

Abstract
Deleterious germline variants in CDKN2A account for around 40% of familial melanoma cases1, while rare variants in CDK4, BRCA2, BAP1, and the promoter of TERT, have also been linked to the disease2-5. Here we set out to identify novel high-penetrance susceptibility genes in unexplained cases by sequencing 184 melanoma patients from 105 pedigrees recruited in the United Kingdom, the Netherlands, and Australia that were negative for variants in known predisposition genes. We identify families where melanoma co-segregates with loss-of-function variants in the protection of telomeres 1 (POT1) gene, a proportion of members presenting with an early age of onset and multiple primaries. We show that these variants either affect POT1 mRNA splicing or alter key residues in the highly conserved oligonucleotide-/oligosaccharide-binding (OB) domains of POT1, disrupting protein-telomere binding, leading to increased telomere length. Thus, POT1 variants predispose to melanoma formation via a direct effect on telomeres.
Cutaneous malignant melanoma accounts for around 75% of skin cancer deaths with around 10% of patients having one first-degree relative, and 1% of patients having two or more first-degree relatives who have had a diagnosis of this disease6. As only around half of cases of familial melanoma can be attributed to variants in known predisposition genes, principally CDKN2A, a significant proportion of genetic risk for melanoma remains elusive. To identify new mediators of germline genetic risk, we sequenced 184 melanoma cases from 105 pedigrees. The patients sequenced came from pedigrees with between two and eleven cases of melanoma (169 cases), or were single cases that presented with either multiple primary melanoma (MPM), multiple primary cancers (one of which was melanoma), and/or an early age of onset (<4th decade) (15 cases) (see Online Methods, Supplementary Tables 1-2). Sequencing of two-case pedigrees was preferentially performed for those families enriched with cases of MPM. All cases were previously found to be negative for pathogenetic variants in CDKN2A and CDK4.
After performing exome (168 samples) or whole genome (16 samples) sequencing we called and filtered variants, keeping only those predicted to affect protein structure or function. Importantly, no known pathogenetic variants in BAP1 or BRCA2 were found, and all samples were re-confirmed as being CDKN2A and CDK4 wildtype. We further filtered the calls taking forward only non-polymorphic variants (Online Methods). Where we had sequenced more than one member of a pedigree we retained only co-segregating variants, whereas all variants were considered from pedigrees in which only one affected member was sequenced. As a result, a total of 23,051 variants remained for downstream analysis. Focusing on the 28 pedigrees for which sequence data was available for three or more members, we found 320 genes carrying co-segregating protein-changing variants (Supplementary Table 3). Of particular interest were five genes that showed novel variants in more than one of these pedigrees (POT1, MPDZ, ACD, SMG1 and NEK10). Analysis of the missense and disruptive variants (nonsense, splice acceptor/donor and frameshift) in these genes led us to identify a 5-case pedigree (id: UF20) carrying a Tyr89Cys change (GRCh37 chr. 7: g. 124503684T>C) in the highly conserved N-terminal oligonucleotide-/oligosaccharide-binding (OB) domain of the POT1 (protection of telomeres 1) gene product7,8, and a 6-case family (id: AF1) carrying a splice acceptor variant in intron 17-18 (g.124465412C>T) of POT1, which was scored as deleterious by the MaxEntScan algorithm9, and shown to affect transcript splicing by RT-PCR and sequencing (Fig. 1, Table 1, Online Methods, Supplementary Fig. 1a-c&2, Supplementary Tables 4-5). Scrutiny of the remaining sequenced pedigrees, including those in which only one member had been sequenced, identified two additional individuals from distinct families with non-synonymous OB domain changes in POT1 (id: UF31, Gln94Glu, g. 124503670G>C and id: UF23, Arg273Leu, g. 124493077C>A) (Fig 1, Table 1, Supplementary Fig. 1d-e&3, Supplementary Tables 1, 4-5). Remarkably, the POT1 codon encoding the Gln94 residue affected in pedigree UF31 has been found to be a target for recurrent somatic mutation (Gln94Arg) in chronic lymphocytic leukemia (CLL), where ~5% of cases carry POT1 mutations that cluster in the OB domains10.
Fig. 1. Rare variants in POT1 found in familial melanoma pedigrees.

a) We identified four pedigrees carrying deleterious variants in the protection of telomeres 1 (POT1) gene. Shown are a 5-(UF20) and a 6-(AF1) member pedigree carrying the disruptive Tyr89Cys OB domain variant and a splice acceptor variant, respectively. Please note that pedigrees have been adjusted to protect the identity of the families without a loss of scientific integrity. Genotypes for all samples available for testing are shown in blue. CMM; cutaneous malignant melanoma. CLL; chronic lymphocytic leukemia. Sp.A; splice acceptor variant. Circles represent females; squares represent males; diamonds represent undisclosed gender. The patients that were sequenced have a red outline. All melanomas were confirmed by histological analysis with the exception of two cases (*). The number of primary melanomas in each patient is indicated; age of onset is shown in parentheses. b) Highly conserved residues of POT1 are mutated in familial melanoma. Shown are the positions of the variants identified on the POT1 protein (top panel), and on an amino acid alignment (missense variants, bottom panel).
Table 1. POT1 variants identified in familial melanoma pedigrees.
| Pedigree | Num. cases in pedigree | Num. of carriers / num. of tested cases | Genomic location | HGVS name* | Exon | Amino acid change | Variant type | Bioinformatic prediction tools | ||
|---|---|---|---|---|---|---|---|---|---|---|
| SIFT23 | PolyPhen 224 | CAROL25 | ||||||||
| UF20 | 5 | 4/4 | g.124503684T>C | c.266A>G | 8 | Tyr89Cys | Missense | Deleterious | Probably damaging | Deleterious |
| AF1 | 6 | 3/3 | g.124465412C>T | c.1687-1G>A | - | - | Splice acceptor (intronic, between ex. 17-18) | - | - | - |
| UF31 | 2 | 1/1 | g.124503670G>C | c.280C>G | 8 | Gln94Glu | Missense | Tolerated | Probably damaging | Deleterious |
| UF23 | 2 | 1/2** | g.124493077C>A | c.818G>T | 10 | Arg273Leu† | Missense | Deleterious | Probably damaging | Deleterious |
The reference transcript, taken from the Ensembl database (release 70) is POT1-001 (ENST00000357628).
A second case within this pedigree had a different clinical presentation (solitary melanoma in situ) in the 6th decade and did not carry the Arg273Leu variant.
This variant was also detected in a melanoma case from population-based case-control series that presented with MPMs and an early age of onset (see Online Methods).
To gather further evidence for an association between POT1 variants and familial melanoma, we compared the representation of POT1 variants in our familial melanoma cases with controls. Importantly, the presence of POT1 variants in 4/105 melanoma families represented a statistically significant enrichment of variants (P = 0.016, excluding a discovery pedigree) compared with a control dataset of 520 exomes from individuals sequenced as part of the UK10K project (see URLs) in which we found only one missense variant located outside the OB domains of POT1 (Online Methods, Supplementary Fig. 4, Supplementary Table 6). Furthermore, none of the four POT1 variants found in our melanoma pedigrees were found by genotyping 2,402 additional population-matched controls (Online Methods, Supplementary Table 6). Interestingly, genotyping of these positions across a matched cohort of 1,739 population-based sporadic melanoma cases identified one individual who carried the POT1 Arg273Leu variant, who presented with early-onset MPM similar to the presenting phenotype of the familial cases (Online Methods).
All of the missense variants identified in POT1 disrupt amino acids completely conserved throughout Eutherian evolution (Fig. 1b) and that are more evolutionarily conserved than the average for other OB domain residues (Online Methods). Co-segregation analysis of POT1 variants identified through genome/exome sequencing, or by targeted PCR re-sequencing of additional members, revealed that all nine POT1 variant carriers from the familial cohort had developed melanoma, presenting with one primary (four cases) to eight melanomas, from 25 to 80 years of age (Fig. 1, Supplementary Fig. 3). One variant carrier from these familial cases also developed breast cancer at 65 years and another small cell lung cancer at 50 years (pedigree UF20). Other malignancies in untested first/second degree relatives of variant carriers included melanoma (pedigrees UF20 and UF31), endometrial cancer (pedigree UF20) and brain tumors (pedigrees UF20 and UF23). Intriguingly, the pedigree with the splice acceptor variant (AF1) had a patient with a history of melanoma and CLL, in keeping with a role for POT1 in CLL development10,11. Collectively, these data suggest a possible role for germline POT1 variants in susceptibility to a range of cancers in addition to melanoma.
To test whether the effect of the identified missense variants might disrupt telomere binding similar to that observed for somatic mutations found in CLL, we examined the structure of POT1 protein bound to a telomere-like polynucleotide (dTUdAdGdGdGdTdTdAdG) (PDB 3KJP)12,13. According to this model, all three mutated residues (Tyr89, Gln94 and Arg273) are amongst 24 residues located in close proximity (<3.5Å) to the telomeric polynucleotide10 (Fig. 2a). Arg273 interacts with the oxygen at position 2 of the telomeric dT7, whereas Gln94 and Tyr89 both interact with the G deoxynucleotide at position 4. Therefore, as described for the somatic mutations in CLL, the POT1 variants we identified are expected to weaken or abolish the interaction between POT1 and telomeres. Analysis of nucleotides coding for these 24 OB domain residues identified one non-synonymous change in 6,498 control exomes14 compared with three in 105 melanoma families, emphasizing a highly significant enrichment of variants in the melanoma cohort (P = 1.54×10−5) (Online Methods, Supplementary Tables 6-7).
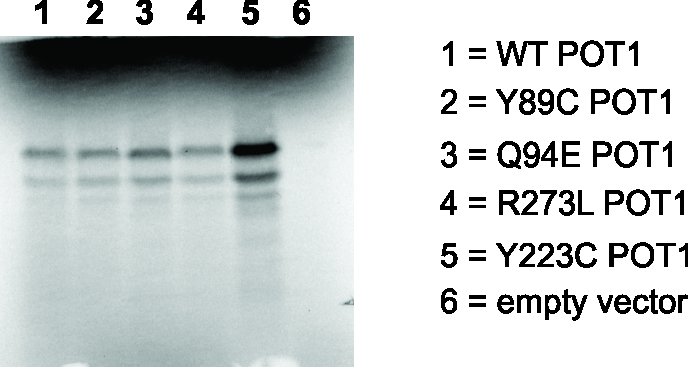
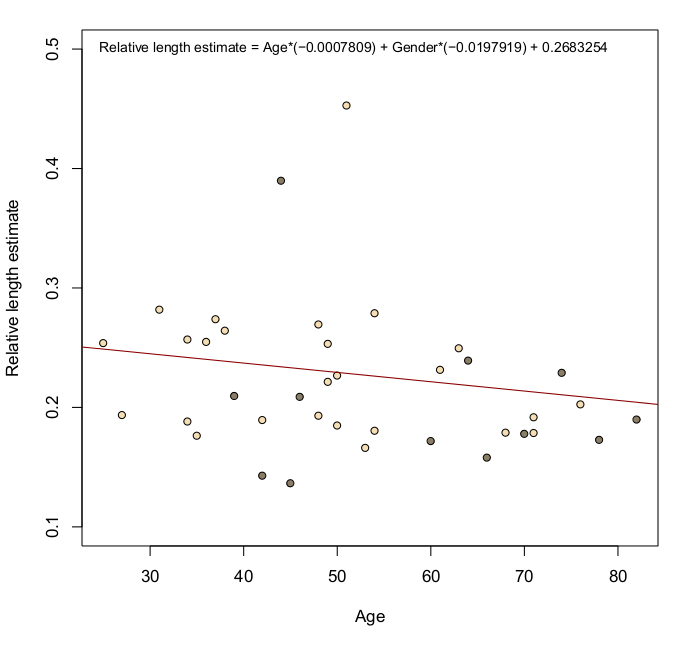
Fig. 2. Missense variants in POT1 disrupt the interaction between POT1 and single-stranded DNA and lead to elongated telomeres.

a) Shown is the location of the POT1 residues Tyr89, Gln94 and Arg273 in the N-terminal two oligonucleotide-/oligosaccharide-binding (OB) domains, in green. A telomere-like polynucleotide sequence is shown in orange. Interacting nucleotides in the telomeric sequence are labeled in gray. All three substitutions are predicted to disrupt the association of POT1 with telomeres. b) Mutant Tyr89Cys, Gln94Glu and Arg273Leu POT1 proteins are unable to bind telomeric (TTAGGG)3 sequences as revealed by an electromobility shift assay. The Tyr223Cys POT1 mutant was used as a positive control representing a known disruptive mutation10. c) Calculation of telomere length from exome sequence data. The method used is analogous to the one described in Ref. 15. Adjusted relative telomere lengths for the three sequenced members of pedigree UF20 are shown alongside the mean telomere length of 38 (all) other melanoma cases who were sequenced alongside, but were wildtype for POT1. The error bar indicates one standard deviation. A Wilcoxon rank sum test was performed comparing the telomere length of the 3 Tyr89Cys cases to the 38 non-carrier controls. d) PCR-based estimate of telomere length. Adjusted mean negative ΔCt values, which correlate positively with telomere length, for POT1 missense variant carriers and non-carrier family controls are shown against a distribution of values from 252 melanoma cases recruited from the Leeds Melanoma cohort that are wildtype at the abovementioned positions (Online Methods). All measurements have been adjusted for age at blood draw and gender. The black line represents a Gaussian kernel density estimate for this set using Silverman’s “rule-of-thumb”22 for bandwidth smoothing. Orange dots represent members of pedigree UF20; pink, UF31; blue, UF23; red, individual CT1663 from the Leeds Melanoma Case-control study carrying the Arg273Leu variant (Supplementary Table 5). The number of biological replicates for each case ranged from 1 to 4, each with two technical replicates for the POT1 missense variant carriers and non-carrier family controls. Two technical replicates were performed for the 252 POT1 non-carrier cases. Error bars indicate the standard error of the mean.
To further test that the OB variants we identified disrupt POT1 function, we assessed the ability of in vitro-translated POT1 Tyr89Cys, Gln94Glu and Arg273Leu proteins to bind to (TTAGGG)3 sequences with electromobility shift assays revealing a complete abolition of mutant POT1-DNA complex formation (Fig. 2b, Online Methods, Supplementary Fig. 5). Importantly, the POT1 Tyr36Asn and Tyr223Cys mutations recently described in CLL (ref. 10), which appear to be functionally analogous to the variants we describe here, promote uncapping of telomeres, telomere length extension, and chromosomal aberrations, and thereby promote tumorigenesis10.
Given the role of POT1 in telomere length maintenance we next asked whether melanoma cases from POT1 pedigrees had telomere lengths that differed from non-carrier melanoma cases. Using exome sequence data from 41 cases, including three members of pedigree UF20, we estimated the telomere length of each patient by counting TTAGGG repeats (ref. 15). This analysis revealed that all three members of pedigree UF20 had telomeres that were significantly longer than POT1 wild-type melanoma cases (P < 0.0002, Fig. 2c, Supplementary Fig. 6, Online Methods). This result was confirmed by telomere length PCRs, which also showed longer telomeres for patients carrying the Gln94Glu and Arg273Leu variants compared with melanoma cases without POT1 variants (P = 3.621×10−5) (Fig. 2d, Supplementary Fig. 7, Online Methods). Thus, missense variants in the OB domains of POT1 not only abolish telomere binding, they are also associated with increased telomere length, a key factor influencing melanoma risk16. Importantly, OB domain variants that disrupt the interaction of POT1 with telomeric ssDNA are thought to function as dominant-negative alleles10,17, yet as we show here are compatible with life, suggesting additional somatic events are required to promote tumorigenesis.
The identification of POT1 mutations in CLL, and the probable susceptibility of our POT1-mutated familial melanoma pedigrees to other tumor types, suggested that POT1 might play a more general role in tumorigenesis. To investigate this we examined pan-cancer data from the COSMIC18 and IntOGen19 databases (TCGA/ICGC data) from 14 cancer types and found that somatic POT1 mutations were more likely to be missense (P < 0.03), to hit residues in close proximity to DNA (P < 0.02), and to have a high functional bias (P < 0.03) (Online Methods). These results suggest that although rare, somatic POT1 mutations may drive tumorigenesis across multiple histologies.
Here we describe germline variants in the gene encoding the telomere-associated protein POT1 in almost 4% of CDKN2A/CDK4-negative familial melanoma pedigrees and in 2/34 (5.8%) 5+ case pedigrees, making POT1 the second most frequently mutated high-penetrance melanoma gene reported to date. This work, and a companion report in this edition of the journal describing germline POT1 mutations in Italian, French and American families20, together with the recently described TERT promoter variant2, significantly extend our understanding of a novel mechanism predisposing to the development of familial melanoma. Since the dysregulation of telomere protection by POT1 has recently been identified as a target for potential therapeutic intervention21, it may be possible that early identification of families with POT1 variants may facilitate better management of their disease in the future.
Online Methods
Patient samples and DNA extraction
The families included in this study were recruited to a UK Familial Melanoma Study directed by the Section of Epidemiology and Biostatistics, University of Leeds, Leeds, U.K; The Leiden University Medical Center, The Netherlands; and the Queensland Familial Melanoma Project (QFMP)26. Informed consent was obtained under MREC: 99/3/045 (UK Familial Melanoma Study cases), Protocol P00.117-gk2/WK/ib (Leiden cases), and from the Human Research Ethics Committee of the QIMR Berghofer Medical Research Institute for QFMP cases. Genomic DNA was extracted from peripheral blood using standard methods.
Pedigrees and clinical presentation
Supplementary Table 1 lists the number of pedigrees in this study by Institute and by sequencing centre. Supplementary Table 2 lists all pedigrees, the number of cases of melanoma in each pedigree, and the number of cases that were whole-genome sequenced. To help anonymize the pedigrees ages were rounded up to the nearest 5-year tier.
Sequence alignment and analysis
DNA libraries were prepared from 5 μg of genomic DNA and exonic regions captured with the Agilent SureSelect Target Enrichment System, 50 Mb Human All Exon Kit. Whole genome libraries were prepared using the standard Illumina library prep protocol. Paired-end reads of between 75-100bp were generated on the HiSeq2000 platform and mapped to the reference GRCh37/ hg19 human genome assembly using BWA27. Reads were filtered for duplicates using Picard28, recalibrated, and re-aligned around indels using the GATK package29 (Familial Melanoma Study and Leiden data). Exome capture and sequencing resulted in an average of 84% of target bases being covered ≥ 10x across the autosomes and sex chromosomes. Whole genomes were sequenced to at least 27x mapped coverage. Data for POT1 variant carriers has been released: EGAS00001000017. Variants were then called using Samtools mpileup30 and filtered for quality. The variant collection was then filtered to remove positions found in Phase 1 of the 1000 Genomes Project October 2011 release31 and the dbSNP 135 release32. Variants were also filtered for positions found in a collection of 805 in-house control exomes. Only variants in exonic regions, as defined in Ensembl release 70, were taken forward for analysis. Positions resulting in protein-altering changes were then identified using the Ensembl Variant Effect Predictor, version 2.8 (Ensembl release 70)33, a combination of VCFTools34, and custom scripts. Variants marked as ‘transcript_ablation’, ‘splice_donor_variant’, ‘splice_acceptor_variant’, ‘stop_gained’, ‘frameshift_variant’, ‘stop_lost’, ‘initiator_codon_variant’, ‘inframe_insertion’, ‘inframe_deletion’, ‘missense_variant’, ‘transcript_amplification’, ‘splice_region_variant’, ‘incomplete_terminal_codon_variant’, ‘mature_miRNA_variant’, ‘TFBS_ablation’, ‘TFBS_amplification’, ‘TF_binding_site_variant’, ‘feature_elongation’ and ‘feature_truncation’ were kept for further analyses. We retained only those variants found in all affected cases of a single pedigree (in order to reduce the impact of systematic mapping errors). An identity-by-descent analysis was performed to confirm that patients from different pedigrees within the study were not related.
Genes with co-segregating variants from the 28 pedigrees for which we had sequence data for three or more members are shown in Supplementary Table 3 with their Gene Ontology (GO) terms. Variants within POT1 identified from this analysis were confirmed by capillary sequencing (Supplementary Fig. 1a,b,d,e).
MaxEntScan scoring of splice site acceptor variant (g.124465412C>T)
We used the MaxEntScan algorithm9 which yielded a score of −3.22 for the mutated splice acceptor site (g.124465412C>T) and 5.53 for the wild-type splice site sequence. To put these values in context we retrieved 10,000 splice acceptor sites from random genes (choosing always the second exon) and obtained a distribution of their scores. The splice acceptor variant lowered the score of the wild-type sequence from the 9.2th to the 0.57th percentile when compared to the score distribution of real splice acceptors (Supplementary Figure 2), and is thus predicted to be highly deleterious.
RT-PCR sequencing of the POT1 product in two individuals carrying the splice acceptor variant (g.124465412C>T)
RNA extracted from the whole blood of two splice acceptor variant carriers was converted to cDNA using Superscript III Reverse Transcriptase (Invitrogen). RT-PCR was then performed to confirm that POT1 g.124465412C>T (ENST00000357628) was indeed disruptive to splicing. M13-tagged forward primer and reverse primer were designed to flank the splice region (GTA AAA CGA CGG CCA GTG TGG CAG AAG CAC TGG GTA T and GCT CAA ACA GGG AAG GTG AG respectively). The product was visualised on a 3% Nusieve Agarose gel and the sequence was verified using standard Sanger sequencing methods. Supplementary Figure 1c shows the sequencing traces for one control and one carrier sample.
Frequency of POT1 variants in a control dataset
All exomes from the UK10K sequencing project (REL 14/03/12) cohorts UK10K_NEURO_MUIR, UK10K_NEURO_IOP_COLLIER and UK10K_NEURO_ABERDEEN (see URLs) were selected as controls because these exomes were captured with the same Agilent SureSelect exome probes as those used for the melanoma cases described above, and were also sequenced on the Illumina HiSeq2000 platform (N=546). One exome was discarded at random from each of three pairs of relatives within this set (N=3). UK10K exomes were aligned, filtered for duplicates, recalibrated, and re-aligned around indels as described above. Variants were then called and filtered for base quality with the same tools and parameters as the melanoma cohort. For 104/105 families we had exome data for at least one individual in the pedigree; for one melanoma family we used whole genome sequence.
To ensure that the controls were ethnically matched to the melanoma cohort, a principal component analysis (PCA) was performed using 1,092 individuals across 14 populations from the 1000 Genomes Project Phase 1 dataset31. A subset of high quality variant positions (quality score > 10, minimum mapping quality > 10, strand bias p-value > 0.0001, end distance bias P-value > 0.0001) that were common to the melanoma cohort and the UK10K controls, as well as the 1000 Genomes dataset, were taken forward for analysis. SNPs with a minor allele frequency < 0.05 or that were in linkage disequilibrium with another SNP (pairwise r2 > 0.1) in the 1000 Genomes dataset, or that had a Hardy-Weinberg P-value < 10−5 in the UK10K controls were excluded. After filtering, 7,196 SNPs remained which were spread across all autosomes. The first ten principal components were estimated using the 1000 Genomes individuals, and then projected onto the melanoma cohort samples and UK10K controls using EIGENSTRAT35.
Controls lying greater than two standard deviations from the mean PC1 or PC2 scores, calculated using only European individuals in the 1000 Genomes dataset, were removed from subsequent analyses (N=20). Supplementary Figure 4 shows this analysis. An identity-by-descent (IBD) analysis was performed to ensure that members of the UK10K cohort were not related. This was performed using the PLINK toolset36, and the same set of variants that were used for the PCA (described above). For each pair of individuals with an estimated IBD > 0.2, one of them was removed at random (N=3). This left 520 exomes for comparison against the melanoma cohort.
Variants in this collection of 520 UK10K control exomes were then filtered as described above (keeping positions with exonic coordinates +/− 100bp and removing all variants in the Phase 1 of the 1000 Genomes Project October 2011 release31, the dbSNP 138 release32,37 and a collection of 805 control exomes). Because we used an updated version of dbSNP for this step, we also checked that the POT1 variants found in this study passed this filter. Consequences were then predicted and filtered as described above. From this analysis we identified one individual in 520 control exomes that a carried rare, potentially disruptive variant in POT1 (a missense variant located outside the OB domains). A two-tailed Fisher’s exact test was performed comparing the 3/104 melanoma families, excluding a discovery pedigree, to the 1/520 controls carrying rare variants in POT1, yielding a P-value of 0.016.
Genotyping in a population-based case-control series (TaqMan)
The Tyr89Cys, Gln94Glu, Arg273Leu and the splice acceptor variants were genotyped in 2,402 control samples belonging to the Leeds Melanoma Case-Control Study. This control set included 499 population matched control DNAs, 370 family controls (family members of melanoma cases without a diagnosis of melanoma), and 1,533 DNAs from the Wellcome Trust Case Control Consortium. All 2,402 samples were wild-type for the abovementioned POT1 variants. We also genotyped these positions in 1,739 population-based melanoma cases that were recruited from across Yorkshire, UK as part of the same study. One case, presenting with MPMs with early onset (48 years old), was found to be a carrier of the Arg273Leu variant (Table 1 and Supplementary Tables 5 & 6). This variant was confirmed by PCR-sequencing.
Protein alignment, structural modeling and characterization of POT1 variants
The amino acid sequences of POT1 from evolutionarily diverse species were gathered from NCBI and aligned with Clustal Omega38. Alignments were displayed using Jalview v2.739. In order to estimate the number of substitutions per site in this amino acid alignment, we used the ProtPars routine from PHYLIP40. This analysis revealed a higher conservation in the three mutated amino acids (2, 2 and 0 substitutions in positions 89, 94 and 273, respectively) than the average for the OB domains (2.42 substitutions per site) and in fact the whole protein (3.49 substitutions per site) across ~450 million years of evolutionary history (from zebrafish to human). If only sequences from Eutherian organisms are taken into account, then no substitutions have occurred in any of the three residues, compared to 0.8 substitutions per site in the OB domains and 1.39 in the whole protein. The OB domain regions were defined as amino acids 8-299 in the human sequence, according to Ensembl’s superfamily domain annotation. The structure of the OB domains of POT1 (Accession 3KJO) was obtained from the Protein Data Bank (PDB) and was rendered with PyMol v0.9941.
Analysis of variants in the nucleotides coding for the 24 key OB domain residues in close proximity (3.5 Å) to telomeric DNA
Ramsay et al10 defined a list of 24 residues that lie closer than 3.5 Å to the telomeric DNA in the crystal structure of POT1 (PDB 3KJP): 31, 33, 36, 39–42, 48, 60, 62, 87, 89, 94, 159, 161, 223, 224, 243, 245, 266, 267, 270, 271 and 273. To assess the statistical significance of finding amino acid substitutions in these residues, we searched 6,503 exomes part of the NHLBI GO ESP14 for substitutions in any of the bases that would cause a change in these amino acids.
Supplementary Table 7 shows the genomic positions that encode these 24 residues. In summary, a minimum of 6,498 exomes had all bases covered at a minimum average coverage of 59x. The variant Asn224Asp was found at an overall allele frequency of 1/13005. No other amino acid-changing variants were found. We compared the number of variants found in the 24 key OB domain residues in controls (one in 6,498) to the number of variants found in all analysed pedigrees (3 in 105), obtaining a P-value of 1.54×10−5 using a two-tailed Fisher’s exact test (Supplementary Table 6).
In vitro translation and G-strand binding assays
Human POT1 in a T7 expression vector (OriGene) was mutated by site-directed mutagenesis to generate cDNAs carrying the POT1 Tyr89Cys, Gln94Glu and Arg273Leu variants. Mutant and control T7 expression vectors were used in an in vitro translation reaction using the TNT coupled reticulocyte lysate kit (Promega) following the manufacturer’s instructions. Briefly, a 50-μl reaction mixture containing 1 μg of plasmid DNA, 2 μl of EasyTag L-[35S]-methionine (1,000 Ci/mmol; PerkinElmer) and 25 μl of rabbit reticulocyte lysate was incubated at 30 °C for 90 min. A 5-μl fraction of each reaction was analyzed by SDS-PAGE, and proteins were visualized and relative amounts quantified using an FLA 7000 phosphorimager system (Fujifilm) (Supplementary Figure 5). DNA binding assays were performed as described previously with minor modifications42. In 20-μl reaction mixtures, 5 μl of each translation reaction was incubated with 10 nM 5-[32P]-labeled telomeric oligonucleotide (GGTTAGGGTTAGGGTTAGGG) and 1 μg of the nonspecific competitor DNA poly(dI-dC) in binding buffer (25 mM HEPES-NaOH (pH 7.5), 100 mM NaCl, 1 mM EDTA and 5% glycerol). Reactions were incubated for 10 min at room temperature, and protein-DNA complexes were analyzed by electrophoresis on a 6% polyacrylamide Tris-borate EDTA gel run at 80 V for 3 h. Gels were visualized by exposure to a phosphorimager screen.
Analysis of telomere length from next-generation sequencing data
Telomere length was determined essentially as described in Ref. 15. The investigator that performed this analysis was blind to the POT1 status of the 41 eligible samples (which are all of those sequenced at the Sanger Institute, as they were the only ones with enough data available [Supplementary Table 2]).
After calculating the relative telomere length, the 38 samples without germline POT1 variants were adjusted for age at blood draw and gender using a linear model (Supplementary Fig. 6). The corresponding values for POT1 variant carriers were estimated based on the same adjustment. A Wilcoxon rank-sum test comparing the adjusted telomere length for the non-carrier melanoma cases with the 3 members of pedigree UF20 (Tyr89Cys variant carriers) (P=0.0001876) supported the increased telomere lengths for variant carriers. For depiction in Fig. 2c, all values are shown relative to the largest sample measurement.
Analysis of telomere length (PCR)
We measured telomere length in melanoma cases recruited from the Leeds Melanoma cohort who did not carry a POT1 variant, seven POT1 missense variant carriers (pedigrees UF20, UF31 and UF23 and the carrier individual from the Leeds Melanoma cohort) and two non-carrier family controls (UF23, individual III-1 and UF20, individual III-1).
The investigator that performed this analysis was blind to the POT1 status of all samples. Relative mean telomere length was ascertained by a SYBR® Green real-time PCR using a version of the published Q-PCR protocols43,44 and modified as described previously45. In brief, genomic DNA was extracted from whole blood and telomere length was ascertained through the ratio of detected fluorescence from the amplification of telomere repeat units (TEL) relative to that of a single-copy reference sequence from the β-Globin gene (CON). Telomere and control reactions were performed separately. For each assay, the PCR cycle at which each reaction crossed a predefined fluorescence threshold was determined (Ct value). The difference in the Ct values, ΔCt = Ct TEL – Ct CON, was the measure of telomere length used in the analysis, as in other published data generated using this assay45,46.
For the analysis, samples with 18 < Ct CON < 27 or Ct CON > 2 standard deviations away from the mean were removed and considered as fails. This left 252 samples from the Leeds Melanoma cohort for further analyses, with no missense variant carriers or non-carrier family controls removed. All samples had between 2 and 8 replicates.
Mean ΔCt values per sample were estimated from all replicates.
The estimated mean values of ΔCt obtained from melanoma cases without germline POT1 variants were adjusted for age at blood draw and gender using a linear model (Supplementary Fig. 7). The corresponding values for POT1 variant carriers were estimated based on the same adjustment. Adjusted mean ΔCt values are plotted (Figure 2d) with the histogram showing the non-carrier melanoma cases compared to the missense variant carriers and the non-carrier family controls plotted above. A Wilcoxon rank-sum test comparing the adjusted mean ΔCt for the 252 non-carrier melanoma cases with the 7 missense variant carriers (P=3.621×10−5) supported the increased telomere lengths for variant carriers.
Analysis of POT1 mutations in cancer databases
Although mutations in POT1 have not been found at a high frequency in the cancer studies deposited in COSMIC18 and IntOGen47 (which integrates only whole-exome data from the ICGC and TCGA as well as other studies), the mutations that have been reported show a tendency to be missense, to hit the residues that are predicted to interact with DNA and to show a high functional impact bias.
To statistically assess the mutational patterns affecting POT1 in cancer, we compiled a list of all residues closer than 3.5Å to the telomeric DNA in the crystal structure of POT110 (PDB 3KJP). We then mined the COSMIC database v66 for confirmed somatic mutations absent from the 1000 Genomes Project affecting the open reading frame of POT1 across 14 cancer types (breast, central nervous system, endometrium, haematopoietic and lymphoid tissue, kidney, large intestine, liver, lung, ovary, parathyroid, prostate, skin, urinary tract and not specified). This yielded 35 somatic mutations, including four that were silent. We also compiled the total frequency of each reference/mutated base pair in the same COSMIC database. Finally, we performed a Monte Carlo simulation with 100,000 groups of 35 mutations at random locations of the POT1 open reading frame. The probability of a given mutation from a reference base (i.e. A to G) was forced to equal the frequency for that pair in the whole COSMIC database.
Of the 100,000 simulations performed with this method, only 2,971 contained four or less silent mutations. Therefore, the COSMIC database contains less silent mutations affecting POT1 than expected by chance (P<0.03). To assess the clustering of mutations in DNA-binding residues (Supplementary Table 7), we only considered missense mutations, since no selection would be expected for nonsense mutations. In the COSMIC database, we found 27 POT1 missense mutations, four of which affected telomere-binding residues. In the Monte Carlo experiment, 9,244 simulations had exactly 27 missense mutations. In only 176 of these were four or more residues identified that were classified as disrupting telomere-binding. This suggests that POT1 missense mutations affect DNA-binding residues at a higher-than-expected rate in the COSMIC database (P<0.02).
In order to assess the functional impact bias of somatic mutations in POT1, we also looked at all mutations in POT1 that are present in the IntOGen database19. We chose this database because it integrates only samples which have been whole-exome sequenced, and thus can provide a valid, non-biased estimate of the functional impact of mutations in POT1 when they are compared with mutations in the rest of the exome. The frequency of POT1 mutations in this dataset is ~0.01 across 9 cancer sites (all of those available in the database, contained in the 14 sites listed above). P-values for the three studies for which the gene passed set thresholds defined in the database (See Ref. 19), calculated with Oncodrive-fm48, were combined to yield a P-value of 0.021, indicating that this gene is biased toward the accumulation of functional mutations.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
D.J.A, C.D.R-E, J.Z.L., J.C.T, M.P and T.M.K were supported by Cancer Research UK and the Wellcome Trust (WT098051). C.D.R-E was also supported by Consejo Nacional de Ciencia y Tecnología of Mexico. K.A.P. and A.M.D. were supported by Cancer Research UK (Grants C1287/A9540; C8197/A10123) and The Isaac Newton Trust.N.K.H. was supported by a fellowship from the National Health and Medical Research Council of Australia (NHMRC). L.G.A was supported by an ANZ Trustees PhD scholarship. A.P is supported by Cure Cancer Australia. The work was funded in part by the NHMRC and the Dutch Cancer Society (UL 2012-5489). M.H. J.A.N-B, D.T.B were supported by Cancer Research UK (Programme Awards C588/A4994, C588/A10589 and the Genomics Initative). C.L-O, A.J.R and V.Q are funded by the Spanish Ministry of Economy and Competitiveness through the Instituto de Salud Carlos III (ISCIII), Red Temática de Investigación del Cáncer (RTICC) del ISCIII and the Consolider-Ingenio RNAREG Consortium. CL-O is an Investigator with the Botín Foundation. We thank the UK10K Consortium (funded by the Wellcome Trust; WT091310) for access to control data.
Footnotes
Accession codes
Sequence data has been deposited at the European Genome-phenome Archive (EGA), hosted at the European Bioinformatics Institute, under accession EGAS00001000017.
URLs
UK10K Sequencing Project, http://www.uk10k.org; European Genome-phenome Archive, http://www.ebi.ac.uk/ega/.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Goldstein AM, et al. Features associated with germline CDKN2A mutations: a GenoMEL study of melanoma-prone families from three continents. J Med Genet. 2007;44:99–106. doi: 10.1136/jmg.2006.043802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horn S, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–61. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 3.The Breast Cancer Linkage Consortium Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91:1310–6. doi: 10.1093/jnci/91.15.1310. [DOI] [PubMed] [Google Scholar]
- 4.Wiesner T, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43:1018–21. doi: 10.1038/ng.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zuo L, et al. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat Genet. 1996;12:97–9. doi: 10.1038/ng0196-97. [DOI] [PubMed] [Google Scholar]
- 6.Law MH, Macgregor S, Hayward NK. Melanoma genetics: recent findings take us beyond well-traveled pathways. J Invest Dermatol. 2012;132:1763–74. doi: 10.1038/jid.2012.75. [DOI] [PubMed] [Google Scholar]
- 7.Loayza D, De Lange T. POT1 as a terminal transducer of TRF1 telomere length control. Nature. 2003;423:1013–8. doi: 10.1038/nature01688. [DOI] [PubMed] [Google Scholar]
- 8.Baumann P, Cech TR. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science. 2001;292:1171–5. doi: 10.1126/science.1060036. [DOI] [PubMed] [Google Scholar]
- 9.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11:377–94. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 10.Ramsay AJ, et al. POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat Genet. 2013 doi: 10.1038/ng.2584. [DOI] [PubMed] [Google Scholar]
- 11.Speedy HE, et al. A genome-wide association study identifies multiple susceptibility loci for chronic lymphocytic leukemia. Nat Genet. 2014;46:56–60. doi: 10.1038/ng.2843. [DOI] [PubMed] [Google Scholar]
- 12.Nandakumar J, Podell ER, Cech TR. How telomeric protein POT1 avoids RNA to achieve specificity for single-stranded DNA. Proc Natl Acad Sci U S A. 2010;107:651–6. doi: 10.1073/pnas.0911099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lei M, Podell ER, Cech TR. Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat Struct Mol Biol. 2004;11:1223–9. doi: 10.1038/nsmb867. [DOI] [PubMed] [Google Scholar]
- 14.Fu W, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–20. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parker M, et al. Assessing telomeric DNA content in pediatric cancers using whole-genome sequencing data. Genome Biol. 2012;13:R113. doi: 10.1186/gb-2012-13-12-r113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burke LS, et al. Telomere Length and the Risk of Cutaneous Malignant Melanoma in Melanoma-Prone Families with and without CDKN2A Mutations. PLoS One. 2013;8:e71121. doi: 10.1371/journal.pone.0071121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kendellen MF, Barrientos KS, Counter CM. POT1 association with TRF2 regulates telomere length. Mol Cell Biol. 2009;29:5611–9. doi: 10.1128/MCB.00286-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forbes SA, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–50. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez-Perez A, et al. IntOGen-mutations identifies cancer drivers across tumor types. Nat Methods. 2013 doi: 10.1038/nmeth.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi J, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014 doi: 10.1038/ng.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacobs JJ. Loss of telomere protection: consequences and opportunities. Front Oncol. 3:88–2013. doi: 10.3389/fonc.2013.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silverman BW. Density Estimation. Chapman and Hall; London: 1986. [Google Scholar]
- 23.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lopes MC, et al. A combined functional annotation score for non-synonymous variants. Hum Hered. 2012;73:47–51. doi: 10.1159/000334984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aitken JF, Green AC, MacLennan R, Youl P, Martin NG. The Queensland Familial Melanoma Project: study design and characteristics of participants. Melanoma Res. 1996;6:155–65. doi: 10.1097/00008390-199604000-00011. [DOI] [PubMed] [Google Scholar]
- 27.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hodis E, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.1000 Genomes Project Consortium et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Database of Single Nucleotide Polymorphisms (dbSNP) National Center for Biotechnology Information, National Library of Medicine; Bethesda (MD): dbSNP Build ID: 135. [Google Scholar]
- 33.McLaren W, et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–70. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Danecek P, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 36.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Database of Single Nucleotide Polymorphisms (dbSNP) National Center for Biotechnology Information, National Library of Medicine; Bethesda (MD): dbSNP Build ID: 138. [Google Scholar]
- 38.Sievers F, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–91. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Felsenstein J. PHYLIP - Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 41.GenoMEL Consortium et al. A variant in FTO shows association with melanoma risk not due to BMI. Nat Genet. 2013;45:428–32. doi: 10.1038/ng.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baumann P, Podell E, Cech TR. Human Pot1 (protection of telomeres) protein: cytolocalization, gene structure, and alternative splicing. Mol Cell Biol. 2002;22:8079–87. doi: 10.1128/MCB.22.22.8079-8087.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGrath M, Wong JY, Michaud D, Hunter DJ, De Vivo I. Telomere length, cigarette smoking, and bladder cancer risk in men and women. Cancer Epidemiol Biomarkers Prev. 2007;16:815–9. doi: 10.1158/1055-9965.EPI-06-0961. [DOI] [PubMed] [Google Scholar]
- 44.Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002;30:e47. doi: 10.1093/nar/30.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pooley KA, et al. Telomere length in prospective and retrospective cancer case-control studies. Cancer Res. 2010;70:3170–6. doi: 10.1158/0008-5472.CAN-09-4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bojesen SE, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45:371–84. 384e1–2. doi: 10.1038/ng.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gundem G, et al. IntOGen: integration and data mining of multidimensional oncogenomic data. Nat Methods. 2010;7:92–3. doi: 10.1038/nmeth0210-92. [DOI] [PubMed] [Google Scholar]
- 48.Gonzalez-Perez A, Lopez-Bigas N. Functional impact bias reveals cancer drivers. Nucleic Acids Res. 2012;40:e169. doi: 10.1093/nar/gks743. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.