

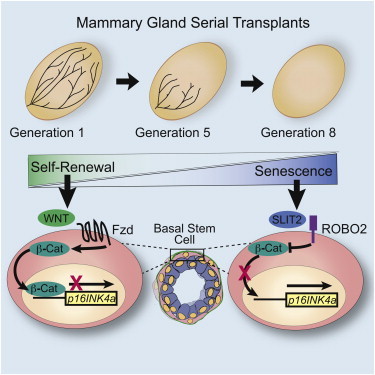
Summary
WNT signaling stimulates the self-renewal of many types of adult stem cells, including mammary stem cells (MaSCs), but mechanisms that limit this activity are poorly understood. Here, we demonstrate that SLIT2 restricts stem cell renewal by signaling through ROBO2 in a subset of basal cells to negatively regulate WNT signaling. The absence of SLIT/ROBO2 signaling leads to increased levels of nuclear β-catenin. Robo2 loss does not increase the number of stem cells; instead, stem cell renewal is enhanced in the absence of SLIT/ROBO2 signaling. This is due to repressed expression of p16INK4a, which, in turn, delays MaSC senescence. Together, our studies support a model in which SLITs restrict the expansion of MaSCs by countering the activity of WNTs and limiting self-renewal.
Graphical Abstract
Highlights
-
•
SLIT2 opposes WNT signaling to act as a mammary stem cell nonrenewal factor
-
•
SLIT/ROBO2 signaling increases p16INK4a expression, promoting cellular senescence
-
•
Loss of SLIT/ROBO2 signaling enhances mammary gland serial transplantability
In this article, Hinck and colleagues show that SLIT2 restricts stem cell renewal by signaling through ROBO2 and opposing WNT signaling. This results in increased expression of p16INK4a, promoting cellular senescence.
Introduction
SLITs are a conserved family of secreted proteins that participate in numerous developmental processes by signaling through ROBO receptors expressed on diverse cell types (Ballard and Hinck, 2012). The bilayered ducts of the mammary gland (MG) comprise an outer layer of basal cells (BCs) and an inner layer of luminal cells (LCs) and grow into a tree-like structure (Macias and Hinck, 2012). SLITs, expressed throughout the gland, regulate the elongation and branching of this structure during puberty by limiting the proliferation of basal cap cells (Macias et al., 2011; Strickland et al., 2006). BCs are capable of generating an entire mammary epithelial tree when transplanted into a cleared fat pad (Shackleton et al., 2006; Stingl et al., 2006). However, this population is heterogeneous, containing a small subset of MaSCs and a majority of differentiated cells. Little is known about mechanisms that regulate the growth, survival, and senescence of the basal MaSCs (bMaSCs).
SLITs regulate cell proliferation in both normal BCs and breast cancer cells by inhibiting AKT, leading to activation of GSK3β, and resulting in reduced nuclear accumulation of β-catenin and decreased expression of WNT-activated genes (Macias et al., 2011; Prasad et al., 2008; Tseng et al., 2010). Inhibition of β-catenin and canonical WNT signaling by SLIT may be important in regulating MaSC numbers because WNT3A enhances the self-renewal capacity of bMaSCs in culture, stimulating their clonal expansion while maintaining their ability to generate functional glands upon transplantation (Zeng and Nusse, 2010). Although expression of WNT1 or activated β-catenin results in an expanded bMaSC population and enhanced tumor formation (Shackleton et al., 2006; Teulière et al., 2005), less is known about the impact of inhibiting WNT signaling. However, treatment of bMaSCs with the WNT inhibitor DKK decreases formation of secondary colonies, presumably by limiting self-renewal (Zeng and Nusse, 2010). Therefore, curbing WNT signaling through SLIT/ROBO signaling may also restrict bMaSC self-renewal.
MaSC numbers must be tightly regulated through a balance of self-renewal, differentiation, and senescence to prevent tumorigenesis, while accommodating high levels of cell proliferation during pregnancy. Several secreted factors such as WNT and Hedgehog promote self-renewal, but little is known about how this process is restricted (Cho et al., 2013; Liu et al., 2006; Zeng and Nusse, 2010). Mechanisms involving p53 signaling have been demonstrated, in which bMaSC self-renewal is limited by either inhibiting symmetric divisions (Cicalese et al., 2009) or increasing senescence (Insinga et al., 2013). Additionally, inhibiting WNT signaling by DKK upregulates senescence-associated cell cycle inhibitor, p16 INK4a (CDKN2A) (Cho et al., 2013), which is directly repressed by β-catenin (Delmas et al., 2007). Here, we hypothesize that SLITs influence the self-renewal of bMaSCs by regulating their senescence. Our investigation identifies SLIT as an extracellular factor that signals through its ROBO2 receptor to inhibit WNT signaling, thereby regulating bMaSC senescence by governing the subcellular localization of β-catenin. As such, SLITs are a class of nonrenewal factors for bMaSCs in the MG.
Results
Loss of Robo2 Enhances bMaSC Self-Renewal In Vitro
To investigate the regulation of bMaSCs by SLIT/ROBO signaling, we examined expression of the Robo1 and Robo2 receptors in flow cytometry (FACS)-purified BCs (Lin-CD24+CD29hi), mature luminal cells (Lin-CD24loCD29+CD61−; ML), and luminal progenitor cells (Lin-CD24loCD29+CD61+; LP), harvested from the mature virgin MG. Quantitative RT-PCR (qRT-PCR) analysis shows expression of Robo1 and Robo2 in BCs, with a significant enrichment of Robo2 expression in BCs as compared to the ML and LP populations (Figures 1A and 1B). We have previously published the localization of ROBO1 on cap/BCs and a subpopulation of LCs (Macias et al., 2011; Marlow et al., 2008). To further confirm ROBO2 expression, we performed immunohistochemistry (IHC) and find ROBO2 expression in subpopulations of BCs and LCs in vivo (Figure 1C) and in vitro (Figure 1D).
Figure 1.
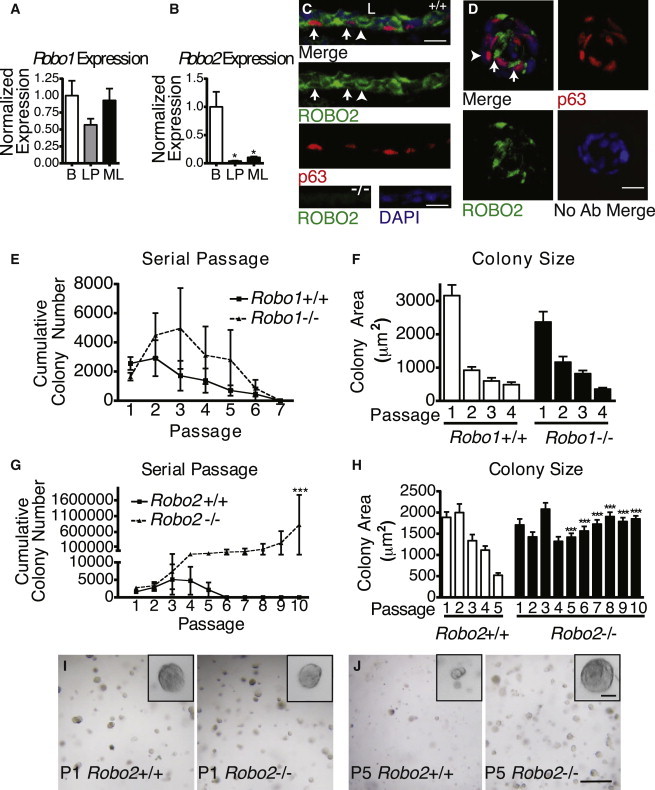
Loss of ROBO2 Enhances BMaSC Self-Renewal
(A and B) (A) qRT-PCR of Robo1 and (B) Robo2 mRNA expression in FACS-purified basal (B), luminal progenitor (LP), and mature luminal cells (MLs). (t test: Robo2 LP ∗p = 0.0229, Robo2 ML ∗p = 0.0286; n = 3).
(C) ROBO2 and p63 IHC on Robo2+/+ duct. Arrows: ROBO2 at the membrane of p63+ cells. Arrowhead: ROBO2 at the membrane of p63- cells touching the basal lamina. Bottom row: Robo2−/− control. L, lumen (scale bar, 50 μm).
(D) ROBO2/p63 IHC on Robo2+/+ basal colony. Arrows: ROBO2 in p63+ cells (scale bar, 50 μm).
(E and G) Matrigel colony numbers from an initial 20K Robo1(E) or Robo2(G) cells over serial passages (G: one-way ANOVA, Bonferroni post hoc test ∗∗∗p = < 0.001; two-way ANOVA, significant interaction genotype and passage ∗∗p < 0.01, n = 3.
(F and H) Robo1(F) and Robo2(H) colony size at each passage (one-way ANOVA, Bonferroni post hoc test ∗∗∗p < 0.001).
(I and J) Representative images of Robo2 colonies at P1(I) and P5 (J) (scale bar, 250 μm; insets, 50 μm).
To determine if loss of SLIT/ROBO signaling leads to changes in the self-renewal capacity of bMaSCs, we examined the ability of Robo1−/− or Robo2−/− FACS-purified BCs to serially passage in 3D Matrigel colony assays. Wild-type (WT) BCs possess a limited capacity for self-renewal (Figures 1E and 1G), with colonies progressively becoming smaller until they senesce at passages 4–5 (Figures 1F and 1H). Loss of Robo1 does not confer an advantage in passaging ability or colony size (Figures 1E and 1F). In contrast, loss of Robo2 leads to an increase in passaging ability (Figure 1G) and reduces the decline in colony size over passage (Figures 1H–1J), suggesting that loss of Robo2 enhances bMaSC self-renewal.
SLIT2 Acts as a Nonrenewal Factor by Inhibiting β-Catenin Activity in bMaSCs
The enhanced self-renewal observed in Robo2−/− BCs suggests that SLIT2 acts through ROBO2 to inhibit self-renewal. To test this, BCs were treated with SLIT2 during the initial plating and then grown in the absence of SLIT2 in subsequent passages (Figures 2A and 2B). Analysis of SLIT2-treated colonies at P1 reveals significantly smaller colonies compared to control (Figure 2B). 5-ethynyl-2′-deoxyuridine (EdU) incorporation was also significantly reduced in SLIT2-treated colonies, compared to control (data not shown). These data are consistent with our previous finding that SLIT2 acts through ROBO1 to inhibit BC proliferation (Macias et al., 2011). We next determined whether bMaSCs continue to self-renew when released from the antiproliferative activity of SLIT2. Despite the absence of exogenous SLIT2, BCs, initially treated with SLIT2, are unable to self-renew and continue to exhibit impaired proliferation as indicated by colony size (Figures 2A and 2B). These data support the hypothesis that SLIT2 acts not only to inhibit BC proliferation, but also as a bMaSC nonrenewal factor.
Figure 2.
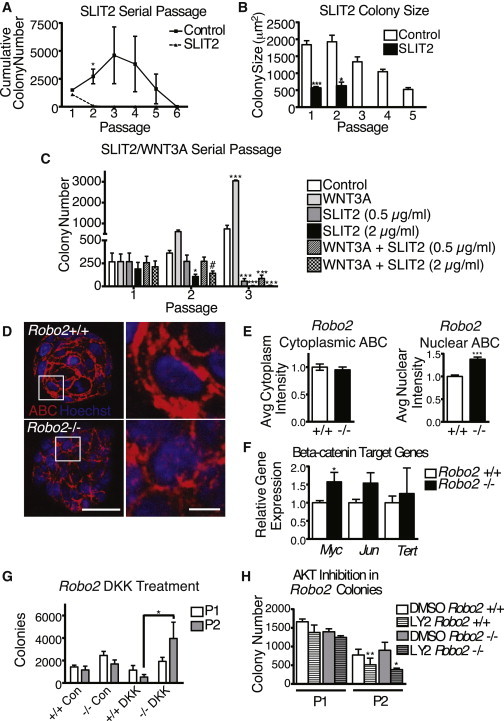
SLIT2 Acts as a Nonrenewal Factor by Inhibiting β-Catenin Activity in bMaSCs
(A and B) (A) Matrigel colony numbers and colony size (B) from an initial 20K control or SLIT2 (2 μg/ml) -treated WT colonies over serial passages (A: paired t test ∗p = 0.0482, n = 3) (B: one-way ANOVA, Bonferroni post hoc test: ∗∗∗p < 0.0001, ∗p = 0.0261).
(C) Colony numbers after treatment with 0.5 μg/ml SLIT2, 2 μg/ml SLIT2, 100 ng/ml WNT3A, or both over serial passages (one-way ANOVA, post hoc t test: P2: C versus 2 μg/ml SLIT2 p = 0.0492, C versus 2 μg/ml SLIT2+ WNT3a: p = 0.0717; P3: ∗∗∗p < 0.001, n = 3).
(D) Representative confocal images of activated-β-catenin IHC (ABC; red) and Hoechst (blue) on Robo2 colony sections (left scale, 50 μm; right scale, 10 μm).
(E) Relative levels of ABC expression detected using ImageJ to determine the average pixel intensity (t test: ∗∗∗p < 0.001, n = 5).
(F) qRT-PCR analysis of relative β-catenin target gene expression in Robo2+/+ and Robo2−/− basal colonies (∗p = 0.0329, n = 3).
(G) Robo2 colony numbers at P1 and P2 of cells treated with 1 μg/ml DKK1 (two-way ANOVA, Bonferroni posttest: ∗p < 0.05, n = 3).
(H) Robo2 colonies were cultured and passaged in the presence of DMSO or the PI3/AKT inhibitor LY294002 (5 μM) (one-tailed t test, ∗p = 0.0497, ∗∗p = 0.0049, n = 3).
Canonical WNT signaling enhances the self-renewal of bMaSCs (Zeng and Nusse, 2010). To determine if SLIT2 acts as a nonrenewal factor by inhibiting the WNT pathway, BCs were treated with SLIT2, WNT3A, or both. Initial colony number, assessed at passage 1 (P1), is similar between all treatment groups, although colonies treated with SLIT, even in the presence of WNT3A, are smaller in size (Figure 2C; data not shown). As previously published, colonies treated with WNT3A alone show a marked increase in colony number (Zeng and Nusse, 2010). Again, colonies treated with SLIT2 alone cannot be passaged past P2, even at a lower SLIT2 concentration. Treatment with both SLIT2 and WNT3A greatly attenuates self-renewal as indicated by reduced cumulative colony numbers. Taken together, these data suggest that SLIT2 inhibits WNT-mediated self-renewal.
Previous research has shown that SLIT2 opposes WNT signaling by regulating the AKT/GSK3β pathway and inhibiting the nuclear localization of β-catenin (Macias et al., 2011; Prasad et al., 2008; Tseng et al., 2010). We next examined whether loss of SLIT/ROBO2 signaling results in enhanced β-catenin activity in BC colonies by examining localization of the activated form of β-catenin (ABC) (Figures 2D and 2E). In Robo2+/+ colonies, ABC is localized primarily to the membrane and throughout the cytoplasm. In contrast, Robo2−/− colonies have a more diffuse localization of ABC. Although levels are similar in the cytoplasm, less ABC is localized to the membrane. Because β-catenin is only transcriptionally active in the nucleus, we also analyzed nuclear ABC, as determined by colabeling with Hoechst, and found a significant increase in Robo2−/− colonies. Although we did not examine whether increased nuclear ABC is localized specifically in BCs, analysis of colony composition using BC marker K14 and LC marker K8 shows the majority of cells (60%–70%) in both Robo2+/+ and Robo2−/− colonies are K14+ (data not shown). This, in conjunction with our finding that ROBO2 colocalizes with BC marker p63 in Robo2+/+ colonies (Figure 1D), suggests that loss of Robo2 is likely regulating β-catenin signaling in at least a subpopulation of BCs. To determine if increased nuclear β-catenin leads to upregulation of stem cell related target genes, we examined transcriptional levels of Myc, Jun, and Tert (Hoffmeyer et al., 2012; Jiao et al., 2010; Moumen et al., 2013). We find that all three genes show increased expression in Robo2−/− BC colonies, although only Myc reaches significance (Figure 2F). Together, these data suggest that loss of Robo2 leads to increased β-catenin signaling in BC colonies.
To determine if increased nuclear ABC in Robo2−/− BC colonies is due to loss of SLIT/ROBO2 signaling and not the result of a compensatory increase in canonical WNT signaling, we examined the consequences of blocking WNT signaling (Figure 2G). DKK1 is a secreted factor that inhibits WNT signaling upstream by interfering with LRP5/6 receptor function, and it has been shown to inhibit BC colony self-renewal (Zeng and Nusse, 2010). Accordingly, we find that treatment of Robo2+/+ colonies with DKK1 leads to reduced secondary colony formation; however, Robo2−/− colonies are resistant to DKK1 treatment and exhibit a significant increase in secondary colony formation (Figure 2G). These results show that inhibition of WNT signaling via DKK1 is insufficient to block self-renewal in bMaSCs that have lost SLIT/ROBO2 signaling. The fact that in the absence of Robo2 there is increased nuclear β-catenin (Figures 2D and 2E), along with enhanced BC colony renewal (Figure 1G), even under DKK1 treatment conditions (Figure 2G), suggests that the loss of SLIT/ROBO2 enhances bMaSC renewal directly by increasing nuclear β-catenin signaling. To further examine if stem cell renewal is increased in Robo2−/− cells through activation of the PI3K/AKT/GSK-β pathway, we treated Robo2+/+ and Robo2−/− colonies with AKT inhibitor LY294002 (Figure 2H). We find that AKT inhibition significantly impedes renewal in Robo2−/− colonies, suggesting that the PI3 kinase pathway is indeed a key pathway promoting β-catenin-mediated self-renewal in the absence of ROBO2.
SLIT2/ROBO2 Signaling Promotes bMaSC Senescence by Derepressing P16 INK4a Expression
The results from the serial passaging assay suggest that one possible mechanism through which SLIT2 acts as a nonrenewal factor is by promoting senescence in bMaSCs and their offspring. P16INK4a is a cell-cycle inhibitor that mediates cellular senescence. Previous studies show that β-catenin immortalizes skin melanocytes by directly binding to and silencing the p16INK4a promoter (Delmas et al., 2007). We first looked at expression of p16 INK4a by qRT-PCR and, although both Robo2+/+ and Robo2−/− colonies have higher levels of p16INK4a at P4 compared to P1, Robo2−/− colonies have significantly attenuated levels (Figure 3A). Next, we evaluated p16INK4a at passages P1 and P4 by IHC (Figures 3B–3D). At P1, little p16INK4a expression is observed (not shown), but at P4, significantly higher levels of p16 are detected in both the cytoplasm and nucleus of WT colonies, whereas its expression is significantly lower in Robo2−/− colonies (Figure 3D). We also examined the expression of p16 INK4a mRNA in colonies that were chronically treated with SLIT2 and find that SLIT2 promotes p16 INK4a expression in Robo2+/+ colonies and loss of Robo2 blocks this increase (Figure 3E). These data show that loss of SLIT/ROBO2 signaling in late passage BC colonies leads to attenuated expression of p16 INK4a and suggest that, similar to melanocytes, increased nuclear β-catenin in Robo2−/− colonies represses p16 INK4a expression and consequently inhibits senescence (Delmas et al., 2007).
Figure 3.
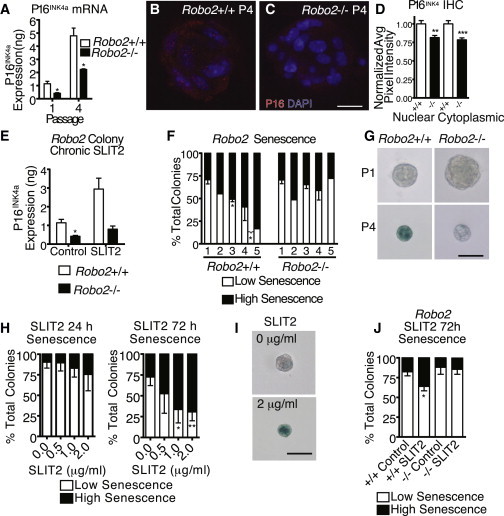
SLIT2/ROBO2 Signaling Promotes bMaSC Senescence by Derepressing p16INK4a Expression
(A) qRT-PCR quantification of p16INK4a mRNA in P1 and P4 Robo2 colonies (t test: P1∗p = 0.0185, P4 ∗p = 0.0119, n = 3).
(B and C) IHC of p16 INK4a (red) and DAPI (blue) in Robo2+/+(B) and (C) Robo2−/− P4 colonies (scale bar, 20 μm).
(D) Quantification of p16 INK4a fluorescent intensity (n = 20–25).
(E) qRT-PCR for p16INK4a mRNA in control or SLIT2- (2 μg/ml) treated (2 weeks) Robo2 colonies (t test:∗p = 0.0354, n = 3).
(F) SA-β-gal in Robo2 colonies over serial passages.
(G) Low senescence P1 (top) and P4 Robo2−/− colonies (bottom, right), high senescence P4 Robo2+/+ colony (bottom, left) (t test∗p < 0.05; scale bar, 50 μm; n = 2–4).
(H) Expression of SA-β-gal in SLIT2-treated colonies (t test, ∗p = 0.0410, ∗∗p = 0.0025, n = 3).
(I) Example of low senescence (≤50% blue) control colony (top) and high senescence (>50% blue) SLIT2-treated colony (bottom) scale bar, 50 μm.
(J) SLIT2 induces SA-β-gal expression in Robo2+/+ colonies, but not Robo2−/− colonies (paired t test ∗p = 0.0142, n = 3).
To determine if loss of SLIT/ROBO2 signaling inhibits senescence, we examined senescence-associated β-galactosidase (SA-β-Gal) expression in Robo2+/+ and Robo2−/− BC colonies. At P1, there is no difference in senescence; however, as colonies are passaged, Robo2+/+ colonies undergo a continual increase in senescence, whereas Robo2−/− colonies maintain a fairly stable rate of senescence similar to P1 (Figures 3F and 3 G). To examine if, conversely, SLIT/ROBO2 signaling promotes senescence in BC colonies, we analyzed SA-β-Gal expression 24 and 72 hr after SLIT2 treatment. By 72 hr, colonies treated with SLIT2 display a significant increase in SA-β-Gal expression (Figures 3H and 3I) that does not occur in Robo2−/− colonies (Figure 3J). Together, these data demonstrate that SLIT/ROBO2 signaling positively regulates p16INK4a expression and promotes cellular senescence.
Loss of SLIT/ROBO2 Signaling Enhances MaSC Self-Renewal In Vivo
To investigate whether loss of SLIT/ROBO2 signaling impairs MaSC self-renewal in vivo, we performed serial MG transplantations. As previously demonstrated (Daniel et al., 1968), WT MG fragments can be serially transplanted four to five times, before no further outgrowths are obtained (Figures 4A, 4C, 4E, and 4G). Similar to our in vitro findings (Figure 1D), loss of Robo1 does not confer a self-renewal advantage in the serial transplantation assay (Figure 4A). In contrast, loss of either Robo2 or Slit2;Slit3 leads to a nearly 2-fold increase in the number of transplant generations (Figures 4C, 4E, and 4G). These results demonstrate that loss of SLIT/ROBO2 signaling enhances MaSC self-renewal.
Figure 4.
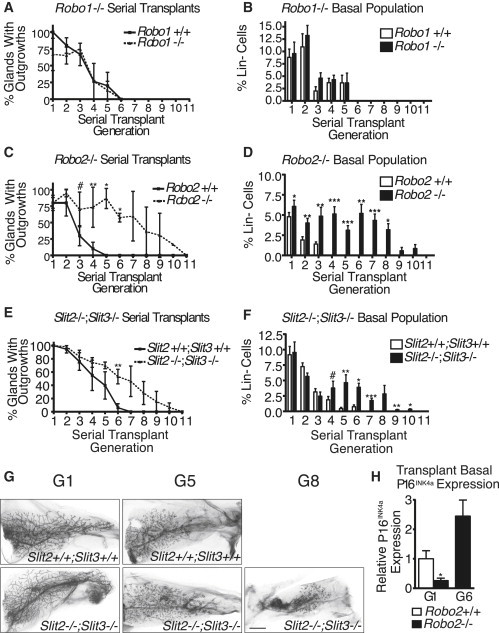
Loss of SLIT/ROBO2 Signaling Enhances MaSC Self-Renewal In Vivo
(A, C, and E) Fragments of littermate +/+, Robo1−/− (A; n = 4), Robo2−/− (C; n = 7), or Slit2−/−;Slit3−/− (E; n = 3) MG were serially transplanted and assessed at 8 week intervals (generations) (C: t test; G3 #p = 0.0699, G4 ∗∗p = 0.0033, G5 ∗p = 0.0403, G6 ∗p = 0.0182; E: paired t test; G6 ∗∗p = 0.0015.
(B, D, and F) FACS analysis of basal cells within the WT, Robo1−/− (B; n = 5–14 MG/time point [tp]), Robo2−/− (D; n = 5–20 MG/tp), or Slit2−/−;Slit3−/− (F; n = 5–20 MG/tp) MG (D: paired t test: G1 ∗p = 0.0199, G2 ∗∗p = 0.0077, G3 ∗∗p = 0.0043, G4 ∗∗∗p = 0.0002, G5 ∗∗∗p < 0.001, G6 ∗∗p = 0.0022, G7 ∗∗∗p < 0.0001, G8 ∗p = 0.0138; F: Paired t test; G4 #p = 0.0796, G5 ∗∗p = 0.0038, G6 ∗p = 0.0184, G7 ∗∗∗p = 0.0002, G9 ∗∗p = 0.0025, G10 ∗p = 0.0154).
(G) Carmine stained whole mounts from WT (top) and Slit2−/−;Slit3−/−(bottom) outgrowths (scale bar, 300 μm).
(H) qRT-PCR of FACS-purified basal cells from Robo2+/+ and Robo2−/− transplant glands at G1 and G6 (t test ∗p = 0.0271, n = 3).
One explanation for the observed increased transplantability of Robo2−/− and Slit2−/−;Slit3−/− tissue is that knockout (KO) tissue contains more bMaSCs. To investigate, we analyzed MG cell populations by FACS at each generation. There is no significant increase in the percent of BCs at generation 1 (G1) in any genotype, showing that transplantation itself does not expand this population. The BC population, however, is retained at higher levels over serial transplantation in KO outgrowths. Moreover, the rate of decline of the BC population is more gradual in KO outgrowths, suggesting that bMaSCs may be retained in this tissue (Figures 4D and 4F). Because the BC population is not pure, but only enriched for bMaSCs, we also performed limiting dilution assays using BCs derived from either G0 Robo2+/+ or Robo2−/− glands, or G4 serially transplanted Robo2+/+ and Robo2−/− outgrowths. We found that the lack of Robo2 in G0 glands does not result in an expansion in bMaSCs, with Robo2+/+ frequency estimated at 1/127 (95% CI 1/76–1/212) and Robo2−/− frequency estimated at 1/145 (95% CI 1/87–1/241). However, we found a remarkable retention of bMaSCs in serially transplanted Robo2−/− glands at G4 (Robo2+/+ frequency 1/1,737 (95% CI 1/753–1/4,008); and Robo2−/− frequency 1/196 (95% CI 1/77–1/498). These studies show that intact Robo2−/− glands do not contain more bMaSCs, but rather that Robo2−/− bMaSCs do not senesce as rapidly as Robo2+/+ bMaSCs. In support of this hypothesis, we examined expression of p16INK4a in FACS-purified cells from Robo2+/+ and Robo2−/− transplant glands from G1 and G6. We find that, similar to P1 Robo2−/− colonies (Figure 3A), Robo2−/− BCs have significantly lower expression of p16INK4a at G1 (Figure 4H). At G6, when Robo2+/+ has already senesced, we find increased levels of p16INK4a in comparison to G1 levels in Robo2−/− BCs, consistent with our hypothesis that loss of Robo2 delays, but does not prevent, senescence. Taken together, the data support a model in which SLIT2 acts as a nonrenewal factor for bMaSCs by acting through ROBO2 to oppose WNT signaling, and that loss of SLIT/ROBO2 signaling delays cellular senescence by increasing the nuclear localization of β-catenin and repressing p16INK4a expression.
Discussion
Here, we show that SLITs act through ROBO2 to inhibit self-renewal of bMaSCs. The actions of SLIT/ROBO signaling on cells, including stem cells, appear to be tissue and context specific, reminiscent of the context-dependent roles this signaling pathway plays as both attractant and repellent in cell migration (Ballard and Hinck, 2012). A recent study found that SLIT2/ROBO1 cooperates with R-spondin in intestinal stem cells to support their division and increase their self-renewal capacity, presumably by enhancing Wnt/β-catenin signaling (Zhou et al., 2013). In contrast, in this study on bMaSCs, we show an opposite effect with SLIT2 signaling through its ROBO2 receptor to limit bMaSC self-renewal by promoting senescence. Thus, although current data support an important role for SLIT signaling in modulating WNT signaling, its regulation appears to be dependent on both the cellular context in which that signal is received and the specific ROBO receptor mediating the signal. Within a single tissue, we have shown that loss of either ROBO1 or ROBO2 increases mammary BC nuclear β-catenin levels, but with very different outcomes: whereas loss of Robo1 leads to increased cell proliferation (Macias et al., 2011), loss of Robo2 results in increased bMaSC self-renewal. During pregnancy a similar dichotomy of function appears to occur, with loss of Robo1 delaying alveologenesis, whereas loss of Robo2 enhances it (J.C. and L.H., unpublished data). Although multiple mechanisms have been shown to increase nuclear β-catenin in response to loss of SLIT/ROBO signaling (Grossmann et al., 2013; Prasad et al., 2008; Tseng et al., 2010), it is not yet understood how this leads to divergent expression of WNT target genes such as those regulating proliferation and self-renewal. Determining the mechanism by which β-catenin activates and represses select WNT target genes remains a question of great interest in both the SLIT/ROBO and WNT signaling fields (Archbold et al., 2012).
Whereas SLITs have previously been characterized as mediators of adhesion, migration, and proliferation, our findings demonstrate another role for SLIT in regulating cellular senescence through its ROBO2 receptor. In identifying this function, our data further elucidate how SLIT acts as a tumor suppressor and how its loss contributes to tumor progression. A recent study found SLIT/ROBO signaling altered in 40.7% of basal, 12.3% of luminal A, and 26.3% of luminal B type tumors (Network, 2012). We show that by increasing the nuclear accumulation of β-catenin, loss of SLIT/ROBO2 signaling inhibits expression of p16 INK4a, itself a potent suppressor of tumors through its regulation of the cell cycle (Rayess et al., 2012). In the breast, silencing of p16 INK4a generates a preclonal phase of tumorigenesis in which cells are positioned for progression to malignancy (Bean et al., 2007). Loss of p16INK4a expression also increases the percentage of cancer stem cells (CD44+/CD24−) in basal-like breast cancer cell lines and reduces their response to chemotherapeutic agents (Arima et al., 2012). Thus, together with previous studies on SLIT signaling in the breast, current data suggest that SLITs inhibit breast tumorigenesis not only by decreasing cellular proliferation through ROBO1 (Macias et al., 2011; Prasad et al., 2008), but also by promoting cellular senescence through ROBO2.
Experimental Procedures
Mouse Strains
Slit2, Slit3, Robo1, and Robo2−/− mice were generated as previously described (Strickland et al., 2006). The study conforms to UCSC animal care committee (IACUC) guidelines.
Statistics
Limiting dilution analysis was performed as previously described (Shackleton et al., 2006). Statistics were performed using Prism software (GraphPad). Error bars in all panels represent the SEM; n = number of independent experiments, unless otherwise noted.
Transplant techniques, antibodies, IHC, SA-β-Gal assay, qRT-PCR, cell preparation, and sorting are described in Supplemental Experimental Procedures (available online).
Acknowledgments
We thank Aviv Spillinger, Kevin Van Houten, and Sharmila Chatterjee for technical assistance, Ben Abrams for support of the UCSC Life Sciences Microscopy Center, and Bari Holm Nazario for support of the IBSC Stem Cell Facility and SCBT for antibodies. This work was supported by CIRM (TG2-01157, FA1-00617-1, CL1-00506-1.2), Komen Postdoctoral Fellowship Award (KG111372 to G.H.), NIH (GM-098897 to L.H.).
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information
References
- Archbold H.C., Yang Y.X., Chen L., Cadigan K.M. How do they do Wnt they do?: regulation of transcription by the Wnt/β-catenin pathway. Acta Physiol. (Oxf.) 2012;204:74–109. doi: 10.1111/j.1748-1716.2011.02293.x. [DOI] [PubMed] [Google Scholar]
- Arima Y., Hayashi N., Hayashi H., Sasaki M., Kai K., Sugihara E., Abe E., Yoshida A., Mikami S., Nakamura S., Saya H. Loss of p16 expression is associated with the stem cell characteristics of surface markers and therapeutic resistance in estrogen receptor-negative breast cancer. Int. J. Cancer. 2012;130:2568–2579. doi: 10.1002/ijc.26271. [DOI] [PubMed] [Google Scholar]
- Ballard M.S., Hinck L. A roundabout way to cancer. Adv. Cancer Res. 2012;114:187–235. doi: 10.1016/B978-0-12-386503-8.00005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean G.R., Bryson A.D., Pilie P.G., Goldenberg V., Baker J.C., Jr., Ibarra C., Brander D.M., Paisie C., Case N.R., Gauthier M. Morphologically normal-appearing mammary epithelial cells obtained from high-risk women exhibit methylation silencing of INK4a/ARF. Clin. Cancer Res. 2007;13:6834–6841. doi: 10.1158/1078-0432.CCR-07-0407. [DOI] [PubMed] [Google Scholar]
- Cho J.H., Dimri M., Dimri G.P. A positive feedback loop regulates the expression of polycomb group protein BMI1 via WNT signaling pathway. J. Biol. Chem. 2013;288:3406–3418. doi: 10.1074/jbc.M112.422931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicalese A., Bonizzi G., Pasi C.E., Faretta M., Ronzoni S., Giulini B., Brisken C., Minucci S., Di Fiore P.P., Pelicci P.G. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138:1083–1095. doi: 10.1016/j.cell.2009.06.048. [DOI] [PubMed] [Google Scholar]
- Daniel C.W., De Ome K.B., Young J.T., Blair P.B., Faulkin L.J., Jr. The in vivo life span of normal and preneoplastic mouse mammary glands: a serial transplantation study. Proc. Natl. Acad. Sci. USA. 1968;61:53–60. doi: 10.1073/pnas.61.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas V., Beermann F., Martinozzi S., Carreira S., Ackermann J., Kumasaka M., Denat L., Goodall J., Luciani F., Viros A. Beta-catenin induces immortalization of melanocytes by suppressing p16INK4a expression and cooperates with N-Ras in melanoma development. Genes Dev. 2007;21:2923–2935. doi: 10.1101/gad.450107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann A.H., Yoo J.H., Clancy J., Sorensen L.K., Sedgwick A., Tong Z., Ostanin K., Rogers A., Grossmann K.F., Tripp S.R. The small GTPase ARF6 stimulates β-catenin transcriptional activity during WNT5A-mediated melanoma invasion and metastasis. Sci. Signal. 2013;6:ra14. doi: 10.1126/scisignal.2003398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmeyer K., Raggioli A., Rudloff S., Anton R., Hierholzer A., Del Valle I., Hein K., Vogt R., Kemler R. Wnt/β-catenin signaling regulates telomerase in stem cells and cancer cells. Science. 2012;336:1549–1554. doi: 10.1126/science.1218370. [DOI] [PubMed] [Google Scholar]
- Insinga A., Cicalese A., Faretta M., Gallo B., Albano L., Ronzoni S., Furia L., Viale A., Pelicci P.G. DNA damage in stem cells activates p21, inhibits p53, and induces symmetric self-renewing divisions. Proc. Natl. Acad. Sci. USA. 2013;110:3931–3936. doi: 10.1073/pnas.1213394110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao X., Katiyar S., Willmarth N.E., Liu M., Ma X., Flomenberg N., Lisanti M.P., Pestell R.G. c-Jun induces mammary epithelial cellular invasion and breast cancer stem cell expansion. J. Biol. Chem. 2010;285:8218–8226. doi: 10.1074/jbc.M110.100792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Dontu G., Mantle I.D., Patel S., Ahn N.S., Jackson K.W., Suri P., Wicha M.S. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–6071. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias H., Hinck L. Mammary Gland Development. Wiley interdisciplinary reviews. Dev. Biol. 2012;1:533–557. doi: 10.1002/wdev.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias H., Moran A., Samara Y., Moreno M., Compton J.E., Harburg G., Strickland P., Hinck L. SLIT/ROBO1 signaling suppresses mammary branching morphogenesis by limiting basal cell number. Dev. Cell. 2011;20:827–840. doi: 10.1016/j.devcel.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlow R., Strickland P., Lee J.S., Wu X., Pebenito M., Binnewies M., Le E.K., Moran A., Macias H., Cardiff R.D. SLITs suppress tumor growth in vivo by silencing Sdf1/Cxcr4 within breast epithelium. Cancer Res. 2008;68:7819–7827. doi: 10.1158/0008-5472.CAN-08-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moumen M., Chiche A., Decraene C., Petit V., Gandarillas A., Deugnier M.A., Glukhova M.A., Faraldo M.M. Myc is required for β-catenin-mediated mammary stem cell amplification and tumorigenesis. Mol. Cancer. 2013;12:132. doi: 10.1186/1476-4598-12-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network C.G.A., Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad A., Paruchuri V., Preet A., Latif F., Ganju R.K. Slit-2 induces a tumor-suppressive effect by regulating beta-catenin in breast cancer cells. J. Biol. Chem. 2008;283:26624–26633. doi: 10.1074/jbc.M800679200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayess H., Wang M.B., Srivatsan E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer. 2012;130:1715–1725. doi: 10.1002/ijc.27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackleton M., Vaillant F., Simpson K.J., Stingl J., Smyth G.K., Asselin-Labat M.L., Wu L., Lindeman G.J., Visvader J.E. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- Stingl J., Eirew P., Ricketson I., Shackleton M., Vaillant F., Choi D., Li H.I., Eaves C.J. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993–997. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- Strickland P., Shin G.C., Plump A., Tessier-Lavigne M., Hinck L. Slit2 and netrin 1 act synergistically as adhesive cues to generate tubular bi-layers during ductal morphogenesis. Development. 2006;133:823–832. doi: 10.1242/dev.02261. [DOI] [PubMed] [Google Scholar]
- Teulière J., Faraldo M.M., Deugnier M.A., Shtutman M., Ben-Ze’ev A., Thiery J.P., Glukhova M.A. Targeted activation of beta-catenin signaling in basal mammary epithelial cells affects mammary development and leads to hyperplasia. Development. 2005;132:267–277. doi: 10.1242/dev.01583. [DOI] [PubMed] [Google Scholar]
- Tseng R.C., Lee S.H., Hsu H.S., Chen B.H., Tsai W.C., Tzao C., Wang Y.C. SLIT2 attenuation during lung cancer progression deregulates beta-catenin and E-cadherin and associates with poor prognosis. Cancer Res. 2010;70:543–551. doi: 10.1158/0008-5472.CAN-09-2084. [DOI] [PubMed] [Google Scholar]
- Zeng Y.A., Nusse R. Wnt proteins are self-renewal factors for mammary stem cells and promote their long-term expansion in culture. Cell Stem Cell. 2010;6:568–577. doi: 10.1016/j.stem.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W.J., Geng Z.H., Spence J.R., Geng J.G. Induction of intestinal stem cells by R-spondin 1 and Slit2 augments chemoradioprotection. Nature. 2013;501:107–111. doi: 10.1038/nature12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.