

Abstract
Live attenuated nonpathogenic Mycobacterium bovis bacillus Calmette-Guérin (BCG) mediates long-lasting immune responses, has been safely administered as a tuberculosis vaccine to billions of humans, and is affordable to produce as a vaccine vector. These characteristics make it very attractive as a human immunodeficiency virus (HIV) vaccine vector candidate. Here, we assessed the immunogenicity of recombinant BCG (rBCG) constructs with different simian immunodeficiency virus (SIV)gag expression cassettes as priming agents followed by a recombinant replication-incompetent New York vaccinia virus (NYVAC) boost in rhesus macaques. Unmutated rBCG constructs were used in comparison to mutants with gene deletions identified in an in vitro screen for augmented immunogenicity. We demonstrated that BCG-SIVgag is able to elicit robust transgene-specific priming responses, resulting in strong SIV epitope-specific cellular immune responses. While enhanced immunogenicity was sustained at moderate levels for >1 year following the heterologous boost vaccination, we were unable to demonstrate a protective effect after repeated rectal mucosal challenges with pathogenic SIVmac251. Our findings highlight the potential for rBCG vaccines to stimulate effective cross-priming and enhanced major histocompatibility complex class I presentation, suggesting that combining this approach with other immunogens may contribute to the development of effective vaccine regimens against HIV.
INTRODUCTION
Diseases related to AIDS are a leading cause for global morbidity and mortality. As there is currently no licensed human immunodeficiency virus (HIV) vaccine available and, so far, only one clinical trial has demonstrated partial efficacy (1), the development of an efficient vaccine is crucial for curtailing the spread of HIV infection. Ideally, an HIV vaccine candidate would elicit durable immune responses and be safe in humans of all ages.
For >90 years, the live attenuated Mycobacterium bovis bacillus Calmette-Guérin (BCG) vaccine has been administered to billions of people to induce protection against tuberculosis (TB), illustrating its broad acceptance as a vaccine. Overall, BCG is recognized to have an exceptional safety track record, although concerns were recently raised about its potential to cause disease in immunocompromised individuals (2). Moreover, production of the BCG vaccine is relatively inexpensive, rendering it affordable in resource-poor geographic regions. As BCG immunity is not inhibited by maternal antibodies, it can be given to newborns and to children. Although the BCG vaccine efficiently prevents childhood TB (3) and provides heterologous protection against leprosy (4), its efficacy in reducing the risk of TB infection varies, averaging around 50% (5, 6).
As an indicator of longevity, not as a correlate of protection against TB, a single dose of BCG sensitizes the vaccinee to tuberculo-protein for decades (7, 8). Durable immune responses are also highly desirable against persistent infections, such as HIV. In several vaccine studies, recombinant BCG (rBCG) carrying different HIV or simian immunodeficiency virus (SIV) transgenes has proven to be an excellent priming vaccine vector for eliciting humoral and cellular immune responses (7, 9, 10). Similarly, we recently showed that an earlier generation of rBCG constructs primed for robust broadly functional SIV-specific T lymphocyte responses in rhesus monkeys (11).
In the present study, we evaluated various replicating rBCG Danish vaccine constructs with SIVgag expression cassettes as priming vectors for their ability to elicit enhanced CD8+ T cell responses in the rhesus macaque/SIV animal model. Mutants with gene deletions affecting the immunogenicity and unchanged parental versions of BCG were used to vector episomal plasmids encoding the antigen 85B (Ag85B) or 19-kDa secretion signal sequences fused to the SIVgag transgene. The mutants were selected from an in vitro screen designed to identify novel BCG strains with higher levels of major histocompatibility complex (MHC) class I presentation of transgenic peptide (M. W. Panas, J. D. Sixsmith, K. White, B. Korioth-Schmitz, S. T. Shields, B. T. Moy, S. Lee, J. E. Schmitz, W. R. Jacobs, Jr., S. A. Porcelli, B. F. Haynes, N. L. Letvin, and G. O. Gillard, unpublished data). A suboptimal dose of replication-incompetent New York vaccinia virus (NYVAC) expressing SIVgag-pol was utilized to boost immune responses, permitting greater resolution of the relative efficacy of the different mycobacterial constructs. Following an interval of >2 years after primary immunization, animals were repeatedly challenged with pathogenic neutralization-resistant SIVmac251 by the intrarectal mucosal route to determine the potential differences in protective efficacy elicited by the priming vectors. Results of the present study demonstrate that the rBCG-SIVgag vectors generated strong and enduring cellular immune responses against Gag. Nevertheless, the combination of the mycobacterial vaccine vector expressing only SIV Gag with a suboptimal boosting dose of a replication-incompetent vaccinia vector was not sufficient to generate protection against repeated mucosal challenges with SIVmac251.
MATERIALS AND METHODS
Mycobacterial vaccine vectors.
The construction of rBCG vectors was performed as previously described (11) using Escherichia coli-Mycobacterium shuttle vectors. Briefly, a multicopy plasmid with a codon-optimized full-length SIVgag (Mac239) insert was transformed into the wild-type BCG Danish (BCG Wt) strain (Staten Serum Institut, Copenhagen). Two expression cassettes (pSL7 or pSL10) that contained the full-length SIVgag sequence plus a hemagglutinin tag driven by a heat shock protein (hsp60) promoter were utilized. Whereas pSL10-SIVgag (in the parental vaccine vector BCG-pSL10) used the Ag85B signal sequence (Panas et al., unpublished), pSL7-SIVgag (in the parental BCG-pSL7) utilized the 19-kDa lipoprotein N-terminal secretion signal. An apramycin resistance cassette was cloned downstream of SIVgag.
Employing the same stably incorporated cassettes for SIV immunogen expression as in the parental vectors, rBCG Danish mutant vaccine vectors were constructed with either a deletion introduced by the specialized transduction of the cmaA2 gene (clone J13), required for mycolic acid cyclopropanation (12), or a deletion of the four-gene BCG_2587-2590 operon (clone AF25) encoding uncharacterized genes (http://genolist.pasteur.fr/BCGList/). The gene deletions were identified using an in vitro screen based on enhanced immunogenicity for MHC class I presentation function (Panas et al., unpublished). This screening assay employed a T cell hybridoma that produces interleukin 2 (IL-2) upon recognition of an immunodominant epitope in the context of MHC class I presentation; the gene deletion mutant strains were selected based on higher levels of IL-2 production in the assay system than those elicited by the parental strain. The mutations were then reconstructed into the vectors using specialized phage transduction to generate precise gene deletions (13). The mutant BCG vaccine vectors J13-pSL10 and AF25-pSL10 employed pSL10-SIVgag, whereas J13-pSL7 contained the pSL7-SIVgag expression cassette.
Expression of the transgene product was confirmed by standard Western blot analysis (11). Processing and secretion of SIV Gag were confirmed by standard p27 antigen enzyme-linked immunosorbent assay (ELISA) (ZeptoMetrix, Buffalo, NY) in the culture supernatants.
Preparation of inocula.
BCG/rBCG vectors were cultured on a rocker, to prevent clumping of the mycobacteria, at 37°C in Middlebrook 7H9 broth (Difco, Becton Dickinson, San Jose, CA) with the addition of 10% albumin-dextrose-saline, 0.5% glycerol, and 0.05% Tween 80 (Sigma-Aldrich, St. Louis, MO). When an optical density of 1 was measured at 600 nm, approximately 1.5 × 108 CFU per ml were present. After centrifugation at 4°C, the mycobacterial pellet was resuspended in injection buffer consisting of phosphate-buffered saline (Life Technologies, Grand Island, NY) with 0.05% Tween 20 (Sigma-Aldrich) and sonicated for 2 min at 4°C before injection.
Animals and immunizations.
Indian rhesus macaques were housed at the New England Primate Research Center (Southborough, MA) or at Bioqual (Rockville, MD) in accordance with the Guide for the Care and Use of Laboratory Animals (14), and approval was provided by the Institutional Care and Use Committees of the Harvard Medical School or Bioqual, respectively. Seven groups of five macaques each were genotypically selected Mamu-A*01 positive, as determined by a PCR-based technique (15), enabling CD8-specific SIV Gag epitope testing. All the rBCG-SIVgag variants (BCG-pSL10, AF25-pSL10, J13-pSL10, BCG-pSL7, and J13-pSL7) were administered once to the macaques by the intravenous route at a dose of 3 × 108 CFU (Fig. 1). The monkeys in a negative-control group received wild-type BCG Danish (BCG Wt) at the same dose and route to account for the potential nonspecific activation effects of BCG. A plasmid DNA-SIVgag-pol-nef (custom-ordered Mac239 pVRC4307 built into a pVR1012x/s backbone; Althea, San Diego, CA) was administered three times by the intramuscular route at a dose of 5 mg at weeks 0, 4, and 8 to generate a positive-control group. At 43 weeks (or at 35 weeks for the BCG-pSL7 group) following the first immunization, all nonhuman primates (NHP) were boosted once with 1 × 107 PFU recombinant NYVAC (rNYVAC)-SIVgag-pol (Mac142, provided by Sanofi Pasteur, Swiftwater, PA) by the intramuscular route.
FIG 1.
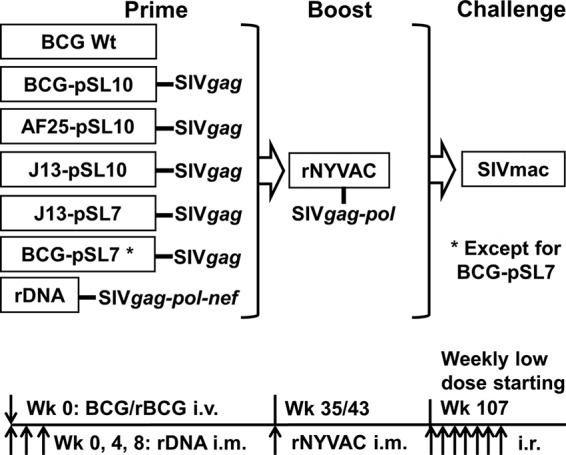
Study design and timeline. Each monkey in the seven groups of 5 Mamu-A*01-positive Indian rhesus macaques was immunized with one of the following vectors: BCG Danish wild-type (BCG Wt, negative control); parental vector BCG-pSL10-SIVgag or BCG-pSL7-SIVgag; mutant rBCG vectors with deletion of the BCG_2587-2590 operon (AF25-pSL10) or the cmaA2 gene (J13-pSL10 or J13-pSL7) with the respective SIVgag expression cassettes; or plasmid DNA (rDNA, positive control). The pSL10-SIVgag expression cassette employed the hsp60 promoter and Ag85B signal sequence, and the pSL7-SIVgag cassette used an hsp60 promoter and 19-kDa signal sequence. All rBCG vectors were administered intravenously (i.v.) once at 3 × 108 CFU. The rDNA was given 3 times by the intramuscular (i.m.) route at 5 mg each. At week 35 (for BCG-pSL7) or week 43 (for all other groups) after primary immunization, all NHP were boosted with a suboptimal dose of rNYVAC-SIVgag-pol at 107 PFU. With the exception of the BCG-pSL7 group and 1 animal from the BCG Wt group (which were unavailable for infection), the macaques were challenged weekly by the intrarectal (i.r.) route with 7.2-log viral copies of SIVmac251 until infection was detected.
SIV challenges and viral load measurements.
SIVmac251 virus (9.0-log copies/ml, SIV2008 Letvin challenge stock; SIVmac RNA branched-DNA [bDNA] assay, performed by Siemens Clinical Laboratory, Berkeley, CA) was diluted 1:60 in 0.5% human serum albumin (Calbiochem, La Jolla, CA) buffer. Anesthetized monkeys that had not received any experimental procedures for >6 months prior to the first challenge, and thus had unmanipulated gastrointestinal mucosa, were placed in sternal recumbency with the hind quarters slightly elevated and the tail raised. Diluted virus (7.2-log copies in 1 ml) was atraumatically administered into the rectal lumen with a 1-ml syringe without a needle. To prevent viral leakage from the anus, the animals were kept in this position for several minutes. Challenges were repeated weekly and discontinued once viremia was detected.
Weekly plasma viral loads were assessed using a QIAsymphony virus/bacterium midi kit (Qiagen, Germantown, MD), the QIAgility platform, and a StepOne real-time PCR (Life Technologies) (CHAVI Viral Core Laboratory, IVQAC, Duke Human Vaccine Institute, Durham, NC) for 12 weeks following infection (16). The following primers and probes were used: reverse primer, 5′-CACTAG GTGTCT CTG CACTAT CTG TTT TG-3′; forward primer, 5′-GTC TGC GTC ATCTGGTGC ATT C-3′; and probe, 5′-/56-FAM-CTT CCT CAGTGT GTT TCA CTT TCT CTT CTG CG-3′-BHQ-1/-3′ (6-FAM, 6-carboxyfluoresein; BHQ-1, black hole quencher 1) (custom synthesis by Integrated DNA Technologies). The sensitivity of this SIV viral load assay has been shown to be 250 copies per ml.
Tetramer staining.
CD8+ T lymphocytes specific for the SIV Gag epitope p11C, which is restricted to the MHC class I allele Mamu-A*01 (17), were quantified by tetramer staining and lymphocyte analysis (11). Briefly, 100 μl of EDTA-anticoagulated whole blood was stained with tetrameric Mamu-A*01/p11C, C-M complex, coupled to phycoerythrin (PE) (17), anti-CD8α-fluorescein isothiocyanate (FITC) (SK1; Becton Dickinson), anti-CD3-allophycocyanin (APC) (SP34, custom conjugate; BD Biosciences, San Jose, CA), and anti-CD4-PerCP Cy5.5 (L200; Becton Dickinson). Fixed cells were collected on a FACSCalibur using CellQuest Pro software (Becton Dickinson). Gated CD8α+ CD3+ T lymphocytes were examined for p11C-specific tetramer binding using FlowJo software (TreeStar, Ashland, OR).
IFN-γ ELISpot.
Standard gamma interferon (IFN-γ) enzyme-linked immunospot (ELISpot) assays were performed to compute cytokine-expressing antigen-specific lymphocytes, as previously described (11). Ficoll gradient-separated peripheral blood mononuclear cells (PBMC) were counted on a Guava easyCyte (Guava Technologies, Hayward, CA) and placed onto coated MultiScreen Immobilon-P microtiter plates (96 wells; Millipore, Bedford, MA) at a density of 2 × 105 cells/well in the presence of SIVmac239 Gag pooled peptides (15-mers overlapping by 11 amino acid residues covering the entire mature Gag polypeptide, 1 μg/ml final concentration; AIDS Reagent Program, Division of AIDS, NIAID, NIH) or p11C peptide (9-mer, 1 μg/ml final concentration; New England Peptide, Gardner, MA). The mitogen phytohemagglutinin (PHA-M) (Sigma-Aldrich) was used as a positive control; the medium served as a negative control. Developed ELISpot plates were read on an ImmunoSpot analyzer (Cellular Technology, Cleveland, OH). The median number of spot-forming cells (SFC) per triplicate with the background from the negative control subtracted served to determine the number of SFC/106 PBMC. Since maximum stimulation can result in high SFC with confluent spots, potentially reducing the accuracy of the automated plate reading (18), the upper limit was set at 4,000 SFC/106 PBMC, similar to what has been reported for the induction of IFN-γ production by PHA stimulation in peripheral blood (19). Responses of more than twice the background and at least 50 SFC/106 PBMC were considered positive.
Antibody-binding and neutralization assays.
Rhesus plasma was tested for SIV Gag-specific antibody binding to SIVmac239 p55 (Protein Sciences, Meriden, CT) and SIVmac251 p27 protein (ImmunoDiagnostics, Woburn, MA) by a custom SIV-binding antibody multiplex assay (SIV-BAMA) (16, 20) before vaccination and at week 4 following primary and boost immunizations in all monkeys that were later challenged with SIV. Serial dilutions of immunoglobulin G (IgG) antibodies were performed starting at a 1:80 dilution, and serum titers were calculated. Responses with at least a 3-fold elevation above baseline per animal, at least a 100 mean fluorescence intensity (MFI), and a 5-fold standard deviation plus the average of baseline MFI preimmunization samples were considered positive.
Prechallenge serum was tested for SIV-specific neutralizing antibodies using a luciferase reporter gene assay in TZM.bl cells based on single-round infection of a molecularly cloned T cell line-adapted (TCLA) neutralization-sensitive strain of SIVmac251 (SIVmac251.TCLA.15) (21). The sera were tested at a primary 1:20 dilution using a 3-fold dilution series.
Statistical analyses.
Data were statistically analyzed with Prism 6 software (GraphPad, San Diego, CA). The nonparametric two-sided Kruskal-Wallis test with corrections for multiple comparisons by Dunn's multiple-comparison test was utilized for one-way analysis of variance by rank between three or more data sets. The two-tailed Mann-Whitney test was applied for rank comparisons between two data sets. Correlations of prechallenge vaccine-elicited immune responses and postchallenge peak or set point viral loads were assessed by two-sided Spearman rank correlation tests. In all cases, a P value of <0.05 was considered significant.
RESULTS
The immunogenicity of rBCG Danish vectors employing either the pSL10 or pSL7 expression cassette for SIVgag (using an Ag85B signal sequence or a 19-kDa secretion signal, respectively) was tested in vivo in rhesus macaques. The immune responses elicited by the parental (BCG-pSL10 and BCG-pSL7) and mutant constructs with a deletion of the four-gene BCG_2587-2590 operon (AF25-pSL10) or the cmaA2 gene (J13-pSL10 and J13-pSL7) were compared (Fig. 1). BCG Wt without a transgene and the plasmid DNA-SIVgag-pol-nef vaccine served as the negative- and positive-control groups, respectively. Two weeks after a single inoculation, the mutant rBCG constructs generated measurable SIV Gag-specific CD8+ T cell peak responses (Fig. 2A). In fact, the J13-pSL7 group developed significantly higher p11C tetramer peak responses than the BCG Wt and BCG-pSL10 groups (Kruskal-Wallis test, P = 0.003; Dunn's multiple-comparison test, P = 0.03 or 0.02, respectively). Direct comparison between the mutant and parental constructs revealed that the monkeys immunized with the mutant construct J13-pSL7-SIVgag developed significantly higher p11C tetramer peak responses than those vaccinated with the parental BCG-pSL7-SIVgag construct (P = 0.008, Mann-Whitney test). After the brief increase, p11C tetramer responses returned to baseline in the rBCG-primed animals. In contrast, three immunizations with ribosomal DNA (rDNA) elicited strong sustained responses against the dominant SIV Gag p11C epitope that waned before boost immunization was initiated.
FIG 2.

Primary immunization with rBCG vectors elicits detectable SIV Gag-specific immune responses. (A) Monkeys in the groups immunized with mutant rBCG-SIVgag developed measurable (>0.05%) peak p11C epitope-specific tetramer responses around weeks 2 to 3 after priming immunization (i.e., 1 NHP in AF25-pSL10 group, 1 in J13-pSL10, and 4 in J13-pSL7; in comparison, there were 4 NHP with strong responses in the rDNA group after 3 immunizations). Kruskal-Wallis test (week 2 postprime), P = 0.003; Dunn's multiple-comparison posttest (significant differences), P = 0.03 (BCG Wt versus J13-pSL7) and P = 0.02 (BCG-pSL10 versus J13-pSL7); Mann-Whitney test, P = 0.008 (BCG-pSL7 versus J13-pSL7). (B) Following priming immunization with parental BCG-pSL7-SIVgag or mutant rBCG constructs, NHP developed robust IFN-γ ELISpot responses upon SIV Gag peptide pool stimulation that were similar to the magnitude of immune responses generated after 3 rDNA vaccinations. IFN-γ ELISpot responses were low or undetectable after immunization with BCG-pSL10 or BCG Wt (total n = 35 [n = 5 in each group]). Kruskal-Wallis test (week 4 postprime), P = 0.003; Dunn's multiple-comparison test (significant differences), P = 0.03 (BCG Wt versus J13-pSL10), P = 0.02 (BCG Wt versus BCG-pSL7), and P = 0.02 (BCG Wt versus J13-pSL7). Arrows indicate immunization time points. Lines connect responses from individual animals over the time course of the study.
Strikingly, the mutant rBCG and the pSL7-SIVgag-expressing parental constructs generated high-frequency and persistent SIV Gag pooled peptide-stimulated IFN-γ ELISpot responses (Fig. 2B). The cellular immune responses induced by these rBCG constructs were clearly superior to those generated by the BCG-pSL10-SIVgag construct that failed to provoke durable and detectable IFN-γ expression. At week 4 postprime, the animals in groups BCG-pSL7, J13-pSL7, and J13-pSL10 developed significantly higher IFN-γ ELISpot peak responses than those in the BCG Wt group (Kruskal-Wallis test, P = 0.003; Dunn's multiple-comparison posttest, P = 0.02, 0.02, and 0.03, respectively). In fact, the SIV Gag-specific peak and plateau responses in the mutant rBCG and BCG-pSL7 groups, as quantified by IFN-γ ELISpot assays, were comparable to those elicited by the plasmid DNA immunogen.
By week 35 or 43 postvaccination, the SIV Gag-specific immune responses had reached a plateau in all the monkeys. All seven groups of primed monkeys were boosted with a suboptimal dose of 1 × 107 PFU NYVAC-SIVgag-pol to enable the differentiation of immune responses between the priming agents. The p11C tetramer responses in the BCG-pSL10-SIVgag-primed group were comparable in magnitude and kinetics with those induced by the empty BCG Danish control vector after rNYVAC vaccination, indicating that limited immunogenicity resulted from priming with the BCG-pSL10-SIVgag construct (Fig. 3A). In contrast, strong CD8+ p11C epitope-specific tetramer responses were measured in monkeys primed with the three mutant rBCG constructs or the parental BCG-pSL7-SIVgag construct following the rNYVAC boost. Immunization with J13-pSL7-SIVgag elicited the highest percentage of p11C+ CD8+ T lymphocytes (median, 4.3%; range, 2.5% to 5.3%). Overall, the p11C tetramer responses signified strong enhancement of CD8+ T lymphocyte-specific immunity by four of the rBCG priming immunogens (BCG-pSL7, AF25-pSL10, J13-pSL10, and J13-pSL7).
FIG 3.

BCG-SIVgag vectors as priming immunogens for strong SIV Gag-specific immune responses following suboptimal rNYVAC boost immunization. (A) Mamu-A*01-positive rhesus macaques primed with one of three mutant rBCG variants (AF25-pSL10, J13-pSL10, or J13-pSL7) or the parental BCG-pSL7-SIVgag developed strong immunodominant SIV Gag p11C epitope-specific immune responses in CD8+ T lymphocytes after a boost with 1 × 107 PFU rNYVAC-SIVgag-pol, comparable to those induced in rDNA-rNYVAC-vaccinated animals but significantly stronger than those primed with BCG Wt or BCG-pSL10-SIVgag. (B) Following rNYVAC boost, cytokine expression levels measured by IFN-γ ELISpot assays in PBMC were considerably enhanced in monkeys immunized with mutant rBCG variants, BCG-pSL7-SIVgag, or plasmid DNA-SIVgag-pol-nef. Postpeak, IFN-γ responses remained at elevated levels. In contrast, the rNYVAC vaccination of BCG-pSL10-primed animals elicited only modest boost responses, similar to those induced in the negative-control group BCG Wt, indicating that the BCG-pSL10 construct used was poorly immunogenic, unlike the other constructs. Arrows indicate the time points of rNYVAC boosts. The dotted lines represent the upper limit of accurate distinction between spots, set at 4,000 SFC/106 PBMC (n = 5 for each of the 7 groups). Kruskal-Wallis test (week 2 postboost), P = 0.003; Dunn's multiple-comparison test (significant differences), P = 0.003 (BCG Wt versus J13-pSL7) and P = 0.02 (BCG-pSL10 versus J13-pSL7). Lines connect responses from individual animals over the time course of the study.
Following the suboptimal boost immunization, the monkeys primed with the mutant BCG-SIVgag strains (AF25-pSL10, J13-pSL10, and J13-pSL7) or with BCG-pSL7-SIVgag showed strong SIV Gag pooled peptide-stimulated IFN-γ ELISpot responses that were equivalent or higher than those elicited by three immunizations with the plasmid DNA-SIVgag-pol-nef vaccine (Fig. 3B). More importantly, monkeys primed with the mutant rBCG or parental BCG-pSL7-SIVgag vector showed enhanced SIV Gag-specific IFN-γ expression after the NYVAC-SIVgag-pol boost compared to that of monkeys primed with the negative-control vector. For the immune responses at week 2 postboost, the Kruskal-Wallis test achieved a P value of 0.003, with significant differences between the J13-pSL7-SIVgag vector and the BCG Wt and the BCG-pSL10-SIVgag vectors in Dunn's multiple-comparison test (P = 0.003 and 0.02, respectively).
The inclusion of CD8 epitope-specific stimulation with p11C peptide in the IFN-γ ELISpot assays provided additional information on whether the cytokine release of the different vaccine modalities was more likely a CD4+ or a CD8+ T lymphocyte response. In the vast majority of the animals, the p11C peptide-stimulated IFN-γ ELISpot responses were low or undetectable following primary immunization (Fig. 4A), even in rBCG-primed animals, with robust IFN-γ ELISpot responses after SIV Gag pooled peptide stimulation. This suggests that the BCG vaccine vectors initially induced bias toward CD4+ T lymphocyte-specific immunity. Only a few monkeys demonstrated IFN-γ ELISpot responses upon p11C peptide stimulation, showing the contribution of CD8+ T cell immunity. In contrast, p11C peptide and SIV Gag peptide pool-stimulated IFN-γ expression levels of PBMC from rDNA-vaccinated monkeys were mostly comparable in range following primary immunization. This and the observation of similarly strong and prolonged immunodominant p11C tetramer responses (Fig. 2A) demonstrate that DNA plasmid immunization of the Mamu-A*01 MHC class I allele-expressing rhesus macaques generated a strong bias toward CD8+ T cell responses. Only one of five rDNA-primed macaques did not induce p11C nonamer-stimulated IFN-γ ELISpot responses. This animal, which also displayed the lowest p11C tetramer responses among the rDNA-primed animals, showed rather modest cytokine expression after SIV Gag peptide pool stimulation following primary immunization, signifying minor CD4+ T lymphocyte responses.
FIG 4.

Dominant p11C epitope-specific IFN-γ expression by CD8+ T lymphocytes. (A) After BCG/rBCG priming immunization, only a few of the MHC class I allele Mamu-A*01-expressing rhesus macaques showed IFN-γ ELISpot responses upon stimulation with the CD8+ T lymphocyte-specific p11C peptide, in contrast to the very strong IFN-γ expression by CD8+ T lymphocytes in plasmid DNA-SIVgag-pol-nef-primed macaques. The data conform to a strong bias of the mycobacterial vaccines toward CD4+ T lymphocyte responses but indicate bias toward CD8 responses in Mamu-A*01-positive monkeys after 3 repeated rDNA vaccinations. (B) Following the rNYVAC-SIVgag-pol boost, IFN-γ expression upon p11C peptide stimulation showed a pattern of responses similar to those measured after SIV Gag pooled peptide stimulation. The results indicate that the boosting viral vector overrides the bias established at prime and changes the bias toward CD8-leaning responses (n = 5 for each of the 7 groups). Lines connect responses from individual animals over the time course of the study. Arrows indicate immunization time points.
After an rNYVAC boost, the MHC class I-specific p11C peptide-stimulated and SIV Gag pool peptide-stimulated IFN-γ ELISpot responses were similar in the groups primed with BCG Wt and BCG-pSL10, indicating that the viral vector induced CD8-biased immune responses with little or no impact by the priming. The IFN-γ secretion levels in the rDNA group were also similar in magnitude between the SIV Gag peptide pool- and p11C peptide-stimulated ELISpot assays. Four groups of monkeys (i.e., three mutant rBCG groups and one parental BCG-pSL7 group) showed robust cytokine expression levels after p11C peptide stimulation following the boost that were only moderately lower than those in the SIV Gag peptide pool-stimulated ELISpot assay, indicating a combination of CD4+ and strong CD8+ T lymphocyte responses after the prime-boost regimen. Since the priming responses were biased toward CD4+ T lymphocyte immunity, the boosting data demonstrate robust augmentation of SIV Gag-specific CD8+ T cell responses after the boost as a direct result of priming with the rBCG constructs. We also explored the humoral immune responses stimulated by the vaccine regimens. SIV Gag-specific antibodies were not detected in the plasma of any of the study animals 4 weeks following primary immunization. After the rNYVAC boost immunization, SIV Gag-specific IgG antibodies were detectable in only 4 of the 35 monkeys, all belonging to the plasmid DNA-SIVgag-pol-nef-primed group. The SIV p27 MFI at 80-fold dilution in the plasma of these four monkeys ranged between 28,129 and 15,170, with half-maximal effective concentration (EC50) values between 1,901 and <80 and areas under the curve (AUC) for the SIV p27 titer between 46,335 and 6,110. For SIV p55, the values ranged between 29,482 and 20,277 MFI at an 80-fold dilution, EC50 values between 704.6 and <80, and AUC for the SIV p55 titer between 31,609 and 9,270. One monkey in the rDNA group that did not have a detectable level of Gag-specific IgG antibodies also lacked robust cellular immune responses. No SIV Gag-specific binding antibodies were detected in any of the rBCG-treated animals. Vaccine-elicited neutralizing antibodies were not detected in any of the study animals.
No immune-related adverse vaccination effects were observed in any of the monkeys. At week 64 following rNYVAC boost vaccination (i.e., week 107 following primary immunization), 29 animals (4 animals from the BCG Wt group and 5 monkeys each from the BCG-pSL10, AF25-pSL10, J13-pSL10, J13-pSL7, and rDNA groups) were challenged weekly with SIVmac251 by the intrarectal route until viremia was confirmed. In peripheral blood samples drawn immediately prior to the challenge, the rBCG mutant variant and rDNA groups showed long-lasting measurable median p11C tetramer responses that trended higher than those in the BCG Wt and BCG-pSL10 groups (Fig. 5A). At the same prechallenge time point, an SIV Gag peptide pool-stimulated IFN-γ ELISpot assay demonstrated sustained robust cytokine expression levels in the mutant rBCG-primed, the rDNA-primed, and the rNYVAC-boosted monkeys in contrast to those in the BCG Wt and BCG-pSL10 groups (Fig. 5B). The long-lasting enhancement of immune responses was statistically significant according to the Kruskal-Wallis test (P = 0.003), with Dunn's multiple-comparison test affirming that monkeys in the J13-pSL7 group had significantly higher secretion levels of IFN-γ than those in the BCG Wt and BCG-pSL10 groups (P = 0.04 and 0.03, respectively). The prechallenge ELISpot responses were approximately one-third the magnitude of those measured at week 16 postboost.
FIG 5.
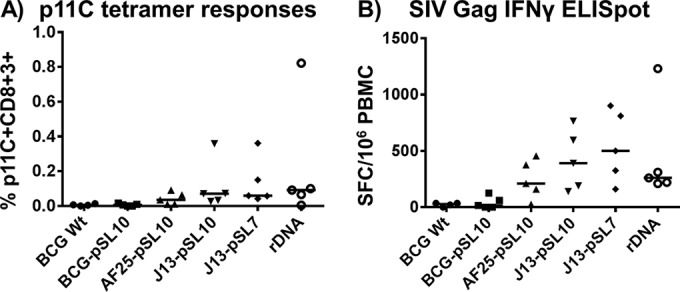
Long-lasting p11C tetramer and SIV Gag pooled peptide-stimulated IFN-γ ELISpot responses. At week 107 postprime (i.e., week 64 postboost), prior to SIV challenge, mutant rBCG- or rDNA-primed groups demonstrated sustained but mostly low levels of p11C tetramer-positive CD8+ T lymphocytes (A) and robust persistent IFN-γ ELISpot responses upon SIV pooled peptide stimulation with variations between groups (B), whereas such responses were lacking in the BCG Wt and BCG-pSL10 cohorts. For BCG Wt, n = 4; for the other groups, n = 5. Kruskal-Wallis test, P = 0.003; Dunn's multiple-comparison test (significant differences), P = 0.04 (BCG Wt versus J13-pSL7) or P = 0.03 (BCG-pSL10 versus J13-pSL7). Bars indicate medians.
Following weekly intrarectal challenges with pathogenic SIVmac251, all the monkeys developed viremia after a median of either one or two challenges (range, one to seven challenges) (Fig. 6A). Thus, the prime-boost vaccination regimens did not show differences in viral acquisition. Moreover, a statistical difference for the peak viral loads between the different vaccinated cohorts was lacking (P = 0.6, Kruskal-Wallis test) (Fig. 6B). Viral loads at the set point displayed more variation than those at the peak, but there was no statistical difference between the groups (P = 0.2, Kruskal-Wallis test) (Fig. 6C). Additional comparisons by the nonparametric Spearman test did not reveal correlations between prechallenge Gag-specific immune responses (as measured by the IFN-γ ELISpot assay or p11C tetramer staining) and viral acquisition or viral loads. SIV Gag-specific antibodies detected in the rDNA-rNYVAC-immunized monkeys also did not have an apparent effect on viral acquisition or viral loads.
FIG 6.
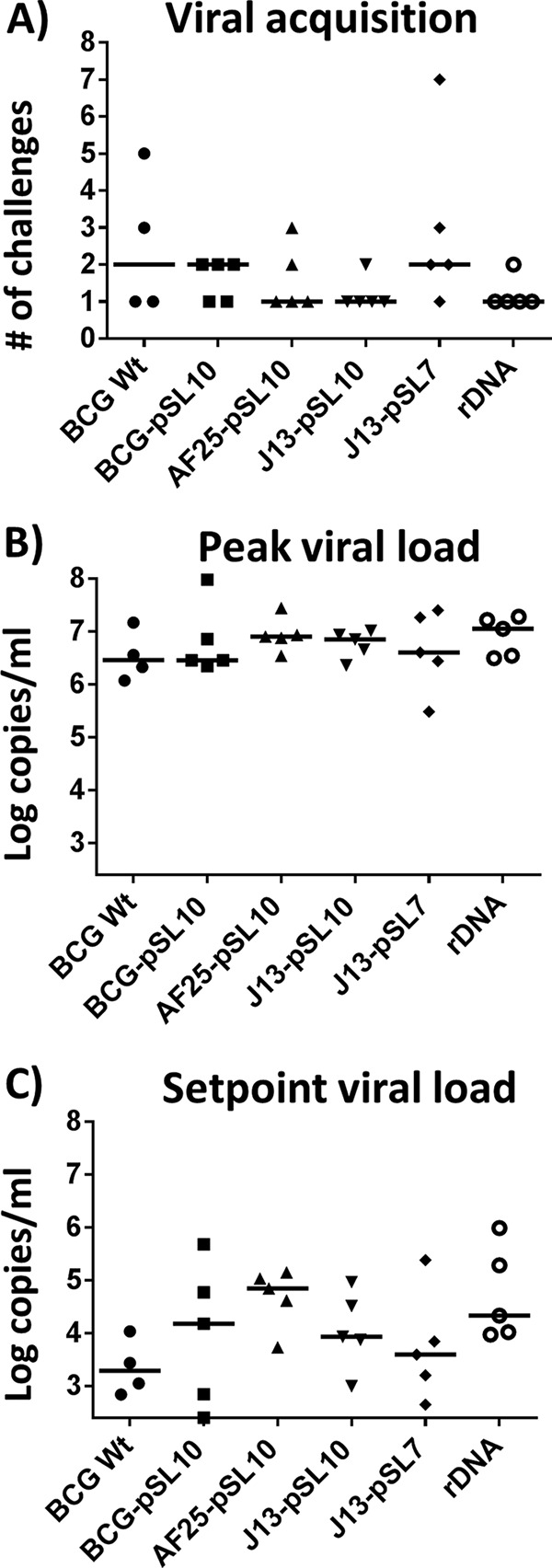
No protective efficacy against SIV rectal infection by any of the prime-boost regimens. At week 64 following a suboptimal boost with 1 × 107 PFU NYVAC-SIVgag-pol, 29 monkeys (for BCG Wt, n = 4; for the other groups, n = 5) were challenged weekly by the intrarectal route until viremia was detectable. (A) Viremia was detected in all monkey groups after a median of either 1 or 2 challenges (range, 1 to 7 challenges). (B) Peak viral loads were similar in all groups, regardless of prechallenge SIV Gag-specific immune responses. (C) Set point viral loads, measured at week 12 following viral acquisition, demonstrated more variation between groups than peak viral loads but lacked any indication of protection by the vaccines. For BCG Wt, n = 4; for the other groups, n = 5. Bars indicate medians.
DISCUSSION
In a previous vaccination study, in which rhesus macaques were immunized with rBCG Pasteur constructs expressing various SIV transgenes under the control of a Mycobacterium tuberculosis α antigen promoter and the use of a 19-kDa signal sequence, we reported robust polyfunctional SIV-specific T lymphocyte responses after a heterologous boost vaccination with a recombinant replication-incompetent adenovirus 5 vector (11). SIV-specific T cell responses were reported to be low or undetectable in contrast to strong anti-vector-specific immunity after two primary immunizations with rBCG (11). Therefore, we sought to improve the immunogenicity by employing rBCG vectors with genetic alterations and modified expression cassettes for SIV Gag as a prototype vaccine for HIV.
In the present study, we demonstrate that rBCG constructs based on the Danish strain with an hsp60 promoter and either a 19-kDa (pSL7) or an Ag85B (pSL10) signal peptide elicited enhanced cellular immune responses to SIV Gag. A primary parenteral immunization with rBCG vectors at a single dose elicited measurable SIV Gag immunodominant epitope-specific CD8+ T lymphocyte responses and robust SIV-specific IFN-γ ELISpot responses that were markedly increased in comparison to the results from our previous study (11).
Our SIVgag-expressing prototype rBCG constructs were aimed at priming for enhanced CD8+ T lymphocyte recall responses, which are crucial for viral control (22, 23). The live attenuated BCG mutants used in the present study, which were selected from an in vitro screen with superior immunogenicity for MHC class I presentation (Panas et al., unpublished), induced measurable immunodominant p11C+ CD8+ T lymphocyte responses and robust CD4+ T lymphocyte-biased IFN-γ secretion levels after primary immunization.
Since mycobacteria are typically phagocytosed by antigen-presenting cells (APCs) and reside in phagolysosomes accessible to MHC class II presentation and CD4+ T lymphocyte activation (24), an important question arises with regard to how rBCG vectors are capable of conveying a transgenic antigen into the MHC class I processing pathway and increasing CD8+ T cell responses. In contrast to pathogenic mycobacteria like Mycobacterium tuberculosis, which is known to access the MHC class I pathway by translocating antigens into the cytosol of APCs and utilizing the type VII protein secretion systems, it has been suggested that bacterial antigens of phagosome-restricted BCG can be cross-presented for CD8+ T lymphocyte priming by trafficking through apoptotic vesicles during programmed cell death (24, 25).
The deletion of the cmaA2 gene results in a reduction of mycolic acid cyclopropanation and is associated with an increased release of proinflammatory cytokines (26, 27) that can also lead to cross-priming and an increased CD8+ T cell response. This is consistent with our finding that rBCG mutants with the cmaA2 gene deleted (J13-pSL7 and J13-pSL10) primed for strong SIV-specific T cellular immunogenicity. The functions of the genes in the BCG_2587-2590 operon (deleted in the immunogenic AF25-pSL10 mutant) are still unknown, so further investigations are necessary.
The strongest CD8+ T cell responses elicited by rBCG constructs employing a SIVgag expression cassette with a 19-kDa secretion signal (in the BCG-pSL7 and J13-pSL7 constructs in the present study) can be explained by efficient cross-priming. In macrophages infected with mycobacteria, the cell wall- or cell membrane-associated and secreted 19-kDa lipoprotein has been demonstrated to mediate apoptosis through the activation of Toll-like receptors via caspase-dependent or -independent pathways (24, 28, 29), independent of TAP (transporter associated with antigen processing) usage (30, 31).
Similar to the 19-kDa glycolipid, the Ag85 protein (used as the signal sequence in the pSL10-SIVgag expression cassettes) is mainly associated with the mycobacterial cell wall (31). This characteristic advocates the partial contribution of these signal peptides to apoptotic vesicle trafficking and cross-presentation of immunogens. In fact, the combination of promoter and signal sequence for specific foreign antigens in rBCG appears to be a determining factor for the strength of immunogenicity (31, 32). Anti-Gag antibody responses were not detected at any time point in rBCG-immunized monkeys, results that are similar to those of a study using hsp70 promoter-regulated rBCG vectors with the SIVgag transgene only (33), which may have been affected by cross-priming and bias toward cellular immune responses.
The BCG-pSL7-SIVgag and BCG-pSL10-SIVgag vectors served as strong priming immunogens for vigorous SIV Gag-specific cellular immunity, including CD8+ T lymphocyte responses and IFN-γ expression levels, despite a reduced dose of the heterologous viral boost. Compared to historical doses, the boosting dose in this study was reduced to a suboptimal level (lowered by 1 order of magnitude, 1 × 107 instead of 1 × 108 PFU) to permit the detection of subtle differences in immune responses elicited by the different priming vectors (34, 35). As enhanced T cell immunogenicity measured in the peripheral blood does not necessarily represent other lymphatic or mucosal compartments, we recently demonstrated in 10 monkeys from the present study (limited to those in the BCG J13-pSL7-SIVgag mutant and rDNA groups at weeks 2 and 10 postboost) (36) that systemic vaccination with the BCG-SIVgag mutant followed by an rNYVAC boost or the rDNA-rNYVAC vaccination regimens induced the expansion of clonally diverse p11C-specific CD8+ T lymphocyte populations in peripheral blood. This was initially shared with the gastrointestinal mucosal and pulmonary compartments but subsequently diverged due to differential trafficking or homing.
It is noteworthy that after the rNYVAC boost, the SIV Gag-specific CD8+ T lymphocytes in peripheral blood and bronchoalveolar lavage samples were mainly central memory cells, whereas effector memory CD8+ T cells were retained over time, with a predominance in the gastrointestinal mucosa (36). The prevailing sustained antigen-specific effector memory CD8+ T lymphocyte population at a major portal of HIV entry is crucial because challenge studies in rhesus macaques vaccinated with persistently replicating rhesus cytomegalovirus vectors that encode most of the SIV proteome were shown to efficiently maintain broad SIV-specific effector memory T cell pools that were able to control pathogenic SIV infection (37, 38).
Long-lasting immune responses were detected in the present study >2 years after primary immunization with rBCG-SIVgag vectors and almost 15 months after a single suboptimal boost vaccination with highly attenuated replication-defective NYVAC. The rBCG- or rDNA-vaccinated monkeys maintained modest levels of MHC class I tetramer responses and robust levels of SIV Gag-specific IFN-γ responses, although markedly lower than at set point following the boost. When the vaccinated macaques were repeatedly challenged by the administration of highly pathogenic SIVmac251 onto the rectal mucosa, they all became infected regardless of the vaccination regimens. Neither the combination of rBCG-SIVgag nor plasmid DNA vaccine with a suboptimal pox boost had a protective effect on viral acquisition, i.e., the numbers of mucosal challenges needed to induce viremia did not substantially differ. The T cell-based prime-boost vaccine regimens also failed to affect viral load dynamics during a 3-month follow-up after infection in rhesus macaques expressing the protective Mamu-A*01 allele (39). Thus, vaccination did not induce viral control.
Potential causes for the lack of protection against infection by the vaccination regimens are multifactorial. T cell-based vaccines in SIV/macaque models have only rarely provided protection against rectally administered pathogenic SIV or simian-human immunodeficiency virus (SHIV) (23, 40). Moreover, the only HIV vaccine with limited protective efficacy in a human trial employed env, gag, and pol immunogens and highlighted the importance of vaccine-induced antibodies. Studies in rhesus macaques confirmed that antibodies to Env conferred resistance against SIV acquisition, which was not achieved in SIV Gag-immunized animals (16). Furthermore, the protective efficacy of rhesus cytomegalovirus vectors that encode numerous SIV proteins, not only SIV Gag, suggests that the inclusion of multiple transgenes may be beneficial (37, 38).
The largely delayed timing of viral exposure in relation to immunization in the present study is crucial, since SIV challenge studies for the assessment of vaccine efficacy usually time the viral exposure relatively close to the last boost immunization when vaccine-elicited responses that have reached plateau are still relatively high. Additionally, a suboptimal boost dose may have induced less durable immune responses than an optimal boosting dose. As time passed, the magnitude of cellular immune responses unsurprisingly decreased, indicating that memory T cell levels waned below critical levels (41). Moreover, at the site of viral exposure in the gastrointestinal mucosa, SIV Gag epitope-specific CD8+ T lymphocytes that preferentially exhibited an effector memory phenotype also slowly decreased over time (36). As CD8+ T effector memory cells have been shown to control and clear viremia in NHP (37, 38), the reduction of vaccine-induced cellular responses below particular levels, codetermined by a challenge dose and additional host, viral, and vaccine factors, is suggested to render them ineffective at controlling the virus and preventing disease (41).
In conclusion, the presented rBCG vaccine constructs employed stable expression cassettes for SIVgag that efficiently primed for cellular immune responses. These responses were markedly augmented after a suboptimal boosting dose of replication-incompetent viral vector. While the prototype vaccine vectors carried only one transgene and did not provide protection against a rectal mucosal challenge with uncloned highly pathogenic SIVmac251, improved rBCG vectors may require modifications to express other HIV/SIV immunogens to provide a greater breadth of immune responses. A shorter time interval between the immunization and the challenge or additional boost immunizations may increase vaccine efficacy. Our presented findings have implications for HIV vaccine design and preclinical vaccine assessment and warrant additional exploration to optimize an rBCG vector-based HIV vaccine approach to successfully prevent the mucosal acquisition of HIV.
ACKNOWLEDGMENTS
We thank the NEPRC and Bioqual for excellent veterinary support and animal care. We also thank Judith Lucas and Shimontini Mitra for their expert technical assistance, and we are grateful to Sampa Santra and Michael S. Seaman for their technical expertise and advice. The SIVmac 239 Gag (15-mer) Peptides–Complete Set was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH. Recombinant NYVAC-SIVgag-pol was provided by Sanofi-Pasteur.
Funding for this study was provided by the Collaboration for AIDS Vaccine Discovery (program grants OPP38614 and OPP1033104) of the Bill & Melinda Gates Foundation, the NIH (grants AI067854, AI100645, AI063537, AI093649, and AI26170), the NEPRC base grant NIH OD011103, and NIH-funded program grant P30 AI060354 to the Harvard University Center for AIDS Research (CFAR). W.R.J. acknowledges generous support from the NIH Centers for AIDS Research (CFAR) at the Albert Einstein College of Medicine (grant AI-051519).
Footnotes
Published ahead of print 30 July 2014
REFERENCES
- 1.Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, Benenson M, Gurunathan S, Tartaglia J, McNeil JG, Francis DP, Stablein D, Birx DL, Chunsuttiwat S, Khamboonruang C, Thongcharoen P, Robb ML, Michael NL, Kunasol P, Kim JH, MOPH-TAVEG Investigators 2009. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 361:2209–2220. 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization, Expert Committee on Biological Standardization. 2011. Recommendations to assure the quality, safety and efficacy of BCG vaccines. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 3.Centers for Disease Control. 1996. The role of BCG vaccine in the prevention and control of tuberculosis in the United States. A joint statement by the Advisory Council for the Elimination of Tuberculosis and the Advisory Committee on Immunization Practices. MMWR Recomm. Rep. 45:1–18. [PubMed] [Google Scholar]
- 4.Merle CS, Cunha SS, Rodrigues LC. 2010. BCG vaccination and leprosy protection: review of current evidence and status of BCG in leprosy control. Expert Rev. Vaccines 9:209–222. 10.1586/erv.09.161. [DOI] [PubMed] [Google Scholar]
- 5.Brewer TF. 2000. Preventing tuberculosis with bacillus Calmette-Guerin vaccine: a meta-analysis of the literature. Clin. Infect. Dis. 31(Suppl 3):S64–S67. 10.1086/314072. [DOI] [PubMed] [Google Scholar]
- 6.Colditz GA, Brewer TF, Berkey CS, Wilson ME, Burdick E, Fineberg HV, Mosteller F. 1994. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. JAMA 271:698–702. [PubMed] [Google Scholar]
- 7.Chapman R, Chege G, Shephard E, Stutz H, Williamson AL. 2010. Recombinant Mycobacterium bovis BCG as an HIV vaccine vector. Curr. HIV Res. 8:282–298. 10.2174/157016210791208686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, Snapper SB, Barletta RG, Jacobs WR, Jr, Bloom BR. 1991. New use of BCG for recombinant vaccines. Nature 351:456–460. 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- 9.Langermann S, Palaszynski SR, Burlein JE, Koenig S, Hanson MS, Briles DE, Stover CK. 1994. Protective humoral response against pneumococcal infection in mice elicited by recombinant Bacille Calmette-Guerin vaccines expressing pneumococcal surface protein A. J. Exp. Med. 180:2277–2286. 10.1084/jem.180.6.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joseph J, Saubi N, Pezzat E, Gatell JM. 2006. Progress towards an HIV vaccine based on recombinant bacillus Calmette-Guerin: failures and challenges. Expert Rev. Vaccines 5:827–838. 10.1586/14760584.5.6.827. [DOI] [PubMed] [Google Scholar]
- 11.Cayabyab MJ, Korioth-Schmitz B, Sun Y, Carville A, Balachandran H, Miura A, Carlson KR, Buzby AP, Haynes BF, Jacobs WR, Letvin NL. 2009. Recombinant Mycobacterium bovis BCG prime-recombinant adenovirus boost vaccination in rhesus monkeys elicits robust polyfunctional simian immunodeficiency virus-specific T-cell responses. J. Virol. 83:5505–5513. 10.1128/JVI.02544-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glickman MS, Cahill SM, Jacobs WR., Jr 2001. The Mycobacterium tuberculosis cmaA2 gene encodes a mycolic acid trans-cyclopropane synthetase. J. Biol. Chem. 276:2228–2233. 10.1074/jbc.C000652200. [DOI] [PubMed] [Google Scholar]
- 13.Bardarov S, Bardarov S, Jr, Pavelka MS, Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR., Jr 2002. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148:3007–3017. [DOI] [PubMed] [Google Scholar]
- 14.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC. [Google Scholar]
- 15.Knapp LA, Lehmann E, Piekarczyk MS, Urvater JA, Watkins DI. 1997. A high frequency of Mamu-A*01 in the rhesus macaque detected by polymerase chain reaction with sequence-specific primers and direct sequencing. Tissue Antigens 50:657–661. 10.1111/j.1399-0039.1997.tb02927.x. [DOI] [PubMed] [Google Scholar]
- 16.Roederer M, Keele BF, Schmidt SD, Mason RD, Welles HC, Fischer W, Labranche C, Foulds KE, Louder MK, Yang ZY, Todd JP, Buzby AP, Mach LV, Shen L, Seaton KE, Ward BM, Bailer RT, Gottardo R, Gu W, Ferrari G, Alam SM, Denny TN, Montefiori DC, Tomaras GD, Korber BT, Nason MC, Seder RA, Koup RA, Letvin NL, Rao SS, Nabel GJ, Mascola JR. 2014. Immunological and virological mechanisms of vaccine-mediated protection against SIV and HIV. Nature 505:502–508. 10.1038/nature12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuroda MJ, Schmitz JE, Barouch DH, Craiu A, Allen TM, Sette A, Watkins DI, Forman MA, Letvin NL. 1998. Analysis of Gag-specific cytotoxic T lymphocytes in simian immunodeficiency virus-infected rhesus monkeys by cell staining with a tetrameric major histocompatibility complex class I-peptide complex. J. Exp. Med. 187:1373–1381. 10.1084/jem.187.9.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Currier JR, Kuta EG, Turk E, Earhart LB, Loomis-Price L, Janetzki S, Ferrari G, Birx DL, Cox JH. 2002. A panel of MHC class I restricted viral peptides for use as a quality control for vaccine trial ELISPOT assays. J. Immunol. Methods 260:157–172. 10.1016/S0022-1759(01)00535-X. [DOI] [PubMed] [Google Scholar]
- 19.Hagiwara E, Abbasi F, Mor G, Ishigatsubo Y, Klinman DM. 1995. Phenotype and frequency of cells secreting IL-2, IL-4, IL-6, IL-10, IFN and TNF-alpha in human peripheral blood. Cytokine 7:815–822. 10.1006/cyto.1995.0098. [DOI] [PubMed] [Google Scholar]
- 20.Tomaras GD, Yates NL, Liu P, Qin L, Fouda GG, Chavez LL, Decamp AC, Parks RJ, Ashley VC, Lucas JT, Cohen M, Eron J, Hicks CB, Liao HX, Self SG, Landucci G, Forthal DN, Weinhold KJ, Keele BF, Hahn BH, Greenberg ML, Morris L, Karim SS, Blattner WA, Montefiori DC, Shaw GM, Perelson AS, Haynes BF. 2008. Initial B-cell responses to transmitted human immunodeficiency virus type 1: virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J. Virol. 82:12449–12463. 10.1128/JVI.01708-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Montefiori DC. 2005. Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. Curr. Protoc. Immunol. Chapter 12:Unit 12 11. 10.1002/0471142735.im1211s64. [DOI] [PubMed] [Google Scholar]
- 22.Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, Racz P, Tenner-Racz K, Dalesandro M, Scallon BJ, Ghrayeb J, Forman MA, Montefiori DC, Rieber EP, Letvin NL, Reimann KA. 1999. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283:857–860. 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- 23.Liu J, O'Brien KL, Lynch DM, Simmons NL, La Porte A, Riggs AM, Abbink P, Coffey RT, Grandpre LE, Seaman MS, Landucci G, Forthal DN, Montefiori DC, Carville A, Mansfield KG, Havenga MJ, Pau MG, Goudsmit J, Barouch DH. 2009. Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature 457:87–91. 10.1038/nature07469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weerdenburg EM, Peters PJ, van der Wel NN. 2010. How do mycobacteria activate CD8+ T cells? Trends Microbiol. 18:1–10. 10.1016/j.tim.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Winau F, Weber S, Sad S, de Diego J, Hoops SL, Breiden B, Sandhoff K, Brinkmann V, Kaufmann SH, Schaible UE. 2006. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity 24:105–117. 10.1016/j.immuni.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 26.Rao V, Gao F, Chen B, Jacobs WR, Jr, Glickman MS. 2006. Trans-cyclopropanation of mycolic acids on trehalose dimycolate suppresses Mycobacterium tuberculosis-induced inflammation and virulence. J. Clin. Invest. 116:1660–1667. 10.1172/JCI27335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barkan D, Hedhli D, Yan HG, Huygen K, Glickman MS. 2012. Mycobacterium tuberculosis lacking all mycolic acid cyclopropanation is viable but highly attenuated and hyperinflammatory in mice. Infect. Immun. 80:1958–1968. 10.1128/IAI.00021-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopez M, Sly LM, Luu Y, Young D, Cooper H, Reiner NE. 2003. The 19-kDa Mycobacterium tuberculosis protein induces macrophage apoptosis through Toll-like receptor-2. J. Immunol. 170:2409–2416. 10.4049/jimmunol.170.5.2409. [DOI] [PubMed] [Google Scholar]
- 29.Sanchez A, Espinosa P, Garcia T, Mancilla R. 2012. The 19 kDa Mycobacterium tuberculosis lipoprotein (LpqH) induces macrophage apoptosis through extrinsic and intrinsic pathways: a role for the mitochondrial apoptosis-inducing factor. Clin. Dev. Immunol. 2012:950503. 10.1155/2012/950503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neyrolles O, Gould K, Gares MP, Brett S, Janssen R, O'Gaora P, Herrmann JL, Prevost MC, Perret E, Thole JE, Young D. 2001. Lipoprotein access to MHC class I presentation during infection of murine macrophages with live mycobacteria. J. Immunol. 166:447–457. 10.4049/jimmunol.166.1.447. [DOI] [PubMed] [Google Scholar]
- 31.Dennehy M, Williamson AL. 2005. Factors influencing the immune response to foreign antigen expressed in recombinant BCG vaccines. Vaccine 23:1209–1224. 10.1016/j.vaccine.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 32.Al-Zarouni M, Dale JW. 2002. Expression of foreign genes in Mycobacterium bovis BCG strains using different promoters reveals instability of the hsp60 promoter for expression of foreign genes in Mycobacterium bovis BCG strains. Tuberculosis (Edinb.) 82:283–291. 10.1054/tube.2002.0374. [DOI] [PubMed] [Google Scholar]
- 33.Yasutomi Y, Koenig S, Haun SS, Stover CK, Jackson RK, Conard P, Conley AJ, Emini EA, Fuerst TR, Letvin NL. 1993. Immunization with recombinant BCG-SIV elicits SIV-specific cytotoxic T lymphocytes in rhesus monkeys. J. Immunol. 150:3101–3107. [PubMed] [Google Scholar]
- 34.Stevceva L, Alvarez X, Lackner AA, Tryniszewska E, Kelsall B, Nacsa J, Tartaglia J, Strober W, Franchini G. 2002. Both mucosal and systemic routes of immunization with the live, attenuated NYVAC/simian immunodeficiency virus SIV(gpe) recombinant vaccine result in gag-specific CD8(+) T-cell responses in mucosal tissues of macaques. J. Virol. 76:11659–11676. 10.1128/JVI.76.22.11659-11676.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hel Z, Nacsa J, Tryniszewska E, Tsai WP, Parks RW, Montefiori DC, Felber BK, Tartaglia J, Pavlakis GN, Franchini G. 2002. Containment of simian immunodeficiency virus infection in vaccinated macaques: correlation with the magnitude of virus-specific pre- and postchallenge CD4+ and CD8+ T cell responses. J. Immunol. 169:4778–4787. 10.4049/jimmunol.169.9.4778. [DOI] [PubMed] [Google Scholar]
- 36.Sircar P, Furr KL, Letvin NL. 2013. Systemic vaccination induces clonally diverse SIV-specific CD8+ T-cell populations in systemic and mucosal compartments. Mucosal Immunol. 6:93–103. 10.1038/mi.2012.52. [DOI] [PubMed] [Google Scholar]
- 37.Hansen SG, Vieville C, Whizin N, Coyne-Johnson L, Siess DC, Drummond DD, Legasse AW, Axthelm MK, Oswald K, Trubey CM, Piatak M, Jr, Lifson JD, Nelson JA, Jarvis MA, Picker LJ. 2009. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat. Med. 15:293–299. 10.1038/nm.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak M, Jr, Lifson JD, Picker LJ. 2011. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473:523–527. 10.1038/nature10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casimiro DR, Wang F, Schleif WA, Liang X, Zhang ZQ, Tobery TW, Davies ME, McDermott AB, O'Connor DH, Fridman A, Bagchi A, Tussey LG, Bett AJ, Finnefrock AC, Fu TM, Tang A, Wilson KA, Chen M, Perry HC, Heidecker GJ, Freed DC, Carella A, Punt KS, Sykes KJ, Huang L, Ausensi VI, Bachinsky M, Sadasivan-Nair U, Watkins DI, Emini EA, Shiver JW. 2005. Attenuation of simian immunodeficiency virus SIVmac239 infection by prophylactic immunization with DNA and recombinant adenoviral vaccine vectors expressing Gag. J. Virol. 79:15547–15555. 10.1128/JVI.79.24.15547-15555.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ami Y, Izumi Y, Matsuo K, Someya K, Kanekiyo M, Horibata S, Yoshino N, Sakai K, Shinohara K, Matsumoto S, Yamada T, Yamazaki S, Yamamoto N, Honda M. 2005. Priming-boosting vaccination with recombinant Mycobacterium bovis bacillus Calmette-Guerin and a nonreplicating vaccinia virus recombinant leads to long-lasting and effective immunity. J. Virol. 79:12871–12879. 10.1128/JVI.79.20.12871-12879.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plotkin SA. 2008. Vaccines: correlates of vaccine-induced immunity. Clin. Infect. Dis. 47:401–409. 10.1086/589862. [DOI] [PubMed] [Google Scholar]