

Summary
Data from the literature indicate that genomic imprint marks are disturbed in human pluripotent stem cells (PSCs). GNAS is an imprinted locus that produces one biallelic (Gsα) and four monoallelic (NESP55, GNAS-AS1, XLsα, and A/B) transcripts due to differential methylation of their promoters (DMR). To document imprinting at the GNAS locus in PSCs, we studied GNAS locus DMR methylation and transcript (NESP55, XLsα, and A/B) expression in human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs) derived from two human fibroblasts and their progenies. Results showed that (1) methylation at the GNAS locus DMRs is DMR and cell line specific, (2) changes in allelic transcript expression can be independent of a change in allele-specific DNA methylation, and (3) interestingly, methylation at A/B DMR is correlated with A/B transcript expression. These results indicate that these models are valuable to study the mechanisms controlling GNAS methylation, factors involved in transcript expression, and possibly mechanisms involved in the pathophysiology of pseudohypoparathyroidism type 1B.
Highlights
-
•
GNAS locus methylation is DMR and cell line specific in human pluripotent stem cells
-
•
Allelic transcript expression can be independent of allele-specific DNA methylation
-
•
A/B transcript expression, a key for PHP1B, is correlated with A/B DMR methylation
GNAS, a complex imprinted locus, produces biallelic (Gsα) and monoallelic (NESP55, GNAS-AS1, XLsα, and A/B) transcripts. In this article, Silve and colleagues show that hESCs and hiPSCs are valuable models to study mechanisms controlling GNAS methylation, regulation of transcript expression, and possibly mechanisms involved in the pathophysiology of PHP1B, a constellation of rare diseases caused by GNAS methylation defects.
Introduction
Human pluripotent stem cells (PSCs) provide invaluable models to study development, human diseases, and regenerative therapies. They can be derived from blastocysts (human embryonic stem cells [hESCs]) or directly reprogrammed from somatic cells (human induced pluripotent stem cells [hiPSCs]) (MacDonald and Mann 2014; Sabour and Schöler 2012; Tobin and Kim 2012). They share the unique property of self-renewal and are both expected to express the paternal and maternal imprints established during gametogenesis and maintained following fertilization. Imprinting maintenance and erasure are essential processes required for the mammalian development (Girardot et al., 2013; Laird 2013; Reik et al., 2001). However, hESCs are derived from a period in mammalian development characterized by global epigenetic remodeling, raising the possibility that the genomic imprint marks may be disturbed in these cells, whereas it is argued that nuclear reprogramming of hiPSCs could erase them (Li and Sasaki 2011; Takikawa et al., 2013). Therefore, it is important to assess if methylation marks at imprinted loci are stable or subject to variation upon derivation technique and subsequent culture.
GNAS is an imprinted locus that produces several transcripts comprising Gsα, the alpha-stimulatory subunit of the G protein; XLsα; A/B (also referred as 1A); NESP55; and the antisense transcript GNAS-AS1. Due to differential methylation of their promoters (DMR), XLsα, A/B, NESP55, and GNAS-AS1 originate from one parental allele only. XLsα, A/B, and GNAS-AS1 are transcribed from the paternal allele; NESP55 is transcribed from the maternal allele only. The promoter of Gsα is not differentially methylated, and therefore, Gsα expression arises from both alleles in most tissues (Figure 1). In a few specific tissues, however, including the renal proximal tubule, the thyroid, the pituitary, and the gonads, Gsα is expressed from the maternal allele only (Bastepe and Jüppner 2005; Hayward et al., 1998a, b; Levine 2012; Linglart et al., 2013; Mantovani et al., 2002; Plagge and Kelsey 2006; Weinstein et al., 2001). Maternally and paternally inherited loss of function of Gsα cause pseudohypoparathyroidism (PHP) type 1A (OMIM 103580) and pseudoPHP, respectively (or progressive osseous heteroplasia). Epigenetic changes at one or several of the promoters of the GNAS locus cause PHP type 1B (PHP1B) (OMIM 603233). All patients affected with PHP1B share a loss of methylation (LOM) at the maternal promoter of A/B, which results in suppressed Gsα transcription in imprinted tissues. LOM can be restricted to the A/B promoter of GNAS, as found in most familial forms of PHP1B (autosomic dominant PHP1B [AD-PHP1B]). Alternatively, A/B DMR LOM can be associated with methylation changes at other DMRs of GNAS on the maternal allele, as found in rare families carrying deletions removing an imprinting control element close to the AS and NESP DMRs, or most frequently in patients with sporadic PHP1B (80%–85% of PHP1B patients) (Bastepe and Jüppner 2005; Hayward et al., 1998a, b; Levine 2012; Linglart et al., 2013; Mantovani et al., 2002; Plagge and Kelsey 2006; Weinstein et al., 2001).
Figure 1.
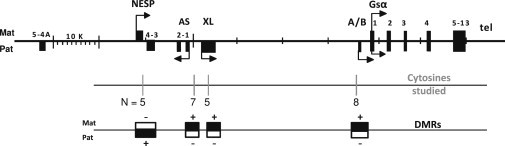
Schematic Drawing of the GNAS Locus
The GNAS locus is scaled, based on HG19. The four differentially methylated regions studied in this report are represented below the genomic line by black boxes (+ or methylated) or white boxes (− or unmethylated) on the paternal (Pat) or maternal (Mat) allele. Exons are indicated as black rectangles and allelic origin of transcription as broken arrows on the Pat or Mat allele. Positions and number of analyzed cytosines regarding methylation analysis are also indicated.
The molecular mechanisms controlling the establishment of imprinting at the GNAS cluster and leading to the methylation defects in PHP1B are mostly unknown, in part because of a paucity of suitable animal models and lack of accessible Gsα-imprinted human tissues. During the murine embryonic development, the differential methylation of exon 1A (A/B in humans) and Nespas/Gnasxl (AS and XL in humans) DMRs is established during the oogenesis (germline DMRs) whereas the differential methylation of Nesp DMR occurs postfertilization (somatic DMR), with a key role played by Nesp transcription in establishing the specific-allele methylation at the Gnas locus (Chotalia et al., 2009; Coombes et al., 2003; Liu et al., 2000). A recent study analyzing a large number of human fetal gonads from gestational weeks 6.5–22 suggested that epigenetic reprogramming in human primordial germ cells (hPGCs) probably involves, as observed in mice but with a different timing, two distinct periods: an early wave of genome-wide demethylation before 7 weeks of gestation and a later wave of imprint erasure and changes in chromatin modifications after 9 weeks of gestation (Gkountela et al., 2013; Laird 2013). Studies in hESCs and hPGCs indicated that allelic silencing of A/B is established during the gametogenesis (Frost et al., 2011) and that of XLsα already established at 5 weeks postfertilization (supporting the gametic specific-allele methylation of both A/B and XL DMRs as observed in the mice) (Crane et al., 2009). The complete allelic silencing of the NESP55 transcript occurs during implantation 5–11 weeks after fertilization (Crane et al., 2009; Rugg-Gunn et al., 2005a, b), in agreement with a somatic DMR. Tissue-specific silencing of paternal Gsα most likely takes place after 11 weeks postfertilization and after tissue differentiation (Turan et al., 2014; Zheng et al., 2001). A genome-wide DNA methylation revealed the maintenance of GNAS methylation in hiPSCs with culture, although hypermethylation and hypomethylation were also observed (Nazor et al., 2012).
In an effort to document imprinting at the GNAS locus and contribute to the development of models allowing its dynamic study and tissue-specific silencing of paternal Gsα in (patho)physiological conditions in humans, we studied methylation at the four GNAS DMRs in hESCs and hiPSCs and their progenies. We also examined the expression of four GNAS transcripts (Gsα, A/B, XLsα, and NESP55) in hiPSCs and derivatives.
Results
Characterization of Cell Lines
Somatic, pluripotent stem, and differentiated cells studied are presented in Table 1. Characterization of all hiPSC and ESC lines revealed a normal karyotype (including the VUBO1P91 cell line, studied at a high passage), expression of pluripotency markers, and expression of markers of the three germ layers upon in vitro embryoid bodies differentiation as illustrated in Figure 2 for hiPSC i90c17 line. In addition, all neural stem cells (NSCs) and mesenchymal stem cells (MSCs) expressed, respectively, the neural markers Nestin and Sox2 and the mesenchymal markers CD29, CD44, CD73, and CD166, as illustrated in Figure 2 for NSCs and MSCs derived from hiPSC i90c17 line.
Table 1.
Somatic, Pluripotent Stem, and Differentiated Cells Studied
| Somatic Cells | PSCs | Differentiated Cells (Progenies) | Allelic Expression | Overall Transcript Levels |
|---|---|---|---|---|
| h fibroblasts IMR90 (XX) | hiPSC i90c01 (retrovirus) | MSC/NSC | Fb/hiPSC/MSC | Fb/hiPSC/MSC/NCS |
| hiPSC i90c17 (episome) | MSC/NSC | Fb/hiPSC/MSC/NSC | Fb/hiPSC/MSC/NSC | |
| h fibroblasts GM04603 (XY) | hiPSC 4603c27 (retrovirus) | MSC/NSC | Fb/hiPSC/MSC/NSC | Fb/hiPSC/MSC/NSC |
| hiPSC 4603 polyF (retrovirus) | Fb/hiPSC | Fb/hiPSC | ||
| hESC VUB01 (XY) | NSC | 0 | Fb/hESC/NSC | |
| hESC W09 (XX) | MSC | 0 | Fb/hESC/MSC | |
| hESC RC9 (XY) | 0 | |||
| hESC SA01 (XY) | NSC | 0 | Fb/hESC/NSC |
For each cell line, whether overall (shown in Figure S1) and allelic (shown in Figure 5) transcript expressions were analyzed is indicated. Methylation at the GNAS DMRs was studied for all cells except hiPSC i90_c01 NSC. Fb, fibroblast; h, human; hESC, human embryonic stem cells; hiPSC, human induced pluripotent stem cells; MSC, mesenchymal stem cells; NSC, neuronal stem cells. “0” indicates homozygosity at the DNA level (all hESC). Only heterozygous cells at the DNA level were analyzed for transcript expression. Overall and allelic expressions were analyzed as described in the Experimental Procedures section.
Figure 2.
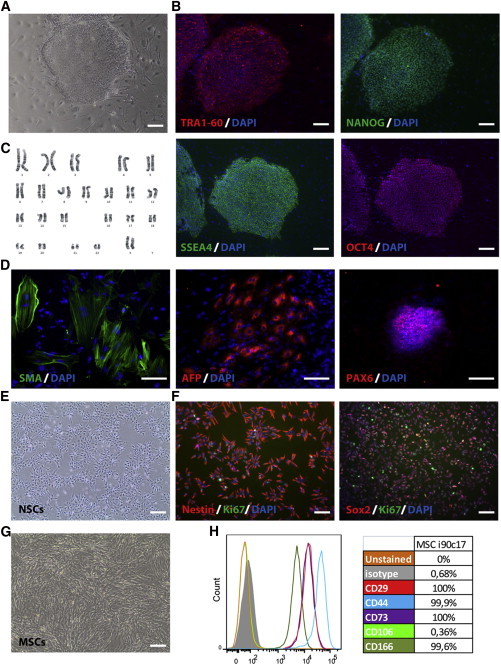
Characterization of i90c17 Human Induced Pluripotent Stem Cells and Neural Stem Cells and Mesenchymal Stem Cells Derived from i90c17 hiPSC Line
(A) Phase-contrast image of an i90c17 hiPSCs colony on feeder cell.
(B) Immunostaining showing the expression of the pluripotency markers TRA1-60, NANOG, SSEA4, and OCT4 in i90c17 hiPSC line.
(C) G-banding chromosome analysis of i90c17 hiPSCs showing a normal karyotype (46, XX).
(D) In vitro embryoid body formation from i90c17 hiPSCs showed three germ layer differentiation as illustrated by the presence of endodermal AFP+ cells, neuroectodermal PAX6+ cells, and mesodermal SMA+ cells.
(E) Phase-contrast image of hiPSCs i90c17-derived NSCs.
(F) Immunostaining showing the expression of the neural (Nestin and Sox2) and proliferating (Ki67) markers in hiPSCs i90c17-derived NSCs.
(G) Phase-contrast image of i90c17 hiPSCs-derived MSCs.
(H) Flow cytometry analysis of CD29, CD44, CD73, CD106, and CD166 expression in i90c17 hiPSCs-derived MSCs.
hiPSC, human pluripotent stem cell; MSC, mesenchymal stem cell; NSC, neural stem cell. The scale bars represent 200 μm. See also Figure S1.
Comparison of overall transcript levels between the various cell types by quantitative real-time PCR (qRT-PCR) indicated similar patterns of expression. When comparing fibroblasts and derived hiPSCs, an increase in all transcripts expression was observed. When comparing PSCs (hESCs and hiPSCs) in both NSC and MSC progenies, a decrease in NESP55, XLsα, and A/B transcripts expression was present, whereas Gsα transcript expression increased upon differentiation (Figure S1 available online).
Methylation at the GNAS Locus DMRs
In order to study the maintenance of imprinting at the GNAS locus, we quantified and compared methylation at the GNAS locus DMRs in eight PSCs (four hESCs and four hiPSCs), their progenies (four MSCs and four NSCs), and the two parental fibroblast cell lines and controls.
hESCs/hiPSCs
We first compared methylation indices at the GNAS DMRs measured in the four hESCs and four hiPSCs (Figures 3 and S2). Results showed that percent of methylation at each DMR was not significantly different when comparing hESCs and hiPSCs (NESP: hESCs: 43.7% ± 2.76%, hiPSC: 46.8% ± 3.07%; AS: hESC: 36.9% ± 16.42%, hiPSC: 34.3% ± 17.33%; XL: hESC: 49.0% ± 7.27%, hiPSC: 48.5% ± 4.6%; A/B: hESC: 38.6% ± 12.57%, hiPSC: 35.7% ± 13.73%; p > 0.05 for each DMR; mean ± SD; n = 4 for all groups).
Figure 3.
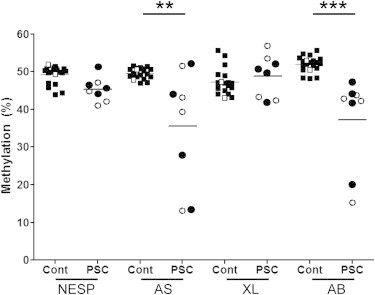
Methylation Quantification at the DMRs of GNAS in PSCs Compared to Parental Fibroblasts
Methylation at each of the four DMRs was similar comparing hESCs (○) and hiPSCs (●) and for NESP and XL DMRs, similar to 20 control subjects (▪) and parental fibroblasts (□). Methylation at the AS and A/B DMRs was significantly lower when comparing PSCs and controls (∗∗p < 0.01; ∗∗∗p < 0.001). Cell cultures, quantification of methylation, and statistical analysis were performed as described in the Experimental Procedures section. See also Figure S2.
PSCs/Controls
Because methylation at the GNAS DMRs was not significantly different in hESCs and hiPSCs, we then compared methylation at each GNAS DMRs comparing all PSCs (hESCs + hiPSCs) to that measured in blood DNA from 20 controls (Figure 3). Results indicated that percent of methylation at the NESP and XL DMRs was not significantly different when comparing PSCs and controls (PSCs, NESP: 45.2% ± 3.15%, XL: 48.8% ± 5.63%; controls, NESP: 49.3% ± 2.34%, XL: 47.28% ± 3.44%; p > 0.05; Figure 3). In contrast, percent of methylation at the AS and A/B DMRs was significantly lower when comparing PSCs and controls (PSCs, AS: 35.6% ± 15.69%, A/B: 37.2% ± 12.29%; controls, AS: 49.5% ± 1.39%, A/B: 51.9% ± 2.07%; p < 0.01 and 0.001, respectively, for AS and A/B; mean ± SD; n = 8 for PSCs and 20 for controls).
Percent of methylation for each DMR in the two parental fibroblast cell lines was within the range of values obtained in genomic DNA from controls (Figure 3).
We also compared the dispersion of GNAS DMR methylation between groups. The methylation scatter at the AS and A/B DMRs, but not at the NESP and XL DMRs, was significantly higher at the AS and A/B DMRs in PSCs compared to that in controls (PSCs, AS: 12.5% ± 8.66%, A/B: 8.5% ± 8.80%; controls, AS: 1.2% ± 0.69%, A/B: 1.6% ± 1.28%; p < 0.001 and p < 0.01, respectively, for AS and A/B; mean ± SD; data not shown). These results further support that the methylation at the AS and A/B DMRs is less stringent than that at the NESP and XL DMRs in PSCs and compared to controls.
PSCs/Progenies
PSCs were differentiated in four MSCs (three from hiPSCs and one from hESCs) and four NSCs (two from hiPSCs and two from hESCs). When comparing NSC progenies to appropriate PSCs, an increase in percent of methylation was observed in 2/4, 4/4, and 2/4, respectively, at the NESP, AS, and A/B DMRs and a decrease in 1/4 and 2/4, respectively, at the XL and A/B DMRs (Figures 4 and S3). Changes in percent of methylation were observed for NSC obtained from both hESCs and hiPSCs (Figures 4 and S3). When comparing MSC progenies to appropriate PSCs, methylation at NESP, XL, and A/B DMRs were similar. As in NSC, an increase in percent of methylation at AS DMR was also observed in MSC compared to appropriate PSCs. Thus, at the AS DMR, percent of methylation was significantly higher in MSC and NSC progenies compared to PSCs (Figure 4) (progenies: 66.1% ± 20.63%; controls: 39.0% ± 13.42%; p < 0.001; mean ± SD; n = 6 for progenies and PSCs).
Figure 4.
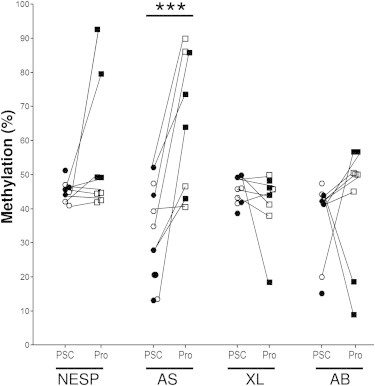
Methylation Quantification at the Four DMRs of GNAS in PSCs Compared to Progenies
Methylation at the GNAS locus is affected both as a function of cell type (NSC ▪ versus MSC □) and DMRs: changes in percent of methylation were observed at the NESP, XL, and A/B DMRs when comparing NSC, but not MSC, to appropriate PSCs (hiPSCs: ●; hESCs: ○); methylation at the AS DMR increases in both MSC and NSC and was significantly higher than that in PSCs (∗∗∗p < 0.001). Cell cultures, quantification of methylation, and statistical analysis were performed as described in the Experimental Procedures section. See also Figure S3.
Allelic Expression in Fibroblasts and Derived hiPSCs and Progenies
We determined that the two parental fibroblasts were heterozygous for the polymorphism rs7121 T/C in exon 5 (GNAS T393C) at the genomic DNA level (%T: 49.7% and 48.2% in IMR90 and GM04603, respectively) and that their derivative cells maintained heterozygosis (data not shown), thereby allowing analysis of GNAS transcript allelic expression as a function of reprogramming and differentiation. All hESCs were homozygous for rs7121 (data not shown).
NESP55 transcripts were detected only in 3/4 hiPSCs and showed monoallelic expression (%T expression: 100% and 98% in hiPSCs 4603_c27 and polyF, respectively; %C: 94% in hiPSC i90_c01) (Figures 5, S4, and S5); they were not detected in fibroblasts or in progenies. As indicated in the Experimental Procedures section, given that NESP55 is essentially maternally expressed, transcript expression is thus expressed as “percent maternal allele ratio.”
Figure 5.
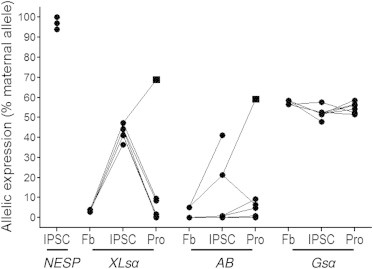
Allelic Transcript Expression in Parental Fibroblasts, hiPSCs, and Progenies
NESP55 transcript, detected in three hiPSC clones, was monoallelic. XLsα expression was monoallelic in the two parental fibroblast (Fb) lines, biallelic in the four hiPSC clones, and returned to monoallelic expression in progenies except one (NSC, ▪). A/B expression was monoallelic in the two parental fibroblast lines, in 2/4 hiPSC clones, and in progenies (Freson et al., 2008) except one (NSC, ▪). Gsα was biallelic in the two parental fibroblast lines, the four hiPSC clones, and progenies. Cell cultures and allelic transcript expression were performed as described in the Experimental Procedures section. Expression for GNAS transcripts are expressed as percent maternal allele ratio, based on the allele expression of the maternally expressed NESP55 transcript (see Experimental Procedures for details). Biallelic expression was defined for maternal allele ratio comprised between 40% and 60% and monoallelic expression for maternal allele ratio comprised between 90% and 100% and 0% and 10%. See also Figures S4 and S5.
As expected, expression of Gsα transcripts was biallelic in the two parental fibroblasts (58.40% and 56.60% maternal allele ratio in fibroblasts i90 and i4603, respectively) (Figures 5 and S3). Gsα remained biallelic after reprogramming in hiPSCs and differentiation in progenies (maternal allele ratio: 47.4% ± 5.8% and 54.8% ± 2.71%, respectively, in hiPSCs and hiPSC progenies; mean ± SD; n = 6) (Figures 5 and S5).
Expression of XLsα transcripts was monoallelic in the two parental fibroblasts and showed, as expected, paternal expression (3.8% and 3.6% maternal expression in i90 and in 4603, respectively) (Figures 5 and S5). Surprisingly, after reprogramming in hiPSCs, XLsα was biallelic (maternal allele ratio 43.1% ± 3.86%; mean ± SD; n = 4) in all cells. In MSC- and NSC-differentiated cells, XLsα expression was again monoallelic (maternal allele ratio 3.9% ± 4.61%; mean ± SD; n = 5), except in one NCS line, in which it stayed biallelic.
As observed for XLsα transcripts, expression of A/B transcripts was also monoallelic in the two parental fibroblasts with paternal expression (5.2% and 0% maternal expression in i90 and in 4603, respectively). Monoallelic expression of A/B transcript was conserved in the two hiPSC i90 clones (maternal allele ratio 0% and 0.8%), but not in the hiPSC 4603 clones, in which expression of A/B transcripts increased and presented partial allelic and biallelic (maternal allele ratio 21.3% and 41.2%) (Figures 5 and S3). In MSC- and NSC-differentiated cells, A/B expression was monoallelic and paternally expressed (maternal allele ratio 4.26% ± 3.89%; mean ± SD; n = 5), except in one NCS line, in which it increased and became biallelic (maternal allele ratio 59.1%). The rs7121 polymorphism is not present in GNAS-AS1 cDNA, therefore precluding GNAS-AS1 allelic expression.
Correlation between DMR Methylation and Allelic Expression
In “normal” conditions, XL and A/B promoters are differentially methylated (i.e., methylated on the maternal allele) and XLsα and A/B transcript expression is described as monoallelic (expression of 90%–100% of a major paternal allele). Changes of methylation are usually associated with changes in parental transcript expression. In order to determine if XLsα and A/B transcript expression was correlated to GNAS DMR methylation, we correlated allelic expression to methylation at the XL and A/B GNAS DMRs in fibroblasts and derivatives. We found that the allelic expression of A/B transcripts, but not that of XLsα with XL DMR, was correlated with A/B DMR methylation (Figures 6A and 6B): the ratio of A/B maternal allele expression decreased when methylation at A/B DMR increased (Pearson r: 0.8974, p = 0.001 and Pearson r: 0.4425, p = ns for A/B and XL, respectively). Gsα allelic expression was not correlated with percent methylation of A/B DMR or with A/B transcript expression (data not shown). Allelic transcript expression analysis was not available for GNAS-AS1 and was available in only three samples for NESP55, precluding any correlation (data not shown).
Figure 6.
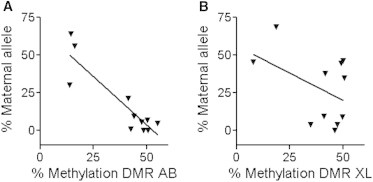
Correlation between Methylation and Allelic Expression for A/B and XLsα
The allelic expression of A/B transcript (A), but not that of XLsα (B), correlates with the methylation at their DMRs (Pearson r: A/B: 0.8974, p = 0.001; XL: 0.4425, p = not significant). Cell cultures, quantification of DMR methylation, and analysis of allelic transcript expression were performed as described in the Experimental Procedures section. GNAS transcripts are expressed as percent maternal allele ratio, based on the allele of the maternally expressed NESP55 transcript (see Experimental Procedures for details).
Discussion
To assess if methylation marks at the GNAS locus were maintained in hESCs and hiPSCs or subjected to variation upon derivation technique and subsequent culture, we quantified and compared methylation at the GNAS locus in hESCs and hiPSCs (four cell lines each). Our results showed that methylation at the four DMRs was similar in hESCs and hiPSCs. These results are consistent with a whole-genome single-base resolution DNA methylome study by Lister et al. (2011) reporting globally similar methylation comparing hESC and hiPSC methylomes. In addition, we found that methylation at the paternally imprinted NESP (maternal expression of the transcript) and maternally imprinted XL (paternal expression of the transcript) DMRs was maintained in all PSCs (hESCs and hiPSCs) and similar to that in controls and parental fibroblasts, in contrast to the two maternally imprinted AS and A/B DMRs.
Two main conclusions can be drawn from these observations. First, previous studies have indicated that epigenetic instability is a rare occurrence in hESCs but, in contrast, that the differential methylation that marks imprinted loci could be erased during nuclear reprogramming of somatic cells (Frost et al., 2011). Analysis of germline methylation imprints in human PSCs has revealed some instability and this independently of the parental origin of the imprint (Lund et al., 2012; Nazor et al., 2012; Rugg-Gunn et al., 2007; Takikawa et al., 2013; Tobin and Kim 2012). In this regard, aberrant DNA methylation at the maternally imprinted H19 and paternally imprinted KCNQOT1 genes in iPSCs has been reported (Lister et al., 2011). Our results indicate that the control of GNAS genomic methylation imprinting stability does not vary specifically as function of the PSC type (hESCs versus hiPSCs) and is independent of the reprogramming procedure. This is further supported by the similar methylation pattern observed for two clones obtained from the same parental fibroblast either by retroviral or episomal reprogramming methods. Second, our results indicate that the control of methylation at the NESP and XL DMRs (paternally and maternally imprinted, respectively) is more stringent than that at AS and A/B DMR (both maternally imprinted). NESP DMR methylation analyzed in two studies was reported differentially methylated in the majority of hESC lines with exceptional loss or gain of methylation (Frost et al., 2011; Huntriss et al., 2011). Methylation of XL DMR reported in only two human in vitro fertilization blastocysts was variable (4.8% and 77.1%) (Huntriss et al., 2011). Our results further document and enrich these observations. In all cases of DMR methylation instability, we observed demethylation and not hypermethylation, indicating that whatever the underlying mechanism, AS and A/B are prone to demethylation during PSC derivation or maintenance. Why the control of methylation at the AS and A/B DMRs is less stringent than that at the NESP and XL DMRs in PSCs is not explained. Methylation at the A/B DMR was low in the polyclonal iPSC04603_polyF cell line and normal in the monoclonal iPSC04603_c27 line, both derived from the same parental fibroblasts, raising the possibility that clonality may affect methylation results.
An important aspect of PSC research is their theoretical ability to be differentiated into any cell type, including cells expressing tissue-specific silencing of paternal Gsα, as described for Gsα. However, for such studies, it is critical to fully control the differentiation of these PSCs into specific cellular types of interest. Thus, our next step was to study methylation marks upon differentiation of PSCs. Our results indicate that methylation at the GNAS locus is affected both as a function of cell type (NSC versus MSC) and DMRs. Indeed, methylation changes at the NESP and A/B DMRs were observed only upon differentiation into NSC, not MSC. In addition, we observed an increase in AS methylation in all progenies (NSC and MSC), reaching hypermethylation levels in 4/8. Few reports, and none for the GNAS locus to our knowledge, have addressed the issue of DMR methylation upon “re”differentiation of PSCs into progenies in human cells. The pattern associating gain of methylation at NESP and loss of methylation at A/B and/or XL DMRs of GNAS is reminiscent to that of patients affected with sporadic PHP1B. Whereas it is tempting to speculate that changes upon reprogramming and epigenetic changes causing PHP1B are connected, the molecular mechanisms causing these changes are not identified. In contrast to our results in PSCs, loss of methylation at the AS DMR of GNAS is common in sporadic PHP1B (Maupetit-Méhouas et al., 2011) as observed for the A/B and XL DMRs, also methylated on the maternal allele). The mechanisms causing the epigenetic changes in PHP1B are under investigation and multiple. Some common mechanism might exist during reprogramming and PHP1B. Further studies analyzing the specific increase in AS methylation as well as the changes in other DMRs methylation observed in progenies from hESCs and hiPSCs may help understand the mechanisms whereby methylation at each DMR is controlled in physiology and the mechanisms leading to methylation defect in PHP1B.
The notion that loss of methylation at the GNAS DMRs controls transcript expression is mostly intuitive, with little available direct evidence. Freson et al. (2008) showed decreased methylation at XL DMR and increased expression of the XL protein in platelets. Loss of methylation at exon A/B is associated with an increase in the levels of the noncoding exon A/B RNA and a loss of Gsα expression (Bastepe and Jüppner 2005; Fröhlich et al., 2010). Studies in mice have shown that paternal deletion of the exon 1A region results in reversal of Gsα allelic silencing with biallelic expression of Gsα (Liu et al., 2000; Williamson et al., 2004). Monoallelic expression of Gsα has been reported in a few studies and mainly mouse studies (for reviews, see Bastepe and Jüppner, 2005; Hayward et al., 2001; Levine, 2012; Linglart et al., 2013; Mantovani et al., 2012; Plagge and Kelsey, 2006; Weinstein et al., 2007). Human tissues expressing paternal Gsα allelic silencing are not easily accessible, and the correlation between transcript expression and DMR methylation is rarely reported. Predominant maternal origin of transcription of Gsα in human thyroid gland and gonads has been reported (Mantovani et al., 2002).
Using the distinguishing parental single-nucleotide polymorphism rs7121, we correlated allelic expression and DMR methylation in hiPSCs and after differentiation for the three imprinted transcripts (A/B, XLsα, and NESP55) and also defined their parental expression as well as that of Gsα. Allelic expression of the maternally imprinted A/B transcripts varied as a function of the cell line. As indicated above, A/B DMR has a maternal-specific germline methylation. It is therefore expected that, in hiPSCs and progenies, the expression of the A/B transcripts originates predominantly from the paternal allele. This was observed in hiPSC clones and progenies derived from one fibroblast line, but not from the other. Importantly, however, we found that A/B transcript expression was correlated with the degree of methylation at the A/B DMR, indicating that allelic-silencing mechanism of A/B expression is methylation dependent.
Evidence from AD-PHP1B patients as well as mouse models indicates that the expression levels of the two transcripts, exon A/B and Gsα, are oppositely regulated in cis in imprinted tissues (Plagge and Kelsey 2006; Williamson et al., 2004). Absence of paternal Gsα transcript expression is attributed at least in part to the prevention of Gsα expression by the expressed A/B transcript. As expected, we detected biallelic expression of Gsα in all hiPSCs and fibroblasts, independently of A/B DMR methylation and transcription. Regarding the results in progenies, tissue-specific silencing of paternal Gsα has been described in brown fat cells (Williamson et al., 2004) of mesenchymal origin) and specific neurons (Chen et al., 2009); however, we do not observe Gsα allelic silencing in the MSC and NSC studied. The MSC and NSC analyzed here have not reached the differentiation status of brown fat cells and imprinted neurons, likely explaining our results and supporting the requirement of “terminal” cell differentiation for Gsα allelic silencing to occur, as previously shown in the kidney (Turan et al., 2014; Zheng et al., 2001).
In most tissues, XL DMR has a maternal-specific germline methylation; thus, XL DMR methylation, absence of maternal XLsα transcription, and monoallelic paternal expression are expected in hiPSCs and progenies. Surprisingly, the paternally expressed imprinted XLsα transcript showed biallelic expression in all hiPSC clones from the two parental fibroblast lines but, as expected, monoallelic expression in all progenies (except one, also unstable for A/B). Intriguingly, this biallelic expression of XLsα in all hiPSC clones was observed in spite of a maintained XL DMR methylation and thus was independent of a change in allele-specific DNA methylation. This indicates that imprinting mechanism of XLsα transcript expression is not methylation dependent (at least mostly).
Correlation between allelic expression of imprinted genes including NESP55 and methylation of identified DMR has been previously reported in hESCs (Adewumi et al., 2007; Kim et al., 2007; Rugg-Gunn et al., 2007). An association between the variability observed in inter-cell line allelic expression status and the DMR DNA methylation was present in one study (Kim et al., 2007), but not others in which monoallelic NESP55 expression associated with maintenance in NESP DMR methylation in hESCs (Adewumi et al., 2007; Rugg-Gunn et al., 2007). We detected the maternally expressed imprinted transcript NESP55 in three hiPSC samples. In contrast to A/B and XLsα transcripts, expression was monoallelic in these hiPSCs. This stability in the monoallelic expression of NESP55 in hiPSCs raises the possibility that the process that maintains methylation at NESP DMR (or protect the unmethylated allele against aberrant methylation) might differ for NESP whose imprint is acquired postfertilization.
In summary, our studies indicate that (1) methylation at the GNAS locus DMRs is DMR and cell line specific, (2) methylation at the A/B DMR is correlated with A/B transcript expression, and (3) changes in allelic transcript expression can be independent of a change in allele-specific DNA methylation. The study of parental, reprogrammed, and differentiated cells should provide a model for studying the mechanisms controlling GNAS methylation, such as hydroxymethylation (Smallwood and Kelsey 2012); factors involved in transcript expression; and possibly mechanisms involved in the pathophysiology of PHP1B. This model will benefit from the possibility of differentiating PSCs in cell types in which Gsα is paternally silenced, such as BAT (Elabd et al., 2009) or proximal tubule (Montserrat et al., 2012) as shown for Angelman and Prader-Willi syndromes, two neurodevelopmental disorders of genomic imprinting (Chamberlain et al., 2010).
Experimental Procedures
Cell Lines
Human Embryonic Stem Cells
Four hESC lines were studied (Table 1). The hESC line VUB01 (XY; passages 80–100) was derived at the Vrije Universiteit Brussels, H9 (XX; passages 50–60; WA09) by the WiCell Institute, and RC9 (XY; passages 20–40) by Roslin Cells. The hESC line SA01 (XY; passages 30–50) is distributed by Cellartis.
Induced Pluripotent Stem Cells
hiPSC lines were obtained by reprogramming of two fibroblast lines (IMR90 and GM04603) obtained from the Coriell Institute either by retroviral or episomal methods as previously reported (Mangeot et al., 2011; Yu et al., 2009). For IMR90 (XX), two clones were studied: one obtained by retroviral methods (i90_c01) and one by episomal methods (i90_c17). For GM04603 (XY), a polyclonal (iPSC04603_polyF) and a clonal (iPSC04603_c27) line were studied (Table 1).
Pluripotent Stem Cell Culture
All PSC lines except RC9 were maintained on a feeder layer of mitomycin-C-inactivated murine embryonic fibroblast cells in a humidified 5% CO2 incubator at 37°C, in KnockOut (KO)-Dulbecco’s modified Eagle’s medium supplemented with 20% KO serum replacement, 1 mM L-glutamine, 1% nonessential amino acids, 0.1 mM β-mercaptoethanol, and 10 ng/ml basic (b)fibroblast growth factor (FGF) (all from Invitrogen). RC9 was maintained on a feeder-free system composed of CellStart matrix and StemPro medium supplemented with 10 ng/ml basic (b)FGF (Invitrogen). Cultures were fed daily and manually passaged every 5–7 days. Quality control of the PSCs was performed as suggested by the International Stem Cell Banking Initiative (2009) and showed normal karyotypes and expression of stemness markers.
Differentiation
Differentiation of PSCs into mesenchymal (MSC) (hESC lines: W09; hiPSC: i90_c01, i90_c17, and 4603_c27) and neural (NSC) (hESC lines: SA01 and VUB01; hiPSC: i90_c17 and 4603_c27) progenies was performed as previously reported (Benchoua and Peschanski 2013; Chambers et al., 2009; Giraud-Triboult et al., 2011; Guenou et al., 2009) (Table 1).
DNA Extraction
Genomic DNA was extracted from parental fibroblasts, undifferentiated and differentiated PSC lines (except one, NSC i90c01; Table 1), and from peripheral blood mononuclear cells from 20 control subjects using Gentra Kit extraction (QIAGEN) according to the manufacturer’s protocol. Informed written consent was obtained from the controls for genomic DNA (gDNA) analysis.
Quantification of DNA Methylation
Quantification of GNAS DMRs DNA methylation was performed by pyrosequencing as described in Supplemental Information and Maupetit-Méhouas et al. (2013). Six hundred nanograms of DNA were bisulfite converted (EZ DNA Methylation-Gold Kit; Zymo Research) according to the manufacturer’s protocol. One microliter of bisulfite-converted DNA was then amplified with specific primers for each DMR (A/B, XL, AS, and NESP). Pyrosequencing reactions were carried out on a PyroMark Q96 ID (QIAGEN) using either one sequencing primer for two distinct bisulfited PCR products (NESP, AS, and XL DMRs) or two different sequencing primers of one bisulfited PCR product (A/B DMR). Primer sequences and PCR conditions are presented in Table S1. The peak heights were determined using the provided software (PyroMark Q24 v2.0.6.20). Results are the mean ± SD of methylation measured at each cytosine for each DMR (NESP and XL 5 cytosines; AS: seven cytosines; A/B: eight cytosines). Replicate differences between <10% and 10% were considered inherent to the technique. In the rare cases of differences >10%, additional analysis pyrosequencing was performed. Five specific DNAs were included in each run and served as internal standards to ensure repeatability: unmethylated DNA (whole-genome amplified control DNA generated using the REPLI-g Mini Kit [QIAGEN]), fully methylated DNA (unmethylated DNA treated with SSI DNA methyltransferase [New England Biolabs]), one DNA prepared from a patient carrying an ∼1.7 Mb 20q paternal deletion comprising the GNAS locus (I. Garin, F.M. Elli, A.L., C.S., L. de Sanctis, P. Bordogna, A. Pereda, J.T.R. Clarke, C. Kannengiesser, R. Coutant, Y. Tenebaum-Rakover, International Clinical Group for PHP, EuroPHP Consortium, G.P. de Nanclares, and G. Mantovani, unpublished data), one DNA obtained from a patient with paternal GNAS duplication (Maupetit-Méhouas et al., 2013), and one control DNA.
Complementary DNA
Total RNA were extracted from 1 × 106 frozen cells pellet using Trizol reagent (Invitrogen) followed by a treatment in the RNeasy MinElute cleanup Kit (QIAGEN) according to the manufacturer’s protocol. Integrity of RNA was verified on 1.5% agarose gel electrophoresis. One microgram total RNA digested by DNaseI (Fermentas) was reverse transcribed using either hexamer random primers (for the transcript allelic expression analysis, see below) or oligo (dT) primers (for the quantitative real-time PCR, see below; RevertedAid H Minus First Strand cDNA synthesis Kit; Fermentas) according to the manufacturer’s protocol.
GNAS rs7121 Polymorphism Analysis in Genomic and cDNA
For gDNA, 200 ng samples were PCR amplified with forward and reverse primers localized in introns 3 and 5, respectively, covering exons 4 and 6 of GNAS (exons common to Gsα, XLsα, A/B, and NESP55) (Table S2). In order to allow analysis of C and T allele ratio by bidirectional pyrosequencing (see below), two PCRs were performed for each product, with either the forward or reverse primer being biotinylated. DNA PCR products were checked by migration on 1.5% agarose gel electrophoresis.
For cDNA, 1 μl of cDNA was amplified with a forward-specific primer for Gsα, XLsα, A/B, and NESP55 transcripts, localized in their respective exon 1, and a reverse primer common of all transcripts except GNAS-AS1 localized in exons 9 and 10 (Table S2). As for gDNA, two PCRs were performed for each product, with either the forward or reverse primer being biotinylated. cDNA PCR products were checked by migration on 1.5% agarose gel electrophoresis.
Heterozygosity at the GNAS Polymorphism rs7121 and Transcript Allelic Expression Analysis
C and T allele ratio was analyzed by bidirectional pyrosequencing using forward- and reverse-sequencing primers localized in exon 5 (Table S2) on PCR products obtained from gDNA and cDNA (described above). Ratios were calculated using the Pyrosequencing software. A similar approach has been reported (Klenke et al., 2011). DNA heterozygosity was defined for C (or T) allele ratio comprised between 40% and 60%.
Comparison of C or T allele ratio of cDNA samples were analyzed only from heterozygous gDNA samples for rs7121 (T393C). When detected, NESP55 transcript expression was monoallelic (>94% C in clone hiPSC90_c01 and >94% T in clone hiPSC 4603_c27 and polyF) (Figure S3; Results). Given that in “physiological” conditions, NESP55 is essentially maternally expressed, expression for GNAS transcripts is thus expressed as percent maternal allele ratio. Biallelic expression was defined for maternal allele ratio comprised between 40% and 60% and monoallelic expression for maternal allele ratio comprised between 90% and 100% and 0% and 10%. Examples of heterozygosity at the DNA level, biallelic (Gsα) and monoallelic (NESP55) transcript expressions quantified by pyrosequencing are shown on Figure S4.
Allelic expression was analyzed for parental IMR90 and GM04603 fibroblasts, all derived hiPSC (i90_c01, i90_c17, 4603_c27, and 4603_polyF), and for MSC (i90_c01, i90_c17, and 4603_c27) and neural (i90_c17 and 4603_c27) progenies (Table 1). Allelic expression was not studied in other samples either because the rs7121 polymorphism was present at the homozygous state (all hESCs) or no RNA was available.
Quantitative Real-Time PCR Analysis
Two qRT-PCR technologies were performed. The Sybr Green technology was used to detect Gsα, A/B, XLsα, NESP55, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) transcripts, and the Taqman technology was used to detect GNAS-AS1. Real-time PCR was carried out in a LightCycler LC480 system (Roche) to amplify Gsα, A/B, XLsα, NESP55, and GAPDH transcripts and an Applied Biosystems 7500 Fast Real-Time PCR System (Life Technologies) to amplify GNAS-AS1.
One microliter of cDNA (see above) was amplified using the LightCycler 480 SYBR Green I Master (Roche) for Gsα, XLsα, NESP55, and GAPDH transcripts, the Luminaris HiGreen qPCR Master Mix (Thermo Scientific) for A/B transcript, and the GoTaq Probe qPCR Master Mix (Promega) for GNAS-AS1. All PCR experiments were performed in triplicate. GNAS-AS1 mRNA level was quantified using a commercially Taqman assay (Hs.PT.58.25851302; Integrated DNA Technologies). Specific pairs of primers were used to amplify other transcripts (Eurofins; primers and PCR conditions available upon request).
Specificity of amplified qRT-PCR products was verified by performing a melting curve analysis at the end of amplification (Sybr Green technology only) and by migration on 1.5% agarose gel electrophoresis. Gene expression was normalized with human GAPDH as endogenous gene control using the formula NE = Ereference CTreference/EtargetCTtarget (Simon 2003), where NE is the normalized expression, E the efficiency of the PCR amplification for the reference (Ereference) and the target (Etarget), and CT the threshold cycle of the transcript detection. Primers used for Gsα amplification were previously described (Mariot et al., 2011). PCR amplification efficiency was comprised between 1.85 and 1.97.
Statistical Analysis
Methylation indices (MI) DMR at each DMR and dispersion of MI were compared by Kruskal-Wallis test followed by Dunn’s multiple comparison test. A p value < 0.05 was considered significant. To calculate the dispersion of MI, the mean of DMR MI for each group was calculated and the deviation of each sample to the mean calculated. Statistical analyses have been performed using Prism software.
Acknowledgments
This work was supported by recurrent INSERM funding. V.G. received a grant from the French Society of Pediatric Endocrinology and Diabetology.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information
References
- Adewumi O., Aflatoonian B., Ahrlund-Richter L., Amit M., Andrews P.W., Beighton G., Bello P.A., Benvenisty N., Berry L.S., Bevan S., International Stem Cell Initiative Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat. Biotechnol. 2007;25:803–816. doi: 10.1038/nbt1318. [DOI] [PubMed] [Google Scholar]
- Bastepe M., Jüppner H. GNAS locus and pseudohypoparathyroidism. Horm. Res. 2005;63:65–74. doi: 10.1159/000083895. [DOI] [PubMed] [Google Scholar]
- Benchoua A., Peschanski M. Pluripotent stem cells as a model to study non-coding RNAs function in human neurogenesis. Front Cell Neurosci. 2013;7:140. doi: 10.3389/fncel.2013.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain S.J., Chen P.F., Ng K.Y., Bourgois-Rocha F., Lemtiri-Chlieh F., Levine E.S., Lalande M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc. Natl. Acad. Sci. USA. 2010;107:17668–17673. doi: 10.1073/pnas.1004487107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers S.M., Fasano C.A., Papapetrou E.P., Tomishima M., Sadelain M., Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M., Wang J., Dickerson K.E., Kelleher J., Xie T., Gupta D., Lai E.W., Pacak K., Gavrilova O., Weinstein L.S. Central nervous system imprinting of the G protein G(s)alpha and its role in metabolic regulation. Cell Metab. 2009;9:548–555. doi: 10.1016/j.cmet.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chotalia M., Smallwood S.A., Ruf N., Dawson C., Lucifero D., Frontera M., James K., Dean W., Kelsey G. Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev. 2009;23:105–117. doi: 10.1101/gad.495809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombes C., Arnaud P., Gordon E., Dean W., Coar E.A., Williamson C.M., Feil R., Peters J., Kelsey G. Epigenetic properties and identification of an imprint mark in the Nesp-Gnasxl domain of the mouse Gnas imprinted locus. Mol. Cell. Biol. 2003;23:5475–5488. doi: 10.1128/MCB.23.16.5475-5488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane J.L., Shamblott M.J., Axelman J., Hsu S., Levine M.A., Germain-Lee E.L. Imprinting status of Galpha(s), NESP55, and XLalphas in cell cultures derived from human embryonic germ cells: GNAS imprinting in human embryonic germ cells. Clin. Transl. Sci. 2009;2:355–360. doi: 10.1111/j.1752-8062.2009.00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elabd C., Chiellini C., Carmona M., Galitzky J., Cochet O., Petersen R., Pénicaud L., Kristiansen K., Bouloumié A., Casteilla L. Human multipotent adipose-derived stem cells differentiate into functional brown adipocytes. Stem Cells. 2009;27:2753–2760. doi: 10.1002/stem.200. [DOI] [PubMed] [Google Scholar]
- Freson K., Izzi B., Labarque V., Van Helvoirt M., Thys C., Wittevrongel C., Bex M., Bouillon R., Godefroid N., Proesmans W. GNAS defects identified by stimulatory G protein alpha-subunit signalling studies in platelets. J. Clin. Endocrinol. Metab. 2008;93:4851–4859. doi: 10.1210/jc.2008-0883. [DOI] [PubMed] [Google Scholar]
- Fröhlich L.F., Mrakovcic M., Steinborn R., Chung U.I., Bastepe M., Jüppner H. Targeted deletion of the Nesp55 DMR defines another Gnas imprinting control region and provides a mouse model of autosomal dominant PHP-Ib. Proc. Natl. Acad. Sci. USA. 2010;107:9275–9280. doi: 10.1073/pnas.0910224107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost J., Monk D., Moschidou D., Guillot P.V., Stanier P., Minger S.L., Fisk N.M., Moore H.D., Moore G.E. The effects of culture on genomic imprinting profiles in human embryonic and fetal mesenchymal stem cells. Epigenetics. 2011;6:52–62. doi: 10.4161/epi.6.1.13361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardot M., Feil R., Llères D. Epigenetic deregulation of genomic imprinting in humans: causal mechanisms and clinical implications. Epigenomics. 2013;5:715–728. doi: 10.2217/epi.13.66. [DOI] [PubMed] [Google Scholar]
- Giraud-Triboult K., Rochon-Beaucourt C., Nissan X., Champon B., Aubert S., Piétu G. Combined mRNA and microRNA profiling reveals that miR-148a and miR-20b control human mesenchymal stem cell phenotype via EPAS1. Physiol. Genomics. 2011;43:77–86. doi: 10.1152/physiolgenomics.00077.2010. [DOI] [PubMed] [Google Scholar]
- Gkountela S., Li Z., Vincent J.J., Zhang K.X., Chen A., Pellegrini M., Clark A.T. The ontogeny of cKIT+ human primordial germ cells proves to be a resource for human germ line reprogramming, imprint erasure and in vitro differentiation. Nat. Cell Biol. 2013;15:113–122. doi: 10.1038/ncb2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenou H., Nissan X., Larcher F., Feteira J., Lemaitre G., Saidani M., Del Rio M., Barrault C.C., Bernard F.X., Peschanski M. Human embryonic stem-cell derivatives for full reconstruction of the pluristratified epidermis: a preclinical study. Lancet. 2009;374:1745–1753. doi: 10.1016/S0140-6736(09)61496-3. [DOI] [PubMed] [Google Scholar]
- Hayward B.E., Kamiya M., Strain L., Moran V., Campbell R., Hayashizaki Y., Bonthron D.T. The human GNAS1 gene is imprinted and encodes distinct paternally and biallelically expressed G proteins. Proc. Natl. Acad. Sci. USA. 1998;95:10038–10043. doi: 10.1073/pnas.95.17.10038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward B.E., Moran V., Strain L., Bonthron D.T. Bidirectional imprinting of a single gene: GNAS1 encodes maternally, paternally, and biallelically derived proteins. Proc. Natl. Acad. Sci. USA. 1998;95:15475–15480. doi: 10.1073/pnas.95.26.15475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward B.E., Barlier A., Korbonits M., Grossman A.B., Jacquet P., Enjalbert A., Bonthron D.T. Imprinting of the G(s)alpha gene GNAS1 in the pathogenesis of acromegaly. J. Clin. Invest. 2001;107:R31–R36. doi: 10.1172/JCI11887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntriss J., Woodfine K., Huddleston J.E., Murrell A., Rutherford A.J., Elder K., Khan A.A., Hemmings K., Picton H. Quantitative analysis of DNA methylation of imprinted genes in single human blastocysts by pyrosequencing. Fertil. Steril. 2011;95:2564–2567. doi: 10.1016/j.fertnstert.2011.04.035. e1–e8. [DOI] [PubMed] [Google Scholar]
- International Stem Cell Banking Initiative Consensus guidance for banking and supply of human embryonic stem cell lines for research purposes. Stem Cell Rev. 2009;5:301–314. doi: 10.1007/s12015-009-9085-x. [DOI] [PubMed] [Google Scholar]
- Kim K.P., Thurston A., Mummery C., Ward-van Oostwaard D., Priddle H., Allegrucci C., Denning C., Young L. Gene-specific vulnerability to imprinting variability in human embryonic stem cell lines. Genome Res. 2007;17:1731–1742. doi: 10.1101/gr.6609207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenke S., Siffert W., Frey U.H. A novel aspect of GNAS imprinting: higher maternal expression of Gαs in human lymphoblasts, peripheral blood mononuclear cells, mammary adipose tissue, and heart. Mol. Cell. Endocrinol. 2011;341:63–70. doi: 10.1016/j.mce.2011.05.032. [DOI] [PubMed] [Google Scholar]
- Laird D.J. Humans put their eggs in more than one basket. Nat. Cell Biol. 2013;15:13–15. doi: 10.1038/ncb2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M.A. An update on the clinical and molecular characteristics of pseudohypoparathyroidism. Curr. Opin. Endocrinol. Diabetes Obes. 2012;19:443–451. doi: 10.1097/MED.0b013e32835a255c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Sasaki H. Genomic imprinting in mammals: its life cycle, molecular mechanisms and reprogramming. Cell Res. 2011;21:466–473. doi: 10.1038/cr.2011.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linglart A., Maupetit-Méhouas S., Silve C. GNAS-related loss-of-function disorders and the role of imprinting. Horm Res Paediatr. 2013;79:119–129. doi: 10.1159/000348516. [DOI] [PubMed] [Google Scholar]
- Lister R., Pelizzola M., Kida Y.S., Hawkins R.D., Nery J.R., Hon G., Antosiewicz-Bourget J., O’Malley R., Castanon R., Klugman S. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Yu S., Litman D., Chen W., Weinstein L.S. Identification of a methylation imprint mark within the mouse Gnas locus. Mol. Cell. Biol. 2000;20:5808–5817. doi: 10.1128/mcb.20.16.5808-5817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund R.J., Närvä E., Lahesmaa R. Genetic and epigenetic stability of human pluripotent stem cells. Nat. Rev. Genet. 2012;13:732–744. doi: 10.1038/nrg3271. [DOI] [PubMed] [Google Scholar]
- MacDonald W.A., Mann M.R. Epigenetic regulation of genomic imprinting from germ line to preimplantation. Mol. Reprod. Dev. 2014;81:126–140. doi: 10.1002/mrd.22220. [DOI] [PubMed] [Google Scholar]
- Mangeot P.E., Dollet S., Girard M., Ciancia C., Joly S., Peschanski M., Lotteau V. Protein transfer into human cells by VSV-G-induced nanovesicles. Mol. Ther. 2011;19:1656–1666. doi: 10.1038/mt.2011.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani G., Ballare E., Giammona E., Beck-Peccoz P., Spada A. The gsalpha gene: predominant maternal origin of transcription in human thyroid gland and gonads. J. Clin. Endocrinol. Metab. 2002;87:4736–4740. doi: 10.1210/jc.2002-020183. [DOI] [PubMed] [Google Scholar]
- Mantovani G., Elli F.M., Spada A. GNAS epigenetic defects and pseudohypoparathyroidism: time for a new classification? Horm. Metab. Res. 2012;44:716–723. doi: 10.1055/s-0032-1314842. [DOI] [PubMed] [Google Scholar]
- Mariot V., Wu J.Y., Aydin C., Mantovani G., Mahon M.J., Linglart A., Bastepe M. Potent constitutive cyclic AMP-generating activity of XLαs implicates this imprinted GNAS product in the pathogenesis of McCune-Albright syndrome and fibrous dysplasia of bone. Bone. 2011;48:312–320. doi: 10.1016/j.bone.2010.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maupetit-Méhouas S., Mariot V., Reynès C., Bertrand G., Feillet F., Carel J.C., Simon D., Bihan H., Gajdos V., Devouge E. Quantification of the methylation at the GNAS locus identifies subtypes of sporadic pseudohypoparathyroidism type Ib. J. Med. Genet. 2011;48:55–63. doi: 10.1136/jmg.2010.081356. [DOI] [PubMed] [Google Scholar]
- Maupetit-Méhouas S., Azzi S., Steunou V., Sakakini N., Silve C., Reynes C., Perez de Nanclares G., Keren B., Chantot S., Barlier A. Simultaneous hyper- and hypomethylation at imprinted loci in a subset of patients with GNAS epimutations underlies a complex and different mechanism of multilocus methylation defect in pseudohypoparathyroidism type 1b. Hum. Mutat. 2013;34:1172–1180. doi: 10.1002/humu.22352. [DOI] [PubMed] [Google Scholar]
- Montserrat N., Ramírez-Bajo M.J., Xia Y., Sancho-Martinez I., Moya-Rull D., Miquel-Serra L., Yang S., Nivet E., Cortina C., González F. Generation of induced pluripotent stem cells from human renal proximal tubular cells with only two transcription factors, OCT4 and SOX2. J. Biol. Chem. 2012;287:24131–24138. doi: 10.1074/jbc.M112.350413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazor K.L., Altun G., Lynch C., Tran H., Harness J.V., Slavin I., Garitaonandia I., Müller F.J., Wang Y.C., Boscolo F.S. Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell. 2012;10:620–634. doi: 10.1016/j.stem.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagge A., Kelsey G. Imprinting the Gnas locus. Cytogenet. Genome Res. 2006;113:178–187. doi: 10.1159/000090830. [DOI] [PubMed] [Google Scholar]
- Reik W., Dean W., Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- Rugg-Gunn P.J., Ferguson-Smith A.C., Pedersen R.A. Epigenetic status of human embryonic stem cells. Nat. Genet. 2005;37:585–587. doi: 10.1038/ng1556. [DOI] [PubMed] [Google Scholar]
- Rugg-Gunn P.J., Ferguson-Smith A.C., Pedersen R.A. Human embryonic stem cells as a model for studying epigenetic regulation during early development. Cell Cycle. 2005;4:1323–1326. doi: 10.4161/cc.4.10.2076. [DOI] [PubMed] [Google Scholar]
- Rugg-Gunn P.J., Ferguson-Smith A.C., Pedersen R.A. Status of genomic imprinting in human embryonic stem cells as revealed by a large cohort of independently derived and maintained lines. Hum. Mol. Genet. 2007;16 Spec No. 2:R243–R251. doi: 10.1093/hmg/ddm245. [DOI] [PubMed] [Google Scholar]
- Sabour D., Schöler H.R. Reprogramming and the mammalian germline: the Weismann barrier revisited. Curr. Opin. Cell Biol. 2012;24:716–723. doi: 10.1016/j.ceb.2012.08.006. [DOI] [PubMed] [Google Scholar]
- Simon P. Q-Gene: processing quantitative real-time RT-PCR data. Bioinformatics. 2003;19:1439–1440. doi: 10.1093/bioinformatics/btg157. [DOI] [PubMed] [Google Scholar]
- Smallwood S.A., Kelsey G. De novo DNA methylation: a germ cell perspective. Trends Genet. 2012;28:33–42. doi: 10.1016/j.tig.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Takikawa S., Ray C., Wang X., Shamis Y., Wu T.Y., Li X. Genomic imprinting is variably lost during reprogramming of mouse iPS cells. Stem Cell Res. (Amst.) 2013;11:861–873. doi: 10.1016/j.scr.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin S.C., Kim K. Generating pluripotent stem cells: differential epigenetic changes during cellular reprogramming. FEBS Lett. 2012;586:2874–2881. doi: 10.1016/j.febslet.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turan S., Fernandez-Rebollo E., Aydin C., Zoto T., Reyes M., Bounoutas G., Chen M., Weinstein L.S., Erben R.G., Marshansky V., Bastepe M. Postnatal establishment of allelic Gαs silencing as a plausible explanation for delayed onset of parathyroid hormone resistance owing to heterozygous Gαs disruption. J. Bone Miner. Res. 2014;29:749–760. doi: 10.1002/jbmr.2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein L.S., Yu S., Warner D.R., Liu J. Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr. Rev. 2001;22:675–705. doi: 10.1210/edrv.22.5.0439. [DOI] [PubMed] [Google Scholar]
- Weinstein L.S., Xie T., Zhang Q.H., Chen M. Studies of the regulation and function of the Gs alpha gene Gnas using gene targeting technology. Pharmacol. Ther. 2007;115:271–291. doi: 10.1016/j.pharmthera.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson C.M., Ball S.T., Nottingham W.T., Skinner J.A., Plagge A., Turner M.D., Powles N., Hough T., Papworth D., Fraser W.D. Acis-acting control region is required exclusively for the tissue-specific imprinting of Gnas. Nat. Genet. 2004;36:894–899. doi: 10.1038/ng1398. [DOI] [PubMed] [Google Scholar]
- Yu J., Hu K., Smuga-Otto K., Tian S., Stewart R., Slukvin I.I., Thomson J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H., Radeva G., McCann J.A., Hendy G.N., Goodyer C.G. Galphas transcripts are biallelically expressed in the human kidney cortex: implications for pseudohypoparathyroidism type 1b. J. Clin. Endocrinol. Metab. 2001;86:4627–4629. doi: 10.1210/jcem.86.10.7940. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.