

Abstract
We propose a class of hierarchical models to investigate the protein functional network of cellular markers. We consider a novel data set from single-cell proteomics. The data are generated from single-cell mass cytometry experiments, in which protein expression is measured within an individual cell for multiple markers. Tens of thousands of cells are measured serving as biological replicates. Applying the Bayesian models, we report protein functional networks under different experimental conditions and the differences between the networks, ie, differential networks. We also present the differential network in a novel fashion that allows direct observation of the links between the experimental agent and its putative targeted proteins based on posterior inference. Our method serves as a powerful tool for studying molecular interactions at cellular level.
Keywords: Bayes, cytometry, graphical model, Markov chain Monte Carlo, network, proteomics, single cell
Introduction
Proteins and their functional interactions play a fundamental role in many biological processes. Measuring and analyzing protein expression is critical to assess their role in cellular functions and in understanding the pathology of diseases like cancer.
Some early attempts to predict protein–protein interactions (PPIs) based themselves on genomic analyses. For example, it was observed that conserved proximity of two coding genes was associated with greater likelihood of protein interaction between the coded proteins. Similarly, phylogenetic association of protein pairs was shown to predict functional linkage. That is, the co-occurrence of a protein pair in many different species suggested that they belong to the complex/pathway.1 Protein fusion events provided yet another evidence of interaction. Marcotte et al.2 showed that if two proteins are sometimes seen in some species fused into one contiguous protein, then these are very likely related in function and, therefore, also more likely to interact.
These methods had the advantage that they only required the simple analysis of a large number of genomes, and they modeled functional association instead of direct protein associations. A disadvantage was that these methods are not very effective in eukaryotic species because they have a more complex genome structure. To complement the genomics approach, structure-based prediction of PPIs became popular. This was made feasible with more complexes being revealed from high-throughput experiments. These methods assume that similar sequences have a similar fold and that domains with a similar fold interact through the same surface.3 They typically incorporate knowledge on binding specificities.4 For example, Shi et al.5 modeled PPIs across species using the statistical distribution of free energy – the biochemical parameter that determines interaction strength. Berg et al.6 constructed a model of evolutionary networks, using the fact that protein–protein binding depended on concentrations, mutations, and gene duplications. Their proposed “link dynamics” equations mathematically described how the network connectivities rapidly declined among proteins encoded by duplicate genes. Structure-based methods though useful for in vitro assays, however, are not sufficient to determine the nature of protein interactions inside the living cell. What determines the latter is a combination of factors, such as expression levels, localization, complex formation (ie, scaffolding), post-translation modifications, splicing forms, and association with small compounds.
It is now a growing consensus that this problem of inferring protein interactions can be tackled only by using integrative probabilistic approaches that (1) weight all different information sources and (2) use the graphs/networks as the intrinsic model parameters. Graphical representation of protein networks was first introduced in Jeong et al.7 Exploiting extraneous information (primarily for grouping genes) emerged via Bayesian frameworks described in Besag.14 This strategy was extended successfully to construct the first probabilistic model for the human protein interactome in Rhodes et al.8 It used genome-wide gene expression data and functional annotation data to predict nearly 40,000 PPIs in humans. This result, based on a purely computational approach, was able to replicate the experimental findings in model organisms. The method, applied to cancer genomics data, identified several interaction subnetworks activated in cancer. Currently, the most existing approaches to modeling protein signaling systems are relying on stochastic network methods.9–11
Motivated by the preliminary success of Bayesian approaches in decoding the human interactome, we develop and apply an integrative Bayesian graphical for functional marker interaction in liver cells. In contrast to previous statistical approaches, our proposed Bayesian graphical models provide a formal interpretation of associations as conditional dependence between markers.12 Apart from that, the approach has several additional advantages. For example, it allows for combining disparate sources of data, eg, different marker distributions and stimulation, borrowing of strength through hierarchical priors, and modeling different distributions by efficient use of latent variables.
To explore the marginal and interaction features, we proposed two variations of the hierarchical network approach. In the first one, we estimated a pair of networks, each representing a set of cells under a specific treatment condition (pre- vs. post-treatment). The model borrowed strengths across two conditions through hierarchical priors. Usually, in biological pathways, there is a fair degree of commonality between two networks pre- and post-treatment by a pharmaceutical agent. The stimulating agent perturbs the network, but does not drastically alter the old topology. The common features between the two networks serve as the basis of sharing information and strengthening the overall inference. For the second analysis, we employed a single graphical model to combine analysis for the stimulated and unstimulated conditions. This analysis produced a single graph containing 19 nodes, 18 of which represented the 18 functional markers, while the 19th node represented the experimental condition (pre- or post-treatment). The presence of an edge from the 19th node to a functional marker implied that stimulation significantly changed the marginal distribution of the marker. The second approach allows for a user-friendly visualization of the differential networks. An important feature of both approaches is the inclusion of a generalized sampling model for the protein expressions. We assumed that the association between protein measurements occurred through a set of latent indicators e denoting their latent activation status. Our approach returned random samples from the joint posterior distribution of the networks.
The rest of the article is organized as follows. In Section 2, we describe the graphical models for joint modeling and the related posterior inference scheme. Section 3 describes some simulation experiments to validate the proposed graphical model. We describe the specific data and the experiment in Section 4. Next, we illustrate the application of our method in Section 4.1 with an application to Mass Cytometry (CyTOF) data for monocytes, a cell type inferred by Qiu et al.13 We conclude with a discussion of our approach in Section 5.
Network Models
Our analysis is centered around a hierarchical Bayesian approach for network inference based on CyTOF data. Treating each cell as an independent and random sample from the cellular population, we have a large sample (tens of thousands of cells) for precise network inference. Also, measurements on individual cells mitigate potential contamination caused by experimental factors for sample-based measurements. We will use i and j to denote proteins, t to denote cells, and k to index networks.
We begin with the simple assumption that the dependence in each of two experimental conditions is characterized by a distinct network. Our goal is to estimate both networks. We denote the two unknown networks by G1 and G2 for the pre- and post-treatment conditions, respectively. Let denote the edge between the nodes i and j in the kth network. The proposed model for the data is now constructed as a hierarchical model, starting with a joint prior distribution on G1 and G2, p(G1, G2), defined as
| (2.1) |
| (2.2) |
Using a common π formalizes the borrowing of strength between the two networks. It allows us to estimate a global similarity parameter. When the data indicates that the networks have common patterns, the graphs become closer toward each other. This is a classical analog of shrinkage effects in univariate analysis, now applied to graphical structures. The remaining layers of the hierarchical model are introduced one at a time. For reference, we state the overall model structure
| (2.3) |
The first factor is the prior on G1 and G2. The second layer of the model is a prior on each latent binary indicator eit for the presence of protein i in cell t. The third, and last layer of the hierarchical model, is a sampling model for the observed protein expression conditional on the latent indicators.
Priors on individual graphs
Each of the two graphs can be expressed as (V, E), where V is a set of vertices and E is a set of undirected edges. For future reference, we define a clique as a set of vertices of which all pairs in the set are connected through edges, ie, {i1, i2} ∈ E for all i1, i2 in the set. The vertices correspond to the proteins, and the absence or presence of edges in the graph denotes the conditional independence (CI)/dependence between them. “CI” between two nodes i and j implies that the random variables i and j are conditionally independent of the remaining variables (nodes). This property can also be restated in terms of the Markov property – each variable is conditionally independent, given its edge neighbors. Here, we emphasize an important distinction between CI and the notions of marginal independence where we average out over the other variables. For example, consider a protein i that simultaneously affects proteins j and k. Although, marginally, the three variables would appear associated, the CI structure (encoded by the graph) would lack an edge between j and k. We follow Mitra et al.12 to construct priors on individual graphs. Details are omitted.
Prior models for indicators
Conditioned on the graphs, we model the joint distribution of latent indicators e through an autologistic model.14 For notational convenience, we first describe the conditional distribution given any single network.
Given this network and a set of coefficients β, the model can be expressed as
| (2.4) |
Tentatively, we drop sample index t for ease of exposition. Also, we impose the restriction that an interaction coefficient is zero if and only if vertices i1,…,ik do not form a clique in the graph G. Henceforth, we use β to denote the vector of all non-zero coefficients For our application, we assume that all interactions of order three and higher are zero, thus ignoring any cliques of size greater than three. This brings up a nice conditional interpretation. Conditional on the other variables, the distribution of a node i turns out to be simply a logistic regression with two-way interaction coefficients. This is a desirable property of the joint distribution and is hugely exploited in the Gibbs sampling of e. It can also be proved that the traditional log odds ratio for eit and ejt is βij conditional on all other parameters. The sign of βij determines how the activation of one protein is enhanced or diminished by those with which it shares an edge. To improve Markov chain Monte Carlo (MCMC) mixing, we employ a centered parametrization of the above autologistic model (2.4). Letting vi = exp(βi)/{l + exp(βi)}, we can restate (2.4) as
| (2.5) |
Following this generic model, we finally write out the conditional distribution of a binary vector et = (eit, i = 1,…,m) for cell t.
This same structure, described for a single network, can now be allowed to vary with the indicator of the covariate Zt = k, k = 0, 1, for cell t. The dependence is borne through the graphical structure Gk and the corresponding parameter set βk
| (2.6) |
Sampling model for [yit]
We complete the model construction with a sampling distribution for the observed counts yit. Figure 1 shows the empirical probability distributions of the counts for the functional marker 166.IkBalpha under the unstimulated condition.
Figure 1.

Histogram of protein expression data. Note that these are not raw expressions but processed data. The background mean effects are subtracted. This explains the negative values.
Motivated by the bell shape of the empirical distribution, and the presence of the long tail to the right, we model the data as a mixture of two Gaussian distributions. The latent states for the mixtures are provided by binary indicators eit. The parameters of the Gaussian mixture are dependent both on the functional marker i and the treatment condition k = 0, 1, ie,
| (2.7) |
We will use θ = (μ1ki, μ2ki, σ1ki, σ2ki, i = 1,…,m, k = 0, 1) to denote the complete parameter vector for the sampling model.
The entire model building process can be summarized as a flowchart. At the very top lies the pair of graphs each having has a CI structure within itself. The pair is connected by the inclusion probability π plays a very important role in the differential graph model. Conditioned on π, the graph edges are independent. However, marginally, the independence gets lost. At the bottom is the data matrix yk. Just above the data matrix, we have a layer of latent indicators ek.
Implementation: posterior inference scheme
Large datasets require an efficient and tractable MCMC scheme to simultaneously search the graphical space and infer protein-specific parameters. The hierarchical structure of the model induces CI among different sets of variables. This makes the model most amenable to a Gibbs sampling scheme, which we implement for our simulations and data analysis. Note that for Gibbs sampling, we require only the full conditionals.
MCMC posterior simulation proceeds by iterating over the following transition probabilities:[θk|βk,Gk,yk], [βk|θk,Gk, yk] for k = 1, 2, [π|G1, G2], [G2, βk|θk, G1, π, y2], [θk, β1|θ1, θ2, β2, G2, π, y1]. Updating the parameters θk of the sampling model is easy. Since we are modeling y|e as mixture of Gaussians, we can find the full conditionals in a closed form. In fact, sampling them is equivalent to sampling the mean and variance parameters in a standard two-component mixture model.
Other than that, there is a scope of huge parallelization on two counts. First, the y’s are independent given the binary indicators e’s. This means that we can update eit, i = 1,…,m, using
and repeat the same loop for each t = 1,…,n. This is just a fallout of the nice property of the autologistic model. (For this part, we have dropped the superscript k and used a general e for notational convenience.) This is just a fallout of the nice property of the autologistic model. Moreover, since the et, t = 1,…,n, are conditionally independent given all other parameters and y, we can run the same conditional loops in parallel for all data points. This parallelization can scale up the computation considerably, especially when we have large number of observations. In our application, we update the binary matrices e1 and e2 in exactly the same way.
Also, note that the graphs are independent given π. This would allow us to distribute the bulk of the computation for p(G1|y…) and p(G2|y…) among parallel nodes. Only for sampling π do we need a combined estimate of the edge similarities in the two networks. This updating of π allows the borrowing of strength among the two networks. This sampling can be done by computing the following. Let m1 = ∑{i,j}∈V×Vδij and m0 = ∑{i,j}∈V×V (1 – δij) denote the number of mismatches and matches between edges of the two graphs G1 and G2. We have . We recognize this as the kernel of Beta(m1 + 1, m0 + 1). Note that sampling this inclusion probability π plays a very important role in the differential graph model. Without the latter, there would be no borrowing of strength across the groups.
Sampling graphs and coefficients were relatively non-trivial because of the presence of normalizing constants in the auto-logistic density. For this, we employed a combination of importance sampling and Reversible jump Markov chain Monte Carlo (RJMCMC) as illustrated in Refs.12,15 In all, 16,000 MCMC simulations for a 400-sized data with seven nodes typically take 22 minutes on the University of Texas at Austin computing cluster. The code is a combination of an R code and a set of C routines. The C routines are included to speed up the updating of Gs and βs.
After completing all MCMC simulations, the marginal posterior probability for an edge in the networks for each possible edge {i, j} in the graph was computed as
substituting the edge set E of the imputed graph for each iteration of the MCMC. To construct a summary graph, we thresholded posterior probabilities where the threshold c is chosen in order to achieve a posterior expected false discovery rate (FDR). The posterior expected FDR for any given threshold c is calculated by
We chose threshold c such that the corresponding FDRc is.01.
Comparisons with other Markov random fields (MRF)s
Some commonly used examples of other MRF models are Gaussian graphical models (GGMs), which uses a multivariate normal distribution to describe the joint distribution of the nodes. Using a GGM would be equivalent to removing the additional layer of e in our model. The non-zero entries of G would then correspond to the non-zero entries of the inverse covariance matrix of the Gaussian distribution. This would be a simpler and a more popular approach.16–19 However, as we just explained, it would misguide us about the form of actual dependence we are interested in.
With the abundance in methods for inferring individual GGMs, the concept of multiple GGMs has also gained ground. Danaher et al.20 and Guo et al.21 developed the idea under frequentist paradigm, where they aimed to estimate multiple-related GGMs from observations belonging to distinct classes. These methods borrowed strength across the classes through appropriate convex penalty functions where the penalty was chosen to encourage similarity across the estimated precision matrices. Some other examples of joint graphical modeling using penalized likelihood appear in Refs.22–26 York et al.27 recently used lasso penalization and GGM assumptions to identify complex protein signaling patterns from reverse phase protein array data in 203 AML patients. However, these techniques require a lot of tuning with ad hoc penalization parameters. We compare our approach with these methods through a series of simulation experiments in Section 3. We further note that the MRFs described above through the prior model do not obey the Gaussian assumptions.
Recently, discrete MRFs for molecular pathways were used in Segal et al.28 for an integrated analysis on gene expression and protein interactions. Their framework forced each gene to belong to precisely one of several pathways. The pathway assignments played the role of latent k-nary random variables that corresponded to the nodes in MRF. Gene expression values are assumed to be conditionally independent given the class. They implemented their method on two Saccharomyces cerevisiae gene expression datasets under various stress conditions. However, our approach is fundamentally different from theirs both in objective and inference. Since they used known protein interactions to predict gene assignment to pathway, they assumed that the edge structure of the MRF is already observed. In contrast to their simplifying assumptions, our approach assumes the node variables as well as the MRF structure to be completely unknown.
Covariate-induced differential graphical model
As an alternative model, we include the treatment condition directly into the graphical model through a binary stimulus covariate Z, taking values in {0, 1} denoting the unstimulated and stimulated experimental conditions, respectively. Generally, any graphical model for markers must depend on our underlying assumption on how the binary covariate Z changes the joint probability model. To represent this differential effect within the graphical model, we regressed the marginal effects of the proteins against the stimulation status. Instead of defining a joint prior on two networks, we now integrated the information into a single network model by adding a new parameter βm+1 that measures the effect of the covariate. We must note that, unlike the other indicator variables, this variable is neither stochastic nor latent. It is treated as a covariate. The main effects are now dependent on the value that covariate z assumes at cell t. Specifically, keeping (2.6) unchanged we assume the intercepts follow a new configuration, given by
| (2.8) |
where I{} is the indicator function, m denotes the number of proteins in the CyTOF data, and m + 1 is used to index the treatment condition as an additional “node.” Since Zt is binary, the main effects βik take two possible values depending on whether the tth cell is stimulated or not. When Zt = 1 or 0, ie, the cell t is stimulated or not, βik = αi + βi,m+1 or αi, respectively, with βi,m+1 describing the edge connecting the (m + 1)th node (or the stimulus) with the ith protein. This edge simply reflects the potential effect of stimulation on protein i. When βi,m+1 is non-zero, the stimulus is believed to have an effect on protein i. The differential graphical model could now be pictorially represented as a graph with an additional node. The node denotes the covariate Zt and is connected to node i if and only if βi,m+1 = 1. Since Zt is not a random variable, the edge connecting nodes m + 1 and i does not have the same interpretation as the edges between other proteins i and j. It simply indicates the presence of an effect of a fixed covariate – the stimulation status. The edges between proteins, on the other hand, represent the conditional dependencies of a graphical model as described before.
Simulation
We set up a simulation experiment to validate the proposed model. For each simulated data set, we carried out inference under (1) the proposed model (differential graph model); (2) a model with two independent priors for G1 and G2, (independent graph model); (3) joint graphical inference by Guo et al.21; (4) joint graphical inference by Danaher et al.20; and (5) independent graphical lasso. The primary objective of these experiments is to investigate whether the differential model provides any advantage over independent analyses and other joint graphical methods in detecting edges of the two networks. We fixed the number of observations for subgroup 1 at 330 and subgroup 2 at 48.
The graph G1 was generated by setting up vertices for m = 7 nodes. For each pair of nodes {i, j}, we included an edge between them with probability π = 0.5. For each imputed edge {i, j}, we generated values of using a discrete uniform prior over three possible values, . Next, we used π to generate G2 from the conditional prior distribution p(G2|G1, ∏). In the simulation truth, we used several choices of π. Values are indicated in the upcoming tables of results.
Conditioned on Gk, we generated latent binary indicators eitk ∈ {0, 1} conditioned on Gk and βk from the autologistic model. Using the imputed ek = {eitk}i,t, we then generated hypothetical data yk from the sampling model. The sampling model, described above, is a mixture of two Gaussian distributions. In our setup, we generated the parameters as μ1ik = N(4, 0.2), μ2ik = N(1, 0.2), and σ1ik = σ2ik = 0.1. We generated 20 hypothetical datasets under this assumed sampling model. We used the same model as the analysis model, ie, we evaluated posterior probabilities under this model. To generate enough variance in sample sizes across groups (as is observed in most of these experiments), we set the number of observations for subgroup 1 at 330 and subgroup 2 at 48.
After inference, we examined the ROC (receiver operating characteristic) curves obtained from each model. The ROC plots the sensitivity versus the false positive rate under different posterior probability thresholds for inferring edges in and . We specifically used the area under the ROC curve (AUC) as a summary of model performance. The curves were smoothed using kernel density estimates of the distribution of for both, true positives and true negatives. For more details, we refer the readers to Lloyd.29
For each dataset, we computed AUCs for all five methods: (1) our proposed differential Bayesian model, (2) independent Bayesian model, (3) joint lasso proposed by Guo et al.21 and (4) Danaher et al,20 and (5) independent graphical lasso. The frequentist lasso methods (3)–(5) required specifying the glasso penalization parameter ρ, which we set to 0.03. (3)–(4) was executed using the glasso package and jgl in R, while (5) was executed with the help of a code obtained from Guo et al.21 For each method, we recorded four measures of model performance (1) AUC for estimating G1 (2) AUC for estimating G2 (3) AUC for estimating the difference graph and (4) the mean of (1) and (2). The fourth measure thus provides a combined summary of how the model jointly estimates the pair of networks.
ROC for the frequentist methods was obtained by thresholding the values of inverse covariance matrix at different cutoff values. Each cutoff yielded a binary matrix of estimated differences, which was then used to compute the corresponding sensitivity and specificity. The average AUCs for all methods along with their standard errors are summarized in Table 1. We observe that the differential prior gains considerably over the independent prior in terms of combined accuracy and the estimation of G2. Figure 2 shows smoothed ROC curves under the differential and independent models for estimating both graphs, for a sample dataset.
Table 1.
Comparing differential prior model against independent priors and other frequentist alternatives under autologistic–Gaussian mixture sampling.
| AUC | PROPOSED MODEL | IND-BAYES | GLASSO | JGL | GUO |
|---|---|---|---|---|---|
| Joint | 0.81 | 0.76 | 0.77 | 0.78 | 0.67 |
| Group 1 | 0.95 | 0.96 | 0.98 | 0.98 | 0.92 |
| Group 2 | 0.91 | 0.80 | 0.77 | 0.80 | 0.72 |
| Difference | 0.72 | 0.73 | 0.76 | 0.77 | 0.62 |
Figure 2.
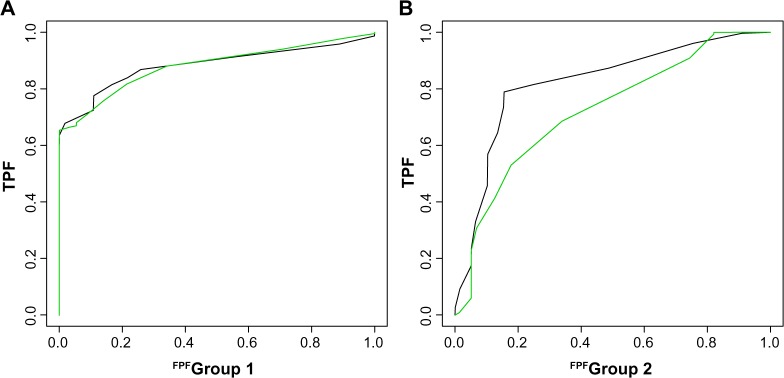
ROC curves for a simulated data set. The green and black curves represent the operating characteristics of the differential graph model and the independent graph model, respectively.
We next varied the penalization parameters of the frequentist methods and found that their performance was very sensitive to these values. Specifying an optimal value of A that works for specific situations remains a challenge. To have a fair comparison, one should however note that these approaches were never specifically intended for a comparison of edges in two graphs and are more focused on the shrinkage of graph coefficients.
Overall, the joint estimation of differential pathways in the differential model allows improved inference on differences across the two graphs. The relative advantage over independent analyses decreases when sample sizes increase (simulations not shown). However, differential prior provided a substantial gain in AUC (and a significantly lower error rate) under unequal and lower sample sizes. Asymptotically, as both sample sizes increase and the data essentially reveal the true graphs, both models achieve an AUC of 100%.
Besides the good performance in simulation experiments, the Bayesian paradigm offers several advantages. First, it would allow the inclusion of prior expert knowledge, when and if they are available. Second, we model the differential structure directly through latent graphs, rather than using features of an assumed sampling model. This makes the approach very flexible. For example, the current sampling could be replaced by any alternative sampling model without substantially changing the implementation of posterior simulation. In fact, we repeated the same simulation experiment by generating data from a binary autologistic model without the mixture component. The results (not shown) are an evidence of the robustness of our prior to different sampling model specifications. Finally, the Bayesian approach includes a full probabilistic description of uncertainties as the posterior distribution.
CyTOF Data and Results
The last few years have seen rapid advances in biotechnology, enabling increasingly precise quantitative measurement of proteins in biological samples. A significant step in this direction was the advent of a single-cell mass cytometry platform called CyTOF.30 The platform was applied successfully to understand protein signaling patterns in human bone marrow cells. For experimental details, we refer the readers to Bendall et al.30 Briefly, the bone marrow cells were targeted by protein specific antibodies, coupled with stable transition element isotopes. The bound cells were sprayed as droplets onto high temperature argon plasma. The extreme plasma temperatures vaporized the cells and induced ionization of its constituent ions. The ions were then fed into a spectrometer where they were separated on the basis of their mass-to-charge ratios. Finally, the spectrometer detected ion signals proportional to the concentrations. This entire process led to the simultaneous measurement of 18 protein markers per individual cell. Based on the assumption that cells at different maturation stages exhibit unique surface marker combination, the marginal expression of the 13 surface markers were analyzed to identify 26 different hematologic cell types. This was achieved by SPADE - an efficient algorithm for agglomerative clustering.13 The analysis yielded a classification of immunological cell types based on a detailed analysis of the marginal protein expressions across different experimental conditions. Table 2 lists the set of 18 markers that we analyzed.
Table 2.
The list of 18 functional markers.
| MARKERS | |
|---|---|
| 1 | 141.pPLCgamma2 |
| 2 | 150.pSTAT5 |
| 3 | 152.Ki67 |
| 4 | 154.pSHP2 |
| 5 | 151.pERK1.2 |
| 6 | 153.pMAPKAPK2 |
| 7 | 156.pZAP70.Syk |
| 8 | 159.pSTAT3 |
| 9 | 164.pSLP.76 |
| 10 | 165.pNFkB |
| 11 | 166.IkBalpha |
| 12 | 168.pH3 |
| 13 | 169.pP38 |
| 14 | 171.pBtk.Itk |
| 15 | 172.pS6 |
| 16 | 174.pSrcFK |
| 17 | 176.pCREB |
| 18 | 175.pCrkL |
The expression data for all markers and all cell types are available from the website http://reports.cytobank.org/l/vl. Note that these are all processed data. For pre-processing steps, we refer the readers to Bendall et al.30
Based on the network models described before, we report differential interactions among these markers for a specific cell type, monocytes under two conditions. The nodes in the graph denote the random variables (the 18 functional markers), and the edges imply statistical associations between pairs of nodes. The conditions are the presence/absence of a lipopolysaccharide (LPS) agent.
Monocytes are a special category of cells produced in the bone marrow from monoblasts. They stay in the spleen, circulate in the bloodstream, and finally diffuse into tissues throughout the body, where they change into macrophages. Macrophages digest pathogens, infectious microbes, and cancer cells. They repair tissues and stimulate immune cells (eg, lymphocytes) to respond to pathogens. Monocytes play multiple influential roles in the immune system of all mammals by replenishing macrophages under normal states. They also respond to inflammation signals by traveling to infection sites in the tissues and differentiating into macrophages. This choice of cell type was motivated by two reasons. First, monocytes were among the mostly populated cell types. The number of stimulated cells and unstimulated monocyte cells in this CyTOF dataset are 25,889 and 33,929, respectively. Second, there is a specific biochemical relationship of monocytes to the stimulating agent LPS. Recent studies have shown how this stimulation is recognized by monocytes and affect the innate immune system. In fact, human monocytes are known to respond extensively to LPS stimulation by expressing numerous inflammatory cytokine markers.31 However, the role of inter-protein-pathways in this marker activation had not been elucidated so far.
Results
As an initial step, we performed some exploratory data analysis to assess the assumptions of our models. In particular, we empirically explored the changes in the marginal distribution of the proteins with respect to stimulus for monocytes. We expected that if a protein’s intensity distribution changed significantly across stimulation status in monocytes, our covariate-induced model would capture that, by assigning an edge to connect the (m + 1)th node, or the stimulation, to that protein. This intuition has been verified. In Figure 3, we show the empirical comparisons for the m = 18 functional markers for monocyte cells under the two conditions (unstimulated vs. stimulation). In Figure 4(A) and (B), we present the posterior estimates of G1 and G2, under the differential network model in (2.6). In Figure 4(C), we present the 19-node graph for monocytes based on the covariate-induced model (2.8). For all proteins connected to the stimulus node, the marker intensity distributions between the two conditions have very different shapes. We make a special note of the subplot representing the marker 166.IkBalpha (node 11 in Table 2). In marked contrast to other plots, the unstimulated marker expression here has a distribution that is stochastically greater than that under stimulation. Our covariate- induced model captures this by a negative edge (colored pink) in Figure 4(C). This strongly affirms the findings in the existing literature, which suggests that LPS stimulation is related to the inhibition of IkBalpha expression in monocytes.32
Figure 3.

Comparison of the empirical densities of the 18 functional markers pre- and post-stimulation. The post-stimulation distributions are shown by the red curve. The unstimulated condition is shown by the black curve. The distributions are for the markers in monocytes. In some of these plots, we see the red curve markedly shifted from the black curve. This implies the effect of stimulation on the marginal mean expression. Interestingly, those cell types that show this effect have edges joining the stimulation node in Figure 4.
Figure 4.
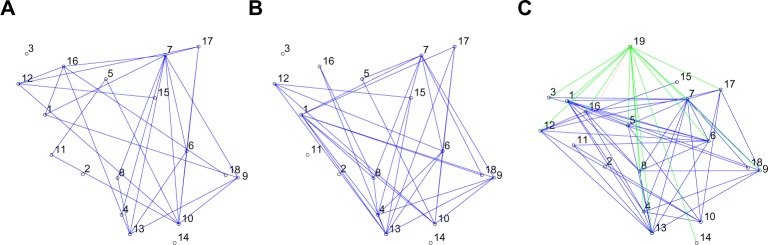
Posterior summaries for the monocyte networks. (A) and (B) show the unstimulated and stimulated networks, respectively, after implanting the joint graphical model. The edges denote presence of relationship between proteins. (C) is the combined network estimated from the covariate-induced model. The 19th node is the stimulation node. Edges from this node to the protein node indicate the effect of stimulation on proteins.
Next, we applied the proposed joint network model and the covariate-induced model on stimulated and unstimulated monocyte cells. Figure 4 shows three estimated networks – two from the joint network model (2.6) and one from the covariate-induced model (2.8). The nodes, representing the functional markers, are indexed by integers from 1 to m = 18. The stimulus node is labeled by the integer m + 1 = 19. The marker indices correspond to the order in which they appear in Table 2. We use solid blue to denote the associations between the protein pairs and a different color coding for the edges connecting the stimulus node. The positive edges from the stimulus are colored green, while the negative ones are colored pink. Overall, the stimulated network is more densely connected than the unstimulated network. As expected, there was a perturbing effect of the stimulus, leading to small and significant differences in topology across two networks. At the same time, the unstimulated and stimulated networks shared a fair number of edges between them. 60% of the edges appearing in unstimulated network appear in the stimulated network as well. The markers 152.Ki67 and 175.pCrkL are unconnected to any other node in all three summary graphs. The protein pairs appearing in the networks provide potentially interesting functional relationships that require future experimental validations. Notably, the model detects a pink edge from the stimulus node to the 11th marker. This formally confirms the well-known inhibiting effect of LPS on 166.IkBalpha in monocytes.
Conclusion
The proliferation of experimental and computational methods to study PPIs has prompted comparative studies under different stimulation conditions, species, and assays. Interestingly, it was demonstrated that interactions that were observed in more than one of the analyses were more likely to be true interactions. From these first efforts, emerged the idea that more meaningful information can be obtained from the combination of different experimental and computational observations. Formally, this demands an integrated joint statistical model for studying interactions. We develop such an approach and apply it to study protein signaling patterns in human bone marrow cells.
These patterns were recently identified using a novel mass cytometry platform called CyTOF. Differential protein signaling across the hematopoietic continuum was observed and analyzed with the help of 18 functional markers. Apart from its novel data-generating potential, the technology has presented both a unique challenge and an excellent opportunity to biostatisticians to efficiently model joint distributions of protein markers. It is an opportunity because single cells act as independent units of observation, allowing us to exploit the assumptions of a statistical model. In particular, the data are free from sample heterogeneity, which could strongly bias the results of network inference. It is a challenge because the protein marker distributions do not follow a conventional pattern. Also, the space of interactions is larger and more complex than marginal protein distributions. The former is important to model because proteins do not function in isolation, but interact with one another and with other cellular entities. The interaction structure can itself change with biochemical perturbations in a complex manner.
We presented a hierarchical Bayesian formulation for joint estimation of protein signaling networks. This introduces a novel perspective to the field of Bayesian networks. Instead of tuning sparsity for a single high-dimensional graph, we now use priors to borrow strengths across multiple graphs. Posterior probabilities from this graphical procedure allowed us to simultaneously construct the relationship between the stimulating agent with the protein markers as well as the protein functional interactions. Our methodology is accompanied by a computationally efficient algorithm for full probabilistic inference. The formulation is general and can be applied to a number of graphical models and other cell types.
Methodologically, this approach can have several natural and useful extensions. The “curse of dimensionality” could emerge when we start including more protein markers. To tackle this, informative priors could be used.12 Instead of assigning equal prior probability to all subgraphs, we might want our inference to be centered around a consensus network obtained from expert knowledge or known databases. Another useful constraint could be that of sparsity. This can be achieved by placing a hyper prior p(ρi,j) = Beta(a, b) on the inclusion probability ρi.j of each edge. The hyper-parameters a, b could be tuned to induce the desired level of sparsity control. Note that both these modifications can be done at the level of the graphical priors, and would not affect the lower levels of the hierarchy.
In the context of informative priors, a potentially successful extension would be the easy inclusion of structural information. As mentioned before, much progress has been made in predicting PPIs on the basis of structures and homology modeling. A Bayesian hierarchical model would be able to naturally distribute the likelihood of protein interaction networks among several structural subnetworks through appropriate prior. Finally, the same approach could be adapted to borrow strength across multiple (more than two) protein networks. This would be highly relevant for comparative graph analysis between the interactomes of different species. In this way, it could strongly enrich the new field of comparative interactomics,33 which would be the protein-pathway parallel to the well-established field of comparative genomics.
The present application focused on healthy bone marrow cells. However, this approach is generalizable to other types of biomolecular data as well. This would have significant implications for drug development. For example, the results from our model identify respondent biomarkers in monocytes and specifically address the molecular basis of LPS recognition by markers. This could directly help in identifying novel therapeutic approaches. Based on such findings, we see considerable scope of application of such models to disease pathways, especially cancer pathways. Such pathways usually demonstrate a high degree of heterogeneity across subgroups. Recent studies have demonstrated that cancer is characterized as much by marginal differences in biomarker expression as by network topologies. Models, like the proposed one, statistically identify complex factors responsible for variation in drug response to cancer. We acknowledge that translating such research into drug development is a long and complicated process requiring several intermediate steps. However, we hope it would play an influential role in the development-targeted therapeutics in the long run.34,35 In general, the ability to accurately estimate stimulus-specific network topology can highly improve research within systems biology, pharmacology, and related disciplines.
Finally, we emphasize that for modeling biological networks across related disease subcategories, related genes, and protein-pathways targeted by the same drug, single independent network inference is no longer adequate. This is a problem that needs to be addressed with the growing relevance of network models in systems biology.
Footnotes
ACADEMIC EDITOR: JT Efird, Editor in Chief
FUNDING: Authors disclose no funding sources.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties.
Author Contributions
Conceived and designed the experiments: RM, PM, PQ, YJ. Analyzed the data: RM, PM, PQ, YJ. Wrote the first draft of the manuscript: RM, PM, PQ, YJ. Contributed to the writing of the manuscript: RM, PM, PQ, YJ. Agree with manuscript results and conclusions: RM, PM, PQ, YJ. Jointly developed the structure and arguments for the paper: RM, PM, PQ, YJ. Made critical revisions and approved final version: RM, PM, PQ, YJ. All authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci U S A. 1999;96:4285–88. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marcotte EM, Pellegrini M, Ng H-L, Rice DW, Yeates TO, Eisenberg D. Detecting protein function and protein–protein interactions from genome sequences. Science. 1999;285:751–3. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- 3.Aloy P, Russell RB. Structural systems biology: modelling protein interactions. Nat Rev Mol Cell Biol. 2006;7:188–97. doi: 10.1038/nrm1859. [DOI] [PubMed] [Google Scholar]
- 4.Linding R, Jensen LJ, Ostheimer GJ, et al. Systematic discovery of in vivo phosphorylation networks. Cell. 2007;129:1415–26. doi: 10.1016/j.cell.2007.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi YY, Miller GA, Qian H, Bomsztyk K. Free-energy distribution of binary protein–protein binding suggests cross-species interactome differences. Proc Natl Acad Sci U S A. 2006;103:11527–32. doi: 10.1073/pnas.0604316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berg J, Lässig M, Wagner A. Structure and evolution of protein interaction networks: a statistical model for link dynamics and gene duplications. BMC Evol Biol. 2004;4:51. doi: 10.1186/1471-2148-4-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeong H, Tombor B, Albert R, Oltvai ZN, Barabási AL. The large-scale organization of metabolic networks. Nature. 2000;407:651–4. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- 8.Rhodes DR, Tomlins SA, Varambally S, et al. Probabilistic model of the human protein–protein interaction network. Nat Biotechnol. 2005;23:951–9. doi: 10.1038/nbt1103. [DOI] [PubMed] [Google Scholar]
- 9.Irish JM, Hovland R, Krutzik PO, et al. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell. 2004;118:217–28. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 10.Sharan R, Suthram S, Kelley RM, et al. Conserved patterns of protein interaction in multiple species. Proc Natl Acad Sci U S A. 2005;102:1974–9. doi: 10.1073/pnas.0409522102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kholodenko BN. Cell-signalling dynamics in time and space. Nat Rev Mol Cell Biol. 2006;7:165–76. doi: 10.1038/nrm1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitra R, Müller P, Liang S, Yue L, Ji Y. A Bayesian graphical model for chip-seq data on histone modifications. J Am Stat Assoc. 2013;108:69–80. [Google Scholar]
- 13.Qiu P, Simonds EF, Bendall SC, et al. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol. 2011;29:886–91. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Besag J. Spatial interaction and the statistical analysis of lattice systems. J R Stat Soc Series B Methodol. 1974;36:192–236. [Google Scholar]
- 15.Mitra R, Mueller P, Ji Y. Bayesian Graphical Models for Differential Pathways. Technical Report, Austin: University of Texas at Austin; 2012. [Google Scholar]
- 16.Yuan M, Lin Y. Model selection and estimation in the Gaussian graphical model. Biometrika. 2007;94:19–35. [Google Scholar]
- 17.Friedman J, Hastie T, Tibshirani R. Sparse inverse covariance estimation with the graphical lasso. Bio Stat. 2008;9:432–41. doi: 10.1093/biostatistics/kxm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scott J, Carvalho C. Feature-inclusion stochastic search for Gaussian graphical models. J Comput Graph Stat. 2008;17:790–808. [Google Scholar]
- 19.Carvalho C, Scott J. Objective Bayesian model selection in Gaussian graphical models. Biometrika. 2009;96:497–512. [Google Scholar]
- 20.Danaher P, Wang P, Witten DM. The joint graphical lasso for inverse covariance estimation across multiple classes. J R Stat Soc Series B Stat Methodol. 2013;76(2):373–97. doi: 10.1111/rssb.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo J, Levina E, Michailidis G, Zhu J. Histone modifications as markers of cancer prognosis: a cellular view. Biometrika. 2011;98:1–15. [Google Scholar]
- 22.Chiquet J, Grandvalet Y, Ambroise C. Inferring multiple graphical structures. Stat Comput. 2011;21:537–53. [Google Scholar]
- 23.Hara S, Washio T. Learning a common substructure of multiple graphical Gaussian models. Neural Networks. 2013;38:23–38. doi: 10.1016/j.neunet.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 24.Yang E, Ravikumar PD, Allen GL, Liu Z. Graphical models via generalized linear models. Adv Neural Inf Process Syst. 2012;25:1367–75. [Google Scholar]
- 25.Mohan K, Chung MJ-Y, Han S, Witten DM, Lee S-L, Fazel M. Advances in Neural Information Processing Systems. 2012. Structured learning of Gaussian graphical models; pp. 629–37. [PMC free article] [PubMed] [Google Scholar]
- 26.Mohan K, London P, Fazel M, Lee S-L, Witten D. Node-based learning of multiple Gaussian graphical models. The Journal of Machine Learning Research. 2014 Jan;15(1):445–88. [PMC free article] [PubMed] [Google Scholar]
- 27.York H, Kornblau SM, Qutub AA. Network analysis of reverse phase protein expression data: characterizing protein signatures in acute myeloid leukemia cytogenetic categories t (8; 21) and inv (16) Proteomics. 2012;12:2084–93. doi: 10.1002/pmic.201100491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Segal E, Wang H, Roller D. Discovering molecular pathways from protein interaction and gene expression data. Bioinformatics. 2003;19:i264–72. doi: 10.1093/bioinformatics/btg1037. [DOI] [PubMed] [Google Scholar]
- 29.Lloyd CJ. Using smoothed receiver operating characteristic curves to summarize and compare diagnostic systems. J Am Stat Assoc. 1998;93:1356–64. [Google Scholar]
- 30.Bendall SC, Simonds EF, Qiu P, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–96. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 32.Jobin C, Haskill S, Mayer L, Panja A, Sartor RB. Evidence for altered regulation of I kappa B alpha degradation in human colonic epithelial cells. J Immunol. 1997;158:226–34. [PubMed] [Google Scholar]
- 33.Cesareni G, Ceol A, Gavrila C, Palazzi LM, Persico M, Schneider MV. Comparative interactomics. FEBS Lett. 2005;579:1828–33. doi: 10.1016/j.febslet.2005.01.064. [DOI] [PubMed] [Google Scholar]
- 34.Lee MJ, Ye AS, Gardino AK, et al. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149:780–94. doi: 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Csermely P, Korcsmáros T, Kiss HJ, London G, Nussinov R. Structure and dynamics of molecular networks: a novel paradigm of drug discovery: a comprehensive review. Pharmacol Ther. 2013;138:333–408. doi: 10.1016/j.pharmthera.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]