

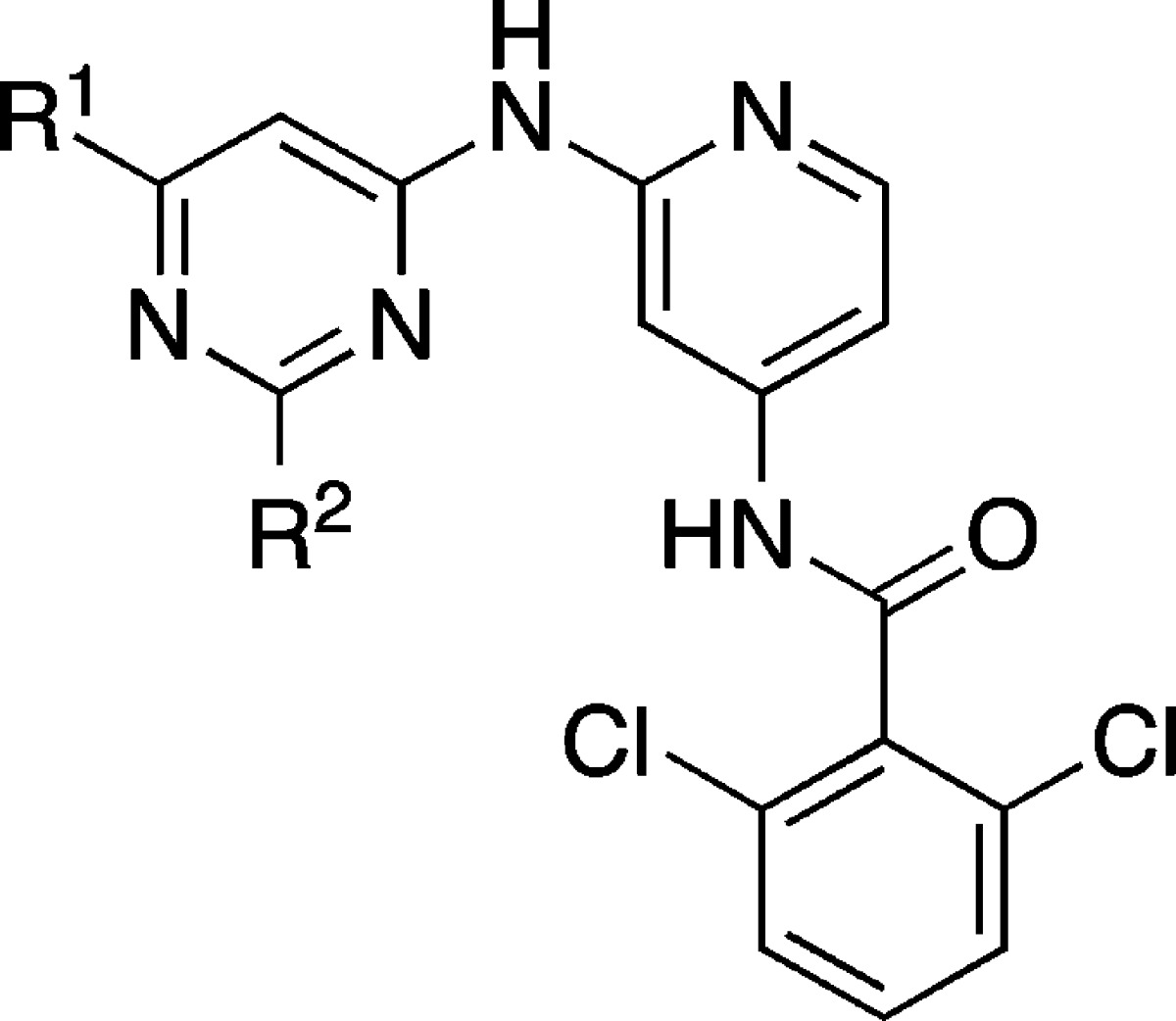
Abstract

Activating mutations within the epidermal growth factor receptor (EGFR) kinase domain, commonly L858R or deletions within exon 19, increase EGFR-driven cell proliferation and survival and are correlated with impressive responses to the EGFR inhibitors erlotinib and gefitinib in nonsmall cell lung cancer patients. Approximately 60% of acquired resistance to these agents is driven by a single secondary mutation within the EGFR kinase domain, specifically substitution of the gatekeeper residue threonine-790 with methionine (T790M). Due to dose-limiting toxicities associated with inhibition of wild-type EGFR (wtEGFR), we sought inhibitors of T790M-containing EGFR mutants with selectivity over wtEGFR. We describe the evolution of HTS hits derived from Jak2/Tyk2 inhibitors into selective EGFR inhibitors. X-ray crystal structures revealed two distinct binding modes and enabled the design of a selective series of novel diaminopyrimidine-based inhibitors with good potency against T790M-containing mutants of EGFR, high selectivity over wtEGFR, broad kinase selectivity, and desirable physicochemical properties.
Introduction
Nonsmall cell lung cancers (NSCLC) harboring mutations in the tyrosine kinase domain of the epidermal growth factor receptor (EGFR) are well-studied examples of oncogene addiction.1 Activating mutations, most commonly the point mutation L858R or deletions within exon 19 (e.g., residues 746–750), increase EGFR-driven cell proliferation and survival.2−5 The first-generation EGFR inhibitors erlotinib and gefitinib have had remarkable success for the treatment of EGFR-mutated NSCLC.6−10 However, the dramatic initial clinical responses to these agents are always followed by an acquired resistance.11−13 Approximately 60% of this acquired resistance arises from a particular secondary mutation within the EGFR kinase domain, leading to the substitution of the gatekeeper residue threonine-790 with methionine (T790M).12−16 This mutation maintains the catalytic function of the enzyme but reduces the activity of gefitinib and erlotinib through two mechanisms. The bulkier side chain of the methionine residue occludes part of the binding site utilized by both quinazoline-based inhibitors and reduces their binding affinity. This is similar to the resistance mechanism observed for Abl tyrosine-kinase inhibitors (TKIs) in CML, which is also the result of a gatekeeper residue substitution (T315I).12,13,17−20 A 2008 report proposed a second contributing mechanism, in which the T790M-containing mutants have an increased affinity for ATP, resulting in reduced cellular potency for the ATP-competitive inhibitors.21
Several second-generation EGFR inhibitors form a covalent bond with Cys-797 within the EGFR active site and have shown preclinical activity against T790M-containing mutants of EGFR. However, their clinical efficacy has been limited by associated skin rash and gastrointestinal toxicity, possibly because of their potency against wild-type EGFR (wtEGFR).22,23 Additionally, there have been reports of acquired resistance to one such covalent inhibitor via the T790M mutation, and it is questionable if drug levels can be achieved to sufficiently inhibit T790M mutant forms of EGFR.24,25 It is therefore desirable to develop a potent inhibitor of T790M-containing EGFR mutants with reduced activity against wtEGFR. Recently, third-generation covalent inhibitors including AZD9291 and CO-1686 have been generated that demonstrate selectivity for T790M-containing EGFR mutants over wtEGFR, and early phase I data indicate promising efficacy and tolerability with this approach.26−30
The compelling nature of T790M EGFR mutants as a drug target and an understanding of the relationship between wtEGFR inhibition and dose-limiting toxicities led us to initiate an effort to identify inhibitors of the major resistance mutations of EGFR, the T790M/L858R mutation (TMLR), and the T790M/del746–750 mutation (TMdel), with selectivity over wtEGFR. It is worth noting that second- and third-generation EGFR inhibitors described to date have been almost exclusively covalent in nature. Due to the low TMLR and TMdel Km for ATP (∼1–2 μM),31 it was expected that exquisite biochemical potency (Ki < 1 nM) against both T790M-containing mutants of EGFR would be required for a robust cellular effect. While covalent inhibitors cannot be displaced by ATP and are able to circumvent this issue, concerns about possible toxicity with covalent inhibitors, both off-target and resulting from time-dependent inhibition of wtEGFR, led us to pursue a noncovalent strategy.32−35 Herein we describe our efforts to identify reversible inhibitors of T790M mutant forms of EGFR beginning with a potent inhibitor of Jak2/Tyk2. X-ray crystal structures revealed two distinct binding modes of these early inhibitors that enabled design of subsequent T790M EGFR inhibitors with high levels of selectivity over wtEGFR, broad kinase selectivity, and with desirable physicochemical properties that render them appealing for further optimization.
Results and Discussion
A high-throughput biochemical screen identified compounds 1–3 as moderately potent inhibitors of both T790M-containing mutant forms of EGFR (Table 1). Interestingly, the acyclic amine 1 has only marginal selectivity for TMLR and TMdel over wtEGFR (5–10 times), whereas piperidine-containing 2 and 3 have a higher level of selectivity (11–53 times). In order to advance these initial hits into a viable lead series, a number of parameters required optimization. First, these compounds were originally identified as potent inhibitors of Jak2 and Tyk2 with Ki values between 1 and 16 nM, and it was desirable to minimize the inhibition of Jak family kinases (see Table S1 of the Supporting Information for the Jak family activity of 1–3).36 Broader kinase selectivity was promising: when tested at 1 μM against a panel of 296 kinases, 3 inhibited only 5 other kinases at >80%.37 Second, compounds 1–3 are highly lipophilic, resulting in both poor solubility and rapid turnover in liver microsomes (Clhep > 15 mL/min/kg determined in human liver microsomes). With the dual goals of increasing potency and decreasing lipophilicity, the lipophilic ligand efficiency (LLE) was a useful parameter for measuring progress during compound optimization.38−40 A low lipophilicity starting point was desired to enhance the likelihood of identifying molecules with high LLE during lead optimization. For advancement of the series, we sought a combined target profile with TMLR and TMdel biochemical potency <10 nM, wtEGFR selectivity >20 times, LogD < 2.5, and kinetic solubility >50 μM.
Table 1. Initial Hits.
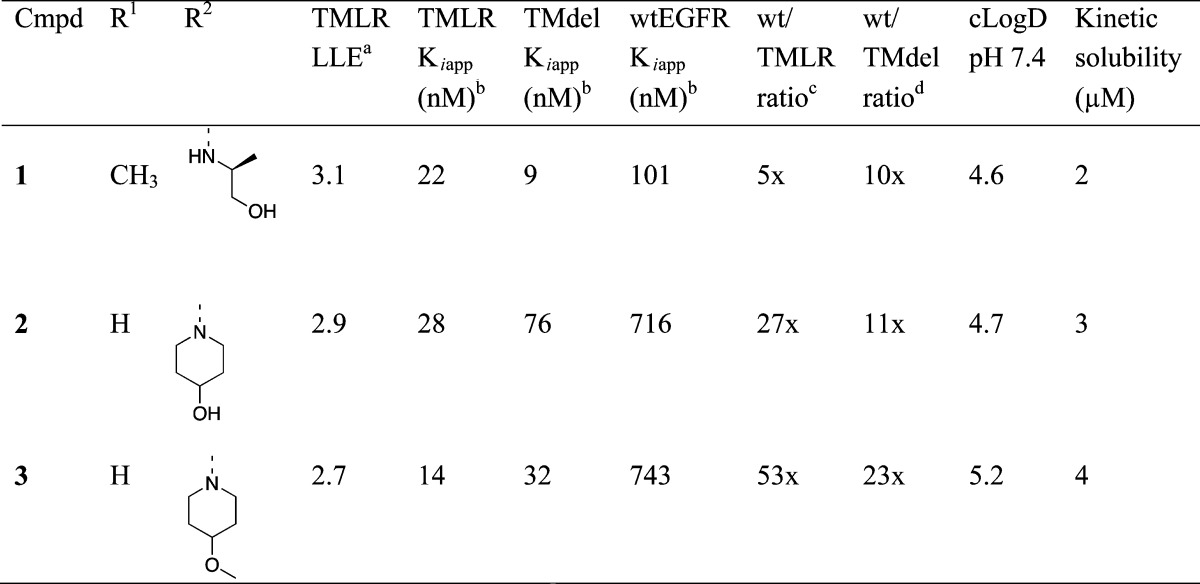
LLE is calculated using cLogD at pH 7.4.
Ki data are an average of at least two independent experiments.
Ratio of wtEGFR Kiapp over EGFR(TMLR) Kiapp.
Ratio of wtEGFR Kiapp over EGFR(TMdel) Kiapp.
An X-ray crystal structure of compound 1 with the TMLR kinase domain revealed a binding mode analogous with those seen for similar compounds in the active site of Tyk2 (Figure 1a).36 The aminopyridine functionality forms hydrogen bonds with the backbone carbonyl and NH of the hinge residue Met793. The hydroxyethyl group extends into the ribose pocket and the oxygen makes an intramolecular hydrogen bond with the amide NH. The carbonyl of the amide group is directed toward the gatekeeper residue Met790, in close proximity (3.2 Å) to the terminal methyl of the methionine side chain. When modeled into the structure of the wtEGFR kinase domain complexed with erlotinib,41 the pyridyl ring of 1 overlays with the quinazoline portion of erlotinib (Figure 1b). The quinazoline N3 of erlotinib is observed to make a water-mediated hydrogen bond with the gatekeeper Thr790 side chain, and we anticipated that the carbonyl of 1 could make a similar water-bridged interaction with the Thr790 of wtEGFR.42,43 Compounds 2 and 3 would not be conformationally restricted by the same intramolecular hydrogen bond, and the observed reduction in potency for these compounds against wtEGFR may be a result of the additional rotational degrees of freedom around the amide bond, with a corresponding increase in population of non-Thr790-interacting conformations.44
Figure 1.

(a) X-ray structure of compound 1 complexed with TMLR and (b) overlay of 1 (green) and erlotinib (magenta) complexed with wtEGFR (PDB ID: 1M17)43
We attempted to modify the dihaloaryl motif found in compounds 1–3, as this group is a significant contributor to the high lipophilicity and poor solubility of these molecules. However, modification of the dihalophenyl group was not productive (see the Supporting Information, Table S2). Nonaromatic amides do improve solubility but lack the potency required for further consideration. Polar substituents on the phenyl ring, as well as heteroaryl replacements, provide no improvement in solubility or potency compared with compounds 1–3.
We next turned our attention to replacement of the amide functionality common to compounds 1–3. Conversion of the amide into a fused heterocyclic ring reduces the degrees of rotational freedom and could therefore improve potency. Furthermore, the crystal structure of 1 suggests additional space in the binding site adjacent to the gatekeeper residue that could accommodate a fused five-membered heterocyclic ring. A series of bicyclic compounds were therefore designed with the general substructure shown in Figure 2. Depending on the choice of heterocycle, this additional ring provides alternative substitution vectors: into the ribose pocket (R1), toward the catalytic lysine (R2), and toward the gatekeeper side chain (R3).
Figure 2.

Design of the bicyclic motif.
Compounds 4 and 5 (Table 2) were synthesized as proof of concept molecules for this design, with the inclusion of a pyrazole substituent at R2 (see Figure 2) designed to interact with catalytic Lys745. We were gratified to see that while there is a slight decrease in potency for 4 and 5 compared to 3, replacement of the dihalophenyl amide results in a significant reduction in lipophilicity and a net increase in LLE. X-ray crystal structures of both compounds 4 and 5 with the TMLR kinase domain revealed that the compounds bind in the active site as designed, with the pendant pyrazole ring interacting with the side chains of both Lys745 and Glu762 (Figure 3). However, the complete loss of selectivity for both compounds against wtEGFR is difficult to rationalize. While the imidazo compound 4 could engage in a water-mediated hydrogen bond with the wtEGFR gatekeeper Thr790 side chain, as described above, and might be expected to have similar TMLR and wtEGFR activity, the pyrrolo compound 5 positions a hydrophobic face toward the gatekeeper residue and could make no such hydrogen-bonding interaction. However, the alternative rotamer of the Thr790 side chain could provide positive lipophilic interactions similar to the lipophilic Met790 side chain.
Table 2. Bicyclic Amide Replacements.

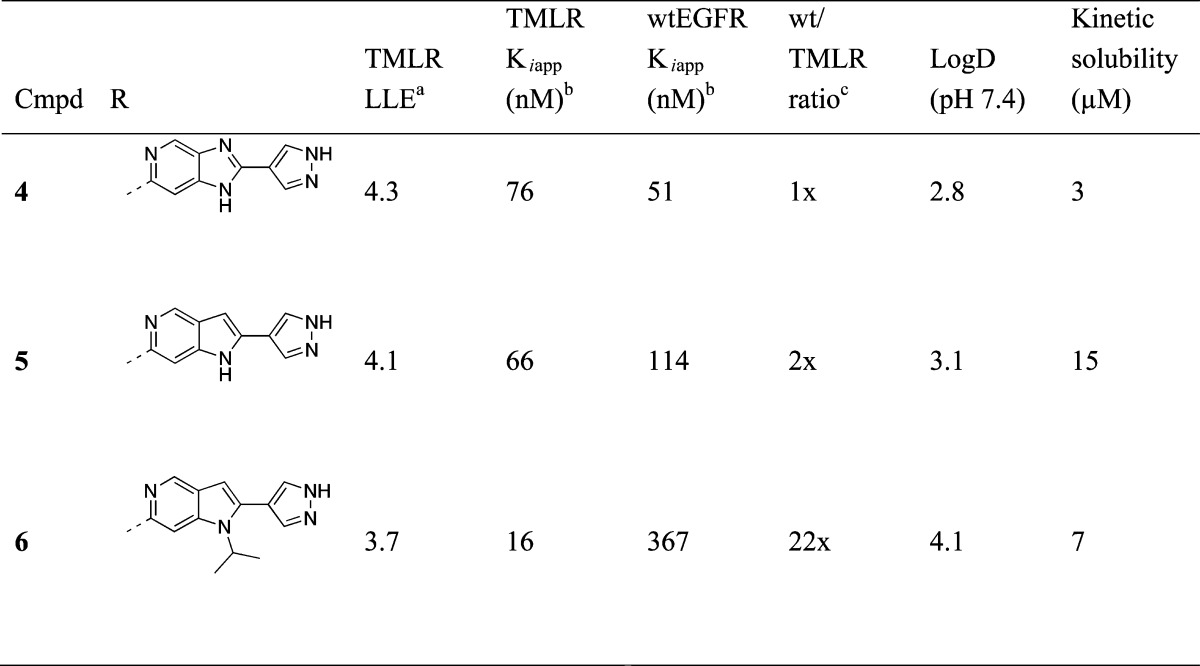
LLE is calculated using LogD measured at pH 7.4.
Ki data are an average of at least two independent experiments.
Ratio of wtEGFR Kiapp over EGFR(TMLR) Kiapp.
Figure 3.
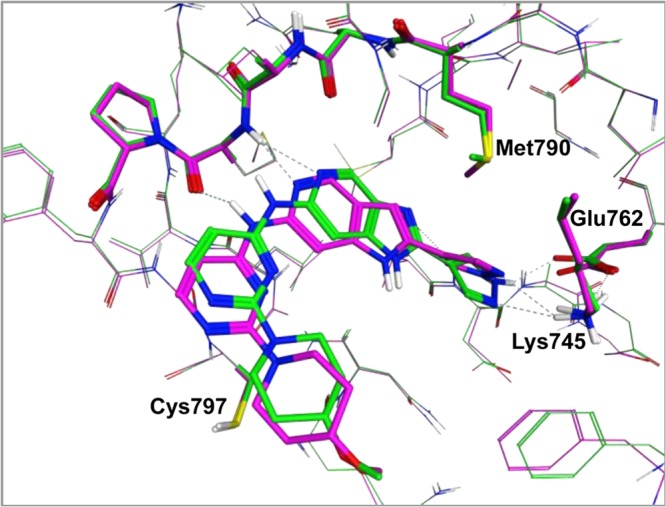
X-ray structures of 4 (green) and 5 (magenta) complexed with TMLR.
While selectivity was not optimized with bicyclic compounds 4 and 5, the structures with TMLR suggest that the unsubstituted N1 of both compounds could provide a useful vector toward the ribose pocket in the center of the binding site (vector R1 in Figure 2). We hypothesized that additional potency for EGFR could be gained by occupying the ribose pocket with lipophilic groups, as this pocket is particularly lipophilic in EGFR with Cys797 forming the floor of the binding site. The addition of an isopropyl substituent to the N1 position (compound 6) does provide an improvement in biochemical potency when compared to the N1-unsubstituted 5 (Table 2). Interestingly, 6 also regains selectivity (22-fold) for TMLR over wtEGFR. We sought a TMLR crystal structure with compound 6 in order to rationalize this selectivity and were surprised to discover that the ligand adopts a different binding mode than previous inhibitors.
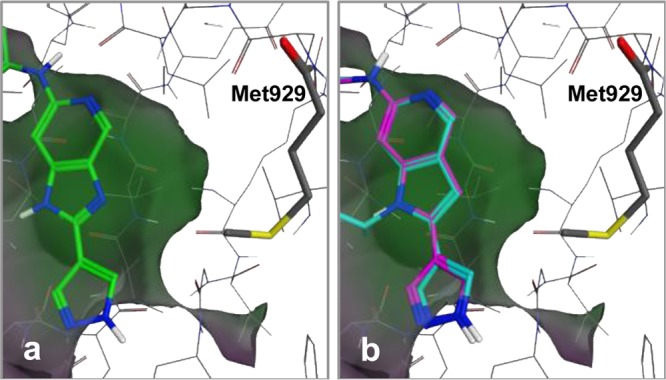
As shown in Figure 4a, relative to compounds 4 and 5 (Figure 3) or 1 (Figure 1a), 6 has flipped 180° horizontally and is positioned deeper within the binding site, with the pyridine nitrogen still maintaining a hydrogen bond with the backbone NH of Met793. However, the NH of the aminopyrimidine in the ligand now forms a hydrogen bond with the backbone carbonyl of Gln791. The pyrimidine ring is positioned underneath the gatekeeper Met790 side chain, providing substantial hydrophobic contact, and the pyrimidine N1 nitrogen achieves a hydrogen bond with the side chain hydroxyl of Thr854. Adding to the back pocket interactions, the oxygen of the methoxypiperidine group is within hydrogen-bonding distance of Lys745. The N-isopropyl group does occupy the ribose pocket as designed, although it approaches from the opposite direction than originally anticipated. In light of what appear to be favorable hydrophobic contacts between the ligand and the Met790 side chain, and the lack of possible hydrogen bonding interactions with Thr790, the enhanced potency of 6 against T790M mutants relative to wtEGFR is understandable.45 While it appears that the original binding mode should be accessible by 6, the isopropyl group may achieve more favorable hydrophobic contacts in the flipped binding mode, serving as the driving force for that conformation. An overlay of the bound conformations of 5 and 6 highlights the dramatic difference in binding mode that is possible with a very minor structural change (NH vs N-iPr) (Figure 4b).
Figure 4.

a) X-ray structure of 6 complexed with TMLR and (b) overlay of ligands 5 (magenta) and 6 (blue).
Methionine–aryl interactions are frequently found in protein structures, and the coplanar interaction of Met790 with the pyrimidine of 6 is similar to the arrangement often observed in the binding sites of adenine rings.46 However, in both the CSD and the PDB, Bissantz et al. have determined that thioether sulfur atoms are preferentially found in the plane of a phenyl ring and not in a coplanar arrangement (see Figure S-8 of ref (46)). The π system···thioether interaction observed in the 6/TMLR structure, as depicted in Figure 5a, is outside of that preferred geometry. To better understand the kinase selectivity of these inhibitors, we were interested in applying a distribution analysis more specifically to kinases. Within the PDB, we found 58 unique examples of methionine-gatekeeper kinases where the sulfur atom of the gatekeeper side chain is within 7 Å of an aromatic ring in the ligand.47 Following the methodology of Bissantz, we plotted the distribution of interaction geometries between ligand aromatic rings and methionine sulfur atoms within these structures (Figure 5b). Interestingly, the methionine interaction geometry observed in the 6/TMLR complex lies in a relatively unpopulated area of the plot. Additional refinement of this analysis to examine phenyl vs heteroaryl ring systems does not change the distribution; the small number of structures in the PDB with a ligand heteroaryl ring in close proximity to a kinase methionine gatekeeper side chain (only 10 structures) further supports the uncommon nature of this binding mode in a kinase active site. Finally, while methionine-gatekeeper residues are common, the combination with a threonine residue (or other hydrogen-bond donor) in a comparable position to Thr854 is relatively unusual.48 Therefore, we believe that the combination of a coplanar pyrimidine interaction with the Met790 side chain and a hydrogen-bonding interaction with Thr854 might provide an opportunity for achieving broader kinase selectivity in this binding mode.
Figure 5.

The centroid shift describes the distance within the plane of a sulfur atom from the center of the aromatic ring. (a) Pyrimidine···Met790 geometry observed in the 6/TMLR complex: centroid shift ≈ 1.375 Å, height ≈ 3.34 Å measured roughly perpendicular to the plane at 92.9°. (b) Distribution of gatekeeper methionine sulfur in the proximity of a ligand aromatic ring for methionine-gatekeeper kinases in the PDB.
The appealing LLE and selectivity over wtEGFR led us to use this new, T790M-selective binding mode as a starting point for structure-based design, and we constructed a series of bicyclic inhibitors, as shown in Table 3. The crystal structure of 6 suggests that the pendant pyrazole ring is pointed toward solvent and is likely not providing additional potency (Figure 4a). Compound 7 (Table 3) seemingly confirms this hypothesis, in which a hydrogen replaces the pyrazole ring and provides a compound of comparable potency for both TMLR and TMdel, as well as similar LLE and selectivity over wtEGFR. Furthermore, a crystal structure of 7 with TMLR recapitulates the binding mode observed with 6, with ligands closely overlapping (data not shown).
Table 3. Heterocycle Modifications.

LLE is calculated using LogD measured at pH 7.4.
Ki data are an average of at least two independent experiments.
Ratio of wtEGFR Kiapp over EGFR(TMLR) Kiapp.
Ratio of wtEGFR Kiapp over EGFR(TMdel) Kiapp.
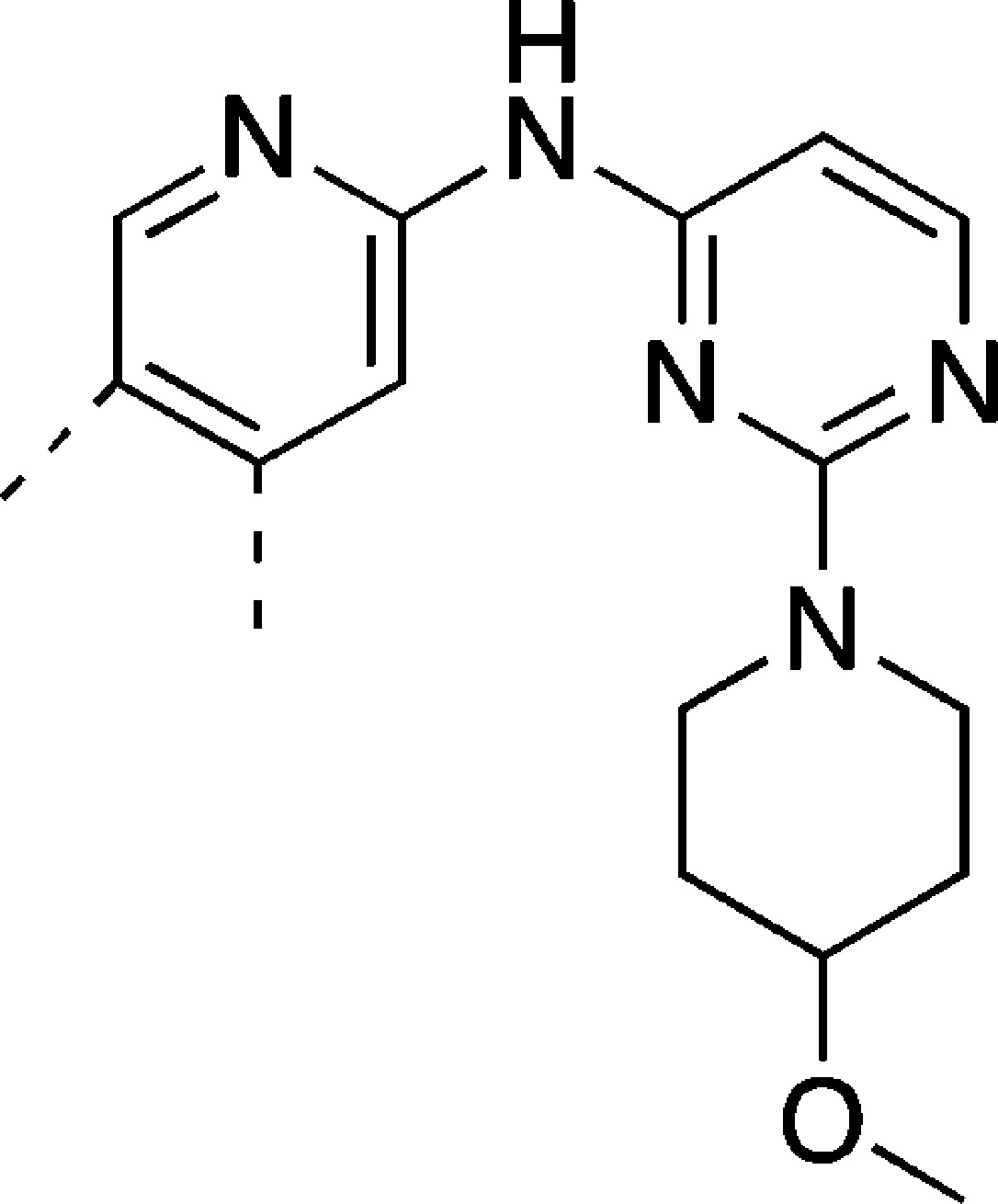
A significant benefit in removing this pyrazole was realized with respect to general selectivity over other kinases. Pyrazole-containing compounds 4–6 exhibit undesirable activity against CDK2, with 6 also showing decreased selectivity against Aurora B and KDR as compared to 3–5 (Table 4). Interestingly, while the imidazopyridine compound 4 ablates the Jak2 and Tyk2 activity exhibited by original hits 1–3 (<50% inhibition of Jak2 or Tyk2 activity observed at 1 μM), both pyrrolopyridine compounds 5 and 6 retain significant Jak family activity. NH-pyrrolopyridine 5 is equipotent against TMLR and Jak2/Tyk2, while N-isopropyl 6 exhibits approximately 10-fold selectivity for TMLR over Jak2. The differential activity against Jak2 for 4 relative to 5 and 6 may be understood using a model of all three compounds bound in the Jak2 active site in the orientation observed for 4 and 5 in TMLR (Figure 6). In this orientation, the five-membered heterocycle is adjacent to the gatekeeper residue and the hydrophobic side chain of Jak2’s Met929 gatekeeper can be expected to preferentially interact with the pyrrolo C3 methines of 5 and 6, while the imidazo N2 of 4 introduces unfavorable polarity in this hydrophobic region. The selectivity of compound 6 can be improved with removal of the terminal pyrazole moiety; as compared to 6, compound 7 exhibits minimal activity against the off-targets Aurora B, KDR, and Jak2 (Table 4). This improvement in broader kinase selectivity is perhaps unsurprising given the alternative hinge-binding motif provided by the unsubstituted pyrazole.
Table 4. Kinase Selectivity Profiles.
Figure 6.
Models of 4 (a, green), 5 (b, magenta) and 6 (b, blue) in the Jak2 active site (PBD ID: 4hge), with the protein surface colored according to lipophilicity.
While 7 has >20-fold selectivity over Aurora B, KDR, and Jak2, we sought to improve its selectivity over CDK2 in order to drive an EGFR-dependent antiproliferative effect. Modification of the ribose-pocket group from an isopropyl to the slightly larger cyclopentyl (compound 8) to fill a greater portion of the lipophilic ribose pocket does improve potency against TMLR and TMdel slightly (∼2-fold). However, this change from isopropyl to cyclopentyl increases lipophilicity and does not substantially affect kinase selectivity. A comparison of X-ray crystal structures of 8 with TMLR and CDK2 (Figures 7a,b) suggested an opportunity to enhance selectivity in subsequent designs. The binding mode of 8 in TMLR is consistent with that observed for 6 (Figure 7a), while in CDK2, the binding mode corresponds to the original binding mode observed for compounds 2, 4, and 5.49 The gatekeeper residue in CDK2 is a phenylalanine (Phe80), and this bulky side chain precludes binding of the ligand in the orientation observed in TMLR. Noting that the phenyl group of the Phe80 side chain provides hydrophobic contacts with the aromatic methines of the pyrrole ring, we hypothesized that introducing polarity in this region would disfavor interactions with CDK2 Phe80 and improve selectivity, similar to the selectivity benefit seen with 4 as compared with 5. Indeed, the addition of an extra ring nitrogen in imidazopyridine compound 9 provides an improvement in selectivity over all key off-targets as compared to 7 and 8 (Table 4), with a 24-fold reduction in activity against CDK2. Aurora B, KDR, and Jak2 also have lipophilic gatekeeper residues (Aurora B = Leu170, KDR = Val914, Jak2 = Met929), and the concurrent reduction in all of these off-target activities suggests that the binding mode observed for 8 in CDK2 may be shared among these other off-targets, consistent with the Jak2 model described in Figure 6. When tested at 1 μM in a broader panel of 219 kinases, 9 inhibits only 5 kinases at >80%.50,51 Compared with 7, compound 9 also has reduced lipophilicity while potency is maintained and thus provides an increased LLE. The improvement in physicochemical properties also results in a significant increase in kinetic aqueous solubility (Table 3).
Figure 7.
Structures of 8 complexed with (a) TMLR and (b) CDK2.
Both pyrazolopyridine compounds 10 and 11 were expected to result in a similar polarity mismatch with the CDK2 binding site, and as shown in Table 4, both of these compounds have good selectivity against all tested off-targets. Additionally, the 3-aminopyrazolopyridine compound 10 is 3–8-fold more potent than 9 with a TMLR Ki of 12 nM and TMdel Ki of 37 nM. This compound reached our initial objective of simultaneously achieving good potency with selectivity against wtEGFR (8–26 times) and with acceptable physicochemical properties that are substantially improved over those of compounds 1–3 (LogD < 2.5, kinetic solubility >50 μM). Critically, 10 has good selectivity over a broad panel of kinases, inhibiting only 5 out of 219 at >80% when tested at 1 μM.51,52
Other bicyclic systems were tested but did not result in further improvement over 10 (see Table 3). 2-Oxoimidazole 12 was similarly anticipated to insult the CDK2 and Jak2 active sites and provide good kinase selectivity; however, compared with 10, this compound has decreased activity against TMLR and was therefore not pursued. The dihydropyrrolo 13 was designed to increase the percentage of sp3 centers in an attempt to improve the solubility of pyrrolopyridine 7. This compound does exhibit an improvement in kinetic aqueous solubility (increased from 8 to 69 μM), but it has significantly reduced TMLR potency. While the structure of 8 with TMLR suggests that the sp3 carbons of 13 should be accommodated sterically, a greater energetic benefit may be provided by the aliphatic–aromatic contacts of pyrroles 7 and 8 sandwiched between Leu718 and Gly796 as compared to the aliphatic–aliphatic contacts afforded by the dihydropyrrole of 13.46 Finally, pyridone 14 was synthesized to evaluate 6–6 bicyclic systems, but the lack of potency and poor solubility led to no additional pursuit.
Further characterization of compounds in Table 3 indicated only modest cellular activity in a pEGFR assay in H1975 cells, an EGFR(TMLR)-driven cell line (8 IC50 = 1.2 μM, 10 IC50 = 1.3 μM). Additionally, the in vitro metabolic stability of all compounds in Table 3 is less than desirable with Clhep determined in human liver microsomes ranging from 11 to 19 mL/min/kg, suggesting directions for further optimization.
Conclusion
In conclusion, beginning with potent inhibitors of Jak2/Tyk2, we identified a novel series of noncovalent diaminopyrimidine-based T790M EGFR inhibitors. Replacement of the biarylamide motif with bicyclic substructures provided a significant reduction in lipophilicity. X-ray crystal structures revealed two distinct binding modes that enabled design of T790M EGFR inhibitors with high levels of selectivity over wtEGFR and with favorable physicochemical properties that provide a desirable starting point for further optimization. The unusual binding mode utilizing interactions with both the gatekeeper Met790 and Thr854 side chains provides a framework for broad kinase selectivity. The bicyclic scaffolds identified in Table 3 (7, 9, and 10) enable a substantial improvement in LLE compared to the initial HTS hits (1–3) and are promising leads in the search for selective and noncovalent inhibitors of T790M-containing EGFR mutants. Further optimization of this scaffold to achieve improved cellular potency and metabolic stability will be the focus of future publications.
Chemistry
Compounds 1–3 were prepared as generally described by Liang et al.36,54 Compounds 4–6 were prepared as described in Scheme 1. 2-Bromo-4,5-diaminopyridine and pyrazole 4-carboxylate were converted to imidazopyridine 15 via a polyphosphoric acid-mediated cyclization. Subsequent bis-SEM protection afforded intermediate 16. The corresponding pyrrolopyridine intermediates were prepared by starting with 2-bromo-4-aminopyridine 17, which was iodinated and N-sulfonylated to provide 18. Annulation with optionally SEM-protected 4-alkynylpyrazoles provided the pyrrolopyridines 19a and 19b. N1 alkylation or SEM protection of 19a,b provided the key intermediates 20a,b. SNAr reaction of 4-methoxypiperidine with 2-chloro-4-aminopyrimidine 21 afforded intermediate 22. Finally, palladium-catalyzed coupling of intermediates 16 or 20a,b with 22 yielded compounds 4–6.
Scheme 1. Preparation of Compounds 4–6.

Reagents and conditions: (a) polyphosphoric acid, 200 °C, 18 h, quant.; (b) sodium hydride, SEM-Cl, THF, DMF, rt, 2 h, 81%; (c) iodine monochloride, NaOAc, HOAc, 70 °C, 18 h, 34%; (d) methanesulfonyl chloride, triethylamine, DCM, 0 °C to rt, 2 h, 46%; (e) 4-ethynyl-1H-pyrazole, Pd(PPh3)2Cl2, CuI, triethylamine, DMF, 50–100 °C, 33%; (f) 4-ethynyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole, Pd(PPh3)2Cl2, CuI, triethylamine, DMF, 50–100 °C, 34%; (g) NaH, SEM-Cl, THF, 0 °C to rt, 5 h, 36%; (h) 2-iodopropane, Cs2CO3, DMF, 90 °C, 48 h, 40%; (i) 4-methoxypiperidine hydrochloride, Cs2CO3, DMF, 120 °C, 18 h, 87%; (j) 16, 20a, or 20b, Pd2dba3, X-Phos or Xantphos, Cs2CO3, 1,4-dioxane, 120 °C, 28–81%; (k) HCl, MeOH, rt to 50 °C, 22–76%.
Schemes 2–4 describe preparation of additional bicyclic pyridine analogs. Pyrrolopyridines 7 and 8 were readily prepared as shown in Scheme 2. Alkylation of commercially available 6-chloro-1H-pyrrolo[3,2-c]pyridine to give 23a,b followed by palladium-catalyzed coupling to aminopyrimidine 22 provided compounds 7 and 8. As described in Scheme 3, a nucleophilic displacement reaction with isopropylamine and 2,4-dichloro-5-nitropyridine provided chloropyridine 24, which was coupled with the aminopyrimidine fragment 22 followed by reduction of the nitro group to provide 25. Closure of the imidazo ring with trimethyl orthoformate yielded 9. Scheme 4 describes the synthesis of 3-aminopyrazolopyridine 10, beginning with compound 2-bromo-5-iodopyridin-4-amine. A Sandmeyer reaction converted the 4-amino functionality to a 4-chloro group (26). Conversion of the 3-iodo group to a methyl ester proceeded via palladium-catalyzed carbonylation. Subsequent reduction to the aldehyde with diisobutylaluminum hydride afforded compound 27. Treatment with hydrazine effected the ring closure to pyrazolopyridine 28. Iodination at the 3-position to 29 followed by N-alkylation provided key intermediate 30. Sequential palladium-catalyzed couplings first at the iodo position and then at the bromo position, followed by deprotection, provided compound 10.
Scheme 2. Preparation of Compounds 7 and 8.

Reagents and conditions: (a) 2-iodopropane or iodocyclopentane, Cs2CO3, DMF, 90 °C, 18–20 h, 56–90%; (b) 22, chloro[[BrettPhos][2-(2-aminoethylphenyl)palladium(II)]]/[BrettPhos], NaOtBu, tBuOH, 90 °C, 5 h, 33–49%.
Scheme 4. Preparation of Compound 10.

Reagents and conditions: (a) CuCl, tert-butyl nitrite, acetonitrile, 50 °C to reflux, 1 h, 72%; (b) palladium(II) acetate, triethylamine, MeOH, CO, 60 °C, 18 h, 47%; (c) diisobutylaluminum hydride, DCM, −78 °C, 1 h, 55%; (d) hydrazine hydrate, triethylamine, dioxane, reflux, 3 d, 34%; (e) iodine, potassium hydroxide, DMF, 40 C, 16 h; (f) 2-iodopropane, sodium hydride, DMF, rt, 16 h, 44% over two steps; (g) diphenylmethanimine, Pd2dba3, Xantphos, Cs2CO3, 1,4-dioxane, 90 °C, 3 h, 58%; (h) 22, Pd2dba3, X-Phos or Xantphos, Cs2CO3, 1,4-dioxane, 100 °C, 2 h, 57%; (i) HCl, dioxane, rt, 0.5 h, 60%.
Scheme 3. Preparation of Compound 9.

Reagents and conditions: (a) isopropylamine, triethylamine, THF, rt, 3 h, quant.; (b) 22, Pd2dba3, X-Phos, Cs2CO3, 1,4-dioxane, reflux, 3 h, 72%; (c) Pd/C, EtOAc, MeOH, H2, rt, 18 h, 92%; (d) trimethyl orthoformate, formic acid, 100 °C, 1 h, 27%.
The preparation of compounds 11–14 is described in the Supporting Information.
Experimental Section
General
Unless otherwise indicated, all reagents and solvents were purchased from commercial sources and used without further purification. Moisture or oxygen sensitive reactions were conducted under an atmosphere nitrogen gas. Unless otherwise stated, 1H NMR spectra were recorded at room temperature using Varian Unity Inova Bruker AVANCE III UltraShield-Plus Digital NMR spectrometer at indicated frequencies. Chemical shifts are expressed in ppm relative to an internal standard, tetramethylsilane (=0.00 ppm). The following abbreviations are used: br = broad signal, s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, m = multiplet. Purification by silica gel chromatography was carried out using a CombiFlash by Teledyne ISCO system with prepacked cartridges. Purification by reverse-phase high-performance liquid chromatography (HPLC) and supercritical fluid chromatography (SFC) was also used. All final compounds were purified to ≥95% chemical purity as determined by HPLC with UV detection at 254 nm. Further details on the analytical conditions used for individual compounds may be found in the Supporting Information.
6-Bromo-2-(1H-pyrazol-4-yl)-3H-imidazo[4,5-c]pyridine (15)
A mixture of 6-bromopyridine-3,4-diamine (100 mg, 0.53 mmol) and 1H-pyrazole-4-carboxylic acid (60 mg, 0.53 mmol) in polyphosphoric acid (1 g) was heated in a sealed vial at 200 °C for 18 h. The reaction mixture was diluted with water and made basic by addition of NaOH (50% aq) and a white precipitate formed. The solid thus formed was collected by filtration and washed with water to afford the title compound (150 mg, quant.). LCMS (ESI): [M + H]+ 264.1.
6-Bromo-3-(2-trimethylsilanylethoxymethyl)-2-[1-(2-trimethylsilanylethoxymethyl)-1H-pyrazol-4-yl]-3H-imidazo[4,5-c]pyridine (16)
To a suspension of 6-bromo-2-(1H-pyrazol-4-yl)-3H-imidazo[4,5-c]pyridine (150 mg, 0.53 mmol) in THF (2 mL) under a nitrogen atmosphere was added NaH (60% dispersion in mineral oil, 63 mg, 1.6 mmol). DMF (2 mL) was added to facilitate dissolution. After effervescence had ceased, 2-(trimethylsilyl)ethoxymethyl chloride (265 mg, 1.6 mmol) was added and stirring was continued for 2 h. The reaction mixture was partitioned between water and EtOAc. The aqueous phase was further extracted with EtOAc (3×), and the combined organic phases were dried (MgSO4) and concentrated in vacuo. The resulting residue was purified by chromatography on silica (solvent gradient of 0–40% EtOAc in cyclohexane) to afford the title compound (a mixture of two regioisomers) as a yellow oil that solidified on standing (224 mg 81%). LCMS (ESI): [M + H]+ 524.2.
2-Bromo-5-iodopyridin-4-amine (18a)
To a solution of 2-bromopyridin-4-amine (15.0 g, 86.7 mmol) and sodium acetate (14.2 g, 173.4 mmol) in glacial acetic acid (150 mL) was added a solution of iodine monochloride (4.9 mL, 95.4 mmol) in glacial acetic acid (70 mL). After heating for 18 h at 70 °C, the reaction was cooled to room temperature and poured into water (800 mL). The aqueous solution was partitioned twice with EtOAc. The combined organic layers were washed with saturated aqueous Na2CO3, saturated aqueous Na2S2O3, and brine and dried over MgSO4. The organic layer was filtered and concentrated in vacuo, and the resulting residue was purified by silica gel chromatography (solvent gradient of 10–60% EtOAc in heptane) to afford the title compound (8.9 g, 34%). LCMS (ESI): [M + H]+ = 300.0. 1H NMR (400 MHz, DMSO-d6): δ 8.16 (s, 1H), 6.77 (s, 1H), 6.50 (s, 2H).
N-(2-Bromo-5-iodopyridin-4-yl)methanesulfonamide (18)
Methanesulfonyl chloride (2.15 mL, 27.3 mmol) in DCM (8 mL) was added dropwise to a cold (0 °C) solution of 2-bromo-5-iodopyridin-4-amine (1.63 g, 5.45 mmol) and triethylamine (3.84 mL, 27.3 mmol) in DCM (20 mL). The reaction mixture was allowed to warm to room temperature and stirred for 2 h. The solvent was removed in vacuo and the resulting residue was purified by silica gel chromatography (solvent gradient of 5–100% EtOAc in heptane) to afford N-(2-bromo-5-iodopyridin-4-yl)-N-(methylsulfonyl)methanesulfonamide (1.1 g, 44% yield) as an off-white solid that was dissolved in aqueous NaOH solution (10%, 15 mL) in THF (15 mL) and stirred at room temperature for 16 h. The reaction mixture was concentrated. Water was added and then the mixture was acidified to pH 4 using an aqueous citric acid solution. The resulting solid was filtered and dried to afford the title compound (0.950 g, 46%). LCMS (ESI): [M + H] = 379.0. 1H NMR (400 MHz, DMSO-d6): δ 8.62 (s, 1H), 7.52 (s, 1H), 3.26 (s, 3H).
6-Bromo-2-(1H-pyrazol-4-yl)-1H-pyrrolo[3,2-c]pyridine (19a)
To a solution of 4-iodo-1H-pyrazole (11.0 g, 56.7 mmol) and ethynyl(trimethyl)silane (22.3 g, 227 mmol) in THF (80 mL) were added diethylamine (80 mL, 758 mmol), bis(triphenylphosphine)palladium(II) dichloride (6.03 g, 8.51 mmol), and copper(I) iodide (1.62 g, 8.51 mmol), and the reaction mixture was stirred at room temperature for 18 h. The solvent was removed in vacuo and the resulting residue was dissolved in Et2O (400 mL) and filtered. The filtrate was concentrated and the residue was purified by silica gel chromatography (solvent gradient of 0–100% Et2O in heptane) to afford 4-((trimethylsilyl)ethynyl)-1H-pyrazole (5.1 g, 55%) as a brown oil. LCMS (ESI): [M + H]+ = 165.2. 1H NMR (400 MHz, DMSO-d6): δ 12.92 (s, 1H), 7.87 (s, 1H), 7.46 (s, 1H), 0.01 (d, J = 6.4 Hz, 9H).
To a solution of 4-((trimethylsilyl)ethynyl)-1H-pyrazole (5.0 g, 30 mmol) in THF (50 mL) was added a solution of lithium hydroxide hydrate (1.9 g, 46 mmol) in water (10 mL). After stirring for 18 h at room temperature, the reaction mixture was neutralized with acetic acid and concentrated. The residue was partitioned between 1-butanol and water, and the combined organic layers were concentrated to yield 4-ethynyl-1H-pyrazole (2.8 g, quantitative). 1H NMR (400 MHz, DMSO-d6): δ 13.08 (s, 1H), 7.84 (s, 2H), 3.93 (s, 1H).
A mixture of N-(2-bromo-5-iodopyridin-4-yl)methanesulfonamide (1500 mg, 4.0 mmol), 4-ethynyl-1H-pyrazole (440 mg, 4.8 mmol), bis(triphenylphosphine)palladium(II) dichloride (140 mg, 0.20 mmol), copper(I) iodide (38 mg, 0.20 mmol), and triethylamine (2.5 mL, 18 mmol) in DMF (35 mL) was heated at 100 °C for 3 h and then cooled to 50 °C. DBU (1.8 mL) was added and stirring continued at 50 °C for 30 min. After cooling to room temperature, the solution was diluted with saturated NH4Cl and extracted twice with EtOAc. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The resulting residue was purified by silica gel chromatography (solvent gradient of 5–100% EtOAc in heptane) to afford the title compound (350 mg, 33%). LCMS (ESI): [M + H]+ = 263.0. 1H NMR (400 MHz, DMSO-d6): δ 13.13 (s, 1H), 11.92 (s, 1H), 8.51 (s, 1H), 8.26 (s, 1H), 8.01 (s, 1H), 7.48 (s, 1H), 6.72 (s, 1H).
6-Bromo-2-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4-yl)-1H-pyrrolo[3,2-c]pyridine (19b)
Into a 1 L three-necked round-bottom flask purged and maintained with nitrogen were added 4-iodo-1H-pyrazole (45.0 g, 232 mmol, 1.00 equiv) and N,N-dimethylformamide (500 mL). This was followed by addition of sodium hydride (60% dispersion in mineral oil, 10.2 g, 2.55 mmol) in several batches at 0 °C. The reaction mixture was stirred for 30 min at 0 °C in an ice/salt water bath. To the reaction mixture was added [2-(chloromethoxy)ethyl]trimethylsilane (42.3 g, 254 mmol) dropwise with stirring. The resulting solution was stirred for an additional 3 h at room temperature. The reaction mixture was quenched by addition of water/ice (250 mL) and extracted with EtOAc (2 × 500 mL), and the organic layers were separated and combined. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate, and concentrated under vacuum to afford 4-iodo-1-[[2-(trimethylsilyl)ethoxy]methyl]-1H-pyrazole (80.0 g) as light yellow oil without further purification.
Into a 1 L three-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen were added 4-iodo-1-[[2-(trimethylsilyl)ethoxy]methyl]-1H-pyrazole (75.0 g, 231 mmol), CuI (880 mg, 4.62 mmol), dichlorobis(triphenylphosphine)palladium(II) (3.25 g, 4.63 mmol), triethylamine (93.5 g, 926 mmol), (trimethylsilyl)acetylene (56.7 g, 578 mmol), and THF (500 mL). After being stirred for 5 h at room temperature, the reaction mixture was diluted with water (300 mL) and EtOAc (1000 mL), filtered, washed with brine, dried over anhydrous sodium sulfate, and concentrated under vacuum to afford 1-[[2-(trimethylsilyl)ethoxy]methyl]-4-[2-(trimethylsilyl)ethynyl]-1H-pyrazole (80.0 g) as brown oil. LCMS (ESI): [M + H]+ = 295.
Into a 100 mL three-necked round-bottom flask purged and maintained with nitrogen were added 1-[[2-(trimethylsilyl)ethoxy]methyl]-4-[2-(trimethylsilyl)ethynyl]-1H-pyrazole (80.0 g, 272 mmol, 1.00 equiv) and THF (100 mL). This was followed by addition of tetrabutylammonium fluoride (1 N solution in THF, 300 mL) dropwise with stirring at room temperature. After being stirred for 4 h at room temperature, the reaction mixture was diluted with EtOAc (300 mL), washed with brine, dried over anhydrous sodium sulfate, and concentrated under vacuum. The residue was purified by chromatography with EtOAc/petroleum ether (1:50) to afford 4-ethynyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole (30.0 g, 50.0%) as yellow oil. LCMS (ESI): [M + H]+ = 223. 1H NMR (300 MHz, CDCl3): 7.68 (s, 1H), 7.59 (s, 1H), 5.34 (s, 2H), 3.50 (m, 2H), 2.99 (s, 1H), 0.85 (m, 2H), 0.03 (s, 9H).
A mixture of N-(2-bromo-5-iodopyridin-4-yl)methanesulfonamide (5.9 g, 16.0 mmol), 4-ethynyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole (4.5 g, 20.0 mmol), bis(triphenylphosphine)palladium(II) dichloride (550 mg, 0.78 mmol), copper(I) iodide (150 mg, 0.78 mmol), and triethylamine (6.6 mL, 47.0 mmol) in DMF (50 mL) was heated at 100 °C for 3 h and then cooled to 50 °C. DBU (7.1 mL) was added and stirring continued for an additional 30 min at 50 °C. After stirring at room temperature for 18 h, the mixture was diluted with NH4Cl aqueous solution and extracted twice with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo. The resulting residue was purified by silica gel chromatography (gradient of 0–100% EtOAc in heptane) to afford the title compound (2.1 g, 34%). LCMS (ESI): [M + H]+ = 395.2. 1H NMR (400 MHz, DMSO-d6): δ 8.57 (s, 1H), 8.41 (s, 1H), 8.09 (s, 1H), 7.53 (s, 1H), 6.80 (s, 1H), 5.51 (s, 2H), 3.67–3.55 (m, 2H), 0.97–0.74 (m, 2H), 0.00 (s, 9H).
6-Bromo-1-((2-(trimethylsilyl)ethoxy)methyl)-2-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4-yl)-1H-pyrrolo[3,2-c]pyridine (20a)
Sodium hydride (60% dispersion in mineral oil, 270 mg, 6.7 mmol) was added slowly to a solution of 6-bromo-2-(1H-pyrazol-4-yl)-1H-pyrrolo[3,2-c]pyridine (350 mg, 1.3 mmol) in THF (10 mL) at 0 °C and stirred for 30 min followed by addition of 2-(chloromethoxy)ethyltrimethylsilane (0.94 mL, 5.3 mmol). The reaction mixture was allowed to warm to room temperature and stirred for an additional 5 h. The reaction was quenched with water (50 mL), and the aqueous layer was extracted twice with EtOAc. The combined organic layers were concentrated in vacuo, and the resulting residue was purified by silica gel chromatography (solvent gradient of 0–50% EtOAc in heptane) to afford the title compound (250 mg, 36%). LCMS (ESI): [M + H]+ = 524.0. 1H NMR (500 MHz, DMSO-d6): δ 8.60 (d, J = 4.1 Hz, 1H), 8.36–8.33 (m, 1H), 8.01 (d, J = 2.9 Hz, 2H), 6.85 (s, 1H), 5.64 (s, 2H), 5.48 (d, J = 8.3 Hz, 2H), 3.61–3.48 (m, 4H), 0.94–0.76 (m, 4H), 0.01–0.00 (m, 18H).
6-Bromo-1-isopropyl-2-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4-yl)-1H-pyrrolo[3,2-c]pyridine (20b)
2-Iodopropane (0.4 mL, 4.0 mmol) was added to a mixture of 6-bromo-2-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4-yl)-1H-pyrrolo[3,2-c]pyridine (500 mg, 1.0 mmol) and cesium carbonate (1000 mg, 4.0 mmol) in DMF (4 mL). After stirring at 90 °C for 48 h, the mixture was diluted with EtOAc (80 mL) and washed with water. The organic layer was concentrated in vacuo and the residue was purified by silica gel chromatography (solvent gradient of 0–40% EtOAc in heptane) to afford the title compound (240 mg, 40%). LCMS (ESI): [M + H]+ = 437.4. 1H NMR (400 MHz, DMSO-d6): δ 8.62 (s, 1H), 8.31 (s, 1H), 7.87 (d, J = 9.9 Hz, 2H), 6.65 (s, 1H), 5.79 (s, 1H), 5.52 (s, 2H), 4.84–4.71 (m, 1H), 3.72–3.59 (m, 2H), 1.58 (d, J = 6.9 Hz, 6H), 0.99–0.85 (m, 2H), 0.00 (s, 9H).
2-(4-Methoxypiperidin-1-yl)pyrimidin-4-ylamine (22)
2-Chloropyrimidin-4-ylamine (3.5 g, 27.0 mmol), 4-methoxypiperidine hydrochloride (4.09 g, 27.0 mmol), and Cs2CO3 (26.4 g, 81.0 mmol) were suspended in DMF (60 mL) and heated at 120 °C for 18 h. The reaction mixture was partitioned between water and EtOAc. The aqueous phase was washed with EtOAc (2×), and the combined organic phases were washed with brine, dried over MgSO4, and concentrated in vacuo affording the title compound as a solid (2.5 g). The aqueous phase was concentrated in vacuo and the slurry was extracted with EtOAc. The volatiles were removed in vacuo, and the resulting residue was purified by silica gel chromatography (solvent gradient of 0–100% EtOAc in cyclohexane) and then triturated with cyclohexane, affording a second batch of the title compound (2.38 g, 87% combining the two batches). 1H NMR (400 MHz, CDCl3): δ 7.94 (1H, d, J = 5.60 Hz), 5.74 (1H, d, J = 5.60 Hz), 4.53 (2H s), 4.33–4.24 (2H, m), 3.47–3.37 (4H, m), 3.33–3.24 (2H, m), 1.98–1.87 (2H, m), 1.60–1.47 (2H, m).
[2-(4-Methoxypiperidin-1-yl)pyrimidin-4-yl]-[2-(1H-pyrazol-4-yl)-3H-imidazo[4,5-c]pyridin-6-yl]amine (4)
A mixture of 6-bromo-3-(2-trimethylsilanylethoxymethyl)-2-[1-(2-trimethylsilanylethoxymethyl)-1H-pyrazol-4-yl]-3H-imidazo[4,5-c]pyridine (120 mg, 0.23 mmol), 22 (53 mg, 0.25 mmol), Pd2dba3 (10 mg, 0.01 mmol), XPhos (22 mg, 0.046 mmol), and Cs2CO3 (273 mg, 0.84 mmol) in dioxane (2 mL) was purged with argon (3×) and heated at 120 °C for 18 h. The cooled reaction mixture was diluted with DCM and filtered through Celite. The filtrate was concentrated in vacuo and the resulting residue was purified by chromatography on silica (solvent gradient of 0–10% MeOH in DCM, followed by 0–15% MeOH in EtOAc and by 0–10% 2 M NH3/MeOH in DCM) to afford [2-(4-methoxypiperidin-1-yl)pyrimidin-4-yl]-[3-(2-trimethylsilanylethoxymethyl)-2-[1-(2-trimethylsilanylethoxymethyl)-1H-pyrazol-4-yl]-3H-imidazo[4,5-c]pyridin-6-yl]amine (120 mg, 81%). LCMS (ESI): [M + H]+ 652.4.
[2-(4-Methoxypiperidin-1-yl)pyrimidin-4-yl]-[3-(2-trimethylsilanylethoxymethyl)-2-[1-(2-trimethylsilanylethoxymethyl)-1H-pyrazol-4-yl]-3H-imidazo[4,5-c]pyridin-6-yl]amine (120 mg, 0.18 mmol) was suspended in MeOH (5 mL) and HCl (6 M aqueous solution, 0.5 mL). The reaction mixture was stirred at room temperature for 3 h and then at 50 °C for a further 3 h. A solid precipitated on standing. The suspension thus obtained was dissolved in MeOH by the use of heat and sonication and the resulting solution was loaded onto an SCX cartridge. The cartridge was washed with MeOH while the product was eluted with 2 M NH3 in MeOH. The product containing fractions were combined and concentrated in vacuo, and the resulting residue was purified by chromatography on silica (solvent gradient of 0–10% 2 M NH3/MeOH in DCM) to afford the title compound as a white solid (55 mg, 76%). LCMS (ESI): [M + H]+ 391.9. 1H NMR (400 MHz, DMSO-d6): δ 13.29 (1H, s), 12.67 (1H, s), 9.61 (1H, s), 8.51 (1H, s), 8.43–8.09 (2H, br s), 8.03 (1H, s), 7.93 (1H, d, J = 5.9 Hz), 6.58–6.50 (1H, m), 4.25–4.14 (2H, m), 3.48–3.30 (3H, m), 3.28 (3H, s), 1.93–1.84 (2H, m), 1.47–1.35 (2H, m).
N-(2-(4-Methoxypiperidin-1-yl)pyrimidin-4-yl)-2-(1H-pyrazol-4-yl)-1H-pyrrolo[3,2-c]pyridin-6-amine (5)
A mixture of 6-bromo-1-((2-(trimethylsilyl)ethoxy)methyl)-2-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4-yl)-1H-pyrrolo[3,2-c]pyridine (125 mg, 0.239 mmol), 22 (64.6 mg, 0.310 mmol), tris(dibenzylideneacetone)dipalladium(0) (10.9 mg, 0.012 mmol), Xantphos (14.2 mg, 0.024 mmol), and cesium carbonate (233 mg, 0.716 mmol) in 1,4-dioxane (1.5 mL) and 1,2-dimethoxyethane (1.5 mL) was added to a microwave tube. After purging with nitrogen for 5 min, the sealed tube was heated at 120 °C in a CEM microwave for 60 min. The reaction mixture was filtered through a pad of Celite and the filtrate was concentrated in vacuo. The resulting residue was purified by silica gel chromatography (solvent gradient of 0–100% EtOAc in heptane) to afford N-(2-(4-methoxypiperidin-1-yl)pyrimidin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-2-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4-yl)-1H-pyrrolo[3,2-c]pyridin-6-amine (43 mg, 28%; LCMS (ESI): [M + H]+ = 652.0) as an off-white solid which was dissolved in MeOH (1 mL) and 10% aqueous NaOH (0.1 mL) and stirred at room temperature for 30 min. The mixture was evaporated and the residue was purified by HPLC (C18 silica on a 20 min gradient of 5–50% acetonitrile/0.1% NH4OH in water) to afford the title compound (5.0 mg, 20%). LCMS (ESI): [M + H]+ = 391.2. 1H NMR (400 MHz, DMSO-d6): δ 9.46 (s, 1H), 8.44 (s, 1H), 8.07 (s, 2H), 7.96–7.87 (m, 2H), 6.63–6.53 (m, 2H), 4.24 (dt, J = 13.0, 4.6 Hz, 2H), 3.52–3.35 (m, 2H), 3.30 (s, 3H), 1.97–1.85 (m, 2H), 1.51–1.36 (m, 2H).
1-Isopropyl-N-(2-(4-methoxypiperidin-1-yl)pyrimidin-4-yl)-2-(1H-pyrazol-4-yl)-1H-pyrrolo[3,2-c]pyridin-6-amine (6)
6-Bromo-1-isopropyl-2-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4-yl)-1H-pyrrolo[3,2-c]pyridine (236 mg, 0.542 mmol), 22 (169 mg, 0.813 mmol), tris(dibenzylideneacetone)dipalladium(0) (24.8 mg, 0.0271 mmol), Xantphos (32.3 mg, 0.054 mmol), cesium carbonate (442 mg, 1.36 mmol), and 1,4-dioxane (4 mL) were added to a pressure tube. After purging with nitrogen for 5 min, the sealed tube was heated at 120 °C for 20 h. The reaction mixture was filtered through a pad of Celite and washed with EtOAc. The filtrate was concentrated in vacuo and the residue was purified by silica gel chromatography (solvent gradient of 0–100% EtOAc in heptane) to afford 1-isopropyl-N-(2-(4-methoxypiperidin-1-yl)pyrimidin-4-yl)-2-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4-yl)-1H-pyrrolo[3,2-c]pyridin-6-amine (110 mg, 36%; LCMS (ESI): [M + H]+ = 563.6) as an off-white solid. This material was dissolved in 4 N HCl in 1,4-dioxane (4 mL) and stirred at room temperature for 1 h. The mixture was concentrated and the residue was purified by preparatory HPLC (C18 silica on a 20 min gradient of 5–50% acetonitrile/0.1% NH4OH in water) to afford the title compound (19 mg, 22%). LCMS (ESI): [M + H]+ = 433.3. 1H NMR (400 MHz, DMSO-d6): δ 13.21 (s, 1H), 9.56 (s, 1H), 8.50 (s, 1H), 8.33 (s, 1H), 7.93 (d, J = 5.7 Hz, 1H), 6.45 (s, 2H), 4.72 (p, J = 7.1 Hz, 1H), 4.33–4.24 (m, 2H), 3.50–3.34 (m, 2H), 3.30 (s, 3H), 1.91 (d, J = 12.2 Hz, 2H), 1.57 (d, J = 7.0 Hz, 6H), 1.43 (dq, J = 8.2, 4.5, 3.9 Hz, 2H).
6-Bromo-1-isopropyl-1H-pyrrolo[3,2-c]pyridine (23a)
A mixture of 6-bromo-1H-pyrrolo[3,2-c]pyridine (250.4 mg, 1.271 mmol), 2-iodopropane (0.50 mL, 5.0 mmol), cesium carbonate (878 mg, 2.67 mmol), and DMF (5.0 mL, 64 mmol) was heated at 90 °C for 20 h. The reaction mixture was cooled to room temperature, diluted with EtOAc, washed with water (2×) and brine, dried over magnesium sulfate, filtered, and evaporated in vacuo. The crude product was purified via flash chromatography on silica gel (12 g silica, solvent gradient of 0–100% EtOAc in DCM) to yield 272.4 mg (90%) of the title compound. LCMS (ESI): [M + H]+ = 239. 1H NMR (400 MHz, DMSO-d6): 8.59 (d, J = 1.3 Hz, 1H), 7.83 (s, 1H), 7.65 (d, J = 3.5 Hz, 1H), 6.65 (dd, J = 3.5, 0.8 Hz, 1H), 4.82 (p, J = 6.6 Hz, 1H), 1.44 (d, J = 6.7 Hz, 6H).
6-Chloro-1-cyclopentyl-1H-pyrrolo[3,2-c]pyridine (23b)
A mixture of 6-chloro-1H-pyrrolo[3,2-c]pyridine (100 mg, 0.65 mmol), iodocyclopentane (254 mg, 1.3 mmol), and Cs2CO3 (422 mg, 1.3 mmol) in DMF (1 mL) was heated at 80 °C for 18 h. The cooled reaction mixture was partitioned between water and EtOAc. The aqueous phase was further extracted with EtOAc, and the combined organic phases were dried (MgSO4) and concentrated in vacuo. The resulting residue was purified by chromatography on silica (solvent gradient of 0–50% EtOAc in cyclohexane) to afford the title compound as a colorless oil (80 mg, 56%). 1H NMR (400 MHz, CDCl3): δ 8.64 (1H, s), 7.31 (1H, s), 7.21 (1H, d, J = 3.3 Hz), 6.57 (1H, d, J = 3.2 Hz), 4.76–4.65 (1H, m), 2.30–2.17 (2H, m), 1.97–1.72 (6H, m).
(1-Isopropyl-1H-pyrrolo[3,2-c]pyridin-6-yl)-[2-(4-methoxypiperidin-1-yl)pyrimidin-4-yl]amine (7)
A mixture of 6-bromo-1-isopropylpyrrolo[3,2-c]pyridine (95.7 mg, 0.400 mmol), 22 (112.4 mg, 0.5397 mmol), chloro[[BrettPhos][2-(2-aminoethylphenyl)palladium(II)]]/[BrettPhos] admixture (31.0 mg, 0.0232 mmol), sodium tert-butoxide (118.1 mg, 1.23 mmol), and tert-butanol (1.5 mL, 16 mmol) was heated under nitrogen at 90 °C for 5 h. The reaction mixture was filtered through Celite, rinsing with 2-propanol, and concentrated. The crude product was purified via flash chromatography on silica gel (4 g of silica, solvent gradient of 0–10% MeOH in DCM) followed by achiral SFC to yield 71.9 mg (49%) of the title compound. LCMS (ESI): [M + H]+ = 361.2. 1H NMR (400 MHz, DMSO-d6): δ 9.60 (s, 1H), 8.52 (d, J = 1.2 Hz, 1H), 8.29 (s, 1H), 7.93 (d, J = 5.6 Hz, 1H), 7.45 (d, J = 3.3 Hz, 1H), 6.52 (dd, J = 3.3, 0.8 Hz, 1H), 6.36 (d, J = 5.7 Hz, 1H), 4.59 (p, J = 6.7 Hz, 1H), 4.31–4.17 (m, 2H), 3.51–3.42 (m, 1H), 3.42–3.33 (m, 2H), 1.98–1.84 (m, 2H), 1.49 (d, J = 6.7 Hz, 6H), 1.46–1.36 (m, 2H).
(1-Cyclopentyl-1H-pyrrolo[3,2-c]pyridin-6-yl)-[2-(4-methoxypiperidin-1-yl)pyrimidin-4-yl]amine (8)
A mixture of 6-bromo-1-cyclopentylpyrrolo[3,2-c]pyridine (98.5 mg, 0.371 mmol), 22 (105.4 mg, 0.5061 mmol), chloro[[BrettPhos][2-(2-aminoethylphenyl)palladium(II)]]/[BrettPhos] admixture (26.1 mg, 0.0195 mmol), sodium tert-butoxide (119.0 mg, 1.24 mmol), and tert-butanol (1.5 mL, 16 mmol) was heated under nitrogen at 90 °C for 5 h. The reaction mixture was filtered through Celite, rinsing with 2-propanol, and concentrated. The crude product was purified via flash chromatography on silica gel (4 g of silica, solvent gradient of 0–10% MeOH in DCM) followed by achiral SFC to yield 48.3 mg (33%) of the title compound. LCMS (ESI): [M + H]+ = 393.2. 1H NMR (400 MHz, DMSO-d6): δ 9.60 (s, 1H), 8.52 (d, J = 0.9 Hz, 1H), 8.29 (s, 1H), 7.93 (d, J = 5.6 Hz, 1H), 7.43 (d, J = 3.3 Hz, 1H), 6.52 (dd, J = 3.4, 0.8 Hz, 1H), 6.36 (d, J = 5.7 Hz, 1H), 4.76–4.67 (m, 1H), 4.28–4.19 (m, 2H), 3.51–3.43 (m, 1H), 3.43–3.33 (m, 2H), 3.29 (s, 3H), 2.19–2.08 (m, 2H), 1.98–1.79 (m, 6H), 1.78–1.65 (m, 2H), 1.51–1.34 (m, 2H).
(2-Chloro-5-nitropyridin-4-yl)isopropylamine (24)
Isopropylamine (1.43 mL, 16.7 mmol) was added dropwise to a solution of 2,4-dichloro-5-nitropyridine (3 g, 13.9 mmol) and triethylamine (3.9 mL, 27.8 mmol) in THF (50 mL) at room temperature, resulting in a mild exotherm. The reaction mixture was stirred at room temperature for 3 h and then partitioned between water and EtOAc. The aqueous phase was washed with EtOAc, and the combined organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo, affording the title compound as a yellow solid (quantitative). 1H NMR (400 MHz, CDCl3): δ 9.02 (1H, s), 8.08 (1H, s), 6.74 (1H, s), 3.88–3.72 (1H, m), 1.37 (6H, d, J = 6.4 Hz).
N4-Isopropyl-N2-[2-(4-methoxypiperidin-1-yl)pyrimidin-4-yl]pyridine-2,4,5-triamine (25)
(2-Chloro-5-nitropyridin-4-yl)isopropylamine (517 mg, 2.4 mmol), 11 (500 mg, 2.4 mmol), XPhos (229 mg, 48 μmol), Pd2(dba)3 (109 mg, 12 μmol), and Cs2CO3 (1.56 g, 4.8 mmol) were suspended in 1,4-dioxane (10 mL). The reaction mixture was degassed with argon, sonicated, and then heated under reflux for 3 h. The cooled reaction mixture was partitioned between water and EtOAc. The aqueous phase was washed with EtOAc (2×), and the combined organic phases were washed with brine, dried over MgSO4, and concentrated in vacuo. The resulting dark brown solid was triturated with diethyl ether, affording N4-isopropyl-N2-[2-(4-methoxypiperidin-1-yl)pyrimidin-4-yl]-5-nitropyridine-2,4-diamine as a bright yellow solid (667 mg, 72%). 1H NMR (400 MHz, CDCl3): δ 9.02 (1H, s), 8.25 (1H, m), 8.11 (1H, d, J = 5.5 Hz), 7.66 (1H, s), 7.30 (1H, s), 6.06 (1H, d, J = 5.5 Hz), 4.33–4.20 (2H, m), 3.94–3.82 (1H, m), 3.56–3.44 (3H, m), 3.41 (3H, s), 2.00–1.89 (2H, m), 1.68–1.57 (2H, m), 1.37 (6H, d, J = 6.38 Hz).
N4-Isopropyl-N2-[2-(4-methoxypiperidin-1-yl)pyrimidin-4-yl]-5-nitropyridine-2,4-diamine (2.18 g, 5.6 mmol) and Pd/C (10% by weight) (300 mg) were suspended in a mixture of EtOAc/MeOH (100 mL/10 mL) and stirred under a hydrogen atmosphere for 18 h. The reaction mixture was filtered and the filtrate was concentrated in vacuo. The resulting residue was purified by silica gel chromatography (solvent gradient of 0–10% 2 M NH3/MeOH in DCM) to afford the title compound (1.84 g, 92%). 1H NMR (400 MHz, CDCl3): δ 7.99 (1H, d, J = 5.7 Hz), 7.61 (1H, s), 7.27 (1H, s), 7.18 (1H, s), 6.14 (1H, d, J = 5.7 Hz), 4.37–4.2 (3H, m), 3.76–3.64 (1H, m), 3.50–3.33 (6H, m), 2.82 (2H, s), 1.99–1.89 (2H, m), 1.65–1.52 (2H, m), 1.28 (6H, d, J = 6.3 Hz).
(1-Isopropyl-1H-imidazo[4,5-c]pyridin-6-yl)-[2-(4-methoxypiperidin-1-yl)pyrimidin-4-yl]amine (9)
To a solution of N4-isopropyl-N2-[2-(4-methoxypiperidin-1-yl)pyrimidin-4-yl]pyridine-2,4,5-triamine (70 mg, 0.19 mmol) in trimethyl orthoformate (2 mL) was added formic acid (5 drops) and the reaction mixture was heated at 100 °C for 1 h. The volatiles were removed in vacuo and the resulting residue was purified by HPLC (C18 silica on a 20 min gradient of 20–60% acetonitrile/0.1% NH4OH in water followed by a 20 min gradient of 5–50% acetonitrile/0.1% HCO2H in water) to afford the title compound as a formate salt (19 mg, 27%). LCMS (ESI): [M + H]+ 367.9. 1H NMR (400 MHz, DMSO-d6): δ 9.74 (1H, s), 8.60 (1H, d, J = 0.9 Hz), 8.35 (1H, s), 8.28 (1H, s), 8.14 (0.4H, s), 7.92 (1H,d, J = 5.7 Hz), 6.33 (1H, d, J = 5.7 Hz), 4.65–4.54 (1H, m), 4.24–4.12 (2H, m), 3.24 (3H, s), 2.46–2.25 (3H, m), 1.91–1.77 (2H, m), 1.51 (6H, d, J = 6.8 Hz), 1.44–1.31 (2H, m).
2-Bromo-4-chloro-5-iodopyridine (26)
To a three-necked round-bottom flask containing acetonitrile (1.5 L), 2-bromo-5-iodopyridin-4-amine (125 g, 418 mmol), and copper(I) chloride (61.0 g, 622 mmol) was added tert-butyl nitrite (86.0 g, 835 mmol) dropwise with stirring at 50 °C. The resulting solution was heated to reflux for 1 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. The crude product was purified via flash chromatography on silica gel (solvent gradient of 1–10% EtOAc in petroleum ether) to afford the title compound (96 g, 72%) as a yellow solid.
6-Bromo-4-chloropyridine-3-carbaldehyde (27)
Into a 2 L pressure tank reactor were placed 2-bromo-4-chloro-5-iodopyridine (30.0 g, 94.2 mmol), MeOH (500 L), triethylamine (28.6 g, 282 mmol), and palladium(II) acetate (2.10 g, 9.35 mmol). The resulting solution was stirred at 60 °C under an atmosphere of CO (1–2 atm). After 18 h, the reaction was cooled to room temperature, filtered, and concentrated in vacuo. The crude product was purified via flash chromatography on silica gel (solvent gradient of 2–20% EtOAc in petroleum ether) to afford methyl 6-bromo-4-chloropyridine-3-carboxylate (11 g, 47%) as a white solid. LCMS (ESI): [M + H]+ = 250. 1H NMR (300 MHz, DMSO-d6): δ 8.14 (s, 1H), 7.64 (s, 1H), 3.98 (s, 3H).
To a three-necked round-bottom flask under nitrogen containing methyl 6-bromo-4-chloropyridine-3-carboxylate (35.0 g, 139 mmol) in DCM (700 mL) was added diisobutylaluminum hydride (1 M in hexanes) (153 mL) dropwise with stirring at −78 °C. The resulting solution was maintained at −78 °C for 1 h and then warmed to room temperature. After 1 h, the reaction was quenched by addition of HCl (2 N in water, 100 mL) followed by water (500 mL). The resulting solution was extracted with DCM (3 × 500 mL), and the combined organic layers were washed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo. The crude product was purified via flash chromatography on silica gel (solvent gradient of 1–10% EtOAc in petroleum ether) to afford the title compound (17 g, 55%) as a white solid. LCMS (ESI): [M + H]+ = 220.
6-Bromo-1H-pyrazolo[4,3-c]pyridine (28)
To a reaction vessel were added 6-bromo-4-chloropyridine-3-carbaldehyde (24.0 g, 109 mmol), 1,4-dioxane (600 mL), triethylamine (22.0 g, 217 mmol), and hydrazine hydrate (9.50 g, 131 mmol), and the reaction mixture was heated to reflux for 3 days. The reaction was cooled to room temperature and concentrated in vacuo. The crude product was purified via flash chromatography on silica gel (solvent gradient of 1–5% MeOH in DCM) to afford the title compound (7.4 g, 34%) as a yellow solid. LCMS (ESI): [M + H]+ = 198. 1H NMR (400 MHz, DMSO-d6): δ 13.62 (br, 1H), 8.93 (s, 1H), 8.35 (s, 1H), 7.81 (s, 1H).
6-Bromo-3-iodo-1H-pyrazolo[4,3-c]pyridine (29)
To a reaction vessel were added a solution of 6-bromo-1H-pyrazolo[4,3-c]pyridine (5.50 g, 27.8 mmol) in DMF (100 mL), potassium hydroxide (3.60 g, 64.2 mmol), and iodine (14.2 g, 55.9 mmol), and the resulting mixture was stirred for 16 h at 40 °C. The reaction was cooled to room temperature and diluted with water (500 mL). The resulting suspension was filtered, and the solids were collected and dried in an oven under reduced pressure to afford the title compound (10.5 g) as a yellow solid. LCMS (ESI): [M + H]+ = 324. 1H NMR (400 MHz, DMSO-d6): δ 14.00 (s, 1H), 8.61 (s, 1H), 7.88 (s, 1H).
6-Bromo-3-iodo-1-isopropyl-1H-pyrazolo[4,3-c]pyridine (30)
To a solution of 6-bromo-3-iodo-1H-pyrazolo[4,3-c]pyridine (10.4 g, 32.1 mmol) in DMF (100 mL) under nitrogen were added sodium hydride (2.10 g, 52.5 mmol) and 2-iodopropane (21.0 g, 123 mmol), and the reaction mixture was stirred at room temperature. After 16 h, the reaction was quenched with water (400 mL) and extracted with EtOAc (2 × 300 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo. The crude product was purified via flash chromatography on silica gel (eluent:of 10% EtOAc in petroleum ether) to afford the title compound (5.2 g, 44%) as a white solid. LCMS (ESI): [M + H]+ = 366. 1H NMR (400 MHz, DMSO-d6): δ 8.57 (s, 1H), 8.13 (s, 1H), 5.07–5.00 (m, 1H), 1.46–1.45 (d, J = 6.8 Hz, 6 H).
6-Bromo-N-(diphenylmethylene)-1-isopropyl-1H-pyrazolo[4,3-c]pyridin-3-amine (31)
A mixture of 6-bromo-3-iodo-1-isopropyl-1H-pyrazolo[4,3-c]pyridine (300 mg, 0.82 mmol), diphenylmethanimine (150 mg, 0.83 mmol), tris(dibenzylideneacetone)dipalladium(0) (75 mg, 0.08 mmol), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (36 mg, 0.08 mmol), and cesium carbonate (540 mg, 1.66 mmol) in 1,4-dioxane (6 mL) was stirred for 3 h at 90 °C in an inert atmosphere of nitrogen. The solution was cooled to room temperature, diluted with water (20 mL), and extracted with EtOAc (2×). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The resulting residue was purified by silica gel chromatography (solvent gradient of 0–60% EtOAc in petroleum ether) to afford the title compound (200 mg, 58%) as a yellow solid. LCMS (ESI): [M + H]+ = 419.
1-Isopropyl-N6-(2-(4-methoxypiperidin-1-yl)pyrimidin-4-yl)-1H-pyrazolo[4,3-c]pyridine-3,6-diamine (10)
A mixture of 6-bromo-N-(diphenylmethylene)-1-isopropyl-1H-pyrazolo[4,3-c]pyridin-3-amine (80 mg, 0.19 mmol), tris(dibenzylideneacetone)dipalladium(0) (17.5 mg, 0.02 mmol), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (9.1 mg, 0.02 mmol), 22 (48 mg, 0.23 mmol), and cesium carbonate (125 mg, 0.39 mmol) in 1,4-dioxane (2 mL) was stirred for 2 h at 100 °C in an inert atmosphere of nitrogen. The resulting mixture was cooled to room temperature and concentrated in vacuo. The resulting residue was purified by silica gel chromatography (solvent gradient 0–65% EtOAc in petroleum) to afford N3-(diphenylmethylene)-1-isopropyl-N6-(2-(4-methoxypiperidin-1-yl)pyrimidin-4-yl)-1H-pyrazolo[4,3-c]pyridine-3,6-diamine (60 mg, 57%) as a yellow solid. LCMS (ESI): [M + H]+ = 547.
To a reaction vessel were added N3-(diphenylmethylene)-1-isopropyl-N6-(2-(4-methoxypiperidin-1-yl) pyrimidin-4-yl)-1H-pyrazolo[4,3-c]pyridine-3,6-diamine (60 mg, 0.11 mmol) and HCl in 1,4-dioxane (4 M, 8 mL). The resulting solution was stirred for 0.5 h at room temperature. The pH value of the solution was adjusted to 8 with sodium bicarbonate (aq) and the mixture was extracted with of EtOAc (2×). The resulting mixture was concentrated in vacuo. The resulting residue was purified via reverse-phase HPLC and lyophilized to afford the title compound (25.3 mg, 60%) as a light yellow solid. LCMS (ESI): [M + H]+ = 383.15. 1H NMR (400 MHz, DMSO-d6): δ 9.76 (s, 1H), 8.63 (s, 1H), 8.05 (s, 1H), 7.97 (d, J = 5.6 Hz, 1H), 6.35 (d, J = 5.6 Hz, 1H), 5.77 (s, 2H), 4.50–4.44 (m, 1H), 4.25–4.20 (m, 2H), 3.49–3.40 (m, 1H), 3.38–3.34 (m, 2H), 3.32 (s, 3H), 1.92–1.90 (m, 2H), 1.50–1.30 (m, 8H).
Enzymatic assays
Experiments were carried out as previously described.31
In Vitro Microsome Metabolic Stability
Experiments were carried out as previously described.55
Acknowledgments
We thank Christopher Hamman, Michael Hayes, Yutao Jiang, Baiwei Lin, Deven Wang, and Mengling Wong for purification and analytical support, Baiwei Lin for conducting kinetic solubility and LogD experiments, and Ning Liu for conducting liver microsome stability experiments. We thank the CCDC56 for their help in the use of Relibase to query the PDB and, in particular, Tjelvar Olsson, who provided scripts for the radial distribution plots. We thank Kristina Grabowski of the Chemical Computing Group57 for her svl script to calculate the geometries specific to gatekeeper methionines. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. Use of the LS-CAT Sector 21 of the Advanced Photon Source (APS) was supported by the Michigan Economic Development Corp. and the Michigan Technology Tri-Corridor (Grant 085P1000817). APS, an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357.
Glossary
Abbreviations Used
- CSD
Cambridge Structural Database
- EtOAc
ethyl acetate
- HOAc
acetic acid
- Jak
Janus kinase
- KDR
kinase insert domain receptor
- LLE
lipophilic ligand efficiency
- MeOH
methanol
- NaOAc
sodium acetate
- NaOtBu
sodium tert-butoxide
- SEM
2-(trimethylsilyl)ethoxymethyl
- TKI
tyrosine kinase inhibitor
- TMdel
epidermal growth factor receptor (T790M/del746–750)
- TMLR
epidermal growth factor receptor (T790M/L858R)
- Tyk2
tyrosine kinase 2
- wtEGFR
wild-type epidermal growth factor receptor.
Supporting Information Available
Tabulated biochemical Jak family activity for compounds 1–3; tabulated structures and data for amide modifications; PDB codes and measurements used for the generation of Figure 5b; experimental procedures for kinetic solubility experiments; preparation and characterization of compounds 1–3 and 11–14; protein expression, purification, and crystallographic methods; data collection and refinement statistics; and HPLC methods used to determine chemical purity. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The PDB codes are as follows: 4RJ7 for 1 complexed with TMLR, 4RJ6 for 4 complexed with TMLR, 4RJ5 for 5 complexed with TMLR, 4RJ4 for 6 complexed with TMLR, 4RJ8 for 8 complexed with TMLR, and 4RJ3 for 8 complexed with CDK2.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sharma S. V.; Settleman J. Oncogene addiction: Setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007, 21, 3214–3231. [DOI] [PubMed] [Google Scholar]
- Sordella R.; Bell D. W.; Haber D. A.; Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167. [DOI] [PubMed] [Google Scholar]
- Tracy S.; Mukohara T.; Hansen M.; Meyerson M.; Johnson B. E.; Janne P. A. Gefitinib induces apoptosis in the EGFRL858R non-small-cell lung cancer cell line H3255. Cancer Res. 2004, 64, 7241–7244. [DOI] [PubMed] [Google Scholar]
- Gazdar A. F.; Minna J. D. Inhibition of EGFR signaling: All mutations are not created equal. PLoS Med. 2005, 2e3771085–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S. V.; Bell D. W.; Settleman J.; Haber D. A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [DOI] [PubMed] [Google Scholar]
- Lynch T. J.; Bell D. W.; Sordella R.; Gurubhagavatula S.; Okimoto R. A.; Brannigan B. W.; Harris P. L.; Haserlat S. M.; Supko J. G.; Haluska F. G.; Louis D. N.; Christiani D. C.; Settleman J.; Haber D. A. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [DOI] [PubMed] [Google Scholar]
- Paez J. G.; Janne P. A.; Lee J. C.; Tracy S.; Greulich H.; Gabriel S.; Herman P.; Kaye F. J.; Lindeman N.; Boggon T. J.; Naoki K.; Sasaki H.; Fujii Y.; Eck M. J.; Sellers W. R.; Johnson B. E.; Meyerson M. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [DOI] [PubMed] [Google Scholar]
- Pao W.; Miller V.; Zakowski M.; Doherty J.; Politi K.; Sarkaria I.; Singh B.; Heelan R.; Rusch V.; Fulton L.; Mardis E.; Kupfer D.; Wilson R.; Kris M.; Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 13306–13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman D. M.; Miller V. A.; Cioffredi L. A.; Yeap B. Y.; Janne P. A.; Riely G. J.; Ruiz M. G.; Giaccone G.; Sequist L. V.; Johnson B. E. Impact of epidermal growth factor receptor and KRAS mutations on clinical outcomes in previously untreated non-small cell lung cancer patients: Results of an online tumor registry of clinical trials. Clin. Cancer Res. 2009, 15, 5267–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosell R.; Moran T.; Queralt C.; Porta R.; Cardenal F.; Camps C.; Majem M.; Lopez-Vivanco G.; Isla D.; Provencio M.; Insa A.; Massuti B.; Gonzalez-Larriba J. L.; Paz-Ares L.; Bover I.; Garcia-Campelo R.; Moreno M. A.; Catot S.; Rolfo C.; Reguart N.; Palmero R.; Sanchez J. M.; Bastus R.; Mayo C.; Bertran-Alamillo J.; Molina M. A.; Sanchez J. J.; Taron M. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 2009, 361, 958–967. [DOI] [PubMed] [Google Scholar]
- Pao W.; Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat. Rev. Cancer 2010, 10, 760–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S.; Boggon T. J.; Dayaram T.; Janne P. A.; Kocher O.; Meyerson M.; Johnson B. E.; Eck M. J.; Tenen D. G.; Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [DOI] [PubMed] [Google Scholar]
- Pao W.; Miller V. A.; Politi K. A.; Riely G. J.; Somwar R.; Zakowski M. F.; Kris M. G.; Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2e73225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balak M. N.; Gong Y.; Riely G. J.; Somwar R.; Li A. R.; Zakowski M. F.; Chiang A.; Yang G.; Ouerfelli O.; Kris M. G.; Ladanyi M.; Miller V. A.; Pao W. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin. Cancer Res. 2006, 12, 6494–6501. [DOI] [PubMed] [Google Scholar]
- Kosaka T.; Yatabe Y.; Endoh H.; Yoshida K.; Hida T.; Tsuboi M.; Tada H.; Kuwano H.; Mitsudomi T. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin. Cancer Res. 2006, 12, 5764–5769. [DOI] [PubMed] [Google Scholar]
- Yu H. A.; Arcila M. E.; Rekhtman N.; Sima C. S.; Zakowski M. F.; Pao W.; Kris M. G.; Miller V. A.; Ladanyi M.; Riely G. J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N. P.; Nicoll J. M.; Nagar B.; Gorre M. E.; Paquette R. L.; Kuriyan J.; Sawyers C. L. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002, 2, 117–125. [DOI] [PubMed] [Google Scholar]
- Kwak E. L.; Sordella R.; Bell D. W.; Godin-Heymann N.; Okimoto R. A.; Brannigan B. W.; Harris P. L.; Driscoll D. R.; Fidias P.; Lynch T. J.; Rabindran S. K.; McGinnis J. P.; Wissner A.; Sharma S. V.; Isselbacher K. J.; Settleman J.; Haber D. A. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 7665–7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintas-Cardama A.; Kantarjian H.; Cortes J. Flying under the radar: The new wave of BCR-ABL inhibitors. Nat. Rev. Drug Discovery 2007, 6, 834–848. [DOI] [PubMed] [Google Scholar]
- Erlotinib activity in biochemical assays: wtEGFR Ki = 0.1 nM, EGFR(L858R) Ki = 0.3 nM, EGFR(d746–750) Ki = 0.1 nM, EGFR(T790M/L858R) Ki = 123 nM, and EGFR(T790M/d746–750) Ki = 95 nM.
- Yun C. H.; Mengwasser K. E.; Toms A. V.; Woo M. S.; Greulich H.; Wong K. K.; Meyerson M.; Eck M. J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janne P. A.; Schellens J. H.; Engelman J. A.; Eckhardt S. G.; Millham R.; Denis L. J.; Britten C. D.; Wong S. G.; Boss D. S.; Camidge D. R.. Preliminary activity and safety results from a phase I clinical trial of PF-00299804, an irreversible pan-HER inhibitor, in patients (pts) with NSCLC. J. Clin. Oncol. 2008, 26, (May 20 suppl; abstr 8027). [Google Scholar]
- Sequist L. V.; Besse B.; Lynch T. J.; Miller V. A.; Wong K. K.; Gitlitz B.; Eaton K.; Zacharchuk C.; Freyman A.; Powell C.; Ananthakrishnan R.; Quinn S.; Soria J. C. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: Results of a phase II trial in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 3076–3083. [DOI] [PubMed] [Google Scholar]
- Sos M. L.; Rode H. B.; Heynck S.; Peifer M.; Fischer F.; Kluter S.; Pawar V. G.; Reuter C.; Heuckmann J. M.; Weiss J.; Ruddigkeit L.; Rabiller M.; Koker M.; Simard J. R.; Getlik M.; Yuza Y.; Chen T. H.; Greulich H.; Thomas R. K.; Rauh D. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR Inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res. 2010, 70, 868–874. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Ko J.; Cui Z.; Abolhoda A.; Ahn J. S.; Ou S. H.; Ahn M. J.; Park K. The EGFR T790M mutation in acquired resistance to an irreversible second-generation EGFR inhibitor. Mol. Cancer Ther. 2012, 11, 784–791. [DOI] [PubMed] [Google Scholar]
- Ranson M.; Pao W.; Kim D. W.; Kim S. W.; Ohe Y.; Felip E.; Planchard D.; Ghiorghiu S.; Cantarini M.; Janne P. A. Preliminary results from a phase I study with AZD9291: An irreversible inhibitor of epidermal growth factor receptor (EGFR) activating and resistance mutations in non-small-cell lung cancer (NSCLC). Eur. J. Cancer 2013, 49Suppl 3S15. [Google Scholar]
- Sequist L. V.; Soria J.-C.; Gadgeel S.; Wakelee H.; Camidge D. R.; Varga A.; Fidias P.; Wozniak A. J.; Neal J. W.; Doebele R. C.; Garon E. B.; Jaw-Tsai S.; Stern J. C.; Allen A.; Goldman J. W. First-in-human evaluation of CO-1686, an irreversible, selective, and potent tyrosine kinase inhibitor of EGFR T790M. J. Clin. Oncol. 2013, 31, 2524. [Google Scholar]
- Cross D. A.; Ashton S. E.; Ghiorghiu S.; Eberlein C.; Nebhan C. A.; Spitzler P. J.; Orme J. P.; Finlay M. R.; Ward R. A.; Mellor M. J.; Hughes G.; Rahi A.; Jacobs V. N.; Red Brewer M.; Ichihara E.; Sun J.; Jin H.; Ballard P.; Al-Kadhimi K.; Rowlinson R.; Klinowska T.; Richmond G. H.; Cantarini M.; Kim D. W.; Ranson M. R.; Pao W. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discovery 2014, 4, 1046–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjin Tham Sjin R.; Lee K.; Walter A. O.; Dubrovskiy A.; Sheets M.; Martin T. S.; Labenski M. T.; Zhu Z.; Tester R.; Karp R.; Medikonda A.; Chaturvedi P.; Ren Y.; Haringsma H.; Etter J.; Raponi M.; Simmons A. D.; Harding T. C.; Niu D.; Nacht M.; Westlin W. F.; Petter R. C.; Allen A.; Singh J. In vitro and In vivo characterization of irreversible mutant-selective EGFR inhibitors that are wild-type sparing. Mol. Cancer Ther. 2014, 13, 1468–1479. [DOI] [PubMed] [Google Scholar]
- Janne P. A.; Ramaligam S. S.; Yang J. C. H.; Ahn M.-J.; Kim D.-W.; Kim S.-W.; Planchard D.; Ohe Y.; Felip E.; Watkins C.; Cantarini M.; Ghiorghiu S.; Ranson M. Clinical activity of the mutant-selective EGFR inhibitor AZD9291 in patients (pts) with EGFR inhibitor-resistant non-small-cell lung cancer (NSCLC). J. Clin. Oncol. 2014, 32Suppl. 58009. [Google Scholar]
- Lee H. J.; Schaefer G.; Heffron T. P.; Shao L.; Ye X.; Sideris S.; Malek S.; Chan E.; Merchant M.; La H.; Ubhayakar S.; Yauch R. L.; Pirazzoli V.; Politi K.; Settleman J. Noncovalent wild-type-sparing inhibitors of EGFR T790M. Cancer Discovery 2013, 3, 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preliminary phase I data suggest that irreversible inhibitors AZD9291 and CO-1686 do not appear to have the toxicities seen with previous irreversible inhibitors of EGFR.
- Evans D. C.; Watt A. P.; Nicoll-Griffith D. A.; Baillie T. A. Drug-protein adducts: An industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chem. Res. Toxicol. 2004, 17, 3–16. [DOI] [PubMed] [Google Scholar]
- Potashman M. H.; Duggan M. E. Covalent modifiers: An orthogonal approach to drug design. J. Med. Chem. 2009, 52, 1231–1246. [DOI] [PubMed] [Google Scholar]
- Kalgutkar A. S.; Dalvie D. K. Drug discovery for a new generation of covalent drugs. Expert Opin. Drug Discovery 2012, 7, 561–581. [DOI] [PubMed] [Google Scholar]
- Liang J.; Tsui V.; Van Abbema A.; Bao L.; Barrett K.; Beresini M.; Berezhkovskiy L.; Blair W. S.; Chang C.; Driscoll J.; Eigenbrot C.; Ghilardi N.; Gibbons P.; Halladay J.; Johnson A.; Kohli P. B.; Lai Y.; Liimatta M.; Mantik P.; Menghrajani K.; Murray J.; Sambrone A.; Xiao Y.; Shia S.; Shin Y.; Smith J.; Sohn S.; Stanley M.; Ultsch M.; Zhang B.; Wu L. C.; Magnuson S. Lead identification of novel and selective TYK2 inhibitors. Eur. J. Med. Chem. 2013, 67, 175–187. [DOI] [PubMed] [Google Scholar]
- Percent inhibition of 296 kinases determined at Invitrogen for compound 3; inhibition of >80% at 1 μM observed for EGFR(T790M/L858R), EGFR(T790M), Jak2, Jak2 JH1/JH2, MAP4K4, Mink1, and Tyk2.
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. [DOI] [PubMed] [Google Scholar]
- Shultz M. D. Setting expectations in molecular optimizations: Strengths and limitations of commonly used composite parameters. Bioorg. Med. Chem. Lett. 2013, 23, 5980–5991. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Keseru G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. [DOI] [PubMed] [Google Scholar]
- PBD accession code 1M17.
- When compound 1 is modeled into the 1M17 wtEGFR structure, if the water molecule adjacent to the gatekeeper side chain is allowed to rotate, the carbonyl oxygen of compound 1 is within hydrogen-bonding distance (<3.0 Å) of that water.
- Stamos J.; Sliwkowski M. X.; Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [DOI] [PubMed] [Google Scholar]
- Conformational searches were carried out in MacroModel 10.0, using the OPLS_2005 forcefield and mixed torsional/low-mode sampling for 1000 steps. Low-energy conformations within 1 kcal/mol of the global minimum for each molecule were compared, and the number of conformers around the amide bond was used as a proxy for the degrees of freedom.
- A structure of compound 6 complexed with wtEGFR was not obtained. Neither of the two binding modes offers a hydrogen-bonding partner for Thr790; if recapitulated in wtEGFR, the binding mode for 6 observed in TMLR would displace the water molecule typically observed to solvate Thr854, while failing to provide a compensating interaction.
- Bissantz C.; Kuhn B.; Stahl M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See Table S3 of the Supporting Information for PDB codes and measurements.
- A total of 34 out of 531 (6%) kinases have equivalent Met + Thr residues and 27 out of 531 (5%) kinases have equivalent Met + Ser residues.
- Electron density suggests that the catalytic Lys of CDK2 is acetylated in the complex with compound 8; however, we believe that this artifact of the crystallization system should not impact the binding mode of this compound.
- Percent inhibition of 219 kinases at 1 μM determined at Invitrogen; kinases inhibited at > 80%: MAP4K4, MELK, Mink1, MST1, Rse.
- While pyrrolopyridine-based inhibitors have been reported to inhibit MPS1, compounds 9 and 10 inhibit MPS1 at only 24–27% when tested at 1 μM (Invitrogen).Naud S.; Westwood I. M.; Faisal A.; Sheldrake P.; Bavetsias V.; Atrash B.; Cheung K. M.; Liu M.; Hayes A.; Schmitt J.; Wood A.; Choi V.; Boxall K.; Mak G.; Gurden M.; Valenti M.; de Haven Brandon A.; Henley A.; Baker R.; McAndrew C.; Matijssen B.; Burke R.; Hoelder S.; Eccles S. A.; Raynaud F. I.; Linardopoulos S.; van Montfort R. L.; Blagg J. Structure-based design of orally bioavailable 1H-pyrrolo[3,2-c]pyridine inhibitors of mitotic kinase monopolar spindle 1 (MPS1). J. Med. Chem. 2013, 56, 10045–10065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percent inhibition of 219 kinases at 1 μM determined at Invitrogen; kinases inhibited at >80%: MAP4K4, Mink1, MST1, MST2, Rse.
- Zak M.; Hurley C. A.; Ward S. I.; Bergeron P.; Barrett K.; Balazs M.; Blair W. S.; Bull R.; Chakravarty P.; Chang C.; Crackett P.; Deshmukh G.; DeVoss J.; Dragovich P. S.; Eigenbrot C.; Ellwood C.; Gaines S.; Ghilardi N.; Gibbons P.; Gradl S.; Gribling P.; Hamman C.; Harstad E.; Hewitt P.; Johnson A.; Johnson T.; Kenny J. R.; Koehler M. F.; Bir Kohli P.; Labadie S.; Lee W. P.; Liao J.; Liimatta M.; Mendonca R.; Narukulla R.; Pulk R.; Reeve A.; Savage S.; Shia S.; Steffek M.; Ubhayakar S.; van Abbema A.; Aliagas I.; Avitabile-Woo B.; Xiao Y.; Yang J.; Kulagowski J. J. Identification of C-2 hydroxyethyl imidazopyrrolopyridines as potent JAK1 inhibitors with favorable physicochemical properties and high selectivity over JAK2. J. Med. Chem. 2013, 56, 4764–4785. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for experimental procedures.
- Halladay J. S.; Wong S.; Jaffer S. M.; Sinhababu A. K.; Khojasteh-Bakht S. C. Metabolic stability screen for drug discovery using cassette analysis and column switching. Drug Metab. Lett. 2007, 1, 67–72. [DOI] [PubMed] [Google Scholar]
- Cambridge Crystallographic Database, Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, United Kingdom.
- Chemical Computing Group, Kaiser-Wilhelm-Ring 11, 50672 Koln, Germany.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.