

Abstract

A series of organometallic ruthenium(II) complexes containing iminophosphorane ligands have been synthesized and characterized. Cationic compounds with chloride as counterion are soluble in water (70–100 mg/mL). Most compounds (especially highly water-soluble 2) are more cytotoxic to a number of human cancer cell lines than cisplatin. Initial mechanistic studies indicate that the cell death type for these compounds is mainly through canonical or caspase-dependent apoptosis, nondependent on p53, and that the compounds do not interact with DNA or inhibit protease cathepsin B. In vivo experiments of 2 on MDA-MB-231 xenografts in NOD.CB17-Prkdc SCID/J mice showed an impressive tumor reduction (shrinkage) of 56% after 28 days of treatment (14 doses of 5 mg/kg every other day) with low systemic toxicity. Pharmacokinetic studies showed a quick absorption of 2 in plasma with preferential accumulation in the breast tumor tissues when compared to kidney and liver, which may explain its high efficacy in vivo.
Introduction
In the search for metal-based chemotherapeutics with improved properties with respect to platinum-based drugs used in the clinic, ruthenium compounds have emerged as promising candidates.1−5 Ruthenium complexes have certain characteristics that make them attractive as potential chemotherapeutics for different diseases.4,5 Ruthenium compounds can easily access three different oxidation states (II, III, and possibly IV) in biological fluids. Ruthenium(III) compounds could potentially behave as pro-drugs as they can be reduced to ruthenium(II) derivatives in solid tumor masses where the low content in oxygen may act as a reducing environment. As platinum-based drugs, ruthenium compounds can exchange N and O-donor molecules with the added advantage of the possibility of forming octahedral complexes (of interest in reactions with DNA). Lastly, ruthenium derivatives probably use transferrin6,7 to accumulate into tumors due to the similarities with iron.
From all the ruthenium compounds reported as potential anticancer agents, there are four main groups (Chart 1) that have been studied in more detail and display important antitumor and/or antimetastatic activities and low toxicity.1−8 The first group corresponds to ruthenium(III) coordination complexes with two compounds currently undergoing clinical trials, NAMI-A4,9 (phase I/II), developed by Sava et al., and the compound KP1019 and its analogue containing Na+ KP1339,4,10 developed by Keppler and co-workers (phase I/II). A number of organometallic ruthenium(II) compounds with arene ligands (piano-stool structure) have also been described as promising candidates.4,6,11−19 Two relevant examples from the groups of Sadler (RM175) and Dyson (RAPTA-T), which have undergone advanced preclinical studies, are depicted in Chart 1. Another important group of ruthenium compounds in the preclinical stage is that of cyclometalated compounds based on pincer C,N ligands (RDC family).4,11,20,21 The recent strategy to bind a drug of well-known therapeutic value (such as curcumin, ketoconazole, clotrimazole, hydroxyflavones, hydroxyquinolinones, letrozole, indolobenzazepins, or aspirin) to ruthenium centers has rendered a number of complexes with improved properties with respect to the parent organic drugs for cancer, e.g., refs (22−29). In this context, ruthenium compounds resembling staurosporine (like DW1/2 in Chart 1) developed by Meggers and co-workers are relevant examples of potential chemotherapeutics targeting protein kinases.4,11,30−32
Chart 1. Selected Ruthenium(III) and (II) Compounds with Important Antitumor and/or Antimetastatic Properties (Refs (4−21,30−32) and Refs Therein).

A simple search on the SciFinder database on the concepts “ruthenium anticancer” since 2010 shows over 800 hits. There are now examples of multinuclear ruthenium compounds,33,34 of ruthenium derivatives which can be activated by light,35 that are thermoresponsive,36 that can be obtained by a combinatorial approach,37 as well as ruthenium compounds that can be delivered to tumor sites more efficiently by binding to polymers,38 nanocarriers,39,40 peptides,41,42 or transport proteins43 to mention a few advances in this field. However, there is still a need to find the ultimate target(s) for these ruthenium compounds as well as to get a better knowledge on the detailed molecular mechanism of action in order to develop more powerful and selective chemotherapeutics.4 In addition, more in vivo data is needed to make more reliable predictions of structure–biological activity correlations.8,18
We have reported that nontoxic iminophosphorane or iminophosphane (IM) compounds (R3P=N-R′, IM) are useful precursors for the preparation of coordination (N,N−) or cyclometalated (C,N−) complexes of d8 metals (Au(III), Pd(II), and Pt(II)) mono- or heterometallic (selected compounds a–g in Chart 2).
Chart 2. Selected Iminophosphorane (IM) d8 Transition Metal Complexes with Significant Anticancer Properties Prepared in Our Group44−49.

These IM metal complexes display high cytotoxicity in vitro (low micromolar to nanomolar) against a variety of human cancer cell lines with different degrees of selectivity.44−49 Organo-gold(III) complexes containing iminophosphorane ligands (e.g., a) exert cell death with pathways involving mitochondrial production of ROS.44,45 We have studied the interaction of the IM metal compounds with (pBR322) DNA and HSA.47−49 We have confirmed that some compounds (f, g) inhibit PARP-1 proteins.48 In most cases (including Pd(II) and Pt(II) derivatives), we have demonstrated that DNA is not the target for these compounds and that the complexes are highly active against cisplatin-resistant cancer cell lines (such as Jurkat sh-Bak, MCF7, or A2780cisR) pointing to a mode of action different from that of cisplatin.45−49
In this context, we aimed to explore the potential of IM complexes containing metals other than d8 transition metals (Au(III), Pd(II), and Pt(II)) as anticancer agents. We report here on the preparation and preliminary biochemical and biological studies (in vitro and in vivo effects on human cancer cells and xenografts) of organometallic ruthenium(II) compounds containing different iminophosphorane ligands.
Results and Discussion
1. Synthesis and Characterization
The synthesis of ruthenium(II) piano-stool complexes containing iminophosphorane ligands has been described by Urriolabeitia and co-workers.50 We have employed here p-cymene as the arene group coordinated to the ruthenium centers. Thus, compounds 1–4 can be easily obtained in high yields by the addition of different IM ligands previously described46−49,51 to [(η6-p-cymene)Ru(μ-Cl)Cl]252 (Scheme 1). 4 is an example of a new heterometallic ruthenium complex containing a ferrocene fragment.
Scheme 1. Preparation of Cationic Ruthenium(II) Compounds Containing IM Ligands.

Ruthenium(II) complexes with cyclometalated IM (pincer C,N−) ligands have been prepared by transmetalation with organomercury derivatives.50 However, the nature of the IM ligand played a crucial role and compounds with semistabilized IM ligands containing carbonyl groups like (2-C6H4)Ph2P=N-CO-Ph could not be obtained due to steric reasons.50
We aimed to prepare cycloruthenated compounds in which the aryl group of the imino fragment is coordinated to the metal center (exo derivatives) as opposed to an aryl group of the phosphine fragment (endo derivatives) in order to be able to incorporate different phosphines into the final molecule to tune electronic/steric properties of the resulting complexes. We followed the strategy for the preparation of Hg(Ph3P=N-CO-2-C6H4)Cl.53 The C–H activation at the N-CO-Ph fragments takes place at a manganese center, and by transmetalation of the resulting cyclometalated iminophosphorane manganese compounds to HgCl2, the organomercury derivatives with PPh3 [Hg(Ph3P=N-CO-2-C6H4)Cl]53 or water-soluble phosphine PTA [Hg(PTA=N-CO-2-C6H4)Cl] (7) are obtained in high yields. Transmetalation reactions of [Hg(Ph3P=N-CO-2-C6H4)Cl]53 and 7 with [(η6-p-cymene)Ru(μ-Cl)Cl]2 afford new cyclometalated compounds [(η6-p-cymene)Ru(IM-k-C,N)Cl] (IM = Ph3P=N-CO-2-C6H48; PTA=N-CO-2-C6H49) in high yields (Scheme 2).
Scheme 2. Preparation of the New Cycloruthenated Compounds 8 and 9 Containing IM Ligands.

All the compounds are obtained as air stable solids in moderate to high yields. Their structures have been proposed on the basis of microanalytical, spectroscopic (IR and NMR), conductivity, and MS data. Some of the compounds are slightly hygroscopic. As previously proposed by Urriolabeitia and co-workers,50 the IM ligand in compounds 1 and 2 is bonded as a chelate giving a fac-Cl,N,O arrangement, while in compounds 3 and 4, the arrangement is fac-Cl,N,N. This can be clearly inferred from the IR data. There is a strong absorption at 1531 cm–1 due to νCO stretch for 1 and 2 shifted to lower frequencies with respect to that of the free ligand at 1598 cm–1. For 3 and 4, the signal that can be assigned to νPN stretch is shifted to lower frequency than that of the free ligands (see Experimental Section). As previously described for compounds containing benzene and some IM ligands,50 the 31P{1H} NMR signals for 1 and 2 resemble that of the free ligand, whereas for 3 and 4, the signals are strongly shifted to low field with respect to the free ligands, indicating iminic N-bonding. In addition, the ortho protons of the quinoline (H2) and pyridine (H6) in the 1H NMR spectra for 3 and pyridine (H6) for 4 are shifted downfield, supporting the idea of N-coordination. Compound 4 shows fluxional behavior at room temperature (giving rise to broad signals in the 1H NMR spectrum in the area of the p-cymene and Cp rings), and its variable temperature NMR spectra are collected in the Supporting Information.
The structure of 1 has been determined by an X-ray analysis, and it is very similar to that of previously reported50 [(η6-C6H6)Ru(k-N,O-Ph3P=N-CO-2-N-C5H4)]PF6 with very close distances and angles. The molecular structure for the cation of 1 is depicted in Figure 1.
Figure 1.
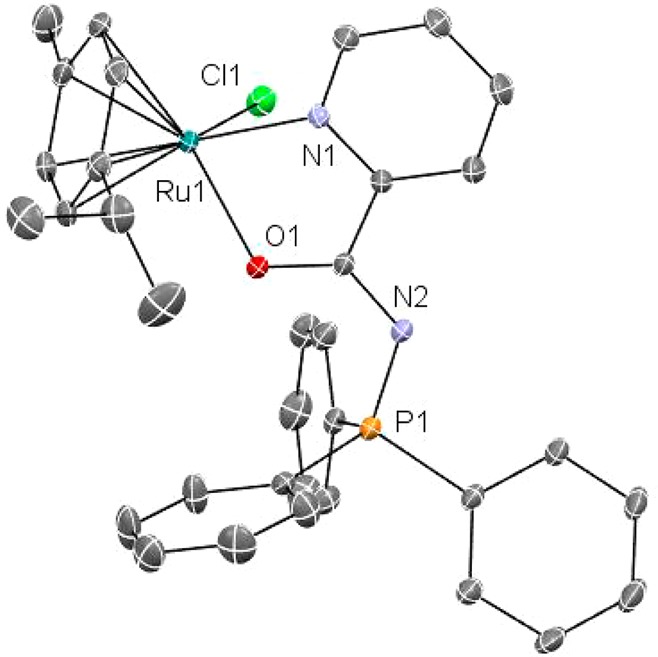
Molecular structure of the cation of compound 1. Selected distances (Å) and angles (deg): Ru(1)–O(1) 2.110(3), Ru(1)–N(1) 2.095(4), Ru(1)–C(10) 2.153(5), Ru(1)–C(6) 2.154(5), Ru(1)–C(7) 2.171(5), Ru(1)–C(9) 2.187(5), Ru(1)–C(11) 2.191(5), Ru(1)–C(8) 2.204(5), Ru(1)–Cl(1) 2.3775(14), O(1)–C(16) 1.266(6), C(1)–N(2) 1.337(7), C(1)–C(2) 1.385(8), N(2)–P(1) 1.619(5); N(1)–Ru(1)–O(1) 76.68(14), O(1)–Ru(1)–Cl(1) 83.61(10), N(1)–Ru(1)–Cl(1) 83.13(12).
The analysis confirms the piano-stool structure around the ruthenium center as well as the coordination of the IM ligand through the N and O atoms. Ru–C distances are on average slightly longer, and the O(1)–Ru(1)–N(1) and N(1)–Ru(1)–Cl(1) angles slightly smaller for 1 than for the compound with (η6-C6H6)50 as expected.
Compounds 8 and 9 are cycloruthenated neutral species with the IM ligand in an exo disposition [(η6-p-cymene)Ru(IM-k-C,N)] (IM = Ph3P=N-CO-2-C6H48, PTA=N-CO-2-C6H49). Their 1H NMR spectra show signals due to η6-arene as well as the C6H4P units. In addition, the 13C{1H} NMR spectrum shows six well-resolved peaks due to the C6H4P unit. The signals in the 31P{1H} NMR spectra are strongly shifted downfield with respect of those for the free ligand as reported before for endo cycloruthenated species.50
The cationic compounds with chloride as counterion (2, 3, and 4) are highly soluble in water (70–100 mg/mL). The cycloruthenated derivative 9 with a water-soluble phosphine PTA (9) is much less soluble in water (1 mg/mL 9). 1 and cyclorutenated 8 and 9 are soluble in micromolar concentrations in 1:99 DMSO:H2O mixtures. All the complexes but 9 are stable for weeks in DMSO-d6 solution (see spectra and stability table in the Supporting Information). The stability of the water-soluble complexes was studied by 31P{1H} and 1H NMR spectroscopy in D2O. The spectrum in D2O for compound [(η6-p-cymene)Ru{Cp-P(Ph2)=N-CH2-2-NC5H4}Fe(Cp)}]Cl (4) does not change for over 3 days, but after that time the compound precipitates. The 31P{1H} NMR spectrum in D2O for 3 (δ = 37.73 ppm) shows an additional signal (δ = 38.34 ppm) that may be assigned to a hydrolyzed species of the type [(η6-p-cymene)Ru(Ph3P=N-8-C9H6N)(OH2)]2+. The integration of these signals is ca. 45:55, and the spectrum does not change much over time (days). In the case of compound 2 (δ in D2O 26.33 ppm), a signal (attributable to [(η6-p-cymene)Ru(Ph3P=N-CO-2-N-C5H4)(OH2)]+ is also visible in D2O (δ = 26.65 ppm) along with another signal (δ = 43.79 ppm) which may correspond to the cyclometalated species [(η6-p-cymene)Ru(IM-k-C,N-C6H4(PPh2=N-CO-2-N-C5H4)Cl] or [(η6-p-cymene)Ru(IM-k-C,N-C6H4(PPh2=N-CO-2-N-C5H4)(OH2)]+ and grows overtime. The 1H NMR spectrum of 2 in D2O after 5 days shows (in addition to those of compound 2) signals due to new species containing η6-arene as well as the C6H4P unit indicating the cyclometalation of the PPh3 ring. In addition, the 13C{1H} NMR spectrum shows most of the six peaks due to this C6H4P unit. The doublet that can be assigned to the C=O peak for these new cyclometalated species appears at 176.06 ppm (versus 178.08 ppm for 2), and it is closer to the chemical shift observed when C=O is not coordinated to the ruthenium center (like in the case of the free ligand which displays a doublet at 175.34). The MS spectra of 2 in H2O overtime shows a peak at m/z = 617 with a pattern fitting that of cationic species [(η6-p-cymene)Ru(IM-k-C,N-C6H4(PPh2=N-CO-2-N-C5H4)]+ (see Supporting Information). We have reported cyclometalation processes at RT in DMSO of arylgroups from PPh3 in IM coordination complexes of Pd and Pt with the Ph3P=N-8-C9H6N IM ligand.49 The half-life for 2 (14.5 mM) in D2O is 2.5 days (although the process slows down for more concentrated samples). The cyclometalation process for 2 proceeds faster in a 100 mM NaCl solution in D2O (half-life ca. 10 h for a concentration of 2 of 14.5 mM) and by increasing the temperature (60% after 1 h at 80 °C). However, as it will be explained in section 2, the biological activity of compound 2 is very fast (in 8 h it induces 80% of apoptosis on Jurkat cells), and thus we believe that the biological activity observed comes mainly from coordination compound 2 or its hydrolysis product.
2. Biological Activity in Vitro
2.1. Antiproliferative Studies in Vitro
The antiproliferative properties of the new ruthenium complexes 1–4, 8, and 9 and of the starting material [(η6-p-cymene)Ru(μ-Cl)Cl]2 were assayed by monitoring their ability to inhibit cell growth using the MTT assay (see Experimental Section). Cytotoxic activity of the compounds was determined as described in the Experimental Section in several human cancer cell lines: leukemia Jurkat-T, lung A549, prostate DU-145, pancreas MiaPaca2, and triple negative breast MDA-MB-231, in comparison to cisplatin. The results are summarized in Table 1. The starting material [(η6-p-cymene)Ru(μ-Cl)Cl]2 is poorly cytotoxic in all tested cell lines (IC50 >125 μM). The IM ligands coordinated to the ruthenium centers are known to be poorly cytotoxic (IC50 in different cell lines >100–500 μM).46−49
Table 1. IC50 (μM) of Metal Complexes 1–4, 8–9, [(η6-p-cymene)Ru(μ-Cl)Cl]2a, and Cisplatin in Human Cell Linesb,c.
| Jurkat | A549 | DU-145 | MiaPaca2 | MDA-MB-231 | HEK-293T | |
|---|---|---|---|---|---|---|
| 1 | 1.1 ± 0.14 | 9.9 ± 1.9 | 1.89 ± 0.64 | 2.4 ± 0.18 | 4.91 ± 2.7 | 2.8 ± 0.2 |
| 2 | 0.78 ± 0.08 | 9.5 ± 2.1 | 1.55 ± 0.21 | 2.9 ± 0.8 | 2.61 ± 1.2 | 2.8 ± 0.2 |
| 3 | 0.9 ± 0.32 | 43.3 ± 8.0 | 6.6 ± 0.85 | 7.0 ± 0.4 | 16.2 ± 0.9 | 2.2 ± 1.1 |
| 4 | 9.3 ± 0.07 | >125 | 148 ± 33 | >125 | >125 | 114.5 ± 14.8 |
| 8 | 2.39 ± 0.27 | 29.9 ± 5.8 | 14.2 ± 4.2 | 8.2 ± 0.98 | 7.1 ± 0.11 | 4.1 ± 0.06 |
| 9 | 17.7 ± 7.5 | >125 | 125.5 ± 28 | 54.5 ± 16 | 75.4 ± 9.8 | 141.9 ± 13.1 |
| cisplatin | 10.8 ± 1.2 | 114.2 ± 9.1 | 112.5 ± 33 | 76.5 ± 7.4 | 131.2 ± 18 | 69.0 ± 6.7 |
IC50 for [(η6-p-cymene)Ru(μ-Cl)Cl]2 >125 μM in all cell lines.
Data are expressed as mean ± SD (n = 4).
All compounds were dissolved in 1% of DMSO and diluted with water before addition to cell culture medium for a 24 h incubation period. Cisplatin was dissolved in H2O.
Compounds with coordinated IM ligands 1–3 and with the IM-PPh3 cyclometalated ligand 8 were considerably more cytotoxic than cisplatin in all the cell lines studied. Compounds 1 and 2 (same cation) display almost identical IC50 values, but 2 is soluble in H2O. The ruthenium compound based on an iminophosphorane ligand containing a ferrocenyl phosphine Fe–Ru 4 was less cytotoxic than cisplatin. The bimetallic compounds Fe–Au and Fe–Pd also showed higher IC50 values when compared to trimetallic derivatives or compounds with different IM ligands. The cycloruthenated compound containing a water-soluble IM ligand (IM-PTA) was more cytotoxic than cisplatin for the pancreas MiaPaca2 and triple negative breast MDA-MB-231 cell lines.
To assess the compounds’ selectivity for cancerous cells with respect to normal cell lines, they were also screened for their antiproliferative effects on the nontumorigenic human embryonic kidney cells HEK293T. In most cases, the cytotoxicity is comparable for the cancerous and HEK293T cells. All compounds are more toxic to leukemia than to HEK293T cell lines (2–8 times) and compounds 1, 2, and 9 are more toxic to the prostate DU-145 cancer cell line than to HEK cell lines. In addition, 9 is more toxic to the pancreas MiaPaca2 cancer cell line and to the breast cancer MDA-MB-231 cell line than to HEK293T although the IC50s for those cell lines are higher than other compounds in the table.
Importantly, as HEK cell lines are immortalized cells that can display a higher sensitivity to chemicals, we measured the effect of compound 2 on human renal proximal tubular cells (RPTC). Renal proximal tubular cells in primary culture have been described as an in vitro model to study nephrotoxicity.54 The IC50 value (XTT assay 24 h, see Experimental Section) for 2 in this cell line was 13.84 ± 1.46 μM (the value obtained for cisplatin as control was 46.42 ± 2.46 μM, similar to a values previously reported55 for 24h in rabbit RPTC). Thus, 2 is more toxic to all the cancer cell lines studied than to the “healthy” human renal cell line and markedly more toxic to the leukemia Jurkat-T (17.7-fold), prostate DU145 (9-fold), triple negative breast cancer MDA-MB-231 (5.3-fold), and pancreas MiaPaca (4.7-fold) cancer cell lines.
2, with an IC50 of 2.64 μM, is 30 times more cytotoxic in MDA-MB-231 breast cancer cell lines than compound RM175 (Chart 1) with an IC50 of 62 μM under the same conditions (MTT, 24 h incubation). The toxicity of RM175 in HBL100 human epithelial cell lines (IC50 = 54 μM) was similar to that in breast cancer cell lines.14
2.2. Mechanism of Cell Death
The mechanism of cell death induced by 2 and 3 was explored in Jurkat cells. Nuclei morphology after 24 h incubation with 1 μM solution of 2 or 3 was analyzed by Hoechst staining. Typical apoptotic features, chromatin condensation and fragmentation, were detected as shown in Figure 2.
Figure 2.
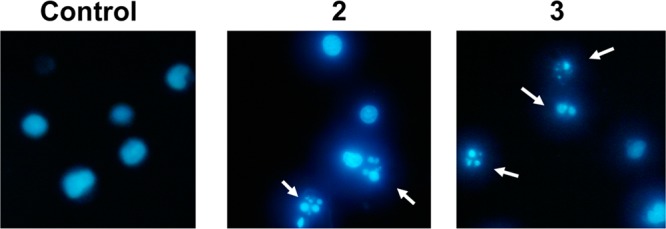
Nuclei morphology after treatment of Jurkat cells with compounds 2 and 3.
We then analyzed other apoptotic parameters such as phosphatidylserine exposure and mitochondrial membrane potential dissipation, using fluorescent probes as indicated in the Experimental Section. Dose–response experiments (Figure 3, right panel) confirmed that 2 is more cytotoxic than 3, as also indicated by MTT assays. Time-course experiments indicated that 2 induced apoptosis in around 80% of cells after 8 h treatment (Figure 3, left panel). Trypan blue staining confirmed that cell death was through apoptosis, with very low secondary necrotic cells at 6 h (data not shown). Phosphatidylserine exposure and loss of transmembrane mitochondrial potential occurred in the same percentage of cells, although at longer incubation periods, there was an apparent decrease in the percentage of AnnexinV positive cells that was in fact due to cell disintegration. Apoptosis induction was slower for 3, with the percentage of both AnnexinV+ and ΔΨmlow cells gradually increasing during the 24 h period of the experiments (Figure 3, left panel).
Figure 3.

Dose–response quantification of PS exposure (A) and time-course analysis of PS exposure and ΔΨm loss (B) caused by 2 and 3 in Jurkat cells.
Proteins of the Bcl-2 family are key regulators of apoptosis, and the levels of some members are modified in early phases of the process. Thus, we analyzed the effect of 2 in the levels of two antiapoptotic proteins of the Bcl-2 family (Bcl-XL and Mcl-1) and three proapoptotic members (Bim, Puma, and Noxa) in Jurkat cells (Supporting Information, Figure 31). Jurkat cells express very low levels of the other antiapoptotic member (Bcl-2). The main changes observed after treatment with 2 were a high increase in the levels of the propapoptotic proteins Noxa, Puma (isoform b), and Bim (isoform β). Also a slight decrease in the levels of the antiapoptotic members Bcl-XL and Mcl-1 was observed in cells treated with 2. The proapoptotic proteins are damage sensors that induce the activation of Bax and Bak and mitochondrial permeabilization to release cytochrome c, the key event in the intrinsic pathway of apoptosis.56
Taken together, these results point to a classical apoptosis mechanism of cell death. This was confirmed by the finding that the cytotoxicity of both compounds was caspase-dependent (Figure 4). The general caspase inhibitor z-VAD-fmk completely abrogated PS exposure but it only partially reduced ΔΨm loss (from 80% to 30% for 2 and from 40% to 20% for 3).
Figure 4.
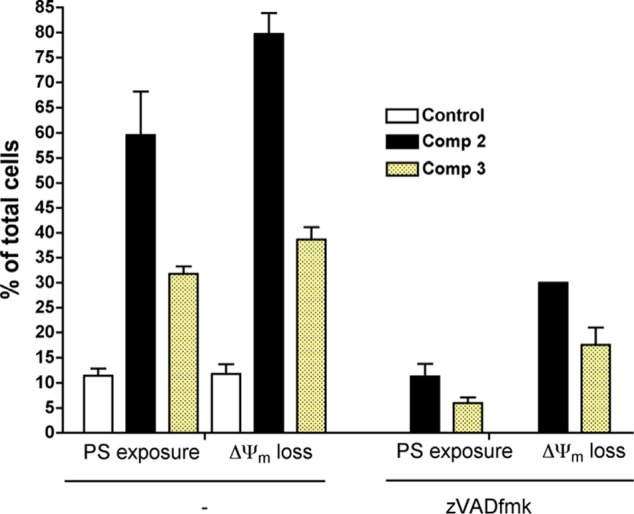
Effect of the general caspase inhibitor z-VAD-fmk in apoptotic features induced by 2 and 3.
Because ΔΨm disruption was not completely inhibited by z-VAD-fmk, we hypothesize that 2 and 3 could activate caspase-independent pathways acting on mitochondria. To determine whether caspase inhibition prevented or just delayed cell death, we performed experiments in which cells were treated with 2 or 3 for 24 h in the presence of z-VAD-fmk and then washed and further cultured in fresh medium for 24 h (Figure 5). PS exposure was analyzed after the first 24 h in the presence of compound+z-VAD-fmk and 24 h after washing. Only around 20% of cells were AnnexinV+ after 24 h incubation with 2+z-VAD-fmk and 24 h in fresh medium, while 70% of cells treated with 2 alone were apoptotic at the end of the experiments. Thus, these results indicate that in the case of compound 2, caspase inhibition prevented death commitment in a very high percentage of cells because only 22% were AnnexinV+ 24 h after washing. However, in the case of 3, the percentage of AnnexinV+ cells 24 h after washing (32%) equaled that of cell treated with 3 alone for 24 h (28%), suggesting that in this case caspase inhibition did not prevent irreversible cell damage leading to cell death. These results suggest that 2 and 3 could be acting through different mechanisms, with 3 causing caspase-independent premitochondrial damage. These differences in the mechanism of 2 and 3 could explain why 3 is less selective than 2 for tumor cell lines (Table 1).
Figure 5.

Analysis of long-term protection by z-VAD-fmk.
Ruthenium compounds have been reported to induce p53-dependent and -independent cytotoxicity.13,20 To determine whether the cytotoxicity of the compounds here is p53-dependent, we analyzed the levels of p53 in A549 cells, bearing wt-p53, after a short-term treatment with 2. As shown in Figure 6, treatment with 2 did not induce the stabilization of p53, and even at 3 and 6 h we observed a slight decline in p53 levels, probably due to cell death and protein loss. These results differ from that reported with other ruthenium organometallic compounds that induce short-term p53 accumulation.13,20 However, although p53 protein is induced by RM175 in HCT116 (colon carcinoma)13 or RDC-9 in A172 (glioblastoma) and HCT116,20 genetic inhibition of p53 does not avoid the cytotoxicity of these compounds, clearly indicating that other p53-independent mechanisms can be activated by ruthenium compounds. Moreover, Gaiddon et al. have shown that a p53–/– cell line (TK6) exhibits the same sensitivity to RDC-9 than its p53+/+ parental cell line (NH32).20 Furthermore, compounds 1–3 showed high toxicity against p53 mutated cell lines (Jurkat, MiaPaca2, DU-145, and MDA-MB-231) as shown in Table 1. Because the activity of cisplatin has been reported to be p53-dependent, new organometallic compounds that actívate p53-independent pathways could be useful in the treatment of tumors with alterations in p53, the most frequently mutated gene in human cancer.
Figure 6.

p53 protein levels after short-term incubation of A549 cells with 2. B-Actin levels were determined in the same membranes as a total protein loading control.
2.3. Reactivity with Biomolecules
2.3.1. Interactions with DNA
Because DNA replication is a key event for cell division, it is among critically important targets in cancer chemotherapy. Most cytotoxic platinum drugs form strong covalent bonds with DNA bases.57 However, a variety of platinum compounds act as DNA intercalators upon coordination to the appropriate ancillary ligands.58 The more thoroughly studied ruthenium antitumor agents (Chart 1) have displayed differences with respect to their interactions with DNA depending on their structure.4 Thus, while NAMI-A is known to have fewer and weaker interactions with DNA than cisplatin,4 indazolium bisindazoletetrachlororuthenate (KP1019) undergoes interactions similar to cisplatin but with a lower intensity in terms of DNA–DNA and DNA–protein cross-links.59 Organometallic piano-stool ruthenium(II) compounds based on biphenyl rings RM175 interact strongly with DNA binding to guanines and by intercalation.60,61 Organometallic ruthenium(II) RAPTA derivatives, characterized by the presence of water-soluble TPA phosphine, exhibit pH-dependent DNA damage: at the pH typical of hypoxic tumor cells DNA was damaged, whereas at the pH characteristic of healthy cells little or no damage was detected.62,63 Cycloruthenated compounds based on pincer C,N ligands (RDC family) displayed a much weaker interaction with plasmid (pBR322) DNA when compared to cisplatin.64,65 Complexes of the type [Ru(Cp)(2,2-bipy)(PR3)][CF3SO3] have shown no observable interaction with DNA.66
In this context, we evaluated the effect of DNA interactions that could, to some extent, contribute to the observed cytotoxicity of compounds 1–4, 8, and 9 and the apoptotic behavior of compounds 2 and 3. We followed the interaction with Calf Thymus DNA (CT DNA) by circular dichroism (CD), and with plasmid pBR322 DNA by electrophoresis in agarose gel. The CD spectral technique is very sensitive to diagnose alterations on the secondary structure of DNA that result from DNA–drug interactions. A typical CD spectrum of CT DNA shows a positive band with a maximum at 275 nm due to base stacking, and a negative band with a minimum at 248 nm due to helipticity, characteristic of the B conformation.67 Therefore, changes in the CD signals can be assigned to corresponding changes in DNA secondary structure. In addition, it is known that simple groove binding or electrostatic interaction of small molecules cause little or no alteration in any of the CD bands when compared to major perturbations induced by covalent binding or intercalation.
CD spectra of CT DNA incubated with compounds 1–4 (see Supporting Information, Figure S32) at 37 °C and pH = 7.30 in Tris/HCl buffer up to molar ratio drug/DNA = 0.5 show no modification of the DNA bands with respect to untreated CT DNA, indicating that drug–DNA interactions, if existing, do not induce any observable perturbation on the DNA secondary structure under our experimental conditions
Higher ratios were also tested, although loss of CD signal was observed due to precipitation of the DNA induced by 1–4, most likely because of phosphate charge neutralization by the cationic compounds, which suggests the existence of an electrostatic attraction. DNA condensation or precipitation by neutralization of backbone charges has been previously described for other ionic ruthenium drugs68 and confirmed by us for compounds 2 and 3 through ICP-MS analysis of metal content in the DNA precipitate. In this experiment, 500 μM concentration DNA solutions were treated with 2 equiv of ruthenium compounds 2 and 3 for 20 h at 37 °C to promote DNA precipitation. The samples were then centrifuged, and the resulting pellets were analyzed for DNA and metal concentration (see Experimental Section for more details). Our results show ruthenium content values of 2.60 ± 0.26 mg Ru/mg DNA for compound 2 and 2.43 ± 0.18 mg Ru/mg DNA for compound 3. This high Ru content in DNA precipitate, especially when compared to similar Ru compounds interacting with DNA through covalent interactions,24 suggests that the key factor promoting the precipitation of DNA is the presence of the ruthenium compound.
Attempts to obtain additional evidence of drug–CT DNA interactions were made by performing thermal denaturation experiments but resulted in cyclometalation of compounds 1 and 2 and hydrolysis of 3 at temperatures above 60 °C, as previously discussed, preventing us from obtaining reliable information through this technique.
To gain further insights on the nature of the compound–DNA interactions, gel electrophoresis studies were also performed with the ruthenium(II) complexes 1–4, 8, and 9 on plasmid (pBR322) DNA (Figure 7).
Figure 7.

Electrophoresis mobility shift assays for cisplatin, [Ru(η6-p-cymene)Cl]2, and compounds 1–4, 8, and 9 (see Experimental Section for details). DNA refers to untreated plasmid pBR322. A, B, C, and D correspond to metal/DNAbp ratios of 0.25, 0.5, 1.0, and 2.0, respectively.
For these experiments, cisplatin, all uncoordinated ligands, and the starting dimeric organometallic ruthenium(II) complex [Ru(η6-p-cymene)Cl]2 were also measured as controls. Plasmid pBR322 presents two main forms, OC (open circular or relaxed) and CCC (covalently closed or supercoiled), which display different electrophoretic mobility. Changes in the electrophoretic mobility of any of the forms upon incubation of the plasmid with a compound are usually interpreted as evidence of interaction. Generally, a drug that induces unwinding of the CCC form will produce a retardation of the electrophoretic mobility, while coiling of the OC form will result in increased mobility. Figure 4 shows the effect of cisplatin and compounds 1–4, 8, and 9 on DNA pBR322 after incubation at 37 °C for 20 h in Tris/HCl buffer up to drug/DNA ratio 2.0. As previously reported, cisplatin is able to both increase and decrease the mobility of the OC and the CCC forms, respectively.69 Interestingly, treatment with increasing amounts of 1 and 2 induce retardation of the mobility of the CCC form of plasmid DNA, while the rest of the compounds, the neutral ligands, and the Ru starting dimer do not seem to induce any alteration on the mobility of the plasmid.
The results of CD, ICP-MS, and gel electrophoresis taken together suggest that compounds 1–4 undergo only electrostatic interactions with DNA. This conclusion is supported by three main facts: (1) results obtained by CD spectroscopy do not show evidence of CT DNA modifications of secondary structure, suggesting that drug–DNA interactions, if any, are of weak nature, but neither covalent nor intercalation; (2) precipitation of CT DNA is observed in CD experiments at high ratios drug/DNA, and it is further confirmed by ICP-MS analysis of metal content in DNA precipitates, suggesting backbone charge neutralization; and (3) retardation of the plasmid DNA electrophoretic mobility is observed also at high drug/DNA ratios for compounds 1 and 2, but only when plasmid DNA is incubated with the cationic metal compounds and not with the neutral ligands or neutral ruthenium starting material under the same conditions, which could also be consistent with charge neutralization or DNA precipitation. Loss of migration in electrophoresis experiments has been previously reported as a consequence of DNA precipitation for other cationic ruthenium compounds.68 The electrophoretic mobility results also suggest that the electrostatic interaction between DNA and compounds 1 and 2 is of larger magnitude than that experienced by 3 and 4 because no mobility retardation is observed for the latter compounds up to drug/DNA ratio of 2.0. Further evidence of this could be found in the fact that lower amount of Ru content in DNA is detected for compound 3 when compared to compound 2, according to ICP-MS results.
Thus, we hypothesize that the antitumor properties observed for compounds 1–4, 8, and 9 are due to non-DNA related mechanisms/factors, as previously observed for other iminophosphorane complexes described by us44−49 and some other ruthenium compounds (refs (4, 5, and 8) and refs therein).
2.3.2. Inhibition of Capthesin B
Cathepsin B (cat B) is an abundant and ubiquitously expressed cysteine peptidase of the papain family, which has turned out to be a prognostic marker for several types of cancers.70 Cathepsin B seems to be involved (along with other cathepsins) in metastasis, angiogenesis, and tumor progression.71 It has been proposed that cat B may be a possible therapeutic target for the control of tumor progression.72 RAPTA Ru compounds which inhibit cat B with IC50 in the low micromolar range can reduce the mass and number of metastases in vivo.73 We therefore, studied the inhibition of Cat B by compound 2 (see Experimental Section for details). Compound 2 does not inhibit the enzymatic activity of cat B at concentrations up to 100 μM, indicating that this protease may not be a target for this type of arene–ruthenium(II) derivatives.
2.3.3. Interactions with HSA
Human serum albumin (HSA) is the most abundant carrier protein in plasma and is able to bind a variety of substrates including metal cations, hormones, and most therapeutic drugs. It has been demonstrated that the distribution, the free concentration, and the metabolism of various drugs can be significantly altered as a result of their binding to the protein.74 HSA possesses three fluorophores, namely tryptophan (Trp), tyrosine (Tyr), and phenylalanine (Phe) residues, with Trp214 being the major contributor to the intrinsic fluorescence of HSA. This Trp fluorescence is sensitive to the environment and binding of substrates, as well as changes in conformation that can result in quenching (either dynamic or static).
Thus, the fluorescence spectra of HSA in the presence of increasing amounts of the compounds 1–5, 8, 9, and cisplatin were recorded in the 300–450 nm range upon excitation of the tryptophan residue at 295 nm. The compounds caused a concentration dependent quenching of fluorescence without changing the emission maximum or the shape of the peak. All these data indicate an interaction of the compounds with HSA. The fluorescence data was analyzed by the Stern–Volmer equation. While a linear Stern–Volmer plot is indicative of a single quenching mechanism, either dynamic or static, the positive deviation observed in the plots of F0/F versus [Q] of compounds 1–4 (Figure 8) suggests the presence of different binding sites in the protein with different binding affinities.75 Of note, a similar behavior was observed in the case of coordination iminophosphorane complexes of d8 metals for which we also reported a concentration dependent fluorescence quenching.46−49 On the other hand, the Stern–Volmer plot for complexes 8 and 9 shows a linear relationship, suggesting the existence of a single quenching mechanism, most likely dynamic, and a single binding affinity. The Stern–Volmer constants for complexes 8 and 9 are 1.81 × 104 and 3.85 × 104 M–1, respectively.
Figure 8.

(A) Fluorescence titration curve of HSA with compound 4. Arrow indicates the increase of quencher concentration (10–100 mM). (B) Stern–Volmer plot for HSA fluorescence quenching observed with compounds 1–4, 8, 9, and cisplatin.
In general, higher quenching by the iminophosphorane complexes was observed compared to that of cisplatin under the chosen conditions, most likely due to the faster reactivity of our compounds with HSA, as compared to cisplatin.
3. Effects on Tumor Growth in Vivo with Compound 2
3.1. Evaluation of the Lethal and Maximum Tolerated Doses
The lethal and maximum tolerated doses of compound 2 were evaluated in C57/Black6 mice (see Experimental Section for details). The lethal dose was determined to be 30 mg/kg/day. No biological samples were collected from those mice. The MTD was determined to be 10 mg/kg/day, at which the mice showed no visible signs of distress over the 7 days course of treatment.
Mice lost weight during the trial in a dose dependent manner where mice treated with 5, 10, or 20 mg/kg/day lost 15%, 19%, or 37% body weight, respectively, while vehicle treated mice gained 3% body weight over the 7 days of treatment. Mice treated with 20 mg/kg/day were euthanized on day 6 of the trial as they had lost too much body weight and looked in distress.
Twenty-four h after the last dose, all the mice used in the MTD study were euthanized and blood plasma, liver, spleen, and kidneys were collected and used for histological analysis. Necropsy and histology indicate that mice treated at 20 mg/kg/day had discolored livers and atrophied spleens; at 10 mg/kg/day, much less atrophy and minor discoloration was observed, while in mice treated at 5 mg/kg/day, there was no detectable liver discoloration and no observable change in spleen size.
We therefore choose the dose of 5 mg/kg/every other day to conduct the subsequent in vivo trial with compound 2.
3.2. Effects of 2 in MDA-MB-231 Mouse Xenografts
Twelve female NOD.CB17-Prkdc SCID/J (nonobese diabetic–severe combined immunodeficiency) were selected for the in vivo trial. The mice were inoculated with MDA-MB-231 cells (see Experimental Section for details) and treated when the tumors were palpable (about 5–6 mm diameter). Each six MDA-MB-231-transplanted animals received compound 2 (5 mg/kg/every other day) or vehicle (0.9% NaCl) intraperitoneally (ip). To palliate the weight loss observed in the MTD study, all the mice used in this trial were fed a 46% fat-adjusted diet (Harlan Teklad, Madison, WI) plus HydroGel (Harlan Teklad, Madison, WI) and received subcutaneous injection of 100 μL normal saline on the off-treatment day.
In the group treated with 2 (see Table 2 and Figure 9), we observed a significant decrease in tumor size (shrinkage) of 56% from the starting volume between day 1 and day 28 of treatment (after a total of 14 doses), while in the control vehicle treated group, we observed 200% increase in tumor volume between day 1 and day 28 of treatment. One 2-treated mouse was removed from the trial because it was not feeding itself and showed other signs of distress, none of which were observed in the other mice on trial. We should note that there was no significant weight loss in mice treated with 5 mg/kg/every other day. Mice treated with 2 gained an average weight of 2.88%, while untreated mice gained an average of 18.67% weight (all groups were fed a 46% fat-adjusted diet).
Table 2. Effects of 2 on the Tumor Growth of MDA-MB-231 Mammary Carcinoma in NOD.CB17-Prkdc scid/J Mice.
| treatment group | primary tumor (mm3) |
|---|---|
| controls | 473.47 ± 45.44 |
| compound 2 (5 mg/kg/eoda × 14) | 59.58 ± 8.66 |
eod = every other day. Tumor measured on day 28, after the 14th dose.
Figure 9.
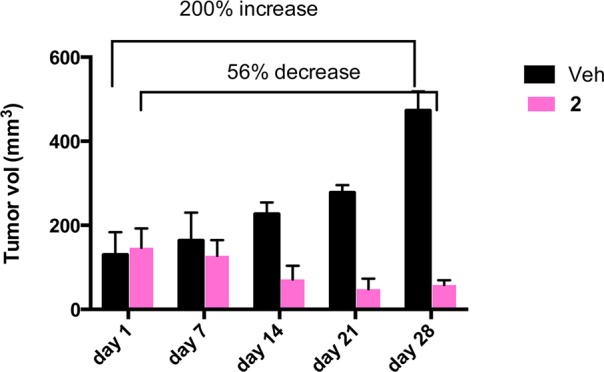
Percent of reduction of tumor burden in a cohort of 12 female NOD.CB17-Prkdc scid/J mice inoculated subcutaneously with 5 × 106 MDA-MB-231 cells. The treatment started when tumors were palpable (5–6 mm diameter). Six mice were treated with compound 2 (pink bars), six were treated with the vehicle 100 μL of normal saline (0.9% NaCl) (black bars). 2 was administered in the amount of 5 mg/kg/every other day.
The results clearly indicate that compound 2 is extremely efficient in vivo because it not only inhibits tumor growth but also results in the decrease in the size of the tumors by 56%. It is interesting to compare these results with those obtained with other arene–ruthenium(II) derivatives. Compound RM175 was reported to have a final primary tumor growth inhibition of 30% at a dose of 7.5 mg/kg/day in an in vivo trial for breast cancer in mice,14 while the compounds RAPTA-C reduced the growth of lung metastases in CBA mice bearing the MCa mammary carcinoma in the absence of a corresponding action at the site of primary tumor growth.16,17 More recently, a ruthenium–arene complex with a perfluoroalkyl-containing amine ligand demonstrated a 90% reduction in the tumor growth in a xenografted ovarian carcinoma tumor (A2780) grown in a chorioallantoic membrane (CAM) assay of chicken embryo.19 As stated above, compound 2 is able to decrease tumor size.
3.3. Pharmacokinetic Study
The pharmacokinetic profile of compound 2 in the NOD.CB17-Prkdc scid/J mice used for the in vivo study described above (Figure 10) is summarized in Table 3. Ruthenium content was determined using inductively coupled plasma–mass spectrometry (ICP-MS). Compound 2 was absorbed quickly into plasma (t1/2 abs = 0.5 h), and the peak plasma concentration was reached within 2 h of dosing. The drug was eliminated slowly from the blood compartment with an elimination half-life (t1/2e) greater than 12 h. This elimination half-life is similar to that reported for compounds RAPTA-C,14 NAMI-A,76 and KP1019.10
Figure 10.
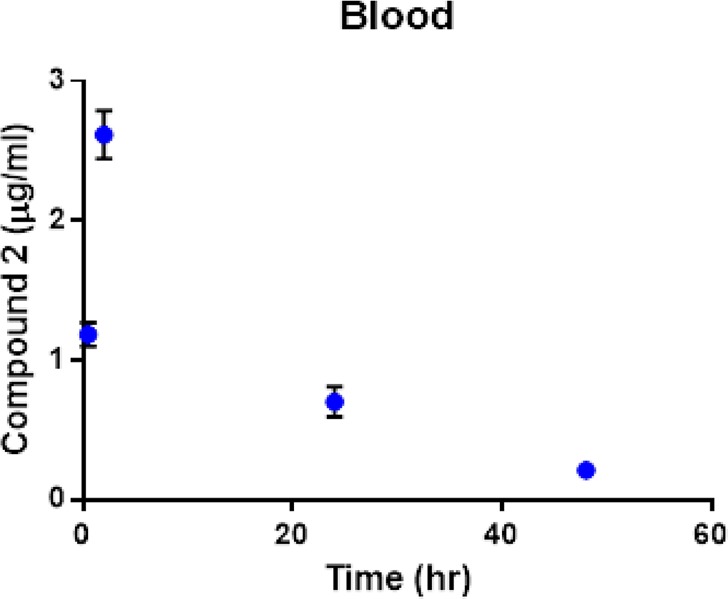
Concentration of compound 2 (ruthenium content) in plasma at various intervals after the first dose.
Table 3. Pharmacokinetic Parameters of Compound 2 after First Injection in NOD.CB17-Prkdc SCID/J Mice.
| pharmacokinetic parameters | values |
|---|---|
| Kabs (h –1) | 1.39 |
| Ke (h –1) | 0.055 |
| t1/2e (h) | 12.67 |
| t1/2abs (h) | 0.50 |
| tmax (h) | 2.00 |
| Cmax (μg/mL) | 2.62 |
| AUCtotal (μg·h/mL) | 54.47 |
| Vapp (mL) | 33.57 |
| CLapp (mL/h) | 1.84 |
During determination of total area under the concentration–time curve (AUC total), only 7% of the AUC was extrapolated from the last time point, suggesting a high confidence in the AUC, Vapp, and apparent clearance (CLapp) determination. Blood concentration, at 6 h after the last dose of compound 2, was 4.2 ± 1.3 μg/mL, which is higher (P < 0.1) than the Cmax after the first dose. This suggests an accumulation of compound 2 after each dose. While it is hard to make comparisons with other ruthenium compounds for which a PK analysis has been performed (e.g., RAPTA-C,16 NAMI-A,76 and KP101910) due to the different structures, oxidation states (see Chart 1), and the amounts employed in these studies (for example 70 mg total of 2 in 14 doses of 5 mg/kg/every other day versus a total of 400 mg or 200 mg for RAPTA-C), there are some differences that can be pointed out. The Vd (volume of distribution) of 2 when compared to that of more structurally related arene–ruthenium(II) RAPTA-C derivative is smaller, which may be due to a higher water solubility of 2 or the possibility that it binds strongly to plasma proteins. Indeed, we have seen in a qualitative way that compound 2 binds faster to HSA than cisplatin (section 2.3). Further studies on the interaction of 2 with plasma proteins are underway.
At the end of the study, ruthenium content in liver, kidney, and tumor was determined (Figure 11). The level of compound 2 in liver and kidney was less than 5 μg/g tissue weight, while the tumor concentration was about 40 μg/g. The high level in tumor suggests enhanced tumor accumulation of compound 2, which may explain the high efficacy observed for this compound in the in vivo studies. The preferential accumulation of some ruthenium complexes in neoplastic masses in comparison with normal tissue has been reported before by different researchers.77,78
Figure 11.
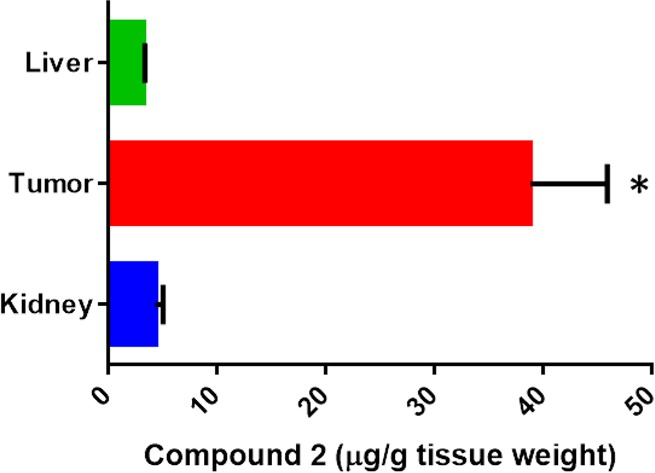
Compound 2 ruthenium content in tissues at the end of efficacy study. Data represents mean ± SD N = 3; * indicates P < 0.05.
Conclusions
To conclude, we have demostrated the potential of a highly water-soluble ruthenium–arene–iminophosphorane compound (2) as an anticancer agent. This compound is active against a number of cisplatin resistant cell lines while being less toxic on human renal proximal tubular cell lines. Initial mechanistic studies indicate that the cell death type for compound 2 is mainly through canonical or caspase-dependent apoptosis. In addition, cell death seems not to be dependent on p53. The interaction of 2 with DNA is weak and electrostatic in nature. Compound 2 does not inhibit protease cathepsin B in concentrations of 100 μM or lower. The efficacy of 2 in vivo has been demostrated on xenografted breast carcinoma MDA-MB-231 tumors grown on NOD.CB17-Prkdc scid/J mice. An impressive tumor reduction (shrinkage) of 56% was observed after 28 days treatment (14 doses of 5 mg/kg every other day) with low systemic toxicity. Pharmacokinetic studies showed a quick absorption of 2 in plasma with an elimination half-life of 12.67 h (similar to that reported for other ruthenium derivatives). Importantly, 2 accumulated preferentially in the breast tumor tissues when compared to kidney and liver, which may explain its high efficacy in vivo. The simple, cheap, and accessible synthesis of compound 2, its high water-solubility, and its encouraging preliminary biological activity in vitro and in vivo makes it therefore a good candidate for further evaluation as a potential chemotherapeutic agent.
Experimental Section
All manipulations involving air-free syntheses were performed using standard Schlenk-line techniques under a nitrogen atmosphere or a glovebox MBraun MOD System. Solvents were purified by use of a PureSolv purification unit from Innovative Technology, Inc. Compounds [PhCH2Mn(CO)5],79 [(η6-p-cymene)Ru(μ-Cl)Cl]2,52 [Hg(2-C6H4C(O)N=PPh3)Cl],53 and IM ligands Ph3P=N-CO-2-N-C5H4,51 Ph3P=N-8-C9H6N,49 [Cp-P(Ph2)=N–CH2-2-NC5H4}Fe(Cp)],48 and Ph3P=N-CO-2-C6H451 were prepared by reported methods. The purity of the compounds, based on elemental analysis, is ≥99.5%. In the case that the compound crystallizes with solvent, the 1H NMR (CDCl3) spectrum is available in the Supporting Information. Elemental analyses were performed by Atlantic Microlab Inc. NMR spectra were recorded in a Bruker AV400 (1H NMR at 400 MHz, 13C NMR at 100.6 MHz, 31P NMR at 161.9 MHz). Chemical shifts (δ) are given in ppm using CDCl3 or DMSO-d6 as solvent, unless otherwise stated. 1H and 13C chemical shifts were measured relative to solvent peaks considering TMS = 0 ppm; 31P{1H} was externally referenced to H3PO4 (85%). Infrared spectra (4000–250 cm–1) were recorded on a Nicolet 6700 FT-IR spectrophotometer from nujol mulls between polyethylene sheets. Mass spectra (electrospray ionization, ESI) were performed on an Agilent Analyzer, a Bruker Analyzer, or a Waters Q-Tof Ultima analyzer. Conductivity was measured in an OAKTON pH/conductivity meter in CH3CN solutions (10–3 M). X-ray collection was performed at room temperature on a Kappa CCD diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Electrophoresis experiments were carried out in a Bio-Rad Mini subcell GT horizontal electrophoresis system connected to a Bio-Rad Power Pac 300 power supply. Photographs of the gels were taken with an Alpha Innotech FluorChem 8900 camera. Fluorescence intensity measurements were carried out on a PTI QM-4/206 SE spectrofluorometer (PTI, Birmingham, NJ) with right angle detection of fluorescence using a 1 cm path length quartz cuvette. Circular dichroism spectra were recorded using a Chirascan CD spectrometer equipped with a thermostated cuvette holder. The inhibition of capthesin B experiments were performed by Reaction Biology Corporation.
Synthesis
[(η6-p-Cymene)Ru{(Ph3P=N-CO-2-N-C5H4)-κ-N,O}Cl](PF6) (1)
[(η6-p-Cymene)Ru(μ-Cl)Cl]2 (0.15 g, 0.25 mmol) was dissolved in MeOH (15 mL), and Ph3P=N(CO)(C5H5-2-N) (0.19 g, 0.5 mmol) was added. To the resulting mixture, KPF6 (0.10g, 0.55 mmol) was added. The mixture was stirred for 4 h. The suspension was then filtered and washed 3 times with Et2O (10 mL). The solution was then concentrated, and the precipitate was collected by filtration and dried in vacuo. Yield: 0.36 g (90%). Anal. Calcd for C34H33N2OP2F6ClRu (798.07): C, 51.17; H, 4.17; N, 3.51. Found: C, 50.95; H, 4.20; N 3.56%. ESI-MS: m/z: 653.10 (100%, [M – PF6]+, calcd 653.11). 31P {1H} NMR (CDCl3): δ 25.72 (s), −148.26 to −140.19 (septet, PF6), (DMSO-d6): 24.27 (s), −152.87 to −130.98 (septet, PF6). 1H NMR (CDCl3): δ 1.10 (6H, dd, J = 6.9, 18.9 Hz, CH3, η6-p-cymene), 1.69 (3H, s, CH3, η6-p-cymene), 1.65 (3H, s, CH3, η6-p-cymene), 2.30 (1H, m, CH, η6-p-cymene), 5.48–5.59 (4H, m, CH, η6-p-cymene), 7.64–7.78 (16H, m, H5, Hm + Ho + Hp), 8.03 (1H, d, J = 7.7 Hz, H4, C5H4N), 8.35 (1H, d, J = 7.5 Hz, H3, C5H4N), 9.21 (1H, d, J = 5.3 Hz, H6, C5H4N). 13C{1H} (CDCl3): δ 18.10 (s, CH3, η6-p-cymene), 21.87 (s, CH3, η6-p-cymene), 22.18 (s, CH3, η6-p-cymene), 30.79 (s, CH, η6-p-cymene), 81.00 (s, CH, η6-p-cymene), 82.09 (d, CH, J = 8.3 Hz, η6-p-cymene), 82.41 (s, CH, η6-p-cymene), 83.92 (s, C, η6-p-cymene), 124.5 (s, C2, η6-p-cymene), 125.5 (d, Cipso, J = 100.4 Hz), 127.7 (s, C3, C5H4N), 129.5 (d, Cm, J = 13.0 Hz), 129.8 (s, C5, C5H4N), 133.2 (d, Co, J = 10.4 Hz), 133.8 (s, Cp), 139.3 (s, C4, C5H4N), 153.7 (s, C6) ppm. The signal due to C2 (C5H4N) and C=O was not observed. IR (cm–1): ν 524 (Ru–N), 834 (v br, PF6–), 1116 (N=P), 1540 (C=O). Conductivity (acetone): 125.5 μS/cm (1:1 electrolyte).
[(η6-p-Cymene)Ru{(Ph3P=N-CO-2-N-C5H4)-κ-N,O}Cl]Cl (2)
[(η6-p-Cymene)Ru(μ-Cl)Cl]2 (0.15 g, 0.25 mmol) and Ph3P=N(CO)(C5H5-2-N) (0.19 g, 0.5 mmol) were stirred in acetone (20 mL) for 3 h. The brown solution was concentrated and 30 mL of Et2O added dropwise. The orange solid that formed was collected by filtration and dried in vacuo. Yield: 0.33 g (94%). Anal. Calcd for C34H33N2OPCl2Ru·2H2O (724.62): C, 56.36; H, 5.15; N, 3.87. Found: C, 56.41; H, 4.99; N 3.87%. ESI-MS: m/z: 653.1 (100%, [M – Cl]+, calcd 653.01), 618.1 (100%, [M – 2Cl]2+, calcd 618.1), 519.0 (100%, [M-p-cymene-Cl]+, calcd 519.0). 31P {1H} NMR (CDCl3): δ 25.18 (s), (DMSO-d6): 28.99 (s). 1H NMR (CDCl3): δ 1.00 (6H, dd, J = 7.0, 24.8 Hz, CH3, η6-p-cymene), 2.01 (3H, s, CH3, η6-p-cymene), 2.25 (1H, m, CH, η6-p-cymene), 5.51–5.71 (4H, m, CH, η6-p-cymene), 7.57 (6H, m, Hm), 7.68 (9H, m, Ho + Hp), 7.87 (1H, d, J = 6.0 Hz, H5, C5H4N), 8.03 (1H, d, J = 7.7 Hz, H4, C5H4N), 8.32 (1H, d, J = 7.4 Hz, H3, C5H4N), 9.62 (1H, d, J = 5.0 Hz, H6, C5H4N); 13C{1H} (CDCl3): δ 18.47 (s, CH3, η6-p-cymene), 21.82 (s, CH3, η6-p-cymene), 22.39 (s, CH3, η6-p-cymene), 30.75 (s, CH, η6-p-cymene), 81.20 (s, CH, η6-p-cymene), 82.60 (d, CH, J = 8.3 Hz, η6-p-cymene), 83.65 (s, CH, η6-p-cymene), 97.87 (s, C, η6-p-cymene), 102.5 (s, C, η6-p-cymene), 125.6 (d, Cipso, J = 101.2 Hz), 127.2 (s, C3, C5H4N), 129.5 (d, Cm, J = 12.8 Hz), 130.3 (s, C5, C5H4N), 133.3 (d, Co, J = 10.2 Hz), 133.9 (s, Cp), 139.0 (s, C4, C5H4N), 151.6 (d, C2, J = 24.0 Hz), 156.3 (s, C6), 176.8 (d, C=O, J = 24.14 Hz) ppm. IR (cm–1): ν 527 (Ru–N), 1114 (N=P), 1535 (C=O). Conductivity (acetone): 124.20 μS/cm (1:1 electrolyte). Solubility: 145.3 mM or 100 mg/mL (H2O). pH (5 × 10–5 M in H2O): 5.76.
[(η6-p-Cymene)Ru{(Ph3P=N-8-C9H6N)-κ-N,N}Cl]Cl (3)
[(η6- p-Cymene)Ru(μ-Cl)Cl]2 (0.092 g, 0.15 mmol) was dissolved in CH2Cl2 (15 mL), and Ph3P=N-8-C9H6N (0.12 g, 0.3 mmol) was added. The mixture was stirred for 3 h. The solution was concentrated to 2 mL and 20 mL of Et2O added to precipitate an orange solid, which was filtered and dried in vacuo. Yield: 0.18 g (84%). Anal. Calcd for C37H35N2PCl2Ru·2.5H2O (755.68): C, 58.81; H, 5.34; N, 3.71. Found: C, 58.95; H, 4.81; N 3.82%. ESI-MS: m/z: 675.13 (100%, [M – Cl]+, calcd 675.13), 540.0 (100%, [M-p-cymene-Cl]+, calcd 541.02). 31P {1H} NMR (CDCl3): δ 37.68 (s), (DMSO-d6): 37.84 (s). 1H NMR (CDCl3): δ 0.73 (3H, d, J = 6.7 Hz, CH3, η6-p-cymene), 1.01 (3H, d, J = 6.6 Hz, CH3, η6-p-cymene), 1.98 (3H, s, CH3, η6-p-cymene), 2.44 (2H, m, CH, η6-p-cymene), 5.43 (2H, s, CH, η6-p-cymene), 5.72 (2H, s, CH, η6-p-cymene), 6.40 (1H, d, J = 8.0 Hz, H7, C9H6N), 6.91 (1H, d, J = 8.1 Hz, H7, C9H6N), 7.15 (1H, d, J = 8.1 Hz, H7, C9H6N), 7.66 (9H, m, Hm + Hp), 7.68 (1H, s, H3, C9H6N), 8.00 (6H, m, Ho), 8.21 (1H, d, J = 8.4 Hz, H4, C9H6N), 8.21 (1H, s, H2, C9H6N). 13C{1H} (CDCl3): δ 19.20 (s, CH3, η6-p-cymene), 21.02 (s, CH3, η6-p-cymene), 22.96 (s, CH3, η6-p-cymene), 31.62 (s, CH, η6-p-cymene), 118.4 (s, C5, η6-p-cymene), 121.7 (d, C7, J = 10.3 Hz, η6-p-cymene), 124.6 (s, C3, η6-p-cymene), 125.8 (s, C6), 129.4 (s, C8), 129.6 (d, Cm, J = 12.7 Hz), 130.0 (s, Cipso), 134.1 (s, Cp), 134.9 (d, Co, J = 9.8 Hz), 138.4 (d, C4), 144.7 (s, C9H6N), 144.9 (s, C9H6N), 149.2 (s, C=NP) ppm. Signals due to the quaternary C atoms were not observed. IR (cm–1): ν 519 (Ru–N), 1268 (N=P). Conductivity (acetone): 125.60 μS/cm (1:1 electrolyte). Solubility: 112.6 mM or 80 mg/mL (H2O).
[(η6-p-Cymene)Ru([{Cp-P(Ph2)=N–CH2-2-NC5H4}Fe(Cp)]-κ-N,N)Cl]Cl (4)
To a solution of [(η6-p-cymene)Ru(μ-Cl)Cl]2 (0.16 g, 0.26 mmol) in CH2Cl2 (10 mL), [{Cp-P(Ph2)=N-CH2-2-NC5H4}Fe(Cp)] (0.25 g, 0.52 mmol) in CH2Cl2 (10 mL) was added and stirred for 40 min. The solvent removed to dryness under reduced pressure. The solid was dissolved in CH2Cl2, and 25 mL of Et2O were added. The solid formed was then filtered and dried in vacuo. Yield: 0.30 g (81%). Anal. Calcd for C38H39N2FePCl2Ru·3.5H2O (845.59): C, 53.98; H, 5.48; N, 3.31. Found: C, 54.02; H, 5.30; N 3.47%. ESI-MS: m/z: 747.0 (100%, [M – Cl]+, calcd 747.1), 613.0 (100%, [M-p-cymene-Cl], calcd 612.98). 31P {1H} NMR (CDCl3): δ 46.40 (s), (DMSO-d6): 45.99 (s), (D2O): 46.95 (s). 1H NMR (CDCl3): δ 0.99 (3H, d, J = 5.4 Hz, CH3, η6-p-cymene), 1.23 (3H, m, CH3 + CH2, η6-p-cymene, NCH2C), 2.03 (3H, s, CH3, η6-p-cymene), 3.51 (1H, m, CH, η6-p-cymene), 3.98 (5H, s, C5H5), 4.42–4.78 (6H, m, CH2 + C5H4), 4.79 (2H, m, Cp), 5.17–5.62 (4H, m, CH, η6-p-cymene), 7.45–7.68 (10H, m, Hm + Ho + Hp), 7.83 (1H, d, J = 7.4 Hz, H5, C5H4N), 7.98 (2H, d, J = 7.7 Hz, H3+4, C5H4N), 9.09 (1H, d, J = 4.7 Hz, H6, C5H4N). 13C{1H} (CDCl3): δ 18.91 (s, CH3, η6-p-cymene), 21.99 (d, CH3, η6-p-cymene), 23.25 (s, CH3, η6-p-cymene), 31.30 (s, CH, η6-p-cymene), 70.55 (s, Cp), 72.24 (d, J = 9.9 Hz, Cp), 73.73 (d, J = 9.9 Hz, Cp), 75.03 (m, Cp), 83.29 (d, CH, J = 14.4 Hz, η6-p-cymene), 85.98 (d, 2CH, J = 8.3 Hz, η6-p-cymene), 87.09 (s, CH, η6-p-cymene), 99.99 (s, C, η6-p-cymene), 103.8 (s, C, η6-p-cymene), 124.9 (s, Ph), 128.4–128.8 (m, Ph), 130.2 (s, Cipso), 133.0–133.3 (s, Ph), 133.9–134.0 (d, C3 + C4, J = 10.1 Hz, C5H4N), 138.8 (s, C4, C5H4N), 155.1 (s, C2, C5H4N), 164.1 (s, C6) ppm. . IR (cm–1): ν 488 (Ru–N), 1116 (N=P). Conductivity (MeCN): 130.37 μS/cm (1:1 electrolyte). Solubility: 89.5 mM or 70 mg/mL (H2O).
[PTA=N-C(O)-2-C6H5] (5)
PTA (0.34 g, 2.18 mmol) and benzamide (0.264 g, 2.18 mmol) were placed in a Schlenk flask under nitrogen. Dry, degassed THF (10 mL) was added and to this solution, tBuDAD (N,N-bis(tert-butyl)1,4-diazabutadiene) (0.503g, 2.18 mmol) in dry and degassed THF (4 mL) was added dropwise at 0 °C. The reaction was left stirring at RT for 2.5 h. After this period, the solvent was removed to dryness under reduced pressure. The white residue was washed three times with Et2O (15 mL), giving a white solid that was filtered and dried in vacuo. Yield: 0.53 g (88%). Anal. Calcd for C13H17N4OP (276.11): C, 56.52; H, 6.20; N, 20.28. Found: C, 55.55; H, 6.16; N 20.69%. ESI-MS: m/z: 277.12.0 (99.6%, [M]+, calcd 276.11). 31P{1H} NMR (CDCl3): δ −30.8 (s). 1H NMR (CDCl3): δ 4.32–4.63 (12H, m, PTA), 7.38 (2H, t, J = 7.6 Hz, H3 + H5, C6H5), 7.44 (1H, t, J = 7.1 Hz, H4, C6H5), 8.07 (2H, d, J = 7.5 Hz, H2 + H6, C6H5). 13C{1H} NMR (CDCl3): δ 55.10 (d, J = 47.2 Hz, PTA), 72.60 (d, J = 8.9 Hz, PTA), 128.08 (s, C3 + C5, C6H4), 129.21 (s,C2 + C6, C6H4), 131.47 (s, C4), 136.74 (d, J = 17.7 Hz, C1), 179.28 (s, J = 9.6 Hz, C=O). Conductivity (acetone): 2.08 μS/cm (neutral).
[(CO)4Mn(2-C6H4C(O)N=PTA)] (6)
PhCH2Mn(CO)5 (0.43 g, 1.5 mmol) and PTA=NC(O)Ph (5) (0.41 g, 1.5 mmol) were refluxed in n-hexane (45 mL) for 4 h. The hot solution was filtered, and the yellow filtrate reduced in volume until signs of crystallization became evident. Storage at −20 °C gave yellow crystals of (CO)4Mn(2-C6H4C(O)N=PTA). Yield: 0.59 g (90%). Anal. Calcd for C17H16N4O4PMn (442.02): C, 46.07; H, 3.65; N, 12.67. Found: C, 45.69; H, 3.56; N 12.72%. ESI-MS: m/z: 443.03 (100%, [M], calcd 443.03), 415.04 (100%, [M – CO], calcd 415.04), 386.01 (100%, [M – 2CO], calcd 387.04), 331.05 (100%, [M – 4CO], calcd 331.05). 31P {1H} NMR (CDCl3): δ −15.27 (s). 1H NMR (CDCl3): δ 4.32–4.58 (12H, m, PTA), 7.16 (1H, t, J = 7.5, 14.8 Hz, H4, C6H4), 7.38 (1H, t, J = 7.5, 14.5 Hz, H3, C6H4), 7.68 (1H, d, J = 7.6 Hz, H2, C6H4), 7.93 (1H, d, J = 7.4 Hz, H5, C6H4). 13C{1H} (CDCl3): δ 53.30 (d, J = 43.3 Hz, PTA) ppm. 72.54 (d, J = 9.8 Hz, PTA), 124.02 (s, C4, C6H4), 128.60 (s, C2, C6H4), 132.55 (s, C3, C6H4), 139.93 (s, C1), 141.25 (s, C5, C6H4), 171.36 (s, C=O), 185.71 (s, C=O), 213.66 (s, C=O), 215.97 (s, C=O) ppm. Conductivity (acetone): 1.11 μS/cm (neutral).
[Hg(2-C6H4C(O)N=PTA)Cl] (7)
(CO)4Mn(2-C6H4C(O)N=PTA) (6) (0.44 g, 1.0 mmol) and HgCl2 (0.54 g, 2.0 mmol) were refluxed in methanol (55 mL) for 5 h, during which time the solution turned yellow and a white solid formed. The mixture was cooled in an ice–water bath and subsequently filtered. The white solid formed was washed well with cold methanol. The solid was redissolved in CH2Cl2 (<200 mL) and filtered through Celite. The resulting clear solution was reduced in volume (<3 mL), and Et2O was added dropwise until the solution became cloudy. Storage at −20 °C gave white crystals of 7, which were filtered off, dried, and used without further purification. Yield: 0.33 g (65%). Anal. Calcd for C13H16N4OPClHg·0.5CH2Cl2 (553.78): C, 29.28; H, 3.09; N, 10.112. Found: C, 29.08; H, 3.02; N, 9.75%. ESI-MS: m/z: 513.05 (100%, [M + H]+, calcd 513.05), 535.03 (100%, [M + Na]+, calcd 535.04). 31P {1H} NMR (CDCl3): δ −25.68 (s, 2JHg–P = 27.4 Hz). 1H NMR (CDCl3): δ 4.40 (12H, m, PTA), 7.39 (2H, t, J = 7.6 Hz, H2 + H4, C6H4), 7.51 (1H, t, J = 7.5 Hz, H3, C6H4), 8.23 (1H, d, J = 8.1 Hz, H5, C6H4). 13C{1H} (CDCl3): δ 52.71 (d, J = 45.9 Hz, TPA), 72.76 (d, J = 9.5 Hz, PTA), 128.33 (s, C4, C6H4) 129.84 (s, C5, C6H4), 131.98 (s, C3, C6H4), 136.31 (s, C2, C6H4), signals corresponding to NC=O, C1and C6 were not observable. Conductivity (acetone): 0.73 μS/cm (neutral) μS/cm.
[(η6-p-Cymene)Ru((Ph3P=N-CO-2-C6H4)-κ-C,N)Cl] (8)
[Hg(2-C6H4C(O)N=PPh3)Cl] (0.12 g, 0.2 mmol) and [(η6-p-cymene)Ru(μ-Cl)Cl]2 (0.13 g, 0.22 mmol) were refluxed in MeCN (20 mL) for 7 days, after which a yellow precipitate formed. The pale-yellow solid was filtered off and discarded, and the orange solution was concentrated to dryness. The solid was dissolved in CH2Cl2 and filtered through Celite. The solvent was removed under reduced pressure to a minimum, followed by addition of Et2O (∼20 mL). The orange solid was filtered off and dried in vacuo. Yield: 0.049 g (37%). Anal. Calcd for C35H33NOPClRu·2H2O (687.18): C, 61.18; H, 5.43; N, 2.04. Found: C, 60.97; H, 5.05; N 2.35%. ESI-MS: m/z: 616.13 (100%, [M – Cl]+, calcd 616.13). 31P {1H} NMR (CDCl3): δ 20.63 (s), (DMSO-d6): 21.15 (s). 1H NMR (CDCl3): δ 1.31 (6H, d, J = 6.9 Hz, CH3, η6-p-cymene), 2.18 (3H, s, CH3, η6-p-cymene), 2.95 (1H, sept, J = 6.9 Hz, CH, η6-p-cymene), 5.37 (2H, d, J = 6.0 Hz, CH, η6-p-cymene), 5.49 (2H, d, J = 6.0 Hz, CH, η6-p-cymene), 7.42–7.48 (2H, m, H3 + H4), 7.49–7.53 (6H, m, Hm), 7.56–7.61 (3H, m, Hp), 7.84–7.89 (6H, m, Ho), 8.36–8.37 (2H, dd, J = 1.4, 8.0 Hz, H2 + H5). 13C{1H} (CDCl3): δ 19.02 (s, CH3, η6-p-cymene), 22.21 (s, 2CH3, η6-p-cymene), 30.68 (s, CH, η6-p-cymene), 80.58 (s, CH, η6-p-cymene), 81.32 (s, CH, η6-p-cymene), 96.77 (s, C, η6-p-cymene), 101.25 (s, C, η6-p-cymene), 127.65 (s, C3 + C4), 127.92 (s, C6), 128.62–128.74 (d, Cm, J = 12.4 Hz), 128.91 (s, Cipso,), 129.51–129.53 (d, J = 3.02 Hz, C2 + C5), 130.67 (s, C3 + C4), 132.19–132.22 (d, Cp, J = 2.9 Hz), 133.13–133.24 (d, Co, J = 9.6 Hz), 138.51–138.72 (d, J = 20.6 Hz, C–Ru), 176.26 (s, C=O) ppm. IR (cm–1): ν 517 (Ru–N), 1162 (N=P), 1593 (C=O). Conductivity (acetone): 3.76 (neutral) μS/cm.
[(η6-p-Cymene)Ru{(PTA=N-CO-2-C6H4)-κ-C,N}Cl] (9)
[Hg(2-C6H4C(O)N=PTA)Cl] (6) (0.1 g, 0.2 mmol) and [(η6-p-cymene)Ru(μ-Cl)Cl]2 (0.13 g, 0.2 mmol) were refluxed in CH2Cl2 (20 mL) for 3 days. Subsequently, the solvent was removed to dryness and the yellow solid obtained was dissolved in CHCl3 and filtered through Celite. The solvent was removed under reduced pressure to a minimum, followed by addition of Et2O. The yellow solid obtained was collected by filtration and dried in vacuo. Yield: 0.14 g (66%). Anal. Calcd for C23H30N4OPClRu (546.01): C, 50.59; H, 5.54; N, 10.26. Found: C, 50.32; H, 5.20; N 10.53%. ESI-MS: m/z: 546.0 (100%, [M – Cl]+, calcd 546.06), 412.0 (100%, [M – p-cymene]−, calcd 412.0). 31P {1H} NMR) (CDCl3): δ −16.3 (s), (DMSO-d6): −11.98 (s). 1H NMR (DMSO-d6): δ 0.61 (3H, d, J = 6.7 Hz, CH3, η6-p-cymene), δ 0.99 (3H, d, J = 6.8 Hz, CH3, η6-p-cymene), 2.34 (3H, s, CH3, η6-p-cymene), 2.24 (1H, sept, J = 7.1 Hz, CH, η6-p-cymene), 4.47–4.67 (12H, m, PTA), 5.81 (2H, d, CH, η6-p-cymene), 6.06 (1H, d, J = 6.5 Hz, CH, η6-p-cymene), 6.74 (1H, d, J = 6.9 Hz, CH, η6-p-cymene), 7.11 (1H, t, J = 7.7 Hz, H4), 7.31 (1H, t, J = 7.5 Hz, H3), 7.37 (1H, d, J = 7.5 Hz, H5), 7.81 (d, 1H, J = 7.5 Hz, H2). 13C{1H} (DMSO-d6): δ 18.70 (s, CH3, η6-p-cymene), 19.97 (s, CH3, η6-p-cymene), 24.13 (s, CH3, η6-p-cymene), 31.00 (s, CH, η6-p-cymene), 51.82 (s, PTA), 52.22 (s, PTA), 71.50 (d, J = 40.5 Hz, PTA), 86.72 (s, CH, η6-p-cymene), 87.22 (s, CH, η6-p-cymene), 95.97 (s, CH, η6-p-cymene), 100.6 (s, C, η6-p-cymene), 127.65 (s, C4), 128.6 (s, C5), 128.6 (s, Cipso,), 132.6 (s, C3), 138.6–138.7 (d, J = 20.6 Hz, C–Ru), 141.1 (s, C2), 183.9 (s, C=O) ppm. IR (cm–1): ν 562 (Ru–N), 1314 (N=P), 1582 (C=O). Conductivity (acetone): 25.4 (neutral) μS/cm. Solubility: 0.79 mM or 0.43 mg/mL (H2O).
X-ray Crystallography
Single crystals of 1 (see details in Table S1 in Supporting Information) were mounted on a glass fiber in a random orientation. Data collection was performed at RT on a Kappa CCD diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Space group assignments were based on systematic absences, E statistics, and successful refinement of the structures. The structures were solved by direct methods with the aid of successive difference Fourier maps and were refined using the SHELXTL 6.1 software package. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were assigned to ideal positions and refined using a riding model. Details of the crystallographic data are given in Table S1 (Supporting Information). These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. (CCDC 1008354) or in the Supporting Information. Crystals of 1 (orange prisms with approximate dimensions 0.26 × 0.24 × 0.21 mm3) were obtained from a solution of 1 in CH2Cl2 by slow diffusion of Et2O at RT.
Cell Culture, Inhibition of Cell Growth, and Cell Death Analysis
Cell Culture
The human T-cell leukemia Jurkat (clone E6.1) and the prostate carcinoma DU-145 were routinely cultured in RPMI 1640 medium supplemented with 5% fetal calf serum (FCS), l-glutamine, and penicillin/streptomycin. A549 (lung carcinoma), MiaPaca2 (pancreatic carcinoma), MDA-MB-231 (Triple negative breast carcinoma), and 293T (nontumoral embryonic kidney cells) were cultured in DMEM medium supplemented with 10% FCS, l-glutamine, and penicillin/streptomycin. Media for A549 cells were also supplemented with 2.2 g/L Na2CO3, 100 μg/mL pyruvate, and 5 mL nonessential amino acids (Invitrogen). All these media will be referred as “complete medium” hereinafter. Cell cultures were maintained in a humidified atmosphere of 95% air/5% CO2 at 37 °C.
MTT Toxicity Assays
For toxicity assays cells (5 × 104 for Jurkat cells and 104 for adherent cell lines) were seeded in flat-bottom 96-well plates (100 μL/well) in complete medium. Adherent cells were allowed to attach for 24 h prior to addition of cisplatin or tested compounds. Compounds were added at different concentrations in triplicate. Cells were incubated with cisplatin or compounds for 24 h, and then cell proliferation was determined by a modification of the MTT-reduction method. Briefly, 10 μL/well of MTT (5 mg/mL in PBS) was added, and plates were incubated for 1–3 h at 37 °C. Finally, formazan crystal was dissolved by adding 100 μL/well iPrOH (0.05 M HCl) and gently shaking. The optical density was measured at 570 nm using a 96-well multiscanner autoreader (ELISA). In some experiments, total cell number and cell viability were determined by the Trypan Blue exclusion test.
Cell Culture and XTT Assay for RPTC Cells
The human renal proximal tubular cells (RPTC), a nontumoral human kidney epithelial cell line (obtained from Lifeline Cell Technology, Frederick, Maryland, USA), were cultured in Lifeline’s RenaLife Medium containing RenaLife LifeFactors with 2.4 mM l-glutamine, 5 Î1/4 g/mL rh insulin, 1.0 Î1/4 M epinephrine, 10 nM triiodothyronine, 0.1 Î1/4 g/mL hydrocortisone hemisuccinate, 10 ng/mL rh EGF, 0.5% FBS, and 5 Î1/4 g/m transferrin PS (all from Lifeline Cell Technology), at 37 °C in a humidified atmosphere of 95% of air and 5% CO2 (University of Hawaii Cancer Center, Honolulu, Hawaii, USA). For evaluation of cell viability, cells were seeded at a concentration of 5 × 103 cells/well in 90 μL of Lifeline’s RenaLife complete medium into tissue culture grade 96-well flat bottom microplates (Thermo Scientific BioLite Microwell Plate, Fisher Scientific, Waltham, Massachusetts, USA) and grown for 24 h. Solutions of the compounds were prepared by diluting a freshly prepared stock solution (in H2O) of the corresponding compound in Lifeline’s RenaLife complete medium. Afterward, the intermediate dilutions of the compounds were added to the wells (10 μL) to obtain a final concentration ranging from 0.1 to 200 μM, and the cells were incubated for 24 h. Following 24 h drug exposure, 50 μL of 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) (Roche Diagnostics, Indianapolis, Indiana, USA) labeling mixture per well was added to the cells at a final concentration of 0.3 mg/mL and incubated for 4 h at 37 °C in a humidified atmosphere of 95% of air and 5% CO2. The optical density of each well (96-well plates) was quantified using EnVision multilabel plate readers (PerkinElmer, Waltham Massachusetts, USA) at 450 nm wavelength to measure absorbance. The percentage of surviving cells was calculated from the ratio of absorbance of treated to untreated cells. The IC50 value was calculated as the concentration reducing the proliferation of the cells by 50% and is presented as a mean (±SE) of at least two independent experiments each with triplicates.
Cell Death Analysis
Apoptosis/necrosis hallmarks of cells treated with compounds 2 and 3 were analyzed by measuring mitochondrial membrane potential and/or exposure of phosphatidylserine. Cells were treated with different concentrations and at different incubation times as indicated in figure legends. In some experiments, the general caspase inhibitor z-VAD-fmk was added at 50 μM 1 h before compounds. For mitochondrial membrane potential determination, cells (2.5 × 105 in 200 μL) after treatment with compounds were incubated at 37 °C for 15 min in ABB (140 mM NaCl, 2.5 mM CaCl2, 10 mM Hepes/NaOH, pH 7,4) containing 60 mM tetramethylrhodamine ethyl ester (TMRE, Molecular Probes). Phosphatidylserine exposure was quantified by labeling cells with AnnexinV-PE or AnnexinV-DY636 (Invitrogen) after treatment with compounds. AnnexinV was added at a concentration of 0.5 μg/mL, and cells were incubated at room temperature for 15 min. In all cases, cells were diluted to 1 mL with ABB to be analyzed by flow cytometry (FACScan, BD Bioscience, Spain).
Intracellular ROS Quantification
Oxidative stress induced by compounds 2 and 3 was analyzed by intracellular staining with the fluorescent probe 2-hydroxiethidium (2-HE, Molecular Probes). After 16 h of culture in the presence of compounds 1–3, cells were incubated with 2 μM 2-HE at 37 °C for 15 min. Red fluorescence produced by reduction of 2-HE to ethidium was quantified in a flow cytometer.
Effect of 2 in the Levels of Proteins of the Bcl-2 Family
Jurkat cells (5 × 105 cells/mL) were treated with 2 (1 μM) for 6 h. At the end of incubations, total protein extracts from 2 × 106 cells were prepared in lysis buffer and samples (50 μg/lane) were resolved by SDS-PAGE and transferred onto nitrocellulose membranes. Then, levels of some members of the Bcl-2 family of proteins were analyzed by Western Blot using specific antibodies: Bcl-XL (Cell Signaling, cat. no. 2764), Bcl-2 (Abcam, cat. no. AB692), Mcl-1 (Santa Cruz Biotech, cat. no. SC819), Bim (Calbiochem, cat. no. 202000), Puma (Cell Signaling, cat. no. 4976), and Noxa (Abcam, cat. no. 114C307). After incubation with primary antibodies, membranes were incubated with appropriate secondary antibodies conjugated with HRP. Finally, membranes were revealed using a chemiluminiscence substrate (Pierce).
Inhibition of Cathepsin B
Cathepsin B, purified from human liver (accession no. P07858) and substrate peptide sequence: Z-FR-AMC [AMC = 7-amino-4-methylcoumarin] were dissolved on a buffer, 25 mM MES pH 6, 50 mM NaCl, 0.005% Brij35, 5 mM DTT, and 1% DMSO, with a final concentration of 10 μM. The enzyme solution was delivered into the reaction well. 2 (1% DMSO solution) was delivered into the enzyme mixture by acoustic technology (Echo550; nanoliter range), incubated for 10 min at room temp. The substrate solution was delivered into the reaction well to initiate the reaction. The enzyme activity was monitored (Ex/Em = 355/460 nm) as a time-course measurement of the increase in fluorescence signal from fluorescently labeled peptide substrate for 120 min at room temperature. The data was analyzed data by taking slope (signal/time) of linear portion of measurement. The slope was calculated by using Excel, and curve fits were performed using Prism software.
Interaction of Compounds 1–4, 8, 9, [(η6-p-Cymene)Ru(μ-Cl)Cl]2, and Cisplatin with Plasmid (pBR322) DNA by Electrophoresis (Mobility Shift Assay)
First, 10 μL aliquots of pBR322 plasmid DNA (20 μg/mL) in buffer (5 mM Tris/HCl, 50 mM NaClO4, pH = 7.39) were incubated with different concentrations of the compounds (1–4, 8, 9, [(η6-p-cymene)Ru(μ-Cl)Cl]2) (in the range 0.25 and 2.0 metal complex:DNAbp) at 37 °C for 20 h in the dark. Samples of free DNA and cisplatin-DNA were prepared as controls. After the incubation period, the samples were then loaded onto the 1% agarose gel. The samples were separated by electrophoresis for 1.5 h at 80 V in Tris-acetate/EDTA buffer (TAE). Afterward, the gel was stained for 30 min with a solution of GelRed nucleic acid stain.
Interaction of Compounds 1–4 with Calf Thymus DNA by Circular Dichroism
Stock solutions (5 mM) of each complex were freshly prepared in water prior to use. The right volume of those solutions was added to 3 mL samples of an also freshly prepared solution of CT DNA (48 μM) in Tris/HCl buffer (5 mM Tris/HCl, 50 mM NaClO4, pH = 7.39) to achieve molar ratios of 0.1, 0.25, 0.5, 1.0, and 2.0 drug/DNA. The samples were incubated at 37 °C for a period of 20 h. All CD spectra of DNA and of the DNA–drug adducts were recorded at 25 °C over a range 220–420 nm and finally corrected with a blank and noise reduction. The final data is expressed in molar ellipticity (millidegrees).
DNA Precipitation with Compounds 2 and 3 and Quantification of Ruthenium by ICP-MS
Stock solutions of compounds 2 and 3 (4 mM in water) and CT DNA (11.56 mM in 5 mM Tris/HCl, 50 mM NaClO4, pH = 7.39) were freshly prepared prior to use. Then 216 μL of DNA stock solution were diluted in 3.53 mL of buffer and 1.25 mL of compound stock solution were then added to achieve 5 mL final volume at concentrations of 500 μM in DNA and 1 mM in metal compound. Each sample was incubated at 37 °C for a period of 20 h, then cooled down to room temperature and centrifuged at 3000 rpm for 15 min and at 4000 rpm for extra 40 min. The supernatant was separated and analyzed for CT DNA concentration by CD spectroscopy. The resulting pellet was washed twice with ice-cold ethanol (1 mL), centrifuged at RT for 1 min at 4000 rpm, dried under high vacuum, and analyzed for Ru content by ICP-MS. The total amount of DNA in each sample was 0.92 mg. Every experiment was run in duplicate.
Interaction of Compounds 1–4, 8, 9, and Cisplatin with HSA by Fluorescence Spectroscopy
A solution of each compound (8 mM) in DMSO was prepared, and 10 aliquots of 2.5 μL were added successively to a solution of HSA (10 μM) in phosphate buffer (pH = 7.4) to achieve final metal complex concentrations in the range 10–100 μM. The excitation wavelength was set to 295 nm, and the emission spectra of HSA samples were recorded at room temperature in the range of 300 to 450 nm. The fluorescence intensities of all the metal compounds, the buffer, and the DMSO are negligible under these conditions. The fluorescence was measured 240 s after each addition of compound solution. The data were analyzed using the classical Stern–Volmer equation F0/F = 1 + KSV[Q].
In Vivo Tests
All animal experiments were performed according to the University of Hawaii Cancer Center regulations and by approval of the responsible authorities (UH IACUC number: A3423–01).
Determination of Lethal Dose (LD) and Maximum Tolerated Dose (MTD) of 2 in Mice
Fourteen female C57/Black 6 mice from Jackson Laboratory (Bar Harbor, ME, and Sacramento, CA, USA), ages 8–14 weeks and weighing 18–26 g, were used for these experiments. Mice were randomized to treatment groups based on their age to ensure equivalent distribution between the groups. At trial end-point, the mice were sacrificed and liver, spleen, kidney, and blood plasma were collected and then processed for further analysis. Gross and microscopic evaluations of liver, spleen, and kidney were conducted. The weight of 2 treated mice compared and that of vehicle-treated mice as measured twice weekly.
The lethal dose (LD) was determined by injecting once mouse ip once 5, 10, 20, 30, or 50 mg/kg/day and one vehicle control with 100 μL of normal saline (0.9% NaCl)). The dose that killed the mice within 24 h was set to be lethal dose. The lethal dose was confirmed by administering that dose to a second mouse. The maximum tolerated dose (MTD) was determined by injecting two mice with 5, 10, or 20 mg/kg/day over 6 days or 20 mg/kg/every other day over 6 days and one vehicle control mouse with 100 μL of normal saline (0.9% NaCl). The MTD was confirmed by administering the determined dose to 3 mice over 7 days and three vehicle control mice with 100 μL of normal saline (0.9% NaCl).
Study of the Effects of 2 in MDA-MB-231 Xenografts in Mice
First, 12 female NOD.CB17-Prkdc scid/J (nonobese diabetic–severe combined immunodeficiency) mice from Jackson Laboratory (Bar Harbor, ME and Sacramento, CA, USA) for the xenograft experiment (ages 8–12 weeks and weighing 19–24 g) were used. Each mouse received 5 × 106 tumor cells subcutaneously without anesthesia. Exponentially growing estrogen-receptor α-negative MDA-MB-231 human breast cancer cells were then suspended in 1:1 ratio 50 μL of phosphate-buffered saline (PBS; pH 7.4) plus 50 μL of matrigel (BD Biosciences, San Jose, CA, USA) were injected subcutaneously on both left and right flank of each mouse. The diameter of the tumors was measured once weekly using an electronic digital caliper, and the tumor volume (TV) was calculated according to the empirical equation TV = (a)(b2) × π/6, where a = longest dimension and b = largest dimension orthogonal to a. The median volumes of each group were normalized to the initial tumor volume, resulting in the relative tumor volume. Each six MDA-MB-231-transplanted animals received compound 2 (5 mg/kg/every other day) or vehicle (0.9% NaCl) intraperitoneally (ip). Treatment started when tumors were palpable (about 5–6 mm diameter). To palliate the weight loss observed in the MTD study, the mice were fed a 46% fat-adjusted diet (Harlan Teklad, Madison, WI) plus HydroGel (Harlan Teklad, Madison, WI) and received subcutaneous injection of 100 μL of normal saline (0.9% NaCl) to improve hydration. Mice were randomized to treatment groups based on their starting tumor burden at 12 weeks of age to ensure equivalent distribution between the two groups. At trial end-point, the mice were sacrificed and tumors measured again after excision and then processed for further analysis. Histological as well as biochemical evaluations of blood, liver, intestine, kidney, and lung were conducted. Tumor volumes were graphed for (2) treated mice compared to vehicle-treated mice based on weekly external digital caliper measurements.
Pharmacokinetic Study: Determination of Ruthenium Content in the Organs, Entire Blood, and Plasma
Female NOD.CB17-Prkdc scid/J nice bearing subcutaneous MDA-MB-231 tumors and treated with compound 2 (5 mg/kg/every other day) intraperitoneally were used for pharmacokinetic evaluation of the drug in blood and other tissues. Blood was collected retroorbitally using a heparin coated glass capillary into heparinized blood collection vials on ice at time intervals of 30 min, 2 h, 6 h, 24 h,and 48 h after the first dose. The blood samples were centrifuged at 2800 rpm at 4 °C for 15 min, and the supernatant plasma was transferred into 1.5 mL microcentrifuge tubes and maintained at −80 °C until analysis.
Ruthenium content was determined using inductively coupled plasma–mass spectrometry (ICP-MS). First, 50 μL of plasma was transferred into a glass vial, and 1 mL of concentrated acid mix (comprising of 75% of 16 N nitric acid and 25% of 12 N hydrochloric acid) was added. The mixture was then heated at 90 °C for 5 h. After cooling, the samples were diluted with water, and 40 ppb of indium internal standard was added and analyzed in a Thermo Scientific XSERIES 2 ICP-MS with ESI PC3 Peltier cooled spray chamber with SC-FAST injection loop and SC-4 autosampler. All the elements were analyzed using He/H2 collision-reaction mode. Plasma from control mice was spiked with the test compound to determine the extraction efficiency.
At the end of the study, liver, kidney, and tumor of the animals were harvested, weighed, and transferred into glass vials. One mL of water was added to each sample and subjected to ultrasonic tissue disruption at 15 W power for 1 min. The tissue homogenates were frozen at −80 °C for 2 h and lyophilized. The lyophilized product was heated at 90 °C with the concentrated acid mix (described above) for 5 h, cooled, diluted with water, and analyzed for ruthenium by ICP-MS. Pharmacokinetic estimates were obtained from the plasma concentration–time profiles by noncompartmental analysis using Phoenix WinNonlin 6.1 (Mountain View, California).
Acknowledgments
Research at Brooklyn College was supported by a grant from the National Cancer Institute (NCI) 1SC1CA182844 (M.C.) and two grants from the National Institute of General Medical Sciences (NIGMS) SC2GM082307 (M.C.) and 1SC1GM089558 (R.A.S.-D.). The Spanish authors thank the support from the Ministerio de Economía y Competitividad project SAF2010-1490 (I.M.), and Joe. W. Ramos thanks the National Institute of General Medical Sciences (NIGMS) grant RO1GM088266-A1. M.C. is very grateful to Mr. and Mrs. Leonard and Claire Tow and the Tow Foundation for a travel fellowship to perform some experiments at the University of Hawaii Cancer Center. We thank Prof. Jayanth Panyam from the Center for Translational Drug Delivery at the University of Minnesota for helpful discussions about the pharmacokinetic studies and Dr. Rick Knurr from the Geochemical Lab in the Department of Earth Sciences at the University of Minnesota for assistance with ICP-MS analysis.
Glossary
Abbreviations Used
- Cat B
cathepsin B
- DMSO
dimethyl sulfoxide
- HSA
human serum albumin
- IM
iminophosphorane
- ITC
isothermal titration calorimetry
- LD
lethal dose
- MTD
maximum tolerated dose
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PARP-1
poly(ADP-ribose) polymerase-1
- PBS
phosphate buffer saline
- RPTC
renal proximal tubular cells
- TAE
Tris-acetate/EDTA buffer
- T-Jurkat
human acute lymphoblastic leukemia cells
- T-Jurkat sh Bak
human acute lymphoblastic leukemia cells which do not express the Bak gene
- XTT
2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
Supporting Information Available
Crystallographic data for compound 1 (CIF); 1H NMR spectra (CDCl3) for compounds 2–4, 7, and 8 (including variable temperature NMR of 4); stability of the ruthenium compounds by 31P{1H} spectroscopy in DMSO-d6 and D2O solution; selected 1H and 31P{1H} NMR spectra for of the ruthenium in DMSO-d6 and/or D2O solution (including 100 mM NaCl/D2O for compound 2); 31P{1H} NMR spectra for 2 and 3 in D2O after heating at 80 °C for 1 h; mass spectra (ESI+) of compound 2 in H2O solution overtime (5 days); study of the effect of 2 in the levels of proteins of the Bcl-2 family, and experiments to assess the interaction of compounds 2–4 with CT DNA by circular dichroism. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Alessio E.Bioinorganic Medicinal Chemistry; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Noffke A. L.; Habtemarian A.; Pizarro A. M.; Sadler P. J. Designing organometallic compounds for catalysis and therapy. Chem. Commun. 2012, 48, 5219–5246. [DOI] [PubMed] [Google Scholar]
- Komeda S.; Casini A. Next-generation anticancer metallodrugs. Curr. Top. Med. Chem. 2012, 12, 219–223. [DOI] [PubMed] [Google Scholar]
- Bergamo A.; Gaiddon C.; Schellens J. H. M.; Beijnen J. H.; Sava G. Approaching tumour therapy beyond platinum drugs: status of the art and perspectives of ruthenium drug candidates. J. Inorg. Biochem. 2012, 106, 90–99. [DOI] [PubMed] [Google Scholar]
- Bergamo A.; Sava G. Ruthenium anticancer compounds: myths and realities of the emerging metal-based drugs. Dalton Trans. 2011, 40, 7817–7823. [DOI] [PubMed] [Google Scholar]
- Mazuryk O.; Kurpiewska K.; Lewinski K.; Stochel G.; Brindell M. Interaction of apo-transferrin with anticancer ruthenium complexes NAMI-A and its reduced form. J. Inorg. Biochem. 2012, 116, 11–18. [DOI] [PubMed] [Google Scholar]
- Pongrazt M.; Schluga P.; Jakupec M. A.; Arion V. B.; Hartinger C. A.; Allmaier G.; Keppler B. K. Transferrin binding and transferrin-mediated cellular uptake of the ruthenium coordination compound KP1019, studied by means of AAS, ESI-MS and CD spectroscopy. J. Anal. Atom. Spectrom. 2004, 19, 46–51. [Google Scholar]
- Nazarov A. A.; Hartinger C. C.; Dyson P. J. Opening the lid on piano-stool complexes: an account of ruthenium(II)–arene complexes with medicinal applications. J. Organomet. Chem. 2104, 751, 251–260. [Google Scholar]
- Rademaker-Lakhai J. M.; van der Bongard D.; Pluim D.; Beijnen J. H.; Schlens J. M. H. A Phase I and Pharmacological Study with Imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a Novel Ruthenium Anticancer Agent. Clin. Cancer Res. 2004, 10, 3717–3727and refs therein. [DOI] [PubMed] [Google Scholar]
- Hartinger C. G.; Zorbas-Seifried S.; Jakupec M. A.; Kynast B.; Zorbas H.; Keppler From bench to bedside—preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 10, 9891–904and refs therein. [DOI] [PubMed] [Google Scholar]
- Hartinger C. G.; Metzler-Nolte N.; Dyson P. J. Challenges and opportunities in the development of organometallic anticancer drugs. Organometallics 2012, 31, 5677–5685. [Google Scholar]
- Morris R. E.; Aird R. E.; Murdoch P.; Chen H.; Cummings J.; Hughes N. D.; Parsons S.; Parkin A.; Boyd G.; Jodrell D. I.; Sadler P. J. Inhibition of Cancer Cell Growth by Ruthenium(II)–Arene Complexes. J. Med. Chem. 2001, 44, 3616–3621. [DOI] [PubMed] [Google Scholar]
- Hayward R. L.; Schornagel Q. C.; Tente R.; MacPherson J. S.; Aird R. E.; Guichard S.; Habtemariam A.; Sadler P. J.; Jodrell D. I. Investigation of the role of Bax, p21/Waf1 and p53 as determinants of cellular responses in HCT116 colorectal cancer cells exposed to the novel cytotoxic ruthenium(II) organo-metallic agent, RM175. Cancer Chemother. Pharmacol. 2005, 577–583. [DOI] [PubMed] [Google Scholar]
- Bergamo A.; Masi A.; Peacock A. F. A.; Habtemariam A.; Sadler P. J.; Sava G. In vivo tumour and metastasis reduction and in vitro effects on invasion assays of the ruthenium RM175 and osmium AFAP51 organometallics in the mammary cancer model. J. Inorg. Biochem. 2010, 104, 79–86and refs therein. [DOI] [PubMed] [Google Scholar]
- Allardyce C. S.; Dyson P. J.; Ellis D. J.; Heath S. L. [Ru(η6-p-cymene)Cl2(pta)] (pta = 1,3,5-triaza-7-phosphatricyclo- [3.3.1.1]decane): a water soluble compound that exhibits pH dependent DNA binding providing selectivity for diseased cells. Chem. Commun. 2001, 15, 1396–1397. [Google Scholar]
- Scolaro C.; Bergamo A.; Brescacin L.; Delfino R.; Cocchietto M.; Laurenczy G.; Gledbach T. J.; Sava G.; Dyson P. J. In Vitro and in Vivo Evaluation of Ruthenium(II)–Arene PTA Complexes. J. Med. Chem. 2005, 48, 4161–4171. [DOI] [PubMed] [Google Scholar]
- Bergamo A.; Masi A.; Dyson P. J.; Sava G. Modulation of the metastatic progression of breast cancer with an organometallic ruthenium compound. Int. J. Oncol. 2008, 33, 1281–1289. [PubMed] [Google Scholar]
- Adhireksan Z.; Davey G. E.; Campomanes P.; Groessl M.; Clavel C. M.; Yu H.; Nazarov A. A.; Fang Yeo C. H.; Ang W. H.; Droge P.; Rothlisberger U.; Dyson P. J.; Davey C. A. Ligand substitutions between ruthenium–cymene compounds can control protein versus DNA targeting and anticancer activity. Nature Commun. 2014, 5, 3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavel C. M.; Paunescu E.; Nowak-Sliwinska P.; Griffioen A. W.; Scopelliti R.; Dyson P. J. Discovery of a higly tumor-selective organometallic ruthenium(II)–arene complex. J. Med. Chem. 2014, 57, 3546–3558. [DOI] [PubMed] [Google Scholar]
- Gaiddon C.; Jeannequin P.; Bischoff P.; Pfeffer M.; Sirlin C.; Loeffler J. P. Ruthenium(II)-derived organometallic compounds induce cytostatatic and cytotoxic effects on mammalian cancer cell lines through p53-dependent and p53-independent mechanisms. Pharmacol. Exp. Ther. 2005, 315, 1403–1411. [DOI] [PubMed] [Google Scholar]
- Meng X.; Leyva M. L.; Jenny M.; Gross I.; Benosman S.; Fricker B.; Harlepp S.; Hebraud P.; Boos A.; Wlosik P.; Bishoff P.; Sirlin C.; Pfeffer M.; Loeffer J. P.; Gaiddon C. A Ruthenium-Containing Organometallic Compound Reduces Tumor Growth through Induction of the Endoplasmic Reticulum Stress Gene CHOP. Cancer Res. 2009, 69, 5458–5466and refs therein. [DOI] [PubMed] [Google Scholar]
- Farhana A.; Hanif M.; Waseeq A.; Ashraf A.; Filak L. K.; Reynisson J.; Sohnel T.; Jamieson S. M. F.; Hartinger C. G. Anticancer ruthenium(η6-pcymene) complexes of nonstereroidal anti-inflammatory drug derivatives. Organometallics 2014, 33, 5546–5553. [Google Scholar]
- Pettinari R.; Marchetti F.; Condello F.; Pettinari C.; Lupidi G.; Scopelliti R.; Mukhopadhyay S.; Riedel T.; Dyson P. J. Ruthenium(II)–arene RAPTA type complexes containing curcumin and bisdemethosycurcumin display potent and selective anticancer activity. Organometallics 2014, 33, 3709–3715. [Google Scholar]
- Robles-Escajeda E.; Martínez A.; Varela-Ramirez A.; Sánchez-Delgado R. A.; Aguilera R. J. Analysis of the cytotoxic effects of ruthenium–ketoconazole and ruthenium–clotrimazole complexes on cancer cells. Cell Biol. Toxicol. 2013, 29, 431–443and refs therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurzwemhart A.; Kandioller W.; Enyedy E. A.; Novak M.; Jakupek M. A.; Keppler B. K.; Hartinger C. G. 3-Hydroxyflavones vs. 3-hydroxyquinolinones: structure–activity relationships and stability studies on RuII(arene) anticancer complexes with biologically active ligands. Dalton Trans. 2013, 42, 6193–6202and refs therein. [DOI] [PubMed] [Google Scholar]
- Kurzwemhart A.; Kandioller W.; Baechler S.; Bartel C.; Martic S.; Buczkowska M.; Muehlgassner G.; Japukec M. A.; Kraatz H.-B.; Bednarski P. J.; Arion V. B.; Marko D.; Keppler B. K.; Hartinger C. G. Structure–activity relationships of targeted Ru(II)(η6-p-cymene) anticancer complexes with flanovol-derived ligands. J. Med. Chem. 2012, 55, 10512–10522. [DOI] [PubMed] [Google Scholar]
- Castonguay A.; Doucet C.; Juhas M.; Maysinger D. New ruthenium(II)–letrozole complexes as anticancer therapeutics. J. Med. Chem. 2012, 55, 8799–8806. [DOI] [PubMed] [Google Scholar]
- Mulgassner G.; Bartel C.; Schmid W. F.; Jakupec M. A.; Arion V. B.; Keppler B. K. Biological activity of ruthenium and osmium arenes complexes with modified paullones in human cancer cells. J. Inorg. Biochem. 2012, 116, 180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubner G.; Bensdorf K.; Wellner A.; K B.; Bergemann S.; Ott I.; Gust R. Synthesis and Biological Activities of Transition Metal Complexes Based on Acetylsalicylic Acid as Neo-Anticancer Agents. J. Med. Chem. 2010, 53, 6889–6898. [DOI] [PubMed] [Google Scholar]
- Debrecnezi J. E.; Bullock A. N.; Atilla G. E.; Williams D. S.; Bregman H.; Knapp S.; Meggers E. Ruthenium half-sandwich complexes bound to protein kinase Pim-1. Angew. Chem., Int. Ed. Engl. 2006, 45, 1580–1585. [DOI] [PubMed] [Google Scholar]
- Smalley K. S.; Contractor R.; Haas N. K.; Kup A. N.; Atilla-Gokcuman G. E.; Williams D. E.; Bregman H.; Flaherty K. T.; Soengas M. S.; Meggers E.; Herlyn M. An organometallic protein kinase inhibitor pharmacologically activates p53 and induces apoptosis in human melanoma cells. Cancer Res. 2007, 67, 209–217. [DOI] [PubMed] [Google Scholar]
- Xie P.; Streu C.; Qin J.; Bregman H.; Pagano N.; Meggers E.; Marmorstein R. The crystal structure of BRAF in complex with an organoruthenium inhibitor reveals a mechanism for inhibition of an active form of BRAF kinase. Biochemistry 2009, 48, 5187–5198and refs therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A recent review: Wang K.; Gao E.. Recent advances in multinuclear complexes as potential anticancer and DNA binding agents. Anti-Cancer Agents Med. Chem. 2014, 14, 147–169. [DOI] [PubMed] [Google Scholar]
- Murray B. S.; Menin L.; Scopelliti R.; Dyson P. J. Conformational control of anticancer activity: the application of arene-linked dinuclear ruthenium(II) organometallics. Chem. Sci. 2014, 5, 2536–2545. [Google Scholar]
- Very recent example: Joshi T.; Pierroz V.; Mari C.; Gemperle L.; Ferrari S.; Gasser G.. A Bis(dipyridophenazine)(2-(2-pyridyl)pyrimidine-4-carboxylic acid)ruthenium(II) complex with anticancer action upon photodeprotection. Angew. Chem., Int. Ed. 2014, 53, 2960–2963and refs therein. [DOI] [PubMed] [Google Scholar]
- Clavel C. M.; Paunescu E.; Nowak-Sliwinska P.; Dyson P. J. Thermoresponsive organometallic arene ruthenium complexes for tumor targeting. Chem. Sci. 2014, 5, 1907–1101. [Google Scholar]
- Chow M. J.; Licona C.; Wong D. Y. Q.; Pastorin G.; Gaiddon C.; Ang W. H. Discovery and investigation of anticancer ruthenium–arene Schiff-base complexes via water-promoted combinatorial three-component assembly. J. Med. Chem. 2014, 57, 6043–6059. [DOI] [PubMed] [Google Scholar]
- Valente A.; Garcia M. H.; Marques F.; Miao Y.; Rousseau C.; Zinck P. J. First polymer “ruthenium–cyclopentadienyl” complex as potential anticancer agent. J. Inorg. Biochem. 2013, 127, 79–81. [DOI] [PubMed] [Google Scholar]
- Nandakumar V.; Vettriselvi V.; Doble M. Toxicity of high glycolic poly(dl-lactic-co-glycolic acid) stabilized ruthenium nanoparticles against human promyelocytic leukemia cells. RSC Adv. 2014, 4, 11438–11443. [Google Scholar]
- Mangiapia G.; D’Errico G.; Simeone L.; Irace C.; Radulescu A.; Di Pascale A.; Colonna A.; Montesarchio D.; Paduano L. Ruthenium-based complex nanocarriers for cancer therapy. Biomaterials 2012, 33, 3770–3782. [DOI] [PubMed] [Google Scholar]
- Meier S. M.; Novak M.; Kandioller W.; Japukec M. A.; Arion V. B.; Metzler-Nolte N.; Keppler B. K.; Hartinger C. G. Identification of the structural determinants for anticancer activity of a ruthenium–arene peptide conjugate. Chem.—Eur. J. 2013, 19, 9297–9307. [DOI] [PubMed] [Google Scholar]
- Gross A.; Huesken N.; Schur J.; Raszeja L.; Ott I.; Metzler-Nolte N. A ruthenocene–PNA bioconjugate-synthesis, characterization, cytotoxicity, and AAS-detected cellular uptake. Bioconjugate Chem. 2012, 23, 1764–1771. [DOI] [PubMed] [Google Scholar]
- Stepanenko I. N.; Casini A.; Edafe F.; Novak M. S.; Arion V. B.; Dyson P. J.; Japukec M. A.; Keppler B. K. Conjugation of Organoruthenium(II) 3-(1H-Benzimidazol-2-yl)pyrazolo[3,4-b] pyridines and Indolo[3,2-d]benzazepines to Recombinant Human Serum Albumin: A Strategy to Enhance Cytotoxicity in Cancer Cells. Inorg. Chem. 2011, 50, 12699–12679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaik N.; Martínez A.; Augustin I.; Giovinazzo H.; Varela A.; Aguilera R.; Sanaú M.; Contel M. Synthesis of Apoptosis-Inducing Iminophosphorane Organogold(III) Complexes and Study of Their Interactions with Biomolecular Targets. Inorg. Chem. 2009, 48, 1577–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vela L.; Contel M.; Palomera L.; Azaceta G.; Marzo I. Iminophosphorane–organogold(III) complexes induce cell death through mitochondrial ROS production. J. Inorg. Biochem. 2011, 105, 1306–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira M.; Calvo-Sanjuán R.; Sanaú M.; Zhao X.; Magliozzo R. S.; Marzo I.; Contel M. Cytotoxic Hydrophilic iminophosphorane coordination compounds of d8 metals. Studies of their Interactions with DNA and HSA. J. Inorg. Biochem. 2012, 116, 204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira M.; Calvo-Sanjuán R.; Sanaú M.; Marzo I.; Contel M. Organometallic Palladium Complexes with a Water-Soluble Iminophosphorane Ligand as Potential Anticancer Agents. Organometallics 2012, 31, 5772–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lease N.; Vasilevski V.; Carreira M.; de Almeida A.; Sanau M.; Hirva P.; Casini A.; Contel M. Potential Anticancer Heterometallic Fe–Au and Fe–Pd Agents: Initial Mechanistic Insights. J. Med. Chem. 2013, 56, 5806–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frik M.; Jimenez J.; Vasilevski V.; Carreira M.; de Almeida A.; Gascon E.; Sanaú M.; Casini A.; Contel M. Luminescent iminophosphorane gold, palladium and platinum complexes as potential anticancer agents. Inorg. Chem. Front. 2014, 1, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar D.; Bielsa R.; Soler T.; Urriolabeitia E. P. Cycloruthenated complexes from iminophosphoranes: synthesis, structure, and reactivity with internal alkynes. Organometallics 2011, 30, 642–648. [Google Scholar]
- Bittner S.; Assaf Y.; Krief P.; Pomerantz M.; Ziennicka B. T.; Smith C. G. Synthesis of N-acyl, N-sulfonyl, and N-phosphinylphospha-λ5-azenes by a redox-condensation reaction using amides, triphenylphosphine, and diethyl azocarboxylate. J. Org. Chem. 1985, 50, 1712–1718. [Google Scholar]
- Bennett M. A.; Huang T. N.; Matheson T. W.; Smith A. K. (η6-Hexamethylbenzene)ruthenium Complexes. Inorg. Synth. 1982, 21, 74–78. [Google Scholar]
- Kilpin K. J.; Linklater R.; Henderson W.; Nicholson B. K. Synthesis and characterization of isomeric cycloaurated complexes derived from the iminophosphorane PP3=NC(O)Ph. Inorg. Chim. Acta 2010, 363, 1021–1030. [Google Scholar]
- Boogard P. J.; Nagelkerke J. F.; Mulder G. J. Renal proximal tubular cells in suspension or in primary culture as in vitro models to study nephrotoxicity. Chem.–Biol. Interact. 1990, 76, 251–292. [DOI] [PubMed] [Google Scholar]
- Cummings B. S.; McHowat J.; Schnellmann R. G. Role of an endoplasmic reticulum Ca2+-independent phospholipase A2 in cisplatin-induced renal cell apoptosis. J. Pharmacol. Exp. Ther. 2004, 308, 921–928. [DOI] [PubMed] [Google Scholar]
- Shamas-Din A. L.; Brahmbhatt H.; Leber B.; Andrews D. W. BH3-only proteins: orchestrators of apoptosis. Biochim. Biophys. Acta 2011, 1813, 508–520. [DOI] [PubMed] [Google Scholar]
- Dabrowiak J. C.Metals in Medicine; John Wiley and Sons, Ltd: Chichester, UK, 2009, Chapter 4, pp 109–114. [Google Scholar]
- Liu H.-K.; Sadler P. Metal complexes as DNA intercalators. Acc. Chem. Res. 2011, 44, 349–359. [DOI] [PubMed] [Google Scholar]
- Hartinger C. H.; Zorbas-Seifried S.; Jakupec M. A.; Kynast B.; Zorbas H.; Keppler B. K. From bench to bedside—preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100, 891. [DOI] [PubMed] [Google Scholar]
- Chen H.; Parkinson J. A.; Parsons S.; Coxall R. A.; Gould R. O.; Sadler P. J. Organometallic ruthenium(II) diamine anticancer complexes: arene-nucleobase stacking and stereospecific hydrogen-bonding in guanine adducts. J. Am. Chem. Soc. 2002, 124, 3064. [DOI] [PubMed] [Google Scholar]
- Chen H.; Parkinson; Morris R. E.; Sadler P. J. Highly selective binding of organometallic ruthenium ethylenediamine complexes to nucleic acids: novel recognition mechanisms. J. Am. Chem. Soc. 2003, 125, 173. [DOI] [PubMed] [Google Scholar]
- Allardyce C. S.; Dyson P. J.; Ellis D. J.; Salter P. A.; Scopelliti R. Synthesis and characterization of some water soluble ruthenium(II) organometallic arene complexes and an investigation of their antibiotic and antiviral properties. J. Organomet. Chem. 2003, 668, 35. [Google Scholar]
- Allardyce C. S.; Dyson P. J.; Ellis D. J.; Heath S. L. [Ru(η6-p-cymene)Cl2(pta)] (pta = 1,3,5-triaza-7-phosphatricyclo-[3.3.3.1]decane): a water soluble compound that exhibits pH dependent DNA binding providing selectivity for diseased cells. Chem. Commun. 2001, 15, 1396. [Google Scholar]
- Klajner M.; Hebraud P.; Sirlin C.; Gaiddon C.; Harlepp P. DNA binding to an anti-cancer organo-ruthenium complex. J. Phys. Chem. B 2010, 114, 14041. [DOI] [PubMed] [Google Scholar]
- Fox K.Drug–DNA Interaction Protocols; Methods in Molecular Biology; Humana Press Inc: Totowa, NJ, 1997. [Google Scholar]
- Corte-Real L.; Mendes F.; Coimbra J.; Morais T. S.; Tomaz A. I.; Valente A.; Garcia M. H.; Santos I.; Bicho M.; Marques F. Anticancer activity of structurally related ruthenium(II) cyclopentadienyl complexes. J. Biol. Inorg. Chem. 2014, 19, 853–867 10.1007/s00775-014-1120-y. [DOI] [PubMed] [Google Scholar]
- Cutts S. M.; Masta A.; Panousis C.; Parsons P. G.; Sturm R. A.; Phillips D. R. A gel mobility assay for probing the effect of drug–DNA adducts on DNA-binding proteins. Methods Mol. Biol. 1997, 90, 95–106. [DOI] [PubMed] [Google Scholar]
- Snelders D. J. M.; Casini A.; Edafe F.; van Koten G.; Klein Gebbink R. J. M.; Dyson P. J. Ruthenium(II)–arene complexes with oligocationic triarylphosphine ligands: synthesis, DNA interactions and in vitro properties. J. Organomet. Chem. 2011, 696, 1108–1116. [Google Scholar]
- Sherman S. E.; Lippard S. J. Structural aspects of platinum anticancer drug interactions with DNA. Chem. Rev. 1987, 87, 1153–1181. [Google Scholar]
- Koblinski J. E.; Ahram M.; Sloane B. F. Unraveling the role of proteases in cancer. Clin. Chim. Acta 2000, 2912113–135. [DOI] [PubMed] [Google Scholar]
- Joyce J. A.; Baruch A.; Chedade K.; Meyer-Morse N.; Giraudo E.; Tsai F.-Y.; Greenbaum D. C.; Hager J. H.; Bogyo M.; Hanahan D. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell 2004, 5, 443–453. [DOI] [PubMed] [Google Scholar]
- Frlan R.; Gobec S. Inhibitors of cathepsin B. Curr. Med. Chem. 2006, 13, 2309–2327. [DOI] [PubMed] [Google Scholar]
- Casini A.; Gabbiani C.; Sorrentino F.; Rigobello M. P.; Bindoli A.; Geldbach T. J.; Marrone A.; Re N.; Hartinger C. G.; Dyson P. J.; Messori L. Emerging protein targets for anticancer metallodrugs: inhibition of thioredoxin reductase and cathepsin B by antitumor ruthenium(II)–arene compounds. J. Med. Chem. 2008, 51, 6773–6781. [DOI] [PubMed] [Google Scholar]
- Timerbaev A. R.; Hartinger C. G.; Aleksenko S. S.; Keppler B. K. Interactions of antitumor metallodrugs with serum proteins: advances in characterization using modern analytical methodology. Chem. Rev. 2006, 106, 2224–2248. [DOI] [PubMed] [Google Scholar]
- Lacowicz J. R.Principles of Fluorescence Spectroscopy; Kluver Academic/Plenum Publishers: New York, 1999, Chapter 8, pp 238–264. [Google Scholar]
- Cocchietto M.; Salerno G.; Alessio E.; Mestroni G.; Sava G. Fate of the antimestatatic ruthenium complex ImH [trans-RuCl4(DMSO)Im] after acute i.v. treatment in mice. Anticancer Res. 2000, 20, 197–202. [PubMed] [Google Scholar]
- Sava G.; Zorzet S.; Giraldi T.; Mestroni G.; Zassinovich G. Antineoplastic activity and toxicity of an organometallic complex of ruthenium(II) in comparison with cis-PDD in mice bearing solid malignant neoplasms. Eur. J. Cancer Clin. Oncol. 1984, 20, 841–847. [DOI] [PubMed] [Google Scholar]
- Srivastava S. C.; Richard P.; Meinken G. E.; Larson S. M.; Grunbaum Z.. Tumor uptake of radioruthenium compounds. In Radiopharmaceuticals: Structure–Activity Relationships; Spencer R. P., Ed.; Grune and Stratton Inc.: New York: 1981; pp 207–223. [Google Scholar]
- Kaesz H. D. Benzylpentacarbonylmanganese. Inorg. Synth. 1989, 26, 172. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.