although immunogenicity was not specifically tested in this study, preclinical studies showed that BAY 94-9027 was less immunogenic than rFVIII in mice with hemophilia A as well as in normal rats and rabbits, perhaps through a mechanism of shielding of antigenic epitopes on the FVIII surface 16,18.
BAY 94-9027 had full coagulation activity in preclinical studies using chromogenic assays and in one-stage coagulation activity assays with ellagic acid as the activator 16. In this study, the BAY 94-9027 half-life and AUC were similar with both assays (differences in mean values between assays were ≤ 2.0% for AUC and ≤ 10.3% for half-life), although Cmax values were higher with the one-stage assay than with the chromogenic assay. Because factor assays with micronized silica as the activator severely underestimate the factor activity of BAY 94-9027 16, ellagic acid was used as the activator in this study.
As predicted by preclinical findings in mouse, rabbit and dog models, this study found that BAY 94-9027 had improved pharmacokinetics in humans, with peak FVIII activity recovery similar to that with rFVIII-FS but with a half-life increased by 32–48% as compared with rFVIII-FS. The longer half-life of BAY 94-9027 is not a result of the longer blood sampling time (up to 168 h vs. 48 h for rFVIII-FS), given that the rFVIII-FS half-life in a previous study with sampling times up to 168 h was similar to that observed here 20. The observed PK properties were sustained after BAY 94-9027 treatment over a period of 8 weeks on two different dosing schedules. The BAY 94-9027 half-life also appears to be substantially longer than that of its parent compound, unmodified BDD-rFVIII, which has PK properties that are bioequivalent to those of plasma-derived FVIII 21 and full-length rFVIII 22 in humans.
The clearance mechanisms of FVIII are complex, and FVIII interactions with VWF further increase this complexity 23. Although free FVIII is cleared via interaction with LRP1 and other ancillary cell surface receptors, most likely a substantial proportion of FVIII is cleared in a complex with VWF that has a half-life of ∼ 15 h. The majority of circulating FVIII molecules are bound in a tight non-covalent complex with VWF; binding of FVIII to VWF inhibits FVIII interaction with clearance receptors, increasing its half-life as compared with free FVIII. However, VWF is cleared with a half-life of ∼ 15 h, and it is hypothesized that FVIII bound to VWF is cleared together with VWF 24. This hypothesis is supported by the observation that the half-life of FVIII:C is shorter in patients with blood type O than in patients with blood type A or B. The survival of FVIII is determined more by the half-life of VWF than by the VWF levels directly 25. The clearance site of the VWF–FVIII complex appears to be the macrophage; FVIII and VWF were codirected to the same macrophages in VWF-deficient mice injected with VWF–FVIII complexes 26. Mei et al. 16 showed that site-specific PEGylated FVIII had a greater prolongation of half-life when injected into mice with von Willebrand deficiency than when injected into mice with hemophilia A, implying that the efficacy of PEGylation is probably attributable to prolongation of the half-life of free FVIII rather than FVIII complexed to VWF. Powell et al. 27 hypothesized that the complex interactions of FVIII with VWF may limit the achievable prolongation of FVIII half-life to ∼ 19 h through modification of the FVIII protein itself, as the half-life of VWF is 16–17 h. The findings from our study support this hypothesis.
Although efficacy in the control or prevention of bleeding episodes was not a specific endpoint of this study, five bleeding episodes were treated with BAY 94-9027, with prompt and complete responses. These results provide preliminary evidence that BAY 94-9027 has clinical coagulant activity in subjects with severe hemophilia A, as predicted by activity in vitro and in preclinical animal models of hemophilia bleeding 16,17. Furthermore, breakthrough bleeding episodes were not seen during prophylaxis with a schedule of 25 IU kg−1 twice weekly. The data on breakthrough bleeding in this study are limited by the fact that bleeding episodes were self-reported and, except for one subject admitted to the hospital with a pelvic muscle bleed, the episodes were not objectively confirmed. Overall, preliminary evidence from this small study suggests that a PEGylated FVIII with a prolonged half-life may provide effective prophylaxis, with longer intervals between doses.
In summary, this study found that BAY 94-9027 had improved pharmacokinetics as compared with rFVIII-FS, and that BAY 94-9027 was well tolerated, with no immunogenicity over an 8-week treatment period. These data support larger clinical trials of BAY 94-9027 to confirm its efficacy and to determine the optimal dose and schedule for prophylaxis of bleeding in hemophilia A. A pivotal phase II/III study is currently underway to further evaluate BAY 94-9027 in patients with severe hemophilia A.
Addendum
T. E. Coyle helped to design the study, assisted with the accrual and treatment of subjects, reviewed results, and assisted with writing of the manuscript. M. T. Reding assisted with the study design, participated as an investigator, reviewed results, and helped to edit the manuscript. J. Powell helped to design the clinical study, participated as an investigator, reviewed results, and assisted in the writing of the manuscript. L. A. Michaels helped with the study design, ensured proper study performance, reviewed results, and assisted with writing of the manuscript. A. Shah helped to design the study, ensured proper study performance, reviewed results, and assisted in writing and editing the manuscript. J. C. Lin participated as an investigator, reviewed results, and assisted in editing the manuscript.
Acknowledgments
This study was funded by Bayer HealthCare AG (Leverkusen, Germany). We would like to thank J. A. Phillips from Complete Healthcare Communications, Inc. (Chadds Ford, PA, USA) for medical writing assistance, which was fully funded by Bayer HealthCare.
Disclosure of Conflict of Interests
T. E. Coyle is an advisory board participant for Bayer. M. T. Reding has served as an advisory board member, consultant, and speaker, and/or has received clinical trial support, from Bayer, Baxter, Biogen Idec, Octapharma, Novo Nordisk, and Pfizer. L. A. Michaels and A. Shah are employees of Bayer HealthCare Pharmaceuticals. J. Powell receives clinical trial support from Bayer, Octapharma, and Biogen Idec. J. C. Lin has no conflicts of interest to report.
References
- 1.Aledort LM, Haschmeyer RH, Pettersson H. A longitudinal study of orthopaedic outcomes for severe factor-VIII-deficient haemophiliacs. The Orthopaedic Outcome Study Group. J Intern Med. 1994;236:391–9. doi: 10.1111/j.1365-2796.1994.tb00815.x. [DOI] [PubMed] [Google Scholar]
- 2.Manco-Johnson MJ, Abshire TC, Shapiro AD, Riske B, Hacker MR, Kilcoyne R, Ingram JD, Manco-Johnson ML, Funk S, Jacobson L, Valentino LA, Hoots WK, Buchanan GR, DiMichele D, Recht M, Brown D, Leissinger C, Bleak S, Cohen A, Mathew P, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535–44. doi: 10.1056/NEJMoa067659. [DOI] [PubMed] [Google Scholar]
- 3.Gringeri A, Lundin B, Mackensen SV, Mantovani L, Mannucci PM the Esprit Study Group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study) J Thromb Haemost. 2011;9:700–10. doi: 10.1111/j.1538-7836.2011.04214.x. [DOI] [PubMed] [Google Scholar]
- 4.Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, Llinas A, Ludlam CA, Mahlangu JN, Mulder K, Poon MC, Street A Treatment Guidelines Working Group on Behalf of The World Federation of Hemophilia. Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1–47. doi: 10.1111/j.1365-2516.2012.02909.x. [DOI] [PubMed] [Google Scholar]
- 5.Feldman BM, Pai M, Rivard GE, Israels S, Poon MC, Demers C, Robinson S, Luke KH, Wu JK, Gill K, Lillicrap D, Babyn P, McLimont M, Blanchette VS. Tailored prophylaxis in severe hemophilia A: interim results from the first 5 years of the Canadian Hemophilia Primary Prophylaxis Study. J Thromb Haemost. 2006;4:1228–36. doi: 10.1111/j.1538-7836.2006.01953.x. [DOI] [PubMed] [Google Scholar]
- 6.Roosendaal G, Lafeber F. Prophylactic treatment for prevention of joint disease in hemophilia – cost versus benefit. N Engl J Med. 2007;357:603–5. doi: 10.1056/NEJMe078098. [DOI] [PubMed] [Google Scholar]
- 7.Brown SA, Aledort LM, Astermark J, Berntorp E, van den Berg M, Blanchette V, Donfield S, Gringeri A, Hilgartner M, Kulkarni R, Leissinger C, Negrier C, Nuss R, Petterson H, Petrini P, Poulios N, Schramm W. Unresolved issues in prophylaxis. Haemophilia. 2002;8:817–21. doi: 10.1046/j.1365-2516.2002.00685.x. [DOI] [PubMed] [Google Scholar]
- 8.du Treil S, Rice J, Leissinger CA. Quantifying adherence to treatment and its relationship to quality of life in a well-characterized haemophilia population. Haemophilia. 2007;13:493–501. doi: 10.1111/j.1365-2516.2007.01526.x. [DOI] [PubMed] [Google Scholar]
- 9.Hacker MR, Geraghty S, Manco-Johnson M. Barriers to compliance with prophylaxis therapy in haemophilia. Haemophilia. 2001;7:392–6. doi: 10.1046/j.1365-2516.2001.00534.x. [DOI] [PubMed] [Google Scholar]
- 10.Kreuz W, Escuriola-Ettingshausen C, Funk M, Schmidt H, Kornhuber B. When should prophylactic treatment in patients with haemophilia A and B start? – The German experience. Haemophilia. 1998;4:413–17. doi: 10.1046/j.1365-2516.1998.440413.x. [DOI] [PubMed] [Google Scholar]
- 11.Berntorp E. Joint outcomes in patients with haemophilia: the importance of adherence to preventive regimens. Haemophilia. 2009;15:1219–27. doi: 10.1111/j.1365-2516.2009.02077.x. [DOI] [PubMed] [Google Scholar]
- 12.Veronese FM, Pasut G. PEGylation, successful approach to drug delivery. Drug Discov Today. 2005;10:1451–8. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- 13.Pipe SW. Go long! A touchdown for factor VIII? Blood. 2010;116:153–4. doi: 10.1182/blood-2010-03-274233. [DOI] [PubMed] [Google Scholar]
- 14.Pipe SW. Hemophilia: new protein therapeutics. Hematology Am Soc Hematol Educ Program. 2010;2010:203–9. doi: 10.1182/asheducation-2010.1.203. [DOI] [PubMed] [Google Scholar]
- 15.Rostin J, Smeds AL, Akerblom E. B-Domain deleted recombinant coagulation factor VIII modified with monomethoxy polyethylene glycol. Bioconjug Chem. 2000;11:387–96. doi: 10.1021/bc990137i. [DOI] [PubMed] [Google Scholar]
- 16.Mei B, Pan C, Jiang H, Tjandra H, Strauss J, Chen Y, Liu T, Zhang X, Severs J, Newgren J, Chen J, Gu J-M, Subramanyam B, Fournel MA, Pierce GF, Murphy JE. Rational design of a fully active, long-acting PEGylated factor VIII for hemophilia A treatment. Blood. 2010;116:270–9. doi: 10.1182/blood-2009-11-254755. [DOI] [PubMed] [Google Scholar]
- 17.Liu T, Lillicrap D, Zhang X, Labelle A, Powell S, Mei B, Murphy JE, Pierce GF, Jiang H. Site-specific PEGylation of factor VIII (PEG-FVIII) preserves full clotting activity and extends therapeutic efficacy in hemophiliaA dogs [abstract] Blood. 2008;112:511. abstract. [Google Scholar]
- 18.Ivens IA, Zierz R, Haaning J. Bay 94-9027, a PEGylated recombinant human FVIII shows less immunogenicity compared to un-PEGylated recombinant FVIII. Blood. 2010;116:2214. abstract. [Google Scholar]
- 19.Ivens IA, Baumann A, McDonald TA, Humphries TJ, Michaels LA, Mathew P. PEGylated therapeutic proteins for haemophilia treatment: a review for haemophilia caregivers. Haemophilia. 2013;19:11–20. doi: 10.1111/j.1365-2516.2012.02931.x. [DOI] [PubMed] [Google Scholar]
- 20.Powell JS, Nugent DJ, Harrison JA, Soni A, Luk A, Stass H, Gorina E. Safety and pharmacokinetics of a recombinant factor VIII with pegylated liposomes in severe hemophilia A. J Thromb Haemost. 2008;6:277–83. doi: 10.1111/j.1538-7836.2008.02856.x. [DOI] [PubMed] [Google Scholar]
- 21.Kessler CM, Gill JC, White GC, 2nd, Shapiro A, Arkin S, Roth DA, Meng X, Lusher JM. B-domain deleted recombinant factor VIII preparations are bioequivalent to a monoclonal antibody purified plasma-derived factor VIII concentrate: a randomized, three-way crossover study. Haemophilia. 2005;11:84–91. doi: 10.1111/j.1365-2516.2005.01068.x. [DOI] [PubMed] [Google Scholar]
- 22.Di Paola J, Smith MP, Klamroth R, Mannucci PM, Kollmer C, Feingold J, Kessler C, Pollmann H, Morfini M, Udata C, Rothschild C, Hermans C, Janco R. ReFacto and Advate: a single-dose, randomized, two-period crossover pharmacokinetics study in subjects with haemophilia A. Haemophilia. 2007;13:124–30. doi: 10.1111/j.1365-2516.2006.01420.x. [DOI] [PubMed] [Google Scholar]
- 23.Lenting PJ, Christophe OD, Gueguen P. The disappearing act of factor VIII. Haemophilia. 2008;16:6–15. doi: 10.1111/j.1365-2516.2008.01864.x. [DOI] [PubMed] [Google Scholar]
- 24.Denis CV, Christophe OD, Oortwijn BD, Lenting PJ. Clearance of von Willebrand factor. Thromb Haemost. 2008;99:271–8. doi: 10.1160/TH07-10-0629. [DOI] [PubMed] [Google Scholar]
- 25.Fischer K, Pendu R, van Schooten CJ, van Dijk K, Denis CV, van den Berg HM, Lenting PJ. Models for prediction of factor VIII half-life in severe haemophiliacs: distinct approaches for blood group O and non-O patients. PLoS One. 2009;4:e6745. doi: 10.1371/journal.pone.0006745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Schooten CJ, Shahbazi S, Groot E, Oortwijn BD, van den Berg HM, Denis CV, Lenting PJ. Macrophages contribute to the cellular uptake of von Willebrand factor and factor VIII in vivo. Blood. 2008;112:1704–12. doi: 10.1182/blood-2008-01-133181. [DOI] [PubMed] [Google Scholar]
- 27.Powell JS, Josephson NC, Quon D, Ragni MV, Cheng G, Li E, Jiang H, Li L, Dumont JA, Goyal J, Zhang X, Sommer J, McCue J, Barbetti M, Luk A, Pierce GF. Safety and prolonged activity of recombinant factor VIII Fc fusion protein in hemophilia A patients. Blood. 2012;119:3031–7. doi: 10.1182/blood-2011-09-382846. [DOI] [PMC free article] [PubMed] [Google Scholar]