

Abstract
Cleft palate following cleft lip may include a developmental disorder during palatogenesis. CL/Fr mice fetuses, which develop cleft lip and palate spontaneously, have less capability for in vivo cell proliferation in palatal mesenchyme compared with CL/Fr normal fetuses. In order to know the changes of signaling molecules contributing to cleft palate morphogenesis following cleft lip, the mRNA expression profiles were compared in palatal shelves oriented vertically (before elevation) in CL/Fr fetuses with or without cleft lip. The changes in mRNA profile of cleft palate morphogenesis were presented in a microarray analysis, and genes were restricted to lists contributing to cleft palate development in CL/Fr fetuses with cleft lip. Four candidate genes (Ywhab, Nek2, Tacc1 and Frk) were linked in a gene network that associates with cell proliferation (cell cycle, MAPK, Wnt and Tgf beta pathways). Quantitative real-time RT-PCR highlighted the candidate genes that significantly changed in CL/Fr fetuses with cleft lip (Ywhab, Nek2 and Tacc1). The results of these molecular contributions will provide useful information for a better understanding of palatogenesis in cleft palate following cleft lip. Our data indicated the genetic contribution to cleft palate morphogenesis following cleft lip.
Keywords: CL/Fr mice, cleft palate, embryology, growth/development, molecular biology
Introduction
Cleft lip and palate (CLP), one of the most common craniofacial defects among live births, is notable for significant lifelong morbidity from early infancy in humans (Sasaki and Fujiwara 2007). Seventy percent of cleft lip only (CL) and CLP are non-syndromic (Jones 1988) and derive from a combination of genetic and environmental factors. In order to know the palatal morphogenesis following cleft lip, it should be noted that CL and CLP may have different genetic causes and should, when feasible, be analyzed separately (Jugessur et al. 2009). A genome screen of polymorphic markers in CL and CLP confirmed the location of the recessive genes in A/WySn, a strain of mice with spontaneous CL and CLP (Juriloff et al. 2001). However, the linkage for cleft palate following cleft lip was unknown because CL and CLP were not distinguished. Furthermore, the mechanism for isolated cleft palate (CP) is not applied to cleft palate following cleft lip because there is strong evidence that in most cases CL and CLP are both developmentally and genetically different from CP (Fraser 1970).
There are three important stages in the development of bilateral palatal shelves: vertical growth down the sides of the tongue, rapid elevation to a horizontal position above the dorsum of the tongue, and fusion of the medial edge epithelia of the approximately palatal shelves (Ferguson 1988). It is clear that disruptions of palatal growth to any one or more of the critical mechanisms during normal palate development results in cleft palate, which is a common occurrence following cleft lip.
A classical observation in A/J mice fetuses with spontaneous CLP showed that the presence of a cleft lip appeared to induce mechanical obstruction by the tongue of palatal shelves, which delayed their palatal elevation and fusion (Trasler and Fraser 1963), whereas palatal shelves dissected from cleft lip A/J mice fetuses (Pourtois 1967) and CL/Fr mice (Sasaki et al. 2004) fused in vitro. These findings indicate that the presence of cleft lip alone did not obviously affect the propensity of the palatal processes to fuse. It was shown that the proliferation of mesenchymal cells play an important role in the appearance of the primordia and during early vertical growth of the palatal shelves (Burdett et al. 1988), and the inadequacy of palatal elevation or delayed development was related to the proliferation of embryonic palate mesenchymal cells (Dixon and Ferguson 1992). The CL/Fr mouse strain is a useful experimental animal model to examine the palatal development following spontaneous cleft lip because it has a spontaneous newborn CLP rate of 15–40% (Millicovsky et al. 1982; Wang et al. 1995), and more than 96% of CL/Fr fetuses with cleft lip subsequently develop cleft secondary palate (Brown et al. 1985). In a recent molecular study with CL/Fr fetuses, in vivo cell proliferation kinetics in palatal shelves were examined at E13.5, when palatal shelves grew vertically down the side of the tongue, and indicated that CL/Fr fetuses with cleft lip have less capability for palatal mesenchymal cell proliferation compared with CL/Fr normal fetuses (Sasaki et al. 2004), whereas in-situ hybridization elucidated a similar gene expression pattern in palatal shelves at E13.5 in CL/Fr fetuses with cleft lip and CL/Fr normal fetuses (Hamachi et al. 2003). The signaling molecules were identified from chromosomal linkage studies in mouse genetic models with CLP (Schutte and Murray 1999; Juriloff and Harris 2008), and the gene expression microarray analysis during epithelial-mesenchymal transformation (LaGamba et al. 2005), among the three stages of palate development (Brown et al. 2003) and between normal and cleft mice derived from the A/J mice head region (E15) (Saito et al. 2002). However, these previous studies did not refer to the palate following cleft lip, it is still unknown whether the signaling molecules change in cleft palate morphogenesis following cleft lip. The present study was undertaken to distinguish the significant genes from a number of genes on cleft palate morphogenesis in CL/Fr and identify their significance and to elucidate the cell kinetics in palatal development in CL/Fr fetuses. Specifically, we have examined the hypothesis that any signaling molecules were up- or downregulated in palatal shelves of CL/Fr fetuses with cleft lip in relation to disruption of cell proliferation in palatal development.
Materials and Methods
Embryos
An origin of CL/Fr mice was derived from the 2nd Department of Oral and Maxillofacial Surgery, Niigata University School of Dentistry, Japan, and was maintained in our department by sibling crossbreeding. Our animal use protocol was reviewed and approved by the Institutional Review Board at Nagasaki University, and the mice were treated in accordance with the NIH guidelines. To collect fetuses of each strain, females were cohabited with one fertile male overnight, and the morning was designated as embryonic day 0 (E0) when a copulatory plug was detected. The fetuses were sampled on the evening of embryonic day 13 (E13.5), when the palatal shelves grow vertically down the sides of the tongue before elevation (Hamachi et al. 2003). Under a dissecting microscope, CL/Fr fetuses were classified into bilateral cleft lip (CL/Fr-BCL) (Fig. 1a) and normal (CL/Fr-N) fetuses (Fig. 1b). The colony of fetuses used in the experiment consisted of 12 pregnant CL/Fr mice with 12 CL/Fr-BCL and 53 CL/Fr-N fetuses, indicating that the frequency of cleft lip and palate equaled 18.5%. Unilateral cleft lip fetuses were not obtained in this study. Palatal shelves were dissected along anterior and posterior axis of the palatal processes on the region which would differentiate to hard and soft palate (Fig. 1a–d). In microarray analysis, 9 CL/Fr-N and 5 CL/Fr-BCL fetuses derived from five pregnant females were used, and eight CL/Fr-N and seven CL/Fr-BCL fetuses derived from the other seven pregnant females were used in real-time reverse transcription-polymerase chain reaction (RT-PCR) experiment. Analysis of craniofacial growth in the CLP affected fetuses of CL/Fr strain indicated no statistical significance in gender difference (Martin et al. 1995; Nonaka et al. 1997). There were no data of gender at E13.5 fetuses in our experiments, and the samples were randomly selected from the litters, resulting in no significant bias of gender.
Figure 1.
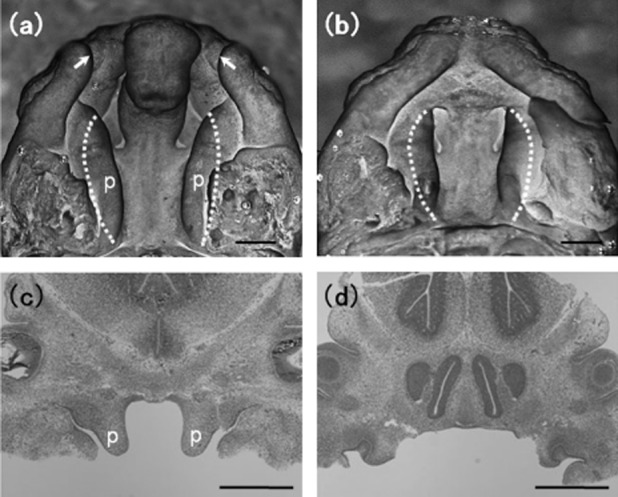
Palatal shelves at embryonic day 13.5. Palatal shelves (p) of CL/Fr-BCL fetuses (a) and CL/Fr-N fetuses (b). Transverse section of palatal shelves (c) and palatal shelves incised (d) of CL/Fr-BCL fetuses. Bilateral cleft lip is indicated by arrows. In (a) and (b), dotted lines indicate the incision line. Scale bars: 0.5 mm.
Microarray analysis
Total RNA from excised palatal shelves was isolated using the RNeasy Micro Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s recommendations. The cDNA was synthesized using the SuperScript II Reverse Transcription Kit (Invitrogen, Carlsbad, CA, USA).
Microarray analysis was performed using GeneChip (Mouse Expression 430 2.0 Array, Affymetrix, Santa Clara, San Francisco, CA, USA) and probe arrays and images from the scanned chips were processed by the manufacturer (Biomatrix, Tokyo, Japan). The GeneChip images of the CL/Fr-BCL samples were normalized to the corresponding CL/Fr-N image across all probe pair sets to analyze the two different embryonic palatal tissue target RNA samples (CL/Fr-BCL vs. CL/Fr-N). Normalized and fold-change values from each of the two palatal samples were exported. The full dataset was obtained using GeneSpring GX (Agilent Technologies, Palo Alto, CA, USA) and filtered for all 45 101 transcripts. The dataset was preprocessed by restricting the list to genes that were up- or downregulated. Gene induction or repression was considered biologically meaningful if the difference in hybridization intensity was greater than two-fold or less than 0.5-fold. The subsets of genes that we selected were based on pathways of Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/). Gene ontology (GO) analysis was performed with Cytoscape (http://cytoscape.org/index.php) and Biological Network Gene Ontology tool (BiNGO; http://www.psb.ugent.be/cbd/papers/BiNGO/Home.html). According to the protocol of BiNGO that was exhibited in the previous reports (Ge et al. 2003; Boyle et al. 2004), the transcripts, which changed between CL/F-BCL and CL/Fr-N, were examined based on normalized intensity. The transcripts were identified by the GO categories that belonged to known biological processes.
Quantitative Real-Time PCR
Total RNA was prepared from palatal shelves at E13.5, and the cDNA was synthesized as described for microarray analysis. Primer sequences were designed for each of the selected genes using Genetyx-Mac (ver.11.2.2, Genetyx, Tokyo, Japan). The primers designed were: (sense primer first): Ywhab, 5′-TCCAGCATTCCCAGGTAGGCCA-3′ and 5′-ACCACACCAGCCAAGTCAGC-3′; Nek2, 5′-GGAGCTGGGTCAGTGTTGAT-3′ and 5′-CACATCCATTTGCAGACCAC-3′; Tacc1, 5′-TGAGCCAGTGAT GGACAG AG-3′ and 5′-CAGGGTCCTCCAGTCTTCAG-3′; Frk, 5′-CCACATTGCCTACCAGGTCT-3′ and 5′-TCCTAGCCTCAGGTGCTGAT-3′. The values were normalized using the housekeeping gene GAPDH (5′-TGC ACCACCAACTGCTTAGC-3′ of sense and 5′-GGATGCAGGGATGATGTTCT-3′ of antisense primer). For all of the four genes analyzed, 0.5 μL of a 5 μmol solution of the forward and reverse primers and cDNA were added to a final volume of 50 μL of SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). Real-time PCR assays were performed with an ABI Prism 7000 System (Applied Biosystems). Cycling parameters were: 50°C for 2 min for initial activation of the probe and primer, 95°C for 10 min for denaturation of the DNA strands, followed by 40 cycles of denaturation at 95°C for 15 s, and primer extension at 58°C for 1 min. Data were acquired and processed with Sequence Detector 1.6.3 software (Applied Biosystems). The data were analyzed by unpaired Student’s t-test for comparison between CL/Fr-N and CL/Fr-BCL.
Results
Gene expression by microarray analysis
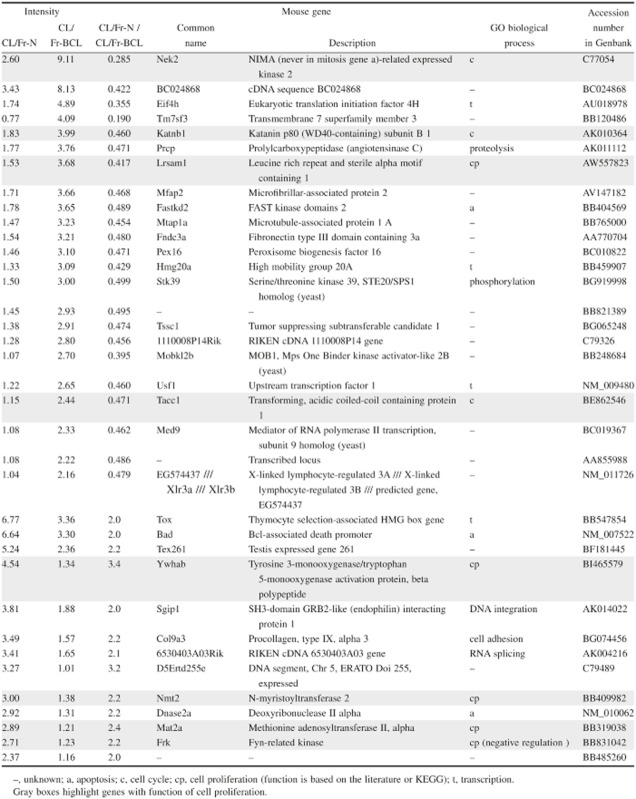
The Affymetrix GeneChip arrays containing oligonucleotide probes represented more than 45 101 transcripts. The expression rates of all 45 101 transcripts (23 366 genes) were investigated. Restricting the list yielded 243 transcripts, of which 112 were upregulated at least two-fold and 131 were downregulated at most 0.5-fold in CL/Fr-BCL fetuses (Fig. 2a). Genes were restricted into 36 genes in excess of intensity (>1.0) in both CL/Fr-BCL and CL/Fr-N (Table 1). Based on their expression profiles, BiNGO was used to classify genes according to the following GO categories: cell cycle, transcription, apoptosis, cell proliferation, others, and unknown. This list includes eight genes with function of cell proliferation in GO categories (cell cycle and cell proliferation).
Figure 2.
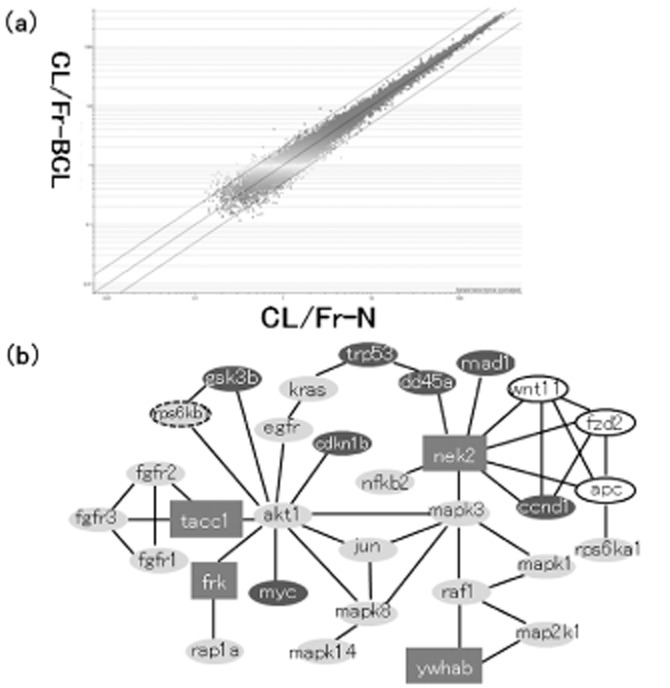
Gene expression analysis between CL/Fr-BCL and CL/Fr-N fetuses. Differential expression between CL/Fr-BCL and CL/Fr-N fetuses (a). Each dot represents a signal intensity measurement for a gene. The lines represent the thresholds for two-fold differential expression. Gene network that associate cell proliferation in CL/Fr-BCL (b). The dataset was preprocessed by restricting the list to genes that were greater than two-fold or less than 0.5-fold in hybridization intensity.  , gene that showed 2-fold;
, gene that showed 2-fold;  , Candidate gen;
, Candidate gen;  , KEGG Cell cycle pathway;
, KEGG Cell cycle pathway;  , KEGG MAPK pathway;
, KEGG MAPK pathway;  , KEGG Wnt pathway;
, KEGG Wnt pathway;  , KEGG Tgfb pathway.
, KEGG Tgfb pathway.
Table 1.
Summary of expression differences between CL/Fr-BCL and CL/Fr-N fetuses
 |
Analysis of gene network and candidate genes
Cytoscape literature research analysis in combination with KEGG pathway showed the gene network that is associated with cell proliferation in CL/Fr-BCL (Fig. 2b). The subsets of genes that we selected were based on KEGG pathways including cell cycle, MAPK, Wnt and Tgf-beta. In detail, four pathways of MAPK signaling, Wnt signaling, Tgf-beta signaling and Cell cycle were extracted by detecting the signal pathways that were registered in the KEGG pathway in KEGG software and associated with the regulation of cell proliferation. All of the genes that constituted these pathways were listed for further analysis. Microarray analysis extracted eight genes associating with cell proliferation from the genes that showed significant difference in hybridization intensity of greater than twofold or less than 0.5-fold. Analysis with molecular network software (Cytoscape plug-in Agilent literature search; http://www.agilent.com/labs/research/litsearch.html) for these eight genes made the molecular network that included the four genes (Ywhab, Nek2, Tacc1 and Frk), thereafter the other four genes were excluded from the candidates. In this network, the genes that were listed from KEGG pathways and associated with cell proliferation remained and the other genes were excluded (Fig. 2b). This final network indicated that the four genes that showed greater than twofold or less than 0.5-fold change were highly connected to each other by either sharing common signaling pathways or through gene–gene interactions that associated with the cell proliferation, and contributed to the cell proliferation of palatal processes.
Using real-time RT-PCR, significant differences of gene expressions were identified in three of the four genes (Ywhab, Nek2 and Tacc1). The amount of mRNA in Nek2 and Tacc1 of CL/Fr-BCL significantly increased compared with that in CL/Fr-N fetuses, while Ywhab significantly decreased in CL/Fr-BCL fetuses (Fig. 3). There was no significant difference in Frk expression between CL/Fr-N and CL/Fr-BCL.
Figure 3.

Real-time polymerase chain reaction (PCR) analysis of mRNA for four genes in CL/Fr-BCL (BCL) and CL/Fr-N (N) fetuses. Real-time PCR analysis of mRNA for four genes in CL/Fr-BCL and CL/Fr–N fetuses. Results are expressed as mean (SD) of the ratio of the specific mRNA to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA. *P < 0.05; ns, not significant.
Discussion
In our experiments, we were fortunate to obtain data for most of the palatal genes that are of interest in the production of cleft secondary palate following cleft lip. We identified the pathways of the possible signaling molecules during palatal morphogenesis in pathway analysis.
In spite of a lot of studies with mouse models of cleft lip with or without cleft palate, molecular event in palatal shelves following spontaneous cleft lip has been poorly understood. Our microarray analysis found that there were only 36 transcripts in 45 101 that showed height intensity of expression (>1) and a twofold upregulation or a 0.5-fold downregulation in affected fetuses of CL/Fr, and these molecules indicated a relatively close expression level in CL/Fr-BCL vs. CL/Fr-N fetuses. These findings strongly support the hypothesis that the cause of spontaneous cleft palate morphogenesis in CL/Fr mice is a multifactorial trait and based on threshold theory involving signaling molecules during palate morphogenesis that occurs subsequent to cleft lip. Nonetheless, eight out of 36 restricted genes (22.2%) were categorized to the molecules that relate to cell proliferation, and its ratio was relatively high in the restricted genes, suggesting that the in vivo signaling molecules associated with cell proliferation might induce a reduction in palatal mesenchymal cell kinetics in CL/Fr-BCL fetuses. This hypothesis is supported by a previous study indicating that palatal shelves from CL/Fr-BCL fetuses have less capability for in vivo mesenchymal cell proliferation than CL/Fr-N fetuses (Sasaki et al. 2004). The downregulation of in vivo cell proliferation kinetics in CL/Fr compared with C57BL mice suggests that cellular susceptibility itself relates to cleft palate in CL/Fr strain of fetuses.
Three restricted molecules (Ywhab, Nek2 and Tacc1) were consistently up- or downregulated using real-time RT-PCR in CL/Fr-BCL. Previous reports with the use of mouse palatal shelves have indicated that genes, associating with cell proliferation in palatal mesenchyme, were Msx1, Pax9, Barx1, Shox2, Lhx8, Gli3, Sall3, p63, Osr2, Bmp2, 4, Fgf10, and PdgfRa et al. (Gritli-Linde 2007). No genes matched the signaling molecules identified from our experiment, because there are objective differences, i.e., no other previous studies have derived from the palatal tissue with spontaneous (nonsyndromic) cleft lip, and we first report the molecules affecting the palatogenesis with spontaneous cleft lip with use of palatal tissue.
The effect of the molecules that we found could be small and indirect on palatal disorder following spontaneous cleft lip. However, regarding the linkage to palatal development, several possibilities may be considered for the GO “biological process” according to the literature. First, the 14-3-3 proteins (Ywhab), which were downregulated in CL/Fr-BCL in our results, are associated with proto-oncogene and oncogene products. Overexpression of Ywhab in NIH 3T3 cells stimulated cell growth and supported anchorage-independent growth in soft agar medium in nude mice (Takihara et al. 2000). Downregulation of Ywhab in CL/Fr-BCL could work as an inhibitory factor for palatal development at the time when cell proliferation is required. Second, immunoblotting has shown an upregulation of Nek2 in all eight breast carcinoma cell lines examined (Barbagallo et al. 2009; Tsunoda et al. 2009). One function of Nek2 kinase activity is to promote the splitting of duplicated centrosomes at the onset of mitosis through phosphorylation of core centriolar proteins (Fry 2002), and overexpression of active Nek2 leads to centrosome splitting and dispersal (Fry et al. 1998). These functions of Nek2 might also contribute to defection of cell cycle during the palatal development in CL/Fr-BCL.
Furthermore, the expression of a newly identified TACC1 isoform, which was upregulated in CL/Fr-BCL in our results, is restricted to brain and gastric cancer tissues, and quantitative RT-PCR revealed a relative overexpression of the gene in cancer tissues (Line et al. 2002). Interestingly, a nationwide study of the occurrence of cancer among Danish oral cleft (Bille et al. 2005) and white patients with cleft lip and cleft palate (Menezes et al. 2009) showed an increased occurrence of cancer in cleft lip and/or cleft palate. We found Wnt11 linked with Nek2 in KEGG Wnt pathway in gene network of cell proliferation of CL/Fr-BCL fetuses. Wnt pathway genes have been suggested as candidate genes for cleft/lip palate based on studies with animal models (Juriloff and Harris 2008) and association studies in humans (Menezes et al. 2009), while WNT11 SNP showed association with human oral squamous cell carcinoma (Andrade Filho et al. 2011) and there was a correlation between Wnt11 expression and odontoblast differentiation in rat dental pulp (Koizumi et al. 2013). Our analysis provides evidence that defection of cell cycle may result in the common etiology of oral cancer and cleft palate with cleft lip in the view of high activity of cell proliferation both in cancer cell and embryonic palatal shelf. The study of these molecular contributions will provide useful information with which to gain a better understanding of palatogenesis in cleft palate following cleft lip.
Acknowledgments
This work was supported by the Japanese Society for the Promotion of Science (grant number 19592358 and 21592603).
References
- Andrade Filho PA, Letra A, Cramer A, et al. Insights from studies with oral cleft genes suggest associations between WNT-pathway genes and risk of oral cancer. J Dent Res. 2011;90:740–746. doi: 10.1177/0022034511401622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbagallo F, Paronetto MP, Franco R, et al. Increased expression and nuclear localization of the centrosomal kinase Nek2 in human testicular seminomas. J Pathol. 2009;217:431–441. doi: 10.1002/path.2471. [DOI] [PubMed] [Google Scholar]
- Bille C, Winther JF, Bautz A, Murray JC, Olsen J, Christensen K. Cancer risk in persons with oral cleft–a population-based study of 8,093 cases. Am J Epidemiol. 2005;161:1047–1055. doi: 10.1093/aje/kwi132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle EI, Weng S, Gollub J, et al. GO:TermFinder–open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20:3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KS, Hetzel SC, Harne LC, Long S. Blebs and hematomas in the lips of CL/Fr and A/J mice. J Craniofac Genet Dev Biol Suppl. 1985;1:313–322. [PubMed] [Google Scholar]
- Brown NL, Knott L, Halligan E, Yarram SJ, Mansell JP, Sandy JR. Microarray analysis of murine palatogenesis: temporal expression of genes during normal palate development. Dev Growth Differ. 2003;45:153–165. doi: 10.1034/j.1600-0854.2004.00686.x. [DOI] [PubMed] [Google Scholar]
- Burdett DN, Waterfield JD, Shah RM. Vertical development of the secondary palate in hamster embryos following exposure to 6-mercaptopurine. Teratology. 1988;37:591–597. doi: 10.1002/tera.1420370608. [DOI] [PubMed] [Google Scholar]
- Dixon MJ, Ferguson MW. The effects of epidermal growth factor, transforming growth factors alpha and beta and platelet-derived growth factor on murine palatal shelves in organ culture. Arch Oral Biol. 1992;37:395–410. doi: 10.1016/0003-9969(92)90024-3. [DOI] [PubMed] [Google Scholar]
- Ferguson MW. Palate development. Development. 1988;(103):41–60. doi: 10.1242/dev.103.Supplement.41. [DOI] [PubMed] [Google Scholar]
- Fraser FC. The genetics of cleft lip and cleft palate. Am J Hum Genet. 1970;22:336–352. [PMC free article] [PubMed] [Google Scholar]
- Fry AM. The Nek2 protein kinase: a novel regulator of centrosome structure. Oncogene. 2002;21:6184–6194. doi: 10.1038/sj.onc.1205711. [DOI] [PubMed] [Google Scholar]
- Fry AM, Meraldi P, Nigg EA. A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J. 1998;17:470–481. doi: 10.1093/emboj/17.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Dudoit S, Speed TP. 2003. Resampling-based multiple testing for microarray data analysis, Technical Report 633, Dept. of Statistics, UC Berkeley. available at : http://statistics.berkeley.edu/tech-reports/633.
- Gritli-Linde A. Molecular control of secondary palate development. Dev Biol. 2007;301:309–326. doi: 10.1016/j.ydbio.2006.07.042. [DOI] [PubMed] [Google Scholar]
- Hamachi T, Sasaki Y, Hidaka K, Nakata M. Association between palatal morphogenesis and Pax9 expression pattern in CL/Fr embryos with clefting during palatal development. Arch Oral Biol. 2003;48:581–587. doi: 10.1016/s0003-9969(03)00104-3. [DOI] [PubMed] [Google Scholar]
- Jones MC. Etiology of facial clefts: prospective evaluation of 428 patients. Cleft Palate J. 1988;25:16–20. [PubMed] [Google Scholar]
- Jugessur A, Farlie PG, Kilpatrick N. The genetics of isolated orofacial clefts: from genotypes to subphenotypes. Oral Dis. 2009;15:437–453. doi: 10.1111/j.1601-0825.2009.01577.x. [DOI] [PubMed] [Google Scholar]
- Juriloff DM, Harris MJ. Mouse genetic models of cleft lip with or without cleft palate. Birth Defects Res A Clin Mol Teratol. 2008;82:63–77. doi: 10.1002/bdra.20430. [DOI] [PubMed] [Google Scholar]
- Juriloff DM, Harris MJ, Brown CJ. Unravelling the complex genetics of cleft lip in the mouse model. Mamm Genome. 2001;12:426–435. doi: 10.1007/s003350010284. [DOI] [PubMed] [Google Scholar]
- Koizumi Y, Kawashima N, Yamamoto M, et al. Wnt11 expression in rat dental pulp and promotional effects of Wnt signaling on odontoblast differentiation. Congenit Anom Kyoto. 2013;53:101–108. doi: 10.1111/cga.12011. [DOI] [PubMed] [Google Scholar]
- LaGamba D, Nawshad A, Hay ED. Microarray analysis of gene expression during epithelial-mesenchymal transformation. Dev Dyn. 2005;234:132–142. doi: 10.1002/dvdy.20489. [DOI] [PubMed] [Google Scholar]
- Line A, Stengrevics A, Slucka Z, Li G, Jankevics E, Rees RC. Serological identification and expression analysis of gastric cancer-associated genes. Br J Cancer. 2002;86:1824–1830. doi: 10.1038/sj.bjc.6600321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DA, Nonaka K, Yanagita K, Nakata M. The effect of dam strain on the craniofacial morphogenesis of CL/Fr mouse fetuses. J Craniofac Genet Dev Biol. 1995;15:117–124. [PubMed] [Google Scholar]
- Menezes R, Marazita ML, Goldstein McHenry T, et al. AXIS inhibition protein 2, orofacial clefts and a family history of cancer. J Am Dent Assoc. 2009;140:80–84. doi: 10.14219/jada.archive.2009.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millicovsky G, Ambrose LJ, Johnston MC. Developmental alterations associated with spontaneous cleft lip and palate in CL/Fr mice. Am J Anat. 1982;164:29–44. doi: 10.1002/aja.1001640104. [DOI] [PubMed] [Google Scholar]
- Nonaka K, Sasaki Y, Watanabe Y, Yanagita K, Nakata M. Effects of fetus weight, dam strain, dam weight, and litter size on the craniofacial morphogenesis of CL/Fr mouse fetuses affected with cleft lip and palate. Cleft Palate Craniofac J. 1997;34:325–330. doi: 10.1597/1545-1569_1997_034_0325_eofwds_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- Pourtois M. Influence of cleft lip upon palatal closure in A/Jax mice. Cleft Palate J. 1967;4:120–123. [PubMed] [Google Scholar]
- Saito K, Fukumoto S, Fukumoto E, et al. Cleft palate- and lip-related gene expression analyzed by cDNA microarray hybridization. Pediatr Dent J. 2002;12:35–42. [Google Scholar]
- Sasaki Y, Fujiwara T. Presurgical growth guidance with naso-alveolar molding in infants with cleft lip and palate. Dent Jpn (Tokyo) 2007;43:112–118. [Google Scholar]
- Sasaki Y, Tanaka S, Hamachi T, Taya Y. Deficient cell proliferation in palatal shelf mesenchyme of CL/Fr mouse embryos. J Dent Res. 2004;83:797–801. doi: 10.1177/154405910408301012. [DOI] [PubMed] [Google Scholar]
- Schutte BC, Murray JC. The many faces and factors of orofacial clefts. Hum Mol Genet. 1999;8:1853–1859. doi: 10.1093/hmg/8.10.1853. [DOI] [PubMed] [Google Scholar]
- Takihara Y, Matsuda Y, Hara J. Role of the beta isoform of 14-3-3 proteins in cellular proliferation and oncogenic transformation. Carcinogenesis. 2000;21:2073–2077. doi: 10.1093/carcin/21.11.2073. [DOI] [PubMed] [Google Scholar]
- Trasler DG, Fraser FC. Role of the tongue in producing cleft palate in mice with spontaneous cleft lip. Dev Biol. 1963;6:45–60. doi: 10.1016/0012-1606(63)90004-6. [DOI] [PubMed] [Google Scholar]
- Tsunoda N, Kokuryo T, Oda K, et al. Nek2 as a novel molecular target for the treatment of breast carcinoma. Cancer Sci. 2009;100:111–116. doi: 10.1111/j.1349-7006.2008.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KY, Juriloff DM, Diewert VM. Deficient and delayed primary palatal fusion and mesenchymal bridge formation in cleft lip-liable strains of mice. J Craniofac Genet Dev Biol. 1995;15:99–116. [PubMed] [Google Scholar]