

Abstract
The prevalence of type 2 diabetes (T2D) is evolving globally at an alarming rate. Prediabetes is an intermediate state of glucose metabolism that exists between normal glucose tolerance (NGT) and the clinical entity of T2D. Relentless β-cell decline and failure is responsible for the progression from NGT to prediabetes and eventually T2D. The huge burden resulting from the complications of T2D created the need of therapeutic strategies in an effort to prevent or delay its development. The beneficial effects of incretin-based therapies, dipeptidyl peptidase-4 inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists, on β-cell function in patients with T2D, together with their strictly glucose-depended mechanism of action, suggested their possible use in individuals with prediabetes when greater β-cell mass and function are preserved and the possibility of β-cell salvage is higher. The present paper summarizes the main molecular intracellular mechanisms through which GLP-1 exerts its activity on β-cells. It also explores the current evidence of incretin based therapies when administered in a prediabetic state, both in animal models and in humans. Finally it discusses the safety of incretin-based therapies as well as their possible role in order to delay or prevent T2D.
Keywords: Type 2 diabetes, Prediabetes, Impaired fasting glucose, Impaired glucose tolerance, Glucagon-like peptide-1, Dipeptidyl peptidase-4 inhibitors, Glucagon-like peptide-1 receptor agonists
Core tip: The beneficial effects of incretin-based therapies on β-cell function in patients with type 2 diabetes (T2D) suggested their possible use in individuals with prediabetes, when greater β-cell mass and function are preserved. Both dipeptidyl peptidase-4 inhibitors and glucagon-like peptide-1 receptor agonists have demonstrated improvements on β-cell function both in preclinical studies and short-term clinical studies. Until future date for their safety are available, large, long term, prevention trials will be required in order to determine whether they can stabilize or reverse β-cell loss and promote a sustained reduction in the development of T2D in this population.
INTRODUCTION
The prevalence of type 2 diabetes (T2D) is evolving globally at an alarming rate[1]. It is estimated that by the year 2030 approximately 366 million people will have diabetes and more than 90% of them T2D[1,2]. Prediabetes is an intermediate state of glucose metabolism that exists between normal glucose tolerance (NGT) and the clinical entity of T2D[3]. It encompasses both impaired fasting glucose (IFG) and impaired glucose tolerance (IGT). IFG is defined by a fasting plasma glucose of 100 mg/dL to 125 mg/dL, while IGT is defined by a 2 h plasma glucose concentration of 140 mg/dL to 199 mg/dL after a 75 g oral glucose tolerance test (OGTT)[3,4]. Furthermore, the American Diabetes Association suggested that glycated hemoglobulin (A1C) between 5.7% and 6.4% can also be used for the diagnosis of prediabetes, considering that A1C test must be performed by a method that is certified by the National Glycohemoglobin Standardization Program and standardized or traceable to the Diabetes Control and Complications Trial reference assay[4]. Approximately 471 million people worldwide (8% of the world’s adult population) are estimated to have IGT by the year 2035[1].
Individuals with IGT have moderate to severe muscle insulin resistance and normal to slightly decreased hepatic insulin sensitivity. They are characterized by defects in both early (0-30 min) and late-phase (60-120 min) of insulin secretion to an oral glucose load[5]. Individuals with IFG have moderate hepatic insulin resistance with normal muscle insulin sensitivity and decreased basal and early phase of insulin secretion[5]. The Veterans Administration Genetic Epidemiology Study and the San Antonio Metabolism (SAM) study have shown a progressive decline in pancreatic β-cell function in individuals with prediabetes[6,7]. The SAM study has demonstrated that when the 2 h plasma glucose during an OGTT was 180-190 mg/dL, β-cell function had already declined by 75% to 80%[6]. Eventually, approximately 20%-34% of the individuals with IFG or IGT progress to T2D over five to six years, while those with combined IFG and IGT have a cumulative incidence of 38%-65%, especially if they have low insulin secretion and severe insulin resistance[8,9]. Relentless β-cell decline and failure is responsible for the progression from NGT to IGT and eventually T2D.
A two to three fold greater increase in plasma insulin response is observed after glucose ingestion compared to a parenteral isoglycemic glucose infusion. This phenomenon was defined as the incretin effect; it accounts for approximately 70%-80% of total insulin release after oral glucose administration[10,11]. Glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are the two major incretins described; they account for approximately 90% of the incretin activity[12]. GLP-1 contributes in the overall maintenance of glucose homeostasis through the reduction of glucagon secretion, slowing of gastric emptying and control of body weight, by its appetite suppressant effect[10,11]. GLP-1 levels are significantly decreased in T2D (approximately 50% compared to healthy individuals)[10,13,14]. GIP levels are found to be elevated in patients with T2D as a result of resistance to its biological effects. Sensitivity of β-cells can be resorted after normoglycemia is established, suggesting that resistance to GIP is a manifestation of glucotoxicity[15].
Impairment in incretin hormone secretion/activity in individuals with prediabetes has been reported, although data are not consistent[16-22]. However, reduced GLP-1 levels were reported in the majority of these studies and mainly in subjects with isolated IGT or combined IFG and IGT; early phase GLP-1 response was found to be severely diminished[17-22]. Interestingly, Toft-Nielsen et al[22] have shown that during the progression from NGT to IGT and eventually T2D, there is a progressive decline in GLP-1 levels. Early GLP-1 therapy was suggested to preserve β-cell function in subjects with IGT or mild T2D[23].
Native GLP-1 is rapidly inactivated (halfe-life of 1-2 min) by the ubiquitously expressed proteolytic enzyme dipeptidyl peptidase-4 (DPP-4)[10]. The DPP-4 inhibitors are a class of oral antidiabetic agents that improve glycemic control, in patients with T2D, by increasing both GLP-1 and GIP concentrations[24]. GLP-1 receptor (GLP-1R) agonists mimic the actions of GLP-1 and are resistant to DPP-4 degradation; they have achieved significantly lower A1C values in patients with T2D that were associated with significant weight reduction[25]. Studies in cell cultures and animal models demonstrated that both DPP-4 inhibitors and GLP-1R agonists have trophic effects on pancreatic β-cells. Specifically they enhance β-cell proliferation, regeneration and differentiation; thus they increase β-cell mass. They also inhibit β-cell apoptosis, including human β-cells, through inhibition of the caspase pathway[24-26]. The identification of their antiapoptotic properties, combined with observations of β-cell function preservation and sustained glycemic control during their administration, suggested their possible use as early in the clinical course of T2D as possible or even earlier in order to prevent the onset of this disease[27]. The present paper summarizes the main molecular intracellular mechanisms through which GLP-1 exerts its activity on β-cells. It also explores the current evidence of incretin-based therapies, DPP-4 inhibitors and GLP-1R agonists, when administered in a prediabetic state both in animal models and in humans. Finally it discusses the safety of incretin-based therapies, as well as their possible role in order to delay or prevent T2D.
MAIN MOLECULAR INTRACELLULAR MECHANISMS OF GLP-1 ACTIVITY ON THE PANCREATIC β-CELL
Increased glucose levels are first transported into the β-cell by the type 2 facillitative glucose transporter (GLUT-2) and are phosphorylated by glucokinase to glucose-6-phosphate, promoting an increased rate of aerobic glycolysis; this in turn generates substrates (mainly pyruvate) for mitochondrial oxidative metabolism. Glycolytic and mitochondrial respiration promotes an increased cytosolic adenosine triphosphate (ATP)/adenosine diphosphate (ADP) concentration[28]. This major cellular metabolic signal provides the link between glucose stimulus and insulin secretion. The increase of ATP/ADP ratio promotes the closure of ATP-sensitive K+ channels (KATP), thereby initiating plasma membrane depolarization, activation of voltage-dependent Ca2+ channels (VDCCs), Ca2+ influx and an increase in the intracellular Ca2+ concentration. This in turn stimulates the granules that contain insulin and promotes their release into the blood compartment. Repolarization of β-cells is mainly mediated by Ca2+-sensitive voltage-depended K+ (KCa) channels and voltage-dependent K+ (Kv) channels. These channels open after glucose-induced membrane depolarization so as to restore the outward flux of K+[29].
GLP-1 is a 30-amino acid peptide produced in the intestinal epithelial L-cells of the distal ileum and colon by differential processing of the proglucagon gene from the prohormone convertase PC1/3[30]. GLP-1 binds to GLP-1R, a class 2 G protein-coupled receptor, in the cell membrane of the pancreatic islets[31]. Through this receptor it mainly exerts its insulinotropic activity, which is strictly glucose-depended. Specifically, it stimulates adenylate cyclase resulting in the production of cyclic adenosine 3’,5’-monophosphate (cAMP). Downstream effectors of cAMP include protein kinase A and the cAMP-regulated guanine nucleotide exchange factor II. Through the activation of these two important cellular pathways GLP-1 enhances and amplifies insulin secretion via its effects on ATP/ADP concentration ratio, KATP channels, Kv and KCa channels, VDCCs, Ca2+ influx and intracellular concentrations and insulin granule exocytosis or priming[32,33]. In this way GLP-1 restores glucose-depended insulin secretion in metabolically compromised β-cells; it promotes the induction of glucose competence (Figure 1)[34,35].
Figure 1.
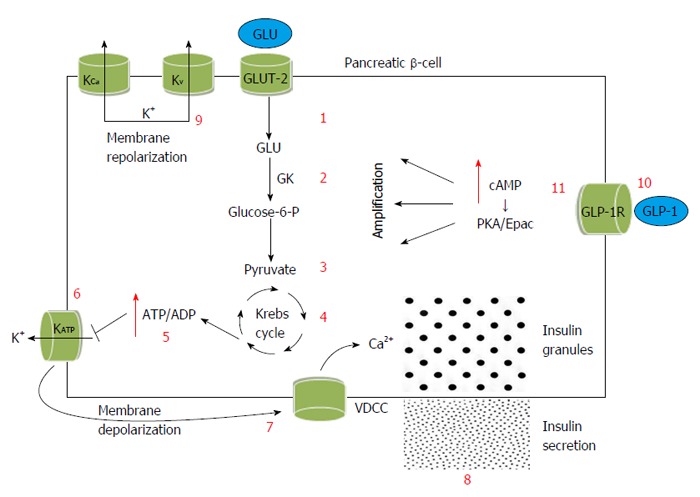
Glucagon-like peptide-1 and the β-cell: Amplification of the glucose-stimulated insulin secretion. Increased glucose levels are transported into the β-cell by GLUT-2. They are phosphorylated by GK to glucose-6-P, promoting an increased rate of aerobic glycolysis. Pyruvate is the main substrate for mitochondrial oxidative metabolism. Increased cytosolic ATP/ADP concentration is the major cellular metabolic signal between the glucose stimulus and insulin secretion. It promotes the closure of KATP channels, thereby initiating plasma membrane depolarization, activation of VDCCs, Ca2+ influx and an increase in the intracellular Ca2+ concentration. This in turn stimulates the granules that contain insulin and promotes their release into the blood compartment. Repolarization of β-cells is mainly mediated by KCa and Kv channels. GLP-1 binds to GLP-1R, a class 2 G protein-coupled receptor, in the cell membrane of the pancreatic cells. Through this receptor it mainly exerts its insulinotropic activity. It promotes increased levels of cAMP through stimulation of adenylate cyclase. Downstream effectors of cAMP are PKA and Epac. Through the activation of these two important cellular pathways GLP-1 amplifies insulin secretion via its effects on ATP/ADP concentration ratio, KATP channels, Kv and KCa channels, VDCCs, Ca2+ influx and insulin granule exocytosis. GLU: Glucose; GLUT-2: Type 2 facillitative glucose transporter; GK: Glucokinase; Glucose-6-P: Glucose-6-phosphate; KATP: ATP-sensitive K+ channels; VDCCs: Voltage-dependent Ca2+ channels; KCa: Ca2+-sensitive voltage-depended K+ channels; Kv: Voltage-dependent K+ channels; GLP-1: Glucagon-like peptide-1; GLP-1R: Glucagon-like peptide-1 receptor; cAMP: Cyclic adenosine 3’,5’-monophosphate; PKA: Protein kinase A; ATP: Adenosine triphosphate; ADP: Adenosine diphosphate.
In addition to its insulinotropic effects, GLP-1 acts as β-cell growth factor. After binding to its receptor, GLP-1 induces the transactivation of the epidermal growth factor receptor, which activates phosphatidylinositol-3 kinase (PI3-K) and its downstream targets protein kinase B (PKB/Akt), extracellular signal-related kinase, p38 mitogen-activated protein kinase (MAPK) and protein kinase Cζ [36,37]. Through these pathways GLP-1 exerts its action on β-cell proliferation and survival. Moreover GLP-1 promotes an increased expression and activity of the pancreatic and duodenal homeobox-1 (PDX-1) gene; hence it increases total PDX-1 levels and promotes its translocation to the nucleus[38]. PDX-1 is of major significance for most of the proliferative, glucoregulatory and cytoprotective actions of GLP-1. It regulates the expression of genes important for β-cell function such as insulin, GLUT-2 and glucokinase. It also replenish β-cell insulin stores and in a long term basis it prevents β-cell exhaustion[38-42]. Moreover, GLP-1 stimulates β-cell proliferation through CREB-mediated Irs2 gene expression, leading to activation of PI3-K/PKB signaling pathway[43]. Its proliferative activity was also related to insulin growth factor (IGF)-1 expression and autocrine IGF-2 secretion by the β-cell[44]. Furthermore, GLP-1 prevents β-cell apoptosis, induced by a variety of cytotoxic stimuli, and enhances β-cell survival[26,45,46].
DPP-4 INHIBITORS IN A PREDIABETIC STATE
Vildagliptin
Studies organized in animal models: Vildagliptin (LAF237) is an oral agent that inhibits DPP-4 and increases both active GLP-1 and GIP levels; it achieved improved glycemic control in patients with T2D[47]. Five-week-old female C57BL/6J mice were fed with a high-fat diet, as a model of IGT and T2D, or a normal diet for 8 wk[48]. After 4 wk, the mice were treated with vildagliptin in their drinking water (approximately 3 μmol per day per mouse). Controls were given only water. All mice were subjected to an OGTT after 4 wk of treatment. In both high-fat diet-fed mice and the normal diet-fed mice, administration of vildagliptin improved glucose tolerance in association with markedly augmented insulin secretion.
Vildagliptin was also administered in anesthetized obese insulin resistant cynomolgus monkeys in a dose of 1 μmol/kg[49]. Each animal received two OGTTs 45 min after oral administration of vildagliptin or vehicle, 3 wk apart. Plasma DPP-4 activity was inhibited by 82% with vildagliptin therapy (P < 0.001) and remained suppressed throughout the duration of the OGTT. Peak plasma GLP-1 levels in the vildagliptin group were significantly higher than those in the vehicle-treated animals, after the glucose load was given (P < 0.001). Vildagliptin reduced glucose excursions during OGTTs compared to the vehicle (P < 0.05). There was also a trend towards an enhanced insulinogenic response to glucose after vildagliptin therapy.
Clinical studies: Although incretins are stimulated during an oral challenge, it was postulated that due to the long half-life of DPP-4 inhibitors, basal levels of active GIP and GLP-1 could play a role in the improvement of β-cell function in individuals with IFG. Vildagliptin was investigated in a single-blind, single-treatment design study, in which 22 individuals with IFG were enrolled. The drug was administered in a dose of 100 mg daily for 6 wk. Two weeks of placebo treatment before (running period) and after (washout period) the 6 wk were also studied[50]. Treatment with vildagliptin resulted in a slight increase in fasting GIP but not GLP-1 levels, while marked increases of both intact GLP-1 and GIP levels during a meal tolerance test were reported. Fasting plasma glucose (FPG) levels were not significantly reduced. Incremental area under the curve (AUC) of glucose and 2 h glucose decreased after a meal tolerance test. Although AUC of C-peptide and insulin responses did not change significantly, when the decrease in glucose levels was taken into consideration, both markers were improved. Since a formal OGTT was not performed in the population enrolled, the possibility that some individuals had combined IFG and IGT could not be excluded. The disposition index (DI) was increased by 69% and insulin sensitivity by 25% after an intravenous glucose tolerance test (IVGTT), suggesting an improvement of β-cell function when no dynamic change in incretin release would be expected to occur. However, after the 2-wk washout period, all the beneficial effects observed returned to baseline levels.
In a multicenter 12-wk double-blind study 179 individuals with IGT were randomized to receive either vildagliptin 50 mg/daily (n = 90) or placebo (n = 89)[51]. Approximately 80% of the patients were IFG and IGT. In individuals receiving vildagliptin there was a marked and sustained increase in active GLP-1 and GIP levels compared to the placebo group (5-fold and almost 2-fold increases in the incremental AUCs for GLP-1 and GIP, respectively). These effects were associated with significant improvements in β-cell function, as estimated by insulin secretion relative to that of glucose (insulin secretory rate AUC0-2 h/glucose AUC0-2 h, mean change between groups 6.1 ± 2.0 pmol/min per meter per millimoles per liter, P = 0.002). Improvements were also reported in α-cell function [glucagon ΔAUC0-2 h, mean change between groups (-3.0 ± 2.0 pmol/L per hour, P = 0.003)]. These beneficial effects contributed approximately to 30% reduction of ΔAUC for glucose. Vildagliptin was well tolerated with a good safety profile and no hypoglycemia was documented.
A three month, double-blind, placebo-controlled study was organized in a population of 48 stable renal transplant recipients, at least six months after transplantation, with newly diagnosed IGT[52]. Participants were randomized to receive 50 mg of vildagliptin, 30 mg of pioglitazone or placebo in a 1:1:1 ratio (16 individuals in each group). There was not any significant difference in corticosteroid therapy between the three groups. Baseline A1C was lowest in the vildagliptin group and higher in the pioglitazone group (P = 0.01). A1C reduction was statistically significant between treatment groups and placebo (placebo vs pioglitazone: -0.17% ± 0.33% vs +0.09% ± 0.26%; P = 0.013; placebo vs vildagliptin: -0.11% ± 0.25% vs +0.09% ± 0.26%; P = 0.049). Vildagliptin and pioglitazone reduced the 2 h plasma glucose at three months compared with baseline (vildagliptin: -20 ± 24 mg/dL; P = 0.002 and pioglitazone: -23 ± 29 mg/dL; P = 0.004), while only pioglitazone slightly reduced FPG.
Sitagliptin
Studies organized in animal models: Sitagliptin is the first DPP-4 inhibitor introduced in clinical practice[53]. Sitagliptin and glyburide were administered in obese prediabetic spontaneously hypertensive rat-obese (SHROB) in order to investigate whether it could reverse the metabolic abnormalities in the secretion of both insulin and glucagon[54]. Sitagliptin was found to normalize glucose tolerance following an OGTT, at least as effective as glyburide, in this rat model of metabolic syndrome and prediabetes. Sitagliptin also restored the first phase of insulin secretion after an OGTT more effectively than glyburide. Fasting glucagon levels, which were elevated in the SHROB model, were normalized after 5 wk of sitagliptin therapy. Fasting insulin and liver glucogen levels were not affected by both drugs. It was suggested that if sitagliptin actions could extend to human prediabetics, then sitagliptin might delay the onset of diabetes[54].
Sitagliptin was also administered in a mouse model of diet-induced obesity with increased FPG and postprandial hyperinsulinemia[55]. It was reported that 12-wk of sitagliptin therapy improved glucose tolerance, reduced FPG, and lowered plasma insulin in randomly fed mice compared with untreated insulin-resistant obese mice. A significant reduction in glucose excursions during an intraperitoneal glucose tolerance test was found. Sitagliptin was also shown to induce a change in the islet size distribution. Specifically, a significantly higher percentage of small islets and a reduced relative percentage of very large islets (due to the very high-fat diet) was demonstrated. This result may explain the better insulin secretory response observed after sitagliptin therapy in response to an in vitro glucose challenge.
An animal model with clinical and metabolic characteristics similar to those of individuals with IGT was recently studied[56]. Fructose administration to normal rats for 21 d induced insulin resistance, IGT, hypertriglyceridemia and decreased β-cell mass, due to an increased percentage of apoptosis. The control group was consistent of rats that were fed with a standard commercial diet. Homeostasis model assessment for insulin resistance (HOMA-IR) and for β-cell function (HOMA-β) decreased to almost control values after sitagliptin therapy. Sitagliptin significantly increased β-cell mass by 68%, attaining values close to those measured in standard commercial diet fed rats; inhibition of β-cell apoptosis was the main cellular mechanism for this effect. These changes were associated with normalization of IGT and liver triacylglycerol content.
Clinical studies: In a double blind placebo-controlled trial 22 individuals with IFG, after a baseline meal study, received sitagliptin 100 mg daily (n = 11) or placebo (n = 11) over an 8-wk treatment period[57]. They underwent a second meal study at the end of the treatment period. Sitagliptin did not alter fasting but increased postprandial intact GLP-1 concentrations, while total postprandial GLP-1 concentrations were reduced. Both fasting and postprandial glucose values were unchanged with sitagliptin therapy. Although sitagliptin resulted in a slight improvement in β-cell function (a slightly increased DI was found), this was not sufficient to alter glucose uptake and production and overcome the defect on insulin action. It was speculated that the limited ability of DPP-4 inhibitors to increase insulin secretion in IFG could be due to their glucose depended mechanism, since glucose concentrations are only modestly elevated in IFG. This speculation can also explain the differing effectiveness of sitagliptin on postprandial concentrations in this study compared to other studies in individuals with IGT, with higher postprandial glucose concentrations.
A four week open-label, parallel group study investigated the effects of sitagliptin on insulin secretion and endogenous glucose production in individuals with IFG and no history of prior antidiabetic therapy[58]. Twenty-three individuals with either IFG (n = 10) or NGT (n = 13) were studied by a fasting glucose test and OGTT. All participants received open-label sitagliptin 100mg once daily for 4 wk. Treatment with sitagliptin resulted in a small but significant decrease in FPG compared to baseline in both groups (P < 0.05). Endogenous glucose production was unchanged after 4 wk of sitagliptin therapy. Administration of sitagliptin did not altered insulin or glucose excursions in the post-intervention OGTT, but did increase AUC for active GLP-1 and C-peptide compared to baseline levels (P < 0.01 for both). Insulin sensitivity and β-cell response indices remained unchanged after administration of sitagliptin.
Beta-cell function in Glucose abnormalities and Acute Myocardial Infarction was a 12-wk multicentre, double-blind, randomized, parallel group study that investigated the effects of sitagliptin 100 mg daily (n = 34) compared to placebo (n = 37) in 71 patients with acute coronary syndrome having IGT or T2D[59]. Investigation of β-cell function was achieved using the insulinogenic index (IGI) derived from an OGTT and acute insulin response to glucose (AIRg) after a frequently sampled IVGTT. At the time of randomization 71% and 62% of the individuals in the sitagliptin and the placebo group had IGT, while 29% and 38% had T2D, respectively. IGI increased significantly, from baseline to 12 wk (9.9 pmol/mmol to 85.0 pmol/mmol) in the sitagliptin group compared to the placebo group (66.4 pmol mmol-1 to 58.1 pmol/mmol, P = 0.013). The AIRg increased significantly in the sitagliptin group compared to the placebo group: 1909 pmol L-1 per minute vs 1043 pmol/L per minute (P < 0.0001). During the OGTT and the frequently sampled IVGTT, glucose levels were significantly lower in the sitagliptin arm compared to the placebo arm. Immediate insulin response was higher after sitagliptin therapy, while it remained unchanged after placebo. By 12 wk, 76%, 18% and 6% of the participants in the sitagliptin group had NGT, IGT and T2D respectively. In the placebo arm 41%, 35% and 24% of the participants had NGT, IGT and T2D respectively.
Other DPP-4 inhibitors
Alogliptin is the newest DPP-4 inhibitor approved for T2D therapy, either alone or in combination with other antidiabetic agents[60]. It was administered alone or in combination with voglibose in prediabetic db/db mice[61]. Specifically, 6 wk old prediabetic db/db mice were fed with a powder CE-2 diet containing 0.001% voglibose alone (equivalent to 1.8 mg/kg per day), 0.03% aloglitpin alone (equivalent to 72.8 mg/kg per day), or combination of both agents (equivalent to alogliptin: 53.8 mg/kg per day + voglibose: 1.8 mg/kg per day) for 27 d. Control db/db and non-diabetic db/+ mice were fed by a drug-free powder CE-2 diet (vehicle). Plasma DPP-4 activity was reduced significantly by 18%, 72% and 80% and plasma active GLP-1 levels were increased significantly by 1.8, 4.5 and 9.1-fold in voglibose, alogliptin and combination treated db/db mice, compared with vehicle treated db/db mice, respectively. Pancreatic insulin content was increased significantly by 3.4, 1.8 and 8.5-fold and A1C was reduced significantly by 1.6%, 0.5% and 2.1% in voglibose, alogliptin and combination treated db/db mice, compared with vehicle treated db/db mice, respectively. Although quantitative analysis was not preformed, combination treatment resulted in an increased pancreatic insulin staining, PDX-1 staining and GLUT2 membrane localization in β-cells. It also maintained normal distribution of β/α-cells in islets; it was suggested that this combination could preserve pancreatic β-cells in db/db mice[61]. The combination of alogliptin and pioglitazone was also found to improve glycemic control and increase pancreatic insulin content in ob/ob mice; however the addition of alogliptin to pioglitazone therapy did not contributed to the prevention or the delay of T2D onset in UCD-T2DM rats[62,63].
The effects of chronic administration of the DPP-4 inhibitor FE 999011 were investigated in both obese and insulin resistant fatty Zucker rats and Zucker diabetic fatty (ZDF) rats[64]. Fatty Zucker rats experience mild glucose intolerance, while ZDF become overtly diabetic after 8 wk of age, if they are fed with a diet containing 6.5% of fat. When administered in the fatty Zucker rats, FE 999011 produced a dose-depended reduction in plasma glucose excursion during the OGTT. During an intraduodenal glucose tolerance test it increased GLP-1 levels, while glucose excursions were indistinguishable from that of lean controls. Chronic treatment with FE 999011 in the fatty Zucker rats significantly improved glucose tolerance, as suggested by the decrease in the insulin-to-glucose ratio. Chronic treatment with FE 999011 twice daily in ZDF rats maintained euglycemia for at least 21 d and delayed the onset of diabetes. Lower basal insulin secretion due to improved insulin sensitivity was reported. It also increased basal GLP-1 levels, stabilized food and water intake to prediabetic levels, reduced hypertriglyceridemia and prevented the rise of circulating non-esterified fatty acids (NEFAs). Up-regulation of pancreatic GLP-1 receptor gene expression was also induced by FE 999011.
The DDP-4 inhibitor isoleucine thiazolidine (P32/98) was orally administered for 3 wk to fatty Zucker rats with incipient IGT (iIGT) and 6 wk in rats with manifest IGT (mIGT) in a dose of 21.61 mg/kg (n = 10 per group)[65]. Control rats received the same amount of placebo. Blood glucose day-night profile was significantly reduced in iIGT Zucker rats achieving values near normalization; it was also improved in mIGT rats. P32/98 tended to reduce food intake and body weight gain, as well as non-fasting plasma insulin levels, only in Zucker rats with iIGT. P32/98 bolus before OGTT increased insulin secretion and reduced glucose load both in iIGT and mIGT Zucker rats, suggesting a broad therapeutic efficacy in animal models of IGT. Treatment of isolated pancreatic islets of mIGT Zucker rats with this agent decreased pancreatic insulin content and increased glucose responsiveness, while the β-cell volume density was not improved.
The DPP-4 inhibitor PFK 275-055, a vildaglitpin analogue, was investigated in obese, insulin resistant prediabetic rats for 4 wk in a dose of 10 mg/kg per day[66]. GLP-1 levels increased after PFK 275-055 therapy. Insulin levels were decreased after therapy with this agent, while glucose levels were not affected; an increased β-cell/α-cell ratio was observed. The DPP-4 inhibitor DA-1229 improved pancreatic insulin content, β-cell function and delayed the onset of diabetes in young db/db mice[67]. Currently, several studies have been launched and are recruiting individuals in order to explore the possible role of alogliptin and saxagliptin in a prediabetic state[68].
GLP-1R AGONISTS IN A PREDIABETIC STATE
Exenatide
Studies organized in animal models: Exenatide is the synthetic form of the naturally occurring exendin-4, a 39-amino-acid peptide hormone secreted by the salivary glands of the venomous lizard Heloderma suspectum, otherwise known as the Gila monster[69]. It shares 53% structural homology with human GLP-1 and resists inactivation by the DPP-4. In an animal model of profound insulin resistance, IGT, hypertriglyceridemia and decreased β-cell mass, exendine-4 significantly increased β-cell mass by 201%[56]. This effect was achieved after a significant decrease in β-cell apoptosis, although the molecular effect for this activity was not studied. HOMA-IR and HOMA-β indexes remained within normal rage. Normalization of IGT and liver triacylglycerol content was also achieved.
In another well-organized study, exendin-4 was administered to obese prediabetic db/db mice at 6 wk of age for 16 d[70]. By the age of 8 wk, vehicle treated mice developed T2D, while mice treated with exendin-4 maintained FPG in the normal range, indicating that this agent delayed the onset of T2D. Improvement in glucose tolerance was also observed with exendin-4. No significant differences were observed between the two groups as far as insulin sensitivity is concerned. Glucose alone induced a two to five-fold increase in insulin secretion in the exendin-4 group, while the pancreas of vehicle-treated mice was unresponsive to the same dose of glucose. A 1.4-fold increase in β-cell mass was observed in exendin-4 mice, which was the result of both increased β-cell proliferation and decreased β-cell apoptosis; these changes were related to higher expression of the protein kinases Akt1 and MAPK.
The ability of exendin-4 to promote β-cell proliferation in young Goto-Kakizaki (GK) rats during the prediabetic state, and therefore prevent the development of T2D when animals become adults, was also explored[71]. Four groups of rats were investigated: two control groups (control GK and control non-diabetic Wistar rats) and two experimental groups. In the two experimental groups, GK rats received either a subcutaneous daily injection of GLP-1 (400 μg/kg of body weight) or exendin-4 (3 μg/kg of body weight) for five days (day’s two to six) after their birth. Animals were killed seven days or two months after birth. Seven days after their birth GK rats showed significantly higher pancreatic insulin content and doubling of β-cell mass compared to the untreated GK group; this effect resulted from both differentiation (neogenesis) and proliferation enhancement of β-cells. Follow up from day seven to the adult age (two months) showed that both treatments decreased postabsorptive basal plasma glucose levels and increased pancreatic insulin content compared to the untreated GK arm. In GK/GLP-1 and GK/exendin-4 groups, β-cell mass was significantly increased and represented 71% and 63% of the β-cell mass of the Wistar group, respectively. Glucose-stimulated insulin release, as evaluated during an IVGTT, was significantly improved in both treated groups. It was concluded that GLP-1 or exendin-4 treatment limited the prediabetic period and delayed the development of T2D in this animal model of prediabetes.
Exendin-4 activity was explored in a rat model of uteroplacental insufficiency[72]. Intrauterine growth retarded (IUGR) rats experience a progressive decline in β-cell mass weeks before the onset of T2D; hence there is a prediabetic neonatal period, which was investigated. At two weeks, exendin-4 significantly decreased body weight in both IUGR and control pups and this effect persisted into adulthood. It also improved glucose tolerance, which was maintained at 7 wk of age. Interestingly, at three months of age, vehicle-treated IUGR rats developed T2D (their β-cell mass declined by almost 80%) whereas exendin-4 treated IUGR rats had NGT and normal β-cell mass. At 18 months of age, exendin-4 treated IUGR rats were normoglycemic, while all vehicle treated IUGR rats had died. Exendin-4 therapy in IUGR rats at 14 d restored PDX-1 mRNA levels, in concentrations similar to controls; this effect persisted for three months.
Clinical studies: One hundred fifty two obese [average body mass index (BMI): 39.6 ± 7.0 kg/m2] individuals with NGT or IGT or IFG were randomized to receive either exenatide (n = 73) (10 μg with a 4-wk 5 μg dose titration period) or placebo (n = 79), along with lifestyle modification for 24 wk[73]. Thirty eight individuals (25%) had IFG or IGT. Exenatide-treated individuals lost 5.1 ± 0.5 kg from baseline vs 1.6 ± 0.5 kg in the placebo group (treatment difference: -3.3%, P < 0.001). An important percentage of individuals with prediabetes returned to NGT after the end of the period (77% compared to 56% in the placebo group). No significant baseline to end point changes was shown for FPG, A1C and OGTT. Diarrhea was reported by 14% and 3% and nausea by 25% and 4% of the exenatide and placebo groups, respectively. Adverse effects were mild or moderate in severity in most cases. It was concluded that exenatide therapy in addition to lifestyle modification is a promising therapeutic approach for obese prediabetic individuals.
In another non randomized study, 105 individuals with IGT and/or IFG were treated with: (1) Lifestyle modification only (n = 18). Participants were advised to achieve 7% body weight loss over three months and to walk 30 min daily, seven days per week; (2) Pioglitazone 15mg daily and metformin 850mg daily (n = 40); and (3) A triple combination of pioglitazone 15mg daily, metformin 850 mg daily and exenatide 10 mcg twice daily (n = 47)[74]. All individuals who received drug therapy had the same advice on lifestyle intervention. Mean follow-up period was 8.9, 6.9, and 5.5 mo in the three groups respectively. Individuals in the lifestyle intervention group achieved only a slight reduction of body weight (82.3 kg to 80.9 kg). No significant change on insulin sensitivity and β-cell function was observed. In the pioglitazone and metformin group FPG was decreased from 109 mg/dL to 102 mg/dL and mean glucose AUC during OGTT was reduced by 12% (P < 0.001). Insulin sensitivity and β-cell function improved by 42% and 50% respectively, while 14% of the individuals with IGT and 36% of the individuals with IFG reverted to NGT. Interestingly, in the triple therapy group, a robust 109% improvement in β-cell function and a 52% increased in insulin sensitivity was observed, while 59% of the individuals with IGT and 56% of the individuals with IFG reverted to NGT. No patient in both double and triple therapy groups developed T2D.
A 24-wk prospective randomized outpatient clinical trial explored the possible role of exenatide (10 μg twice daily) and metformin (1000 mg twice daily), alone or in combination, on menstrual cyclicity and metabolic and endocrinological parameters in 60 overweight/obese women with polycystic ovary syndrome (PCOS)[75]. Forty two participants (70%), 14 in each arm completed the study protocol. Weight loss was more profound in the exenatide arms compared to metformin (P = 0.003). Combination treatment promoted a dramatic improvement in central adiposity. At the end of the study, the combination arm experienced weight loss of 6 ± 0.5 kg, the exenatide arm 3.2 ± 0.1 kg, and the metformin arm 1.6 ± 0.2 kg. Eighteen women with PCOS had glucose intolerance and 11 of them completed the study. Seven (64%) of them had NGT at the end of the trial (three of three in the combination arm, three of five on the metformin arm and one of three on the exenatide arm). Insulin sensitivity and HOMA-IR were significantly improved in all treatment groups. Insulin secretion, as measured by the corrected insulin response at glucose peak, was significantly reduced in the exenatide and combination arms (P < 0.016). The insulin secretion-sensitivity index increased progressively from metformin arm (232 ± 116) to the exenatide arm (395 ± 112) and the combination arm (516 ± 117) (P < 0.005), suggesting an improved β-cell function with enhanced insulin sensitivity.
The role of exenatide in order to improve postprandial endothelial function in individuals with IGT (n = 16) and patients with recent T2D with optimal glycemic control (n = 12) was investigated in a double-blinded randomized crossover study[76]. Endothelial function was estimated by reactive hyperemia peripheral arterial tonometry (PAT). In individuals with IGT, PAT index tended to increase after exenatide and was higher compared to the placebo period. Exenatide reduced postprandial rises in insulin, glucose and triglycerides concentrations. Postprandial PAT index was inversely correlated only with mean postprandial concentrations of triglycerides, possibly due to the high fat content of the meal administered. Change in postprandial triglycerides after exenatide accounted for 64% of the estimated effect of exenatide on postprandial endothelial function. Exenatide also reduced the postprandial elevation of triglycerides, apolipoprotein B-48, apolipoprotein CIII, remnant lipoprotein cholesterol and remnant lipoprotein triglyceride in individuals with IGT (n = 20) and patients with recent onset T2D (n = 15)[77]. These effects were not affected either with statin therapy or by glucose tolerance status. Both studies suggested an additional cardiovascular benefit of this agent beyond the improved glycemic control in this population[76,77]. Another randomized 3-wk head-to-head study examined the effects of exenatide vs metformin on microvascular endothelial function in 50 individuals with abdominal obesity and prediabetes[78]. Similar effects of both agents were shown on microvascular endothelial function, vascular activation, oxidative stress and markers inflammation. Exenatide did not demonstrate any beneficial effect on postprandial function in individuals with IGT. It was suggested that the reason for this observation was the administration of a glucose-only meal instead of a high fat meal, which would be expected to increase postprandial triglycerides[76,78].
Liraglutide
Studies organized in animal models: Liraglutide is a long acting analog with 97% homology to human GLP-1. It has an additional 16-carbon fatty acid and a small amino acid-spacer that promotes reversible binding to albumin and enhances resistance to DPP-IV degradation, providing a half-life of approximately 13 h[79]. The possible role of chronic liraglutide therapy in prediabetic UCD-T2D rats, in order to prevent or delay T2D, was investigated in a well organized study[80]. The UCD-T2D rat model develops polygenic adult-onset obesity and insulin resistance, followed by inadequate β-cell compensation and eventually T2D. UCD-T2D rats develop diabetes in a later age than other animal models of T2D; thus they are highly suitable for diabetes prevention studies[81]. At two months of age male sibling rats were divided in three groups (n = 32 per group): a control group (higher energy intake, body weight and adiposity compared to the other groups), a food-restricted group and a liraglutide group (0.2 mg/kg sc for 15 mo). Restricted rats were food restricted to 9% less energy per kg of body weight compared to the liraglutide group, in order to equalize body weights between these two groups. Half of the animals in each group were killed at 6.5 mo for tissue collection, while the remaining half continued treatment until T2D onset. FPG and A1C were lower in the liraglutide and food-restricted groups. Liraglutide treatment delayed T2D onset by 4.1 ± 0.8 mo compared to controls (P < 0.0001) and by 1.3 ± 0.8 mo compared to restricted animals (P < 0.05). Liraglutide-treated animals had lower fasting plasma triglycerides, glucagon and leptin levels, as well as body fat (despite similar body weight), compared to both groups. Decreased body fat could be the result of an increased lipid oxidation. Rats in the liraglutide group had significantly lower fasting plasma insulin compared to the other groups (P < 0.001), starting from one month and lasting throughout the 6 mo period, suggesting that this effect was not solely related to reduced body weight. Liraglutide treatment and energy restriction equally preserved pancreatic insulin content and islet morphology, possibly due to the lower weight gain and delayed hyperglycemia. Pancreatic insulin content in the control group was approximately one-third of that of the two other groups.
In another study, 12-wk old Otsuka-Long-Evans-Tokushima fatty (OLETF) rats (n = 8) were treated with three doses of liraglutide (50, 100, and 200 μg/kg twice a day) or 0.9% saline intraperitoneally (n = 8), twice daily for 12 wk. Eight Long-Evans-Tokushima-Otsuka rats with saline injection served as normal controls[82]. At the end of the 12 wk of treatment, all rats were euthanized and pancreatic tissues were used for histopathological and immunohistochemical analysis; only in the liraglutide 100 μg/kg group an analysis was performed, since this dose can be converted to a human equivalent dose. OLETF rats experienced obesity, IFG, hyperinsulinemia, insulin resistance, increased cholesterol levels, and a high inflammatory state. Although liraglutide treatment had only an acute effect on food intake, its beneficial effect on weight loss was sustained independently of feeding. All three doses of liraglutide suppressed IFG, IGT and insulin resistance. At the end of the 12-wk intervention period, 87.5% of the vehicle-treated OLETF progressed to T2D. On the contrary, 42.9% of IFG rats were reversed to NGT, while none of the liraglutide-treated OLETF rats progressed to T2D compared to vehicle-treated animals (P < 0.0001). Liraglutide improved both triglyceridemia and the inflammatory state observed. It also preserved islet morphology. Up-regulation of the anti-apoptotic Bcl-2 protein and down-regulation of the pro-apoptotic Bax factor were reported, which may contribute to the improvement of pancreatic islet function and structure.
When liraglutide was administered in a dose of 150 mg/kg twice daily for 6 wk in prediabetic rats, it strongly attenuated T2D development[83]. Approximately 53% of the antihyperglycemic effect observed was mediated by a reduction in food intake. In the experiments with 60% pancreatectomized rats, liraglutide significantly reduced glucose excursions after an OGTT. Furthermore, when NGT status was established, no increase in β-cell proliferation and mass was observed in both models of β-cell deficiencies. It was suggested that the influence of GLP-1 agonism on β-cell mass dynamics in vivo was strongly related to the glycemic state observed.
Clinical studies: In a 20-wk prospective multicentre study, 564 nondiabetic obese individuals (31% of whom had prediabetes) were randomized to receive either one of four doses of liraglutide (1.2 mg, 1.8 mg, 2.4 mg, or 3.0 mg, n: 95, 90, 93 and 93, respectively) or placebo (n = 98) administered once daily subcutaneously or open label orlistat 120 mg three times daily (n = 95)[84]. All individuals increased their physical activity using pedometers and were advised to adhere a low fat diet with about to 500 kcal per day deficit. Sixty-one percent of the individuals in the liraglutide groups lost at least 5% of body weight from baseline, which was significantly more than the placebo arm. The proportion of individuals who lost more than 10% of baseline weight was dose depended and was greater in the 3 mg liraglutide arm than in the placebo arm (28% vs 2%). Systolic/diastolic blood pressure was reduced by 5.7/3.7 mmHg. The incidence of metabolic syndrome was reduced by more than 60% in those treated with liraglutide 2.4 mg and 3.0 mg. The prevalence of prediabetes was decreased by 84-96% with liraglutide 1.8 mg, 2.4 mg and 3 mg. Mean FPG was decreased by 7%-8% in the liraglutide arm, while no visible effect was described in the two other arms. Mean A1C was slightly reduced in a dose depended fashion in individuals treated with liraglutide compared to that in the two other groups. Mean change in plasma glucose during OGTT was reduced in all liraglutide groups compared to that of orlistat and placebo. Liraglutide therapy did not have any effect on insulin resistance as estimated by HOMA. However, median β-cell function was decreased with orlistat and placebo by 21% and 17% respectively, but increased in the liraglutide arm by 5%-24%. Fasting insulin levels initially increased, but as body weight and glucose concentrations gradually decreased, insulin levels were reduced, suggesting the glucose-depended activity of liraglutide on insulin secretion.
The two-year results from the extension of this 20-wk trial were recently reported[85]. Three hundred ninety eight individuals entered the extension and 268 (67%) completed the two-year trial. All participants continued on randomization treatment for one year, after which liraglutide or placebo individuals switched initially to liraglutide 2.4 mg and then 3 mg (based on 20-wk and one-year results, respectively). After two years, individuals on liraglutide 2.4/3.0 mg lost 3.0kg (1.3-4.7 kg) more weight than those on orlistat (P < 0.001). Approximately 70% of the individuals on liraglutide 2.4/3.0mg maintained weight loss more than 5% of screening weight after two years, 43% maintained more than 10% loss and 25% maintained more than 15% loss. Estimated weight loss of 7.8 kg and mean systolic blood pressure reduction of 12.5 mmHg was sustained with liraglutide 2.4/3.0 mg in completers from screening. Between 52%-62% of liraglutide-treated individuals with prediabetes at randomization achieved NGT after two years compared to 26% in the orlistat arm. Mean FPG and A1C concentrations were also reduced. The two year prevalence of prediabetes and metabolic syndrome in the liraglutide 2.4/3.0 mg group was decreased by 52% and 59% respectively. The most frequent liraglutide-associated adverse effects were gastrointestinal, mainly nausea and vomiting, as expected from T2D trials. However, most nausea/vomiting episodes were transient; more than 90% were mild or moderate in intensity.
Recently, a 14-wk double blind, randomized placebo-controlled study was launched in order to investigate the possible role of liraglutide 1.8 mg treatment in 68 older (mean age: 58 ± 8 years) overweight/obese (mean BMI: 31.9 kg/m2) individuals with prediabetes (IFG and/or IGT)[86]. Participants were also advised to eat a moderate carbohydrate diet and decrease total caloric intake by 500 kcal/d. Twenty four (68%) individuals randomized in the liraglutide group and 27 (82%) individuals in the placebo group completed testing at the end of the trial. Participants randomized to liraglutide arm lost twice as much weight as those assigned to placebo (6.8 kg vs 3.3 kg; P < 0.001). More individuals in the liraglutide arm finally lost 7% of baseline weight compared to the placebo arm (54% vs 4%); 10% weight loss was only observed in the liraglutide arm (17%). Weight loss after liraglutide therapy was associated with significant reduction of insulin resistance. Steady state plasma glucose concentrations were reduced by 29% in the liraglutide arm compared with no change in the placebo arm; FPG (-0.5 mmol/L vs 0 mmol/L), systolic blood pressure (-8.1 mmHg vs -2.6 mmHg), and triglyceride levels (-0.4 mmol/L vs -0.1 mmol/L) were also significantly decreased in the liraglutide arm compared to the placebo arm respectively (P ≤ 0.04). In addition, 75% of the participants in the liraglutide arm achieved normal FPG. The most common adverse effect in the liraglutide arm was nausea (67% vs 26% in the placebo arm). It was suggested that the improvement of glycemia in the liraglutide group appeared to be better than reported with weight loss alone in this population.
Indeed, the effects of GLP-1R agonists on insulin secretion are not a simple phenomenon. These medications can increase glucose secretion in a glucose-depended manner after acting directly on the β-cell; they can also decrease insulin secretion secondary to weight loss and enhancement of insulin sensitivity. In this view, it is unclear what the net effect would be when they are administered in individuals with prediabetes. In order to investigate this observation, a parallel study was organized in order to evaluate the relative impact of the indirect effect of weight loss and increase insulin sensitivity compared to the direct effect of GLP-1R agonists on β-cell function[86,87]. In this recent double-blind, randomized, placebo-controlled, parallel-group study 49 individuals (mean age: 58 years, mean BMI: 32.9 kg/m2) with prediabetes (isolated IFG, isolated IGT and combined IFG/IGT) received either liraglutide 1.8 mg daily (n = 24) or placebo (n = 25). All participants were instructed to decrease total energy intake by 500 kcal per day and to continue their baseline physical activity[87]. There was a little overlap in the degree of weight loss between the two arms since 88% of the individuals in the liraglutide arm lost more than 5% of baseline body weight compared to 22% in the placebo arm. Weight loss promoted a significant improvement on insulin resistance in the liraglutide arm compared to the placebo arm (-7.7% vs -3.9%, P < 0.001). Insulin response, after intravenous glucose infusion, was decreased by 7% in the placebo arm whereas it increased by 34% in the liraglutide arm. C-peptide AUC was increased by 29% in individuals receiving liraglutide and NEFAs concentration was reduced. Placebo treatment had no effect on these two parameters. Regression analyses suggested that weight loss was not associated with any changes in pancreatic β-cell function. Despite weight loss and reduction of insulin resistance in the liraglutide arm, the insulin secretion rate was significantly increased and there was no association between weight loss and changes on insulin secretion. It was concluded that changes following liraglutide treatment in patients with prediabetes are not those that are described after weight loss and improved insulin sensitivity, but rather similar effects after an acute GLP-1 infusion[87,88].
SAFETY OF INCRETIN-BASED THERAPIES
An acceptable safety profile is of major importance for every intervention administered in order to prevent or delay T2D. As far as GLP-1R agonists are concerned, the most common adverse effects are gastrointestinal, including nausea, vomiting and diarrhea[89]. However, they occur early on during treatment and tend to be transient. For DPP-4 inhibitors, adverse effects resemble that of placebo, with nasopharyngitis and headache being the most common described[90]. Moreover, discontinuation of therapy because of side effects was similar to placebo[91].
Small preclinical studies, as well as some post-marketing reports, raised the possibility of an increased risk of pancreatitis with incretin based therapies[92-96]. In a study that data were collected from the Food and Drug Administration (FDA) adverse event reporting system database, GLP-1 based therapies were associated with pancreatitis and pancreatic cancer[97]. Another case-control study reported an increased risk for hospitalization for acute pancreatitis with GLP-1 based therapies (after combining exenatide and sitaglitpin treatments) and adjusting for potential confounders[98]. Concerns were also raised after the results of a study organized in organ donors with T2D, who received either sitagliptin or exenatide. A possible expansion of endocrine and exocrine pancreatic compartments after incretin-based therapy, the former being associated by α-cell hyperplasia with the potential progression to neuroendocrine tumors and the latter with an enhanced proliferation and dysplasia, was described[99]. Furthermore, a recent case-control analysis, based on the French pharmacovigilance database, suggested an association of all incretin-based therapies with pancreatitis[100]. A trend towards a slightly elevated risk of pancreatitis, only with GLP-1R agonists, was also shown in a recent pooled analysis of phase III trials, although the number of cases was very small and the statistical power was limited[101].
However larger preclinical studies did not established an association of incretin-based therapies with pancreatitis[102-109]. Interestingly in three of these studies, GLP-1R activation or DPP-4 inhibition had a beneficial effect on exocrine pancreatic function and structure[103,104]. A recent study also suggested that pancreatic findings attributed to incretin-based therapies in rodents are commonly observed background findings, without any drug treatment and independent of diet or glycemic status[110]. Moreover large retrospective population studies and recent meta-analysis suggested a negative association of incretin-based therapies with either pancreatitis or pancreatic cancer[111-120]. Recently the FDA reevaluated more than 250 toxicology studies, organized in nearly 18000 healthy animals, and found no association with pancreatitis or any pancreatic toxicity. The European Medicines Agency conducted a same review and reported no pancreatic tumors in mice and rats treated with incretin-based drugs, even at doses that greatly exceed the level of human clinical exposure[121].
A higher expression of GLP-1Rs in rodent calcitonin-producing thyroid C cells, (mainly in rats and mice) combined with sustained GLP-1R activation can result in stimulation of calcitonin secretion, hyperplasia, adenoma and eventually medullary thyroid cancer[122,123]. Indeed, both liraglutide and exenatide were shown to promote the development of thyroid C cell cancer after chronic therapy in rodents[122]. An elevated risk for thyroid carcinoma was described in one study[97]. However, thyroid C cells in humans and monkeys express lower levels of GLP-1Rs[124]. Long-term treatment with high doses liraglutide did not produced thyroid C cell proliferation in monkeys, while no association between calcitonin levels and liraglutide, up to 3 mg daily, was established in large numbers of patients with T2D[125].
Retrospective analysis of phase III clinical trials, in which major cardiovascular events were reported as adverse events, have been published for exenatide, liraglutide, vildagliptin, sitagliptin, alogliptin, saxagliptin, and linagliptin[126]. In all of these studies the relative risk for a major cardiovascular event (acute myocardial infarction, stroke and cardiovascular death) was reduced relative to placebo or a comparator therapy to a value below one. However, the 95%CI was more than one in most of these studies, thus the number of events was too small so as to extract definite conclusions. Both the Saxagliptin Assessment of Vascular Outcomes Recorded (SAVOR)-Thrombolysis in Myocardial Infarction (TIMI) 53 (SAVOR-TIMI 53) and the Examination of Cardiovascular Outcomes with Alogliptin vs Standard of Care (EXAMINE) trials met the FDA criteria for non inferiority of saxagliptin and alogliptin over placebo respectively, but unfortunately they did not demonstrated any positive evidence on cardiovascular risk reduction[127,128]. Two recent meta-analysis suggested that DPP-4 inhibitors may have a neutral effect or reduce the risk of cardiovascular events and all-cause mortality in patients with T2D[129,130]. As far as GLP-1R agonists are concerned two recent meta-analysis reported that these agents do not appear to increase cardiovascular morbidity in comparison with placebo or other active drugs[131,132].
Hospitalization for heart failure among T2D who received saxagliptin in the SAVOR-TIMI 53 was increased by 27% compared to the placebo group (3.5% vs 2.8%; HR = 1.27; 95%CI: 1.07-1.51; P = 0.007), while no association of alogliptin with heart failure was found in the EXAMINE study[133]. Two recent meta-analysis suggested a possible increased risk of developing heart failure after DPP-4 therapy[134,135]. Currently, a large number of long-term cardiovascular outcome trials in patients with T2D are being performed in order to clarify the cardiovascular safety and efficacy of incretin-based therapies[136].
In addition to safety and efficacy of incretin-based therapies, cost is another significant issue that must be taken into consideration. Although the cost of incretin-based therapies is greater compared to other glucose-lowering therapies, long term effectiveness of these agents can be associated with a decreased in the cost of management of T2D and its complications compared to other therapies[137].
CONCLUSIONS-PERSPECTIVES
During the last two decades there has been an immense investigation in order to understand the pathophysiology of the early stages of hyperglycemia, which very often progress to overt T2D within a few years, as β-cell decline and failure progresses. The huge burden resulting from the complications of T2D created the need of novel therapeutic strategies in an effort to prevent its development[8]. The beneficial effects of incretin-based therapies on β-cell function in patients with T2D, together with their strictly glucose-depended mechanism of action, suggested their possible use in individuals with prediabetes, when greater β-cell mass and function are preserved and the possibility of β-cell salvage is higher[138]. The main results of the most important clinical studies of incretin-based therapies in individuals with prediabetes are shown in Tables 1 and 2.
Table 1.
Main clinical studies of dipeptidyl peptidase-4 inhibitors in a prediabetic state
| Ref. | Study population | Study design | Main results |
| Utzschneider et al[50] | 22 individuals with IFG | VILDA was administered in a dose of 100 mg daily for 6 wk. Two weeks of placebo treatment before (running period) and after (washout period) 6 wk was studied | FPG levels were not significantly reduced. AUC GLU and 2-h GLU decreased after a MTT. DI was increased by 69% and insulin sensitivity by 25% after an IVGTT These effects were not sustained in the washout period |
| Rosenstock et al[51] | 179 individuals with IGT (80%: IFG + IGT) | Multicenter 12-wk double-blind study 90 participants received VILDA 50 mg/daily and 89 received placebo therapy | Improvements in β-cell function as estimated by insulin secretion relative to that of GLU. Improvements were also reported in α-cell function. These beneficial effects contributed to approximately 30% reduction in prandial GLU excursions |
| Werzowa et al[52] | 48 IGT renal transplant recipients | 3-mo, double-blind, placebo-controlled study. Participants were randomized to receive 50 mg of VILDA, 30 mg of PIO or placebo in a 1:1:1 ratio (n = 16 in each arm) | A1C reduction was statistically significant between treatment groups and placebo. VILDA and PIO reduced the 2 h plasma GLU at three months compared with baseline, while only PIO reduced FPG |
| Bock et al[57] | 22 individuals with IFG | 8-wk double blind placebo-controlled study Participants received SITA 100 mg daily (n = 11) or placebo (n = 11) | SITA increased postprandial intact GLP-1 concentrations. Both fasting and postprandial GLU values were unchanged with SITA therapy. A slightly increased DI was reported |
| Perreault et al[58] | 23 individuals with either IFG (n = 10) or NGT (n = 13) | 4-wk open-label, parallel group study. All participants received SITA 100 mg once daily | SITA resulted in a small, but significant decrease in FPG compared to baseline in both groups (P < 0.05) Administration of SITA did not altered insulin or GLU excursions in the post-intervention OGTT, but did increase AUC for active GLP-1 and C-peptide compared to baseline levels (P < 0.01 for both) |
GLP-1: Glucagon-like peptide 1; IFG: Impaired fasting glucose; IGT: Impaired glucose tolerance; NGT: Normal glucose tolerance; FPG: Fasting plasma glucose; AUC: Area under the curve; DI: Disposition index; IVGTT: Intravenous glucose tolerance test; MTT: Meal tolerance test; A1C: Glycated hemoglobulin; VILDA: Vildagliptin; SITA: Sitagliptin; PIO: Pioglitazone; GLU: Glucose; OGTT: Oral glucose tolerance test.
Table 2.
Main clinical studies of glucagon-like peptide-1 receptor agonists in a prediabetic state
| Ref. | Study population | Study design | Main results |
| Rosenstock et al[73] | 152 obese individuals of whom 38 had IGT or IFG | Participants were randomized to receive either EXE (n = 73) (10 μg with a 4-wk 5 μg dose titration period) or placebo (n = 79) along with lifestyle modification for 24 wk | EXE-treated individuals lost 5.1 ± 0.5 kg from baseline vs 1.6 ± 0.5 kg in the placebo group (P < 0.001). An important percentage of individuals with prediabetes returned to NGT after the end period (77% compared to 56% in the placebo group) |
| Armato et al[74] | 105 individuals with IGT and/or IFG. Mean follow-up period was 8.9, 6.9, and 5.5 mo in the three groups respectively | Participants were treated with: (1) Lifestyle modification only (n = 18); (2) PIO 15 mg daily and MET 850 mg daily (n = 40); and (3) PIO 15 mg daily, MET 850 mg daily and EXE 10 mcg twice daily (n = 47) | A robust 109% improvement in β-cell function and 52% increased in insulin sensitivity was observed in the EXE group, while 59% of individuals with IGT and 56% individuals with IFG reverted to NGT. No patient in both double and triple therapy groups developed T2D |
| Astrup et al[84] | 564 obese individuals (31% had prediabetes) | 20 wk double-blind prospective multicentre study. Participants were randomized to receive either one of four doses of LIRA (1.2 mg, 1.8 mg, 2.4 mg, or 3.0 mg, n: 95, 90, 93 and 93) or placebo (n = 98) or open label orlistat 120 mg three times a day (n = 95) | 61% of the individuals in the LIRA groups lost at least 5% of body weight from the baseline, which was significantly more than in the placebo arm. The prevalence of prediabetes was decreased by 84%-96% with LIRA 1.8 mg, 2.4 mg and 3 mg. Mean FPG was decreased by 7%-8% only in the LIRA arm. Mean change in plasma GLU during OGTT were reduced in all LIRA groups compared with that of orlistat and placebo. Median β-cell function increased in the LIRA arm by 5%-24% |
| Kim et al[86] | 68 overweight/obese individuals with IFG and/or IGT | 14 wk double blind randomized placebo-controlled study. 24 individuals received LIRA 1.8 mg daily and 27 placebo therapy | Participants randomized to LIRA arm lost twice as much weight as those assigned to placebo (P < 0.001). Steady sate plasma GLU was reduced by 29% in the LIRA arm compared with no change in the placebo arm. 75% of the participants in the LIRA arm achieved normal FPG |
| Kim et al[87] | 49 individual with isolated IFG, isolated IGT and combined IFG/IGT | 14 wk double-blind, randomized, placebo-controlled, parallel-group study. Participants received LIRA 1.8 mg daily (n = 24) or placebo (n = 25) | Weight loss promoted a significant improvement in insulin resistance in the LIRA arm compared to the placebo arm (-7.7% vs -3.9%, P < 0.001). Insulin response, after intravenous GLU infusion, was decreased by 7% in the placebo arm whereas it increased by 34% in the LIRA arm. Despite weight loss and reduction of insulin resistance in the LIRA arm, the insulin secretion rate was significantly increased and there was no association between weight loss and changes in insulin secretion |
IFG: Impaired fasting glucose; IGT: Impaired glucose tolerance; NGT: Normal glucose tolerance; FPG: Fasting plasma glucose; EXE: Exenatide; LIRA: Liraglutide; PIO: Pioglitazone; MET: Metformin; T2D: Type 2 diabetes; GLU: Glucose; OGTT: Oral glucose tolerance test.
DPP-4 inhibitors have shown beneficial effects on β-cell mass and function in preclinical models of prediabetes. However short-term clinical studies (maximum duration of 12 wk) have only demonstrated a modest effect on glucose homeostasis, which was lost after treatment discontinuation[50]. Whether longer periods of DPP-4 inhibition in individuals with prediabetes can measurably alter β-cell function, in a way that is sustained even after treatment discontinuation, remains unproven. One year treatment with vildagliptin in drug-naïve patients with T2D and mild hyperglycemia initially increased β-cell secretory capacity, but this effect was not maintained after the washout period[139]. However, when vildagliptin was administered in drug-naïve patients with T2D and mild hyperglycemia (A1C: 6.2%-7.2%) for two years, β-cell function tended to be greater after two years than after one year of treatment[140].
GLP-1R agonists have also shown significant improvements on β-cell mass and function in preclinical studies. Important improvements on β-cell function and insulin sensitivity were also reported in short term clinical studies, in which an important percentage of individuals with prediabetes returned to NGT. Weigh reduction in overweight and obese individuals with prediabetes was also shown, as well as improvements of endothelial function and lipid profile. Whether GLP-1R agonists can prevent or delay the transition to T2D needs further investigation in well-designed long term studies. The Restoring Insulin Secretion consortium will examine whether medication, including liraglutide, or surgical intervention strategies can reduce the progressive β-cell dysfunction in adults and youth with prediabetes or early T2D[141]. The duration of GLP-1R agonists therapy in order to promote sustained β-cell improvements is also an issue of investigation. Interestingly, when exenatide was administered in patients with T2D for one year, the treatment related improvement of β-cell function was lost after a four-week drug cessation[142]. However, the three-year data of exenatide treatment suggested a small but statistically significant effect on DI following a four-week off therapy period[143].
Recent evidence also demonstrates the presence of genetically induced GLP-1 resistance both in prediabetic and diabetic states. Whether pharmacogenomic studies are needed in order to identify responders and non-responders to incretin based therapies regarding glucose metabolism, is an issue of future research[144].
The safety of incretin-based therapies remains a topic of scientific discussion and exploration[126,145,146]. Currently, precise estimates for the risk of possible serious adverse effects associated with incretin-based therapies cannot be estimated. Future data from cardiovascular outcome studies and ongoing clinical studies, which will improve the statistical power of prospective studies and facilitate larger meta-analyses, are crucially anticipated in order to clarify their long-term safety. Until these data are available, large, long term, well designed future diabetes prevention trials of incretin-based therapies will be required in order to determine whether they can stabilize or reverse β-cell loss and promote a sustained reduction in the development of T2D in this population.
Footnotes
P- Reviewer: Cheng TH, Efstathiou S, Martinez-Costa OH S- Editor: Tian YL L- Editor: A E- Editor: Liu SQ
References
- 1.Papaetis GS, Papakyriakou P, Panagiotou TN. Central obesity, type 2 diabetes and insulin: exploring a pathway full of thorns. Arch Med Sci. 2015:In press. doi: 10.5114/aoms.2015.52350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papaetis GS, Orphanidou D, Panagiotou TN. Thiazolidinediones and type 2 diabetes: from cellular targets to cardiovascular benefit. Curr Drug Targets. 2011;12:1498–1512. doi: 10.2174/138945011796818243. [DOI] [PubMed] [Google Scholar]
- 3.Aponte J. Prevalence of normoglycemic, prediabetic and diabetic A1c levels. World J Diabetes. 2013;4:349–357. doi: 10.4239/wjd.v4.i6.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.American Diabetes Association. Standards of medical care in diabetes--2014. Diabetes Care. 2014;37 Suppl 1:S14–S80. doi: 10.2337/dc14-S014. [DOI] [PubMed] [Google Scholar]
- 5.Abdul-Ghani MA, Tripathy D, DeFronzo RA. Contributions of beta-cell dysfunction and insulin resistance to the pathogenesis of impaired glucose tolerance and impaired fasting glucose. Diabetes Care. 2006;29:1130–1139. doi: 10.2337/diacare.2951130. [DOI] [PubMed] [Google Scholar]
- 6.Gastaldelli A, Ferrannini E, Miyazaki Y, Matsuda M, DeFronzo RA. Beta-cell dysfunction and glucose intolerance: results from the San Antonio metabolism (SAM) study. Diabetologia. 2004;47:31–39. doi: 10.1007/s00125-003-1263-9. [DOI] [PubMed] [Google Scholar]
- 7.Abdul-Ghani MA, Jenkinson CP, Richardson DK, Tripathy D, DeFronzo RA. Insulin secretion and action in subjects with impaired fasting glucose and impaired glucose tolerance: results from the Veterans Administration Genetic Epidemiology Study. Diabetes. 2006;55:1430–1435. doi: 10.2337/db05-1200. [DOI] [PubMed] [Google Scholar]
- 8.Sherwin RS, Anderson RM, Buse JB, Chin MH, Eddy D, Fradkin J, Ganiats TG, Ginsberg HN, Kahn R, Nwankwo R, et al. Prevention or delay of type 2 diabetes. Diabetes Care. 2004;27 Suppl 1:S47–S54. doi: 10.2337/diacare.27.2007.s47. [DOI] [PubMed] [Google Scholar]
- 9.Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia. 2003;46:3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- 10.Creutzfeldt W. The incretin concept today. Diabetologia. 1979;16:75–85. doi: 10.1007/BF01225454. [DOI] [PubMed] [Google Scholar]
- 11.Nauck MA, Homberger E, Siegel EG, Allen RC, Eaton RP, Ebert R, Creutzfeldt W. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab. 1986;63:492–498. doi: 10.1210/jcem-63-2-492. [DOI] [PubMed] [Google Scholar]
- 12.Holst JJ. Glucagon-like peptide-1: from extract to agent. The Claude Bernard Lecture, 2005. Diabetologia. 2006;49:253–260. doi: 10.1007/s00125-005-0107-1. [DOI] [PubMed] [Google Scholar]
- 13.Nauck M, Stöckmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia. 1986;29:46–52. doi: 10.1007/BF02427280. [DOI] [PubMed] [Google Scholar]
- 14.Knop FK, Vilsbøll T, Højberg PV, Larsen S, Madsbad S, Vølund A, Holst JJ, Krarup T. Reduced incretin effect in type 2 diabetes: cause or consequence of the diabetic state? Diabetes. 2007;56:1951–1959. doi: 10.2337/db07-0100. [DOI] [PubMed] [Google Scholar]
- 15.Højberg PV, Vilsbøll T, Rabøl R, Knop FK, Bache M, Krarup T, Holst JJ, Madsbad S. Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia. 2009;52:199–207. doi: 10.1007/s00125-008-1195-5. [DOI] [PubMed] [Google Scholar]
- 16.Smushkin G, Sathananthan A, Man CD, Zinsmeister AR, Camilleri M, Cobelli C, Rizza RA, Vella A. Defects in GLP-1 response to an oral challenge do not play a significant role in the pathogenesis of prediabetes. J Clin Endocrinol Metab. 2012;97:589–598. doi: 10.1210/jc.2011-2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rask E, Olsson T, Söderberg S, Holst Jj Jj, Tura A, Pacini G, Ahrén B. Insulin secretion and incretin hormones after oral glucose in non-obese subjects with impaired glucose tolerance. Metabolism. 2004;53:624–631. doi: 10.1016/j.metabol.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 18.Fritsche A, Stefan N, Hardt E, Häring H, Stumvoll M. Characterisation of beta-cell dysfunction of impaired glucose tolerance: evidence for impairment of incretin-induced insulin secretion. Diabetologia. 2000;43:852–858. doi: 10.1007/s001250051461. [DOI] [PubMed] [Google Scholar]
- 19.Zhang F, Tang X, Cao H, Lü Q, Li N, Liu Y, Zhang X, Zhang Y, Cao M, Wan J, et al. Impaired secretion of total glucagon-like peptide-1 in people with impaired fasting glucose combined impaired glucose tolerance. Int J Med Sci. 2012;9:574–581. doi: 10.7150/ijms.4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muscelli E, Mari A, Natali A, Astiarraga BD, Camastra S, Frascerra S, Holst JJ, Ferrannini E. Impact of incretin hormones on beta-cell function in subjects with normal or impaired glucose tolerance. Am J Physiol Endocrinol Metab. 2006;291:E1144–E1150. doi: 10.1152/ajpendo.00571.2005. [DOI] [PubMed] [Google Scholar]
- 21.Tura A, Muscelli E, Gastaldelli A, Ferrannini E, Mari A. Altered pattern of the incretin effect as assessed by modelling in individuals with glucose tolerance ranging from normal to diabetic. Diabetologia. 2014;57:1199–1203. doi: 10.1007/s00125-014-3219-7. [DOI] [PubMed] [Google Scholar]
- 22.Toft-Nielsen MB, Damholt MB, Madsbad S, Hilsted LM, Hughes TE, Michelsen BK, Holst JJ. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab. 2001;86:3717–3723. doi: 10.1210/jcem.86.8.7750. [DOI] [PubMed] [Google Scholar]
- 23.Byrne MM, Gliem K, Wank U, Arnold R, Katschinski M, Polonsky KS, Göke B. Glucagon-like peptide 1 improves the ability of the beta-cell to sense and respond to glucose in subjects with impaired glucose tolerance. Diabetes. 1998;47:1259–1265. doi: 10.2337/diab.47.8.1259. [DOI] [PubMed] [Google Scholar]
- 24.Crepaldi G, Carruba M, Comaschi M, Del Prato S, Frajese G, Paolisso G. Dipeptidyl peptidase 4 (DPP-4) inhibitors and their role in Type 2 diabetes management. J Endocrinol Invest. 2007;30:610–614. doi: 10.1007/BF03346357. [DOI] [PubMed] [Google Scholar]
- 25.Meier JJ. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8:728–742. doi: 10.1038/nrendo.2012.140. [DOI] [PubMed] [Google Scholar]
- 26.Farilla L, Bulotta A, Hirshberg B, Li Calzi S, Khoury N, Noushmehr H, Bertolotto C, Di Mario U, Harlan DM, Perfetti R. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology. 2003;144:5149–5158. doi: 10.1210/en.2003-0323. [DOI] [PubMed] [Google Scholar]
- 27.Wajchenberg BL. beta-cell failure in diabetes and preservation by clinical treatment. Endocr Rev. 2007;28:187–218. doi: 10.1210/10.1210/er.2006-0038. [DOI] [PubMed] [Google Scholar]
- 28.Ronner P, Naumann CM, Friel E. Effects of glucose and amino acids on free ADP in betaHC9 insulin-secreting cells. Diabetes. 2001;50:291–300. doi: 10.2337/diabetes.50.2.291. [DOI] [PubMed] [Google Scholar]
- 29.Dukes ID, Philipson LH. K+ channels: generating excitement in pancreatic beta-cells. Diabetes. 1996;45:845–853. doi: 10.2337/diab.45.7.845. [DOI] [PubMed] [Google Scholar]
- 30.Dhanvantari S, Izzo A, Jansen E, Brubaker PL. Coregulation of glucagon-like peptide-1 synthesis with proglucagon and prohormone convertase 1 gene expression in enteroendocrine GLUTag cells. Endocrinology. 2001;142:37–42. doi: 10.1210/endo.142.1.7870. [DOI] [PubMed] [Google Scholar]
- 31.Mayo KE, Miller LJ, Bataille D, Dalle S, Göke B, Thorens B, Drucker DJ. International Union of Pharmacology. XXXV. The glucagon receptor family. Pharmacol Rev. 2003;55:167–194. doi: 10.1124/pr.55.1.6. [DOI] [PubMed] [Google Scholar]
- 32.Holz GG, Leech CA, Habener JF. Activation of a cAMP-regulated Ca(2+)-signaling pathway in pancreatic beta-cells by the insulinotropic hormone glucagon-like peptide-1. J Biol Chem. 1995;270:17749–17757. [PMC free article] [PubMed] [Google Scholar]
- 33.Kang G, Chepurny OG, Holz GG. cAMP-regulated guanine nucleotide exchange factor II (Epac2) mediates Ca2+-induced Ca2+ release in INS-1 pancreatic beta-cells. J Physiol. 2001;536:375–385. doi: 10.1111/j.1469-7793.2001.0375c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holst JJ, Gromada J. Role of incretin hormones in the regulation of insulin secretion in diabetic and nondiabetic humans. Am J Physiol Endocrinol Metab. 2004;287:E199–E206. doi: 10.1152/ajpendo.00545.2003. [DOI] [PubMed] [Google Scholar]
- 35.Holz GG, Kühtreiber WM, Habener JF. Pancreatic beta-cells are rendered glucose-competent by the insulinotropic hormone glucagon-like peptide-1(7-37) Nature. 1993;361:362–365. doi: 10.1038/361362a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buteau J, Foisy S, Joly E, Prentki M. Glucagon-like peptide 1 induces pancreatic beta-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes. 2003;52:124–132. doi: 10.2337/diabetes.52.1.124. [DOI] [PubMed] [Google Scholar]
- 37.Buteau J, Foisy S, Rhodes CJ, Carpenter L, Biden TJ, Prentki M. Protein kinase Czeta activation mediates glucagon-like peptide-1-induced pancreatic beta-cell proliferation. Diabetes. 2001;50:2237–2243. doi: 10.2337/diabetes.50.10.2237. [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Cao X, Li LX, Brubaker PL, Edlund H, Drucker DJ. beta-Cell Pdx1 expression is essential for the glucoregulatory, proliferative, and cytoprotective actions of glucagon-like peptide-1. Diabetes. 2005;54:482–491. doi: 10.2337/diabetes.54.2.482. [DOI] [PubMed] [Google Scholar]
- 39.Wang X, Zhou J, Doyle ME, Egan JM. Glucagon-like peptide-1 causes pancreatic duodenal homeobox-1 protein translocation from the cytoplasm to the nucleus of pancreatic beta-cells by a cyclic adenosine monophosphate/protein kinase A-dependent mechanism. Endocrinology. 2001;142:1820–1827. doi: 10.1210/endo.142.5.8128. [DOI] [PubMed] [Google Scholar]
- 40.Buteau J, Spatz ML, Accili D. Transcription factor FoxO1 mediates glucagon-like peptide-1 effects on pancreatic beta-cell mass. Diabetes. 2006;55:1190–1196. doi: 10.2337/db05-0825. [DOI] [PubMed] [Google Scholar]
- 41.Stoffers DA, Kieffer TJ, Hussain MA, Drucker DJ, Bonner-Weir S, Habener JF, Egan JM. Insulinotropic glucagon-like peptide 1 agonists stimulate expression of homeodomain protein IDX-1 and increase islet size in mouse pancreas. Diabetes. 2000;49:741–748. doi: 10.2337/diabetes.49.5.741. [DOI] [PubMed] [Google Scholar]
- 42.Kaneto H, Matsuoka TA. Down-regulation of pancreatic transcription factors and incretin receptors in type 2 diabetes. World J Diabetes. 2013;4:263–269. doi: 10.4239/wjd.v4.i6.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jhala US, Canettieri G, Screaton RA, Kulkarni RN, Krajewski S, Reed J, Walker J, Lin X, White M, Montminy M. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev. 2003;17:1575–1580. doi: 10.1101/gad.1097103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cornu M, Modi H, Kawamori D, Kulkarni RN, Joffraud M, Thorens B. Glucagon-like peptide-1 increases beta-cell glucose competence and proliferation by translational induction of insulin-like growth factor-1 receptor expression. J Biol Chem. 2010;285:10538–10545. doi: 10.1074/jbc.M109.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buteau J. GLP-1 receptor signaling: effects on pancreatic beta-cell proliferation and survival. Diabetes Metab. 2008;34 Suppl 2:S73–S77. doi: 10.1016/S1262-3636(08)73398-6. [DOI] [PubMed] [Google Scholar]
- 46.Portha B, Tourrel-Cuzin C, Movassat J. Activation of the GLP-1 receptor signalling pathway: a relevant strategy to repair a deficient beta-cell mass. Exp Diabetes Res. 2011;2011:376509. doi: 10.1155/2011/376509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mari A, Sallas WM, He YL, Watson C, Ligueros-Saylan M, Dunning BE, Deacon CF, Holst JJ, Foley JE. Vildagliptin, a dipeptidyl peptidase-IV inhibitor, improves model-assessed beta-cell function in patients with type 2 diabetes. J Clin Endocrinol Metab. 2005;90:4888–4894. doi: 10.1210/jc.2004-2460. [DOI] [PubMed] [Google Scholar]
- 48.Winzell MS, Ahrén B. The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes. 2004;53 Suppl 3:S215–S219. doi: 10.2337/diabetes.53.suppl_3.s215. [DOI] [PubMed] [Google Scholar]
- 49.Dardik B, Valentin M, Schwartzkoph C, Gutierrez C, Stevens D, Russell M, Villhauer E, Hughes T. NVP-LAF237, a dipeptidyl peptidase IV inhibitor, improves glucose tolerance and delays gastric emptying in obese insulin resistant cynomolgus monkeys. Diabetes. 2003;52 Suppl 1:A322. [Google Scholar]
- 50.Utzschneider KM, Tong J, Montgomery B, Udayasankar J, Gerchman F, Marcovina SM, Watson CE, Ligueros-Saylan MA, Foley JE, Holst JJ, et al. The dipeptidyl peptidase-4 inhibitor vildagliptin improves beta-cell function and insulin sensitivity in subjects with impaired fasting glucose. Diabetes Care. 2008;31:108–113. doi: 10.2337/dc07-1441. [DOI] [PubMed] [Google Scholar]
- 51.Rosenstock J, Foley JE, Rendell M, Landin-Olsson M, Holst JJ, Deacon CF, Rochotte E, Baron MA. Effects of the dipeptidyl peptidase-IV inhibitor vildagliptin on incretin hormones, islet function, and postprandial glycemia in subjects with impaired glucose tolerance. Diabetes Care. 2008;31:30–35. doi: 10.2337/dc07-1616. [DOI] [PubMed] [Google Scholar]
- 52.Werzowa J, Hecking M, Haidinger M, Lechner F, Döller D, Pacini G, Stemer G, Pleiner J, Frantal S, Säemann MD. Vildagliptin and pioglitazone in patients with impaired glucose tolerance after kidney transplantation: a randomized, placebo-controlled clinical trial. Transplantation. 2013;95:456–462. doi: 10.1097/TP.0b013e318276a20e. [DOI] [PubMed] [Google Scholar]
- 53.Dhillon S. Sitagliptin: a review of its use in the management of type 2 diabetes mellitus. Drugs. 2010;70:489–512. doi: 10.2165/11203790-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 54.Chen B, Moore A, Escobedo LV, Koletsky MS, Hou D, Koletsky RJ, Ernsberger P. Sitagliptin lowers glucagon and improves glucose tolerance in prediabetic obese SHROB rats. Exp Biol Med (Maywood) 2011;236:309–314. doi: 10.1258/ebm.2010.010161. [DOI] [PubMed] [Google Scholar]
- 55.Dobrian AD, Ma Q, Lindsay JW, Leone KA, Ma K, Coben J, Galkina EV, Nadler JL. Dipeptidyl peptidase IV inhibitor sitagliptin reduces local inflammation in adipose tissue and in pancreatic islets of obese mice. Am J Physiol Endocrinol Metab. 2011;300:E410–E421. doi: 10.1152/ajpendo.00463.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maiztegui B, Borelli MI, Madrid VG, Del Zotto H, Raschia MA, Francini F, Massa ML, Flores LE, Rebolledo OR, Gagliardino JJ. Sitagliptin prevents the development of metabolic and hormonal disturbances, increased β-cell apoptosis and liver steatosis induced by a fructose-rich diet in normal rats. Clin Sci (Lond) 2011;120:73–80. doi: 10.1042/CS20100372. [DOI] [PubMed] [Google Scholar]
- 57.Bock G, Dalla Man C, Micheletto F, Basu R, Giesler PD, Laugen J, Deacon CF, Holst JJ, Toffolo G, Cobelli C, et al. The effect of DPP-4 inhibition with sitagliptin on incretin secretion and on fasting and postprandial glucose turnover in subjects with impaired fasting glucose. Clin Endocrinol (Oxf) 2010;73:189–196. doi: 10.1111/j.1365-2265.2009.03764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perreault L, Man CD, Hunerdosse DM, Cobelli C, Bergman BC. Incretin action maintains insulin secretion, but not hepatic insulin action, in people with impaired fasting glucose. Diabetes Res Clin Pract. 2010;90:87–94. doi: 10.1016/j.diabres.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 59.Hage C, Brismar K, Efendic S, Lundman P, Rydén L, Mellbin L. Sitagliptin improves beta-cell function in patients with acute coronary syndromes and newly diagnosed glucose abnormalities--the BEGAMI study. J Intern Med. 2013;273:410–421. doi: 10.1111/joim.12032. [DOI] [PubMed] [Google Scholar]
- 60.Seino Y, Yabe D. Alogliptin benzoate for the treatment of type 2 diabetes. Expert Opin Pharmacother. 2014;15:851–863. doi: 10.1517/14656566.2014.898750. [DOI] [PubMed] [Google Scholar]
- 61.Moritoh Y, Takeuchi K, Hazama M. Combination treatment with alogliptin and voglibose increases active GLP-1 circulation, prevents the development of diabetes and preserves pancreatic beta-cells in prediabetic db/db mice. Diabetes Obes Metab. 2010;12:224–233. doi: 10.1111/j.1463-1326.2009.01156.x. [DOI] [PubMed] [Google Scholar]
- 62.Moritoh Y, Takeuchi K, Asakawa T, Kataoka O, Odaka H. The dipeptidyl peptidase-4 inhibitor alogliptin in combination with pioglitazone improves glycemic control, lipid profiles, and increases pancreatic insulin content in ob/ob mice. Eur J Pharmacol. 2009;602:448–454. doi: 10.1016/j.ejphar.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 63.Cummings BP, Bettaieb A, Graham JL, Stanhope K, Haj FG, Havel PJ. Administration of pioglitazone alone or with alogliptin delays diabetes onset in UCD-T2DM rats. J Endocrinol. 2014;221:133–144. doi: 10.1530/JOE-13-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sudre B, Broqua P, White RB, Ashworth D, Evans DM, Haigh R, Junien JL, Aubert ML. Chronic inhibition of circulating dipeptidyl peptidase IV by FE 999011 delays the occurrence of diabetes in male zucker diabetic fatty rats. Diabetes. 2002;51:1461–1469. doi: 10.2337/diabetes.51.5.1461. [DOI] [PubMed] [Google Scholar]
- 65.Augstein P, Berg S, Heinke P, Altmann S, Salzsieder E, Demuth HU, Freyse EJ. Efficacy of the dipeptidyl peptidase IV inhibitor isoleucine thiazolidide (P32/98) in fatty Zucker rats with incipient and manifest impaired glucose tolerance. Diabetes Obes Metab. 2008;10:850–861. doi: 10.1111/j.1463-1326.2007.00813.x. [DOI] [PubMed] [Google Scholar]
- 66.Huisamen B, Genis A, Marais E, Lochner A. Pre-treatment with a DPP-4 inhibitor is infarct sparing in hearts from obese, pre-diabetic rats. Cardiovasc Drugs Ther. 2011;25:13–20. doi: 10.1007/s10557-010-6271-7. [DOI] [PubMed] [Google Scholar]
- 67.Kim MK, Chae YN, Kim HD, Yang EK, Cho EJ, Choi SH, Cheong YH, Kim HS, Kim HJ, Jo YW, et al. DA-1229, a novel and potent DPP4 inhibitor, improves insulin resistance and delays the onset of diabetes. Life Sci. 2012;90:21–29. doi: 10.1016/j.lfs.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 68. Available from: http: //clinicaltrials.gov/show/NCT01960205.
- 69.Göke R, Fehmann HC, Linn T, Schmidt H, Krause M, Eng J, Göke B. Exendin-4 is a high potency agonist and truncated exendin-(9-39)-amide an antagonist at the glucagon-like peptide 1-(7-36)-amide receptor of insulin-secreting beta-cells. J Biol Chem. 1993;268:19650–19655. [PubMed] [Google Scholar]
- 70.Wang Q, Brubaker PL. Glucagon-like peptide-1 treatment delays the onset of diabetes in 8 week-old db/db mice. Diabetologia. 2002;45:1263–1273. doi: 10.1007/s00125-002-0828-3. [DOI] [PubMed] [Google Scholar]
- 71.Tourrel C, Bailbe D, Lacorne M, Meile MJ, Kergoat M, Portha B. Persistent improvement of type 2 diabetes in the Goto-Kakizaki rat model by expansion of the beta-cell mass during the prediabetic period with glucagon-like peptide-1 or exendin-4. Diabetes. 2002;51:1443–1452. doi: 10.2337/diabetes.51.5.1443. [DOI] [PubMed] [Google Scholar]
- 72.Stoffers DA, Desai BM, DeLeon DD, Simmons RA. Neonatal exendin-4 prevents the development of diabetes in the intrauterine growth retarded rat. Diabetes. 2003;52:734–740. doi: 10.2337/diabetes.52.3.734. [DOI] [PubMed] [Google Scholar]
- 73.Rosenstock J, Klaff LJ, Schwartz S, Northrup J, Holcombe JH, Wilhelm K, Trautmann M. Effects of exenatide and lifestyle modification on body weight and glucose tolerance in obese subjects with and without pre-diabetes. Diabetes Care. 2010;33:1173–1175. doi: 10.2337/dc09-1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Armato J, DeFronzo RA, Abdul-Ghani M, Ruby R. Successful treatment of prediabetes in clinical practice: targeting insulin resistance and β-cell dysfunction. Endocr Pract. 2012;18:342–350. doi: 10.4158/EP11194.OR. [DOI] [PubMed] [Google Scholar]
- 75.Elkind-Hirsch K, Marrioneaux O, Bhushan M, Vernor D, Bhushan R. Comparison of single and combined treatment with exenatide and metformin on menstrual cyclicity in overweight women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93:2670–2678. doi: 10.1210/jc.2008-0115. [DOI] [PubMed] [Google Scholar]
- 76.Koska J, Schwartz EA, Mullin MP, Schwenke DC, Reaven PD. Improvement of postprandial endothelial function after a single dose of exenatide in individuals with impaired glucose tolerance and recent-onset type 2 diabetes. Diabetes Care. 2010;33:1028–1030. doi: 10.2337/dc09-1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schwartz EA, Koska J, Mullin MP, Syoufi I, Schwenke DC, Reaven PD. Exenatide suppresses postprandial elevations in lipids and lipoproteins in individuals with impaired glucose tolerance and recent onset type 2 diabetes mellitus. Atherosclerosis. 2010;212:217–222. doi: 10.1016/j.atherosclerosis.2010.05.028. [DOI] [PubMed] [Google Scholar]
- 78.Kelly AS, Bergenstal RM, Gonzalez-Campoy JM, Katz H, Bank AJ. Effects of exenatide vs. metformin on endothelial function in obese patients with pre-diabetes: a randomized trial. Cardiovasc Diabetol. 2012;11:64. doi: 10.1186/1475-2840-11-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Agersø H, Jensen LB, Elbrønd B, Rolan P, Zdravkovic M. The pharmacokinetics, pharmacodynamics, safety and tolerability of NN2211, a new long-acting GLP-1 derivative, in healthy men. Diabetologia. 2002;45:195–202. doi: 10.1007/s00125-001-0719-z. [DOI] [PubMed] [Google Scholar]
- 80.Cummings BP, Stanhope KL, Graham JL, Baskin DG, Griffen SC, Nilsson C, Sams A, Knudsen LB, Raun K, Havel PJ. Chronic administration of the glucagon-like peptide-1 analog, liraglutide, delays the onset of diabetes and lowers triglycerides in UCD-T2DM rats. Diabetes. 2010;59:2653–2661. doi: 10.2337/db09-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cummings BP, Digitale EK, Stanhope KL, Graham JL, Baskin DG, Reed BJ, Sweet IR, Griffen SC, Havel PJ. Development and characterization of a novel rat model of type 2 diabetes mellitus: the UC Davis type 2 diabetes mellitus UCD-T2DM rat. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1782–R1793. doi: 10.1152/ajpregu.90635.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guo N, Sun J, Chen H, Zhang H, Zhang Z, Cai D. Liraglutide prevents diabetes progression in prediabetic OLETF rats. Endocr J. 2013;60:15–28. doi: 10.1507/endocrj.ej12-0094. [DOI] [PubMed] [Google Scholar]
- 83.Sturis J, Gotfredsen CF, Rømer J, Rolin B, Ribel U, Brand CL, Wilken M, Wassermann K, Deacon CF, Carr RD, et al. GLP-1 derivative liraglutide in rats with beta-cell deficiencies: influence of metabolic state on beta-cell mass dynamics. Br J Pharmacol. 2003;140:123–132. doi: 10.1038/sj.bjp.0705397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Astrup A, Rössner S, Van Gaal L, Rissanen A, Niskanen L, Al Hakim M, Madsen J, Rasmussen MF, Lean ME. Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet. 2009;374:1606–1616. doi: 10.1016/S0140-6736(09)61375-1. [DOI] [PubMed] [Google Scholar]
- 85.Astrup A, Carraro R, Finer N, Harper A, Kunesova M, Lean ME, Niskanen L, Rasmussen MF, Rissanen A, Rössner S, et al. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int J Obes (Lond) 2012;36:843–854. doi: 10.1038/ijo.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim SH, Abbasi F, Lamendola C, Liu A, Ariel D, Schaaf P, Grove K, Tomasso V, Ochoa H, Liu YV, et al. Benefits of liraglutide treatment in overweight and obese older individuals with prediabetes. Diabetes Care. 2013;36:3276–3282. doi: 10.2337/dc13-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim SH, Liu A, Ariel D, Abbasi F, Lamendola C, Grove K, Tomasso V, Reaven G. Pancreatic beta cell function following liraglutide-augmented weight loss in individuals with prediabetes: analysis of a randomised, placebo-controlled study. Diabetologia. 2014;57:455–462. doi: 10.1007/s00125-013-3134-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kjems LL, Holst JJ, Vølund A, Madsbad S. The influence of GLP-1 on glucose-stimulated insulin secretion: effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes. 2003;52:380–386. doi: 10.2337/diabetes.52.2.380. [DOI] [PubMed] [Google Scholar]
- 89.Garber AJ. Long-acting glucagon-like peptide 1 receptor agonists: a review of their efficacy and tolerability. Diabetes Care. 2011;34 Suppl 2:S279–S284. doi: 10.2337/dc11-s231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dicker D. DPP-4 inhibitors: impact on glycemic control and cardiovascular risk factors. Diabetes Care. 2011;34 Suppl 2:S276–S278. doi: 10.2337/dc11-s229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gooßen K, Gräber S. Longer term safety of dipeptidyl peptidase-4 inhibitors in patients with type 2 diabetes mellitus: systematic review and meta-analysis. Diabetes Obes Metab. 2012;14:1061–1072. doi: 10.1111/j.1463-1326.2012.01610.x. [DOI] [PubMed] [Google Scholar]
- 92.Matveyenko AV, Dry S, Cox HI, Moshtaghian A, Gurlo T, Galasso R, Butler AE, Butler PC. Beneficial endocrine but adverse exocrine effects of sitagliptin in the human islet amyloid polypeptide transgenic rat model of type 2 diabetes: interactions with metformin. Diabetes. 2009;58:1604–1615. doi: 10.2337/db09-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nachnani JS, Bulchandani DG, Nookala A, Herndon B, Molteni A, Pandya P, Taylor R, Quinn T, Weide L, Alba LM. Biochemical and histological effects of exendin-4 (exenatide) on the rat pancreas. Diabetologia. 2010;53:153–159. doi: 10.1007/s00125-009-1515-4. [DOI] [PubMed] [Google Scholar]
- 94.Ahmad SR, Swann J. Exenatide and rare adverse events. N Engl J Med. 2008;358:1970–1971; discussion 1971-1972. [PubMed] [Google Scholar]
- 95.Kunjathaya P, Ramaswami PK, Krishnamurthy AN, Bhat N. Acute necrotizing pancreatitis associated with vildagliptin. JOP. 2013;14:81–84. doi: 10.6092/1590-8577/1203. [DOI] [PubMed] [Google Scholar]
- 96.Sue M, Yoshihara A, Kuboki K, Hiroi N, Yoshino G. A case of severe acute necrotizing pancreatitis after administration of sitagliptin. Clin Med Insights Case Rep. 2013;6:23–27. doi: 10.4137/CCRep.S10856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Elashoff M, Matveyenko AV, Gier B, Elashoff R, Butler PC. Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology. 2011;141:150–156. doi: 10.1053/j.gastro.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Singh S, Chang HY, Richards TM, Weiner JP, Clark JM, Segal JB. Glucagonlike peptide 1-based therapies and risk of hospitalization for acute pancreatitis in type 2 diabetes mellitus: a population-based matched case-control study. JAMA Intern Med. 2013;173:534–539. doi: 10.1001/jamainternmed.2013.2720. [DOI] [PubMed] [Google Scholar]
- 99.Butler AE, Campbell-Thompson M, Gurlo T, Dawson DW, Atkinson M, Butler PC. Marked expansion of exocrine and endocrine pancreas with incretin therapy in humans with increased exocrine pancreas dysplasia and the potential for glucagon-producing neuroendocrine tumors. Diabetes. 2013;62:2595–2604. doi: 10.2337/db12-1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Faillie JL, Babai S, Crépin S, Bres V, Laroche ML, Le Louet H, Petit P, Montastruc JL, Hillaire-Buys D. Pancreatitis associated with the use of GLP-1 analogs and DPP-4 inhibitors: a case/non-case study from the French Pharmacovigilance Database. Acta Diabetol. 2014;51:491–497. doi: 10.1007/s00592-013-0544-0. [DOI] [PubMed] [Google Scholar]
- 101.Meier JJ, Nauck MA. Risk of pancreatitis in patients treated with incretin-based therapies. Diabetologia. 2014;57:1320–1324. doi: 10.1007/s00125-014-3231-y. [DOI] [PubMed] [Google Scholar]
- 102.Koehler JA, Baggio LL, Lamont BJ, Ali S, Drucker DJ. Glucagon-like peptide-1 receptor activation modulates pancreatitis-associated gene expression but does not modify the susceptibility to experimental pancreatitis in mice. Diabetes. 2009;58:2148–2161. doi: 10.2337/db09-0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tatarkiewicz K, Smith PA, Sablan EJ, Polizzi CJ, Aumann DE, Villescaz C, Hargrove DM, Gedulin BR, Lu MG, Adams L, et al. Exenatide does not evoke pancreatitis and attenuates chemically induced pancreatitis in normal and diabetic rodents. Am J Physiol Endocrinol Metab. 2010;299:E1076–E1086. doi: 10.1152/ajpendo.00479.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mizukami H, Inaba W, Takahashi K, Kamata K, Tsuboi K, Yagihashi S. The effects of dipeptidyl-peptidase-IV inhibitor, vildagliptin, on the exocrine pancreas in spontaneously diabetic Goto-Kakizaki rats. Pancreas. 2013;42:786–794. doi: 10.1097/MPA.0b013e318287c9b5. [DOI] [PubMed] [Google Scholar]
- 105.Engel SS, Williams-Herman DE, Golm GT, Clay RJ, Machotka SV, Kaufman KD, Goldstein BJ. Sitagliptin: review of preclinical and clinical data regarding incidence of pancreatitis. Int J Clin Pract. 2010;64:984–990. doi: 10.1111/j.1742-1241.2010.02382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vrang N, Jelsing J, Simonsen L, Jensen AE, Thorup I, Søeborg H, Knudsen LB. The effects of 13 wk of liraglutide treatment on endocrine and exocrine pancreas in male and female ZDF rats: a quantitative and qualitative analysis revealing no evidence of drug-induced pancreatitis. Am J Physiol Endocrinol Metab. 2012;303:E253–E264. doi: 10.1152/ajpendo.00182.2012. [DOI] [PubMed] [Google Scholar]
- 107.Tatarkiewicz K, Belanger P, Gu G, Parkes D, Roy D. No evidence of drug-induced pancreatitis in rats treated with exenatide for 13 weeks. Diabetes Obes Metab. 2013;15:417–426. doi: 10.1111/dom.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nyborg NC, Mølck AM, Madsen LW, Knudsen LB. The human GLP-1 analog liraglutide and the pancreas: evidence for the absence of structural pancreatic changes in three species. Diabetes. 2012;61:1243–1249. doi: 10.2337/db11-0936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gotfredsen CF, Mølck AM, Thorup I, Nyborg NC, Salanti Z, Knudsen LB, Larsen MO. The human GLP-1 analogs liraglutide and semaglutide: absence of histopathological effects on the pancreas in nonhuman primates. Diabetes. 2014;63:2486–2497. doi: 10.2337/db13-1087. [DOI] [PubMed] [Google Scholar]
- 110.Chadwick KD, Fletcher AM, Parrula MC, Bonner-Weir S, Mangipudy RS, Janovitz E, Graziano MJ, Roy D, Reilly TP. Occurrence of spontaneous pancreatic lesions in normal and diabetic rats: a potential confounding factor in the nonclinical assessment of GLP-1-based therapies. Diabetes. 2014;63:1303–1314. doi: 10.2337/db13-1268. [DOI] [PubMed] [Google Scholar]
- 111.Garg R, Chen W, Pendergrass M. Acute pancreatitis in type 2 diabetes treated with exenatide or sitagliptin: a retrospective observational pharmacy claims analysis. Diabetes Care. 2010;33:2349–2354. doi: 10.2337/dc10-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dore DD, Seeger JD, Arnold Chan K. Use of a claims-based active drug safety surveillance system to assess the risk of acute pancreatitis with exenatide or sitagliptin compared to metformin or glyburide. Curr Med Res Opin. 2009;25:1019–1027. doi: 10.1185/03007990902820519. [DOI] [PubMed] [Google Scholar]
- 113.Dore DD, Bloomgren GL, Wenten M, Hoffman C, Clifford CR, Quinn SG, Braun DK, Noel RA, Seeger JD. A cohort study of acute pancreatitis in relation to exenatide use. Diabetes Obes Metab. 2011;13:559–566. doi: 10.1111/j.1463-1326.2011.01376.x. [DOI] [PubMed] [Google Scholar]
- 114.Wenten M, Gaebler JA, Hussein M, Pelletier EM, Smith DB, Girase P, Noel RA, Braun DK, Bloomgren GL. Relative risk of acute pancreatitis in initiators of exenatide twice daily compared with other anti-diabetic medication: a follow-up study. Diabet Med. 2012;29:1412–1418. doi: 10.1111/j.1464-5491.2012.03652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Engel SS, Round E, Golm GT, Kaufman KD, Goldstein BJ. Safety and tolerability of sitagliptin in type 2 diabetes: pooled analysis of 25 clinical studies. Diabetes Ther. 2013;4:119–145. doi: 10.1007/s13300-013-0024-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Monami M, Dicembrini I, Mannucci E. Dipeptidyl peptidase-4 inhibitors and pancreatitis risk: a meta-analysis of randomized clinical trials. Diabetes Obes Metab. 2014;16:48–56. doi: 10.1111/dom.12176. [DOI] [PubMed] [Google Scholar]
- 117.Faillie JL, Azoulay L, Patenaude V, Hillaire-Buys D, Suissa S. Incretin based drugs and risk of acute pancreatitis in patients with type 2 diabetes: cohort study. BMJ. 2014;348:g2780. doi: 10.1136/bmj.g2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Romley JA, Goldman DP, Solomon M, McFadden D, Peters AL. Exenatide therapy and the risk of pancreatitis and pancreatic cancer in a privately insured population. Diabetes Technol Ther. 2012;14:904–911. doi: 10.1089/dia.2012.0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Giorda CB, Picariello R, Nada E, Tartaglino B, Marafetti L, Costa G, Gnavi R. Incretin therapies and risk of hospital admission for acute pancreatitis in an unselected population of European patients with type 2 diabetes: a case-control study. Lancet Diabetes Endocrinol. 2014;2:111–115. doi: 10.1016/S2213-8587(13)70147-5. [DOI] [PubMed] [Google Scholar]
- 120.Li L, Shen J, Bala MM, Busse JW, Ebrahim S, Vandvik PO, Rios LP, Malaga G, Wong E, Sohani Z, et al. Incretin treatment and risk of pancreatitis in patients with type 2 diabetes mellitus: systematic review and meta-analysis of randomised and non-randomised studies. BMJ. 2014;348:g2366. doi: 10.1136/bmj.g2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Egan AG, Blind E, Dunder K, de Graeff PA, Hummer BT, Bourcier T, Rosebraugh C. Pancreatic safety of incretin-based drugs--FDA and EMA assessment. N Engl J Med. 2014;370:794–797. doi: 10.1056/NEJMp1314078. [DOI] [PubMed] [Google Scholar]
- 122.Bjerre Knudsen L, Madsen LW, Andersen S, Almholt K, de Boer AS, Drucker DJ, Gotfredsen C, Egerod FL, Hegelund AC, Jacobsen H, et al. Glucagon-like Peptide-1 receptor agonists activate rodent thyroid C-cells causing calcitonin release and C-cell proliferation. Endocrinology. 2010;151:1473–1486. doi: 10.1210/en.2009-1272. [DOI] [PubMed] [Google Scholar]
- 123.Madsen LW, Knauf JA, Gotfredsen C, Pilling A, Sjögren I, Andersen S, Andersen L, de Boer AS, Manova K, Barlas A, et al. GLP-1 receptor agonists and the thyroid: C-cell effects in mice are mediated via the GLP-1 receptor and not associated with RET activation. Endocrinology. 2012;153:1538–1547. doi: 10.1210/en.2011-1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rosol TJ. On-target effects of GLP-1 receptor agonists on thyroid C-cells in rats and mice. Toxicol Pathol. 2013;41:303–309. doi: 10.1177/0192623312472402. [DOI] [PubMed] [Google Scholar]
- 125.Hegedüs L, Moses AC, Zdravkovic M, Le Thi T, Daniels GH. GLP-1 and calcitonin concentration in humans: lack of evidence of calcitonin release from sequential screening in over 5000 subjects with type 2 diabetes or nondiabetic obese subjects treated with the human GLP-1 analog, liraglutide. J Clin Endocrinol Metab. 2011;96:853–860. doi: 10.1210/jc.2010-2318. [DOI] [PubMed] [Google Scholar]
- 126.Nauck MA. A critical analysis of the clinical use of incretin-based therapies: The benefits by far outweigh the potential risks. Diabetes Care. 2013;36:2126–2132. doi: 10.2337/dc12-2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, Ohman P, Frederich R, Wiviott SD, Hoffman EB, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317–1326. doi: 10.1056/NEJMoa1307684. [DOI] [PubMed] [Google Scholar]
- 128.White WB, Cannon CP, Heller SR, Nissen SE, Bergenstal RM, Bakris GL, Perez AT, Fleck PR, Mehta CR, Kupfer S, et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369:1327–1335. doi: 10.1056/NEJMoa1305889. [DOI] [PubMed] [Google Scholar]
- 129.Monami M, Ahrén B, Dicembrini I, Mannucci E. Dipeptidyl peptidase-4 inhibitors and cardiovascular risk: a meta-analysis of randomized clinical trials. Diabetes Obes Metab. 2013;15:112–120. doi: 10.1111/dom.12000. [DOI] [PubMed] [Google Scholar]
- 130.Wu D, Li L, Liu C. Efficacy and safety of dipeptidyl peptidase-4 inhibitors and metformin as initial combination therapy and as monotherapy in patients with type 2 diabetes mellitus: a meta-analysis. Diabetes Obes Metab. 2014;16:30–37. doi: 10.1111/dom.12174. [DOI] [PubMed] [Google Scholar]
- 131.Monami M, Marchionni N, Mannucci E. Glucagon-like peptide-1 receptor agonists in type 2 diabetes: a meta-analysis of randomized clinical trials. Eur J Endocrinol. 2009;160:909–917. doi: 10.1530/EJE-09-0101. [DOI] [PubMed] [Google Scholar]
- 132.Monami M, Cremasco F, Lamanna C, Colombi C, Desideri CM, Iacomelli I, Marchionni N, Mannucci E. Glucagon-like peptide-1 receptor agonists and cardiovascular events: a meta-analysis of randomized clinical trials. Exp Diabetes Res. 2011;2011:215764. doi: 10.1155/2011/215764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Standl E. Saxagliptin, alogliptin, and cardiovascular outcomes. N Engl J Med. 2014;370:483. doi: 10.1056/NEJMc1313880. [DOI] [PubMed] [Google Scholar]
- 134.Monami M, Dicembrini I, Mannucci E. Dipeptidyl peptidase-4 inhibitors and heart failure: a meta-analysis of randomized clinical trials. Nutr Metab Cardiovasc Dis. 2014;24:689–697. doi: 10.1016/j.numecd.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 135.Wu S, Hopper I, Skiba M, Krum H. Dipeptidyl peptidase-4 inhibitors and cardiovascular outcomes: meta-analysis of randomized clinical trials with 55,141 participants. Cardiovasc Ther. 2014;32:147–158. doi: 10.1111/1755-5922.12075. [DOI] [PubMed] [Google Scholar]
- 136.Petrie JR. The cardiovascular safety of incretin-based therapies: a review of the evidence. Cardiovasc Diabetol. 2013;12:130. doi: 10.1186/1475-2840-12-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gaebler JA, Soto-Campos G, Alperin P, Cohen M, Blickensderfer A, Wintle M, Maggs D, Hoogwerf B, Han J, Pencek R, et al. Health and economic outcomes for exenatide once weekly, insulin, and pioglitazone therapies in the treatment of type 2 diabetes: a simulation analysis. Vasc Health Risk Manag. 2012;8:255–264. doi: 10.2147/VHRM.S28744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cernea S, Raz I. Therapy in the early stage: incretins. Diabetes Care. 2011;34 Suppl 2:S264–S271. doi: 10.2337/dc11-s223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Foley JE, Bunck MC, Möller-Goede DL, Poelma M, Nijpels G, Eekhoff EM, Schweizer A, Heine RJ, Diamant M. Beta cell function following 1 year vildagliptin or placebo treatment and after 12 week washout in drug-naive patients with type 2 diabetes and mild hyperglycaemia: a randomised controlled trial. Diabetologia. 2011;54:1985–1991. doi: 10.1007/s00125-011-2167-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Scherbaum WA, Schweizer A, Mari A, Nilsson PM, Lalanne G, Wang Y, Dunning BE, Foley JE. Evidence that vildagliptin attenuates deterioration of glycaemic control during 2-year treatment of patients with type 2 diabetes and mild hyperglycaemia. Diabetes Obes Metab. 2008;10:1114–1124. doi: 10.1111/j.1463-1326.2008.00875.x. [DOI] [PubMed] [Google Scholar]
- 141.RISE Consortium. Restoring Insulin Secretion (RISE): design of studies of β-cell preservation in prediabetes and early type 2 diabetes across the life span. Diabetes Care. 2014;37:780–788. doi: 10.2337/dc13-1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bunck MC, Diamant M, Cornér A, Eliasson B, Malloy JL, Shaginian RM, Deng W, Kendall DM, Taskinen MR, Smith U, et al. One-year treatment with exenatide improves beta-cell function, compared with insulin glargine, in metformin-treated type 2 diabetic patients: a randomized, controlled trial. Diabetes Care. 2009;32:762–768. doi: 10.2337/dc08-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bunck MC, Cornér A, Eliasson B, Heine RJ, Shaginian RM, Taskinen MR, Smith U, Yki-Järvinen H, Diamant M. Effects of exenatide on measures of β-cell function after 3 years in metformin-treated patients with type 2 diabetes. Diabetes Care. 2011;34:2041–2047. doi: 10.2337/dc11-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Herzberg-Schäfer S, Heni M, Stefan N, Häring HU, Fritsche A. Impairment of GLP1-induced insulin secretion: role of genetic background, insulin resistance and hyperglycaemia. Diabetes Obes Metab. 2012;14 Suppl 3:85–90. doi: 10.1111/j.1463-1326.2012.01648.x. [DOI] [PubMed] [Google Scholar]
- 145.Butler PC, Elashoff M, Elashoff R, Gale EA. A critical analysis of the clinical use of incretin-based therapies: Are the GLP-1 therapies safe? Diabetes Care. 2013;36:2118–2125. doi: 10.2337/dc12-2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Devaraj S, Maitra A. Pancreatic safety of newer incretin-based therapies: are the “-tides” finally turning? Diabetes. 2014;63:2219–2221. doi: 10.2337/db14-0545. [DOI] [PubMed] [Google Scholar]