

Abstract
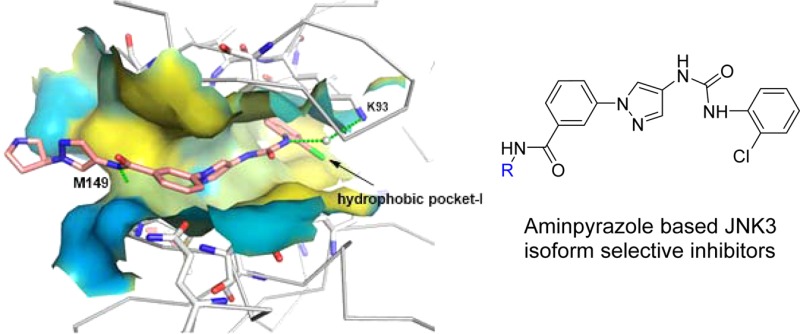
The c-jun N-terminal kinase 3 (JNK3) is expressed primarily in the brain. Numerous reports have shown that inhibition of JNK3 is a promising strategy for treatment of neurodegeneration. The optimization of aminopyrazole-based JNK3 inhibitors with improved potency, isoform selectivity, and pharmacological properties by structure–activity relationship (SAR) studies utilizing biochemical and cell-based assays, and structure-based drug design is reported. These inhibitors had high selectivity over JNK1 and p38α, minimal cytotoxicity, potent inhibition of 6-OHDA-induced mitochondrial membrane potential dissipation and ROS generation, and good drug metabolism and pharmacokinetic (DMPK) properties for iv dosing. 26n was profiled against 464 kinases and was found to be highly selective hitting only seven kinases with >80% inhibition at 10 μM. Moreover, 26n showed good solubility, good brain penetration, and good DMPK properties. Finally, the crystal structure of 26k in complex with JNK3 was solved at 1.8 Å to explore the binding mode of aminopyrazole based JNK3 inhibitors.
Introduction
As a member of the mitogen-activated protein kinase (MAPK) family, the c-Jun N-terminal kinases (JNKs) regulate the serine/threonine phosphorylation of several transcription factors1 when they are activated in response to various stimuli such as oxidative stress, neurotoxins, cytokines, and fatty acids.2−8 There are three human JNK isoforms: JNK1, JNK2, and JNK3.9−12 JNK1 and JNK2 are ubiquitously expressed in most tissues, while JNK3 is primarily expressed in the brain and, to a lesser extent, in the heart and testes.1,10−17 Recent studies have shown that JNK3 plays a central role in the brain to mediate neurodegeneration, such as β-amyloid processing and neuronal apoptosis in Alzheimer’s disease,18 as well as the mediation of neurotoxicity in rodent models of Parkinson’s disease.3,19−22 The selective expression of JNK3 in the brain, along with findings that JNK3 knockout mice exhibit amelioration of neurodegeneration in animal models of Parkinson and Alzheimer’s disease, makes inhibiting this isoform a particularly promising therapeutic target for neurodegenerative diseases.18,23,24
Identifying potent and selective inhibitors of JNK3 may contribute toward neuroprotective therapies with reduced untoward side effect profiles if JNK1 inhibition has such negative effects. To date, however, the development of specific small molecule inhibitors with high isoform selectivity for JNK3, especially against JNK1, is still a relatively untapped area as the three JNK isoforms share more than 90% sequence identity in the ATP pocket. Many published JNK3 inhibitors are also potent for JNK1, JNK2, and some inhibit p38α as well, because of their high degree of amino acid sequence similarity, which might lead to potential side effect profiles on immune and inflammatory systems.25,26 Thus, developing isoform-specific JNK3 inhibitors as therapeutics has gained considerable interest over the past few years despite most reports being centered on pan-JNK inhibitors.25,27−45 Our previous effort toward the development of isoform selective JNK3 inhibitors led to the identification of a class of aminopyrazole compounds. These aminopyrazole-based JNK3 inhibitors not only had high selectivity against p38α but also showed noticeable isoform selectivity against JNK1 (compound SR-4326, 18.5-fold, Figure 1).36 Herein, we describe extensive SAR studies in a continuing effort to develop highly isoform selective, efficacious, and pharmacologically viable JNK3 inhibitors from this novel aminopyrazole scaffold. The focused medicinal chemistry efforts led to the discovery of several highly potent and isoform selective JNK3 inhibitors with an isoform selectivity of >50-fold over JNK1. In addition, these JNK3 inhibitors generally had high selectivity against the closely related protein kinase p38α, were potent in protecting against ROS generation and mitochondrial dysfunction, and were optimized to have good DMPK properties for topical use and/or in iv dosing.
Figure 1.
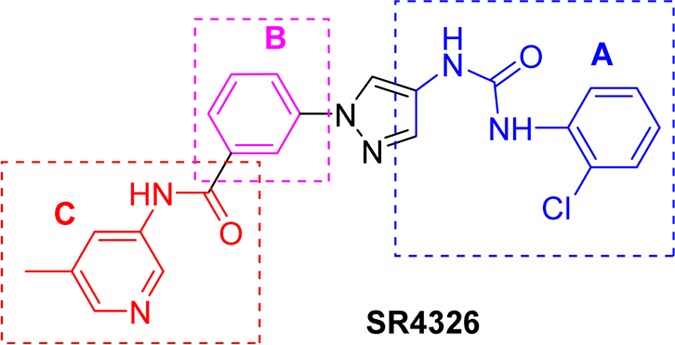
A previously disclosed isoform selective JNK3 inhibitor SR-4326.
Chemistry
Several short routes were used to synthesize inhibitors 6–8 which possessed different substituents on the urea moiety (Scheme 1). Ullman coupling of 4-nitro-1H-pyrazole 1 with 3-bromobenzoic acid 2 yielded acid 3. Amide formation to 3 gave intermediate 4, followed by a Pd/C mediated hydrogenation to afford amine 5. This intermediate was directly subjected to amide formation to furnish inhibitor 6. Inhibitor 7 was formed via a one-step coupling reaction from amine 5 under microwave reaction conditions. Inhibitors 8a–l were synthesized by treating amine 5 with different isocyanates or with triphosgene and a secondary amine, respectively.
Scheme 1. Synthesis of Inhibitors 6–8.

Reagents and conditions: (a) CuI, trans-N,N-dimethylcyclohexane-1,2-diamine, Cs2CO3, DMF, 90 °C; (b) 6-methylpyridin-3-amine, EDC, HOBt, DIEA, CH2Cl2, 25 °C; (c) Pd/C, MeOH, H2; (d) K2CO3, THF or EDC, HOBt, DIEA, CH2Cl2, 25 °C; (e) 2-chloro-1H-benzo[d]imidazole, EtOH, HCl, microwave, 140 °C; (f) isocyanate, CH2Cl2, 25 °C, or secondary amine; triphosgene, 50 °C, or CDI, THF, microwave, 150 °C.
The preparation of inhibitor 12 is described in Scheme 2. Treatment of ethyl 1H-pyrazole-4-carboxylate 9 with 3-bromobenzoic acid 2 followed by amide formation gave amide 10. Amide 10 was hydrolyzed under basic conditions to yield acid 11, which was then subjected to an amide formation reaction using EDC as the coupling reagent to furnish the final product 12.
Scheme 2. Synthesis of Inhibitors 12.

Reagents and conditions: (a) CuI, trans-N,N-dimethylcyclohexane-1,2-diamine, Cs2CO3, DMF, 90 °C; (b) 6-methylpyridin-3-amine, EDC, HOBt, DIEA, CH2Cl2, 25 °C; (c) LiOH, THF/H2O (1:1), 100 °C; (d) 2-chloroaniline, EDC, HOBt, DIEA, CH2Cl2, 25 °C.
Inhibitors 16a–d, all of which have a fluoro substitution on the middle phenyl ring, were synthesized following procedures shown in Scheme 3. The commercially available 4-nitro-1H-pyrazole 1 was N-arylated with ethyl 3-bromobenzoate 13 using standard Ullman procedures, followed by ester hydrolysis under basic conditions to form acid 14. Amide formation followed by Pd/C mediated reduction of 14 was applied to yield amine 15. Finally, intermediate 15 was subjected to a urea formation to furnish product 16.
Scheme 3. Synthesis of Inhibitors 16.

Reagents and conditions: (a) CuI, trans-N,N-dimethylcyclohexane-1,2-diamine, Cs2CO3, DMF, 90 °C; (b) LiOH, THF/H2O (1:1), 100 °C; (c) 6-methylpyridin-3-amine, EDC, HOBt, DIEA, CH2Cl2, 25 °C; (d) Pd/C, MeOH, H2; (e) 1-chloro-2-isocyanatobenzene, CH2Cl2, 25 °C.
The preparation of inhibitors 20–22 is outlined in Scheme 4. Ullman coupling of 4-nitro-1H-pyrazole 1 with 1-bromo-3-iodobenzene (for synthesis of compounds 20) or 3-bromobenzonitrile (for synthesis of compounds 21) or 3-bromobenzoic acid (for synthesis of compounds 22), followed by a Pd/C mediated reduction, afforded the intermediate amine 18. These amines were reacted with 1-chloro-4-isocyanobenzene in DCM to furnish intermediates 19a−c. Target compounds 20 were prepared through a Suzuki coupling reaction between intermediate 19a and a boronic acid pinacol ester catalyzed by Pd(PPh3)4. Target compounds 21a and 21b were prepared through treatment of intermediate 19b with hydrazine monohydrate, followed by a cyclization reaction with acetimidamide or acetic acid. Target compounds 22 were prepared through amide formation in the presence of HATU in DMF. All final inhibitors were purified by reverse-phase preparative HPLC.
Scheme 4. Synthesis of Inhibitors 20–22.

Reagents and conditions: (a) CuI, trans-N,N-dimethylcyclohexane-1,2-diamine, Cs2CO3, DMF, 100 °C; (b) Pd/C, MeOH, H2; (c) 1-chloro-2-isocyanatobenzene, CH2Cl2, 25 °C; (d) R1B(OH)2, Ph(PPh3)4, K2CO3, dioxane/H2O (3:1), 95 °C; (e) N2H4·xH2O, EtOH, reflux ; (f) acetimidamide, EtOH, NaOMe or acetic acid, propylphosphonic anhydride, TEA, DMF, 110 °C; (g) R3NH2, EDC, HOBt, DIEA, CH2Cl2, 25 °C.
Applications of N-substituted pyrazole amines led to inhibitors 26. As shown in Scheme 5, the N1 alkylation of 3-nitropyrazole with a primary iodo- or bromoalkane using potassium carbonate as the base was first used to furnish N-substituted pyrazoles, followed by reduction of the nitro group to give pyrazole amines 24. Amide formation between 24 and acid 3, followed by a Pd/C mediated reduction furnished amine 25. A one-pot urea formation of 25 with 1-chloro-4-isocyanobenzene in DCM afforded the target inhibitor 26 which was purified by reverse-phase preparative HPLC.
Scheme 5. Synthesis of Inhibitors 26.

Reagents and conditions: (a) K2CO3, DMF, 40 °C; (b) Pd/C, MeOH, H2; (c) EDC, HOBt, DIEA, CH2Cl2, 25 °C; (d) Pd/C, MeOH, H2; (e) 1-chloro-2-isocyanatobenzene, CH2Cl2, 25 °C.
Results and Discussion
In the crystal structures published in our previous work, the aniline–urea moiety (Figure 1) binds to the hydrophobic selectivity pocket (hydrophobic pocket I) and is responsible for the isoform selectivity of JNK3 over JNK1.36 We therefore started our SAR investigations by replacing the aniline–urea moiety in region A (Figure 1) with selected bioisosters. As shown in Figure 2, very few replacements to the urea moiety were tolerated at region A, as all compounds had JNK3 IC50 values of >10 μM. For example, replacement of the aniline NH group with a carbon (6a) or oxygen (6b) or a direct heterocyclic ring connection (6c,d) all resulted in complete loss of JNK3 inhibition. Similarly, structural changes to the other NH group of urea were not tolerated either. For example, amide compound 12a and pyrazolo[4,3-c]pyridine compound 12b were completely inactive. Some other replacements of the urea moiety were also investigated (such as compound 7; for more data see Supporting Information), but no improvement was observed. The observed results for modifications to the urea moiety suggested that the urea core was essential for achieving a high JNK3 affinity. Therefore, this urea moiety will be retained in all further optimizations.
Figure 2.
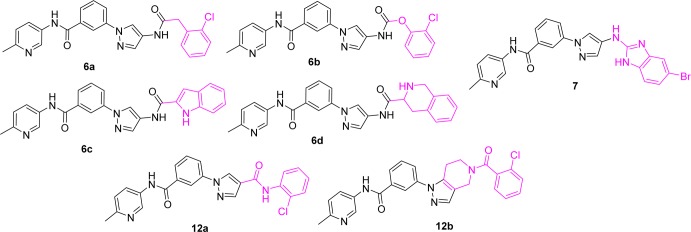
Compounds with low JNK3 inhibition activity with IC50 > 10 μM.
We next investigated a series of replacements and substitutions on the phenyl group of the urea moiety. As shown in Table 1, replacing the aromatic group with an alkyl group was not tolerated, as indicated by the significant loss of JNK inhibition activity for 8a. Compound 8b, which had no substitutions on the urea phenyl ring, showed a JNK3 inhibition activity similar to the lead compound SR-4326 but with almost no isoform selectivity. In the case of chloro substitutions, both the meta- and the para-Cl substitutions (8d and 8e) led to a decrease in JNK affinity, which could be due to space limitation inside the binding pocket. However, when a Cl substituent was introduced at the ortho-position of the phenyl ring (8c), where there should be enough space to accommodate a Cl group based on the crystal structures,36 higher JNK3 potency was obtained probably because of the extra hydrophobic interactions from the Cl group. More interestingly, some isoform selectivity (JNK3 over JNK1) was achieved as well (4.4-fold). On the other hand, fluoro substitutions on this phenyl ring exhibited a significantly different SAR profile. Compared to the lead compound SR-4326 and compound 8c, a mono-F-substitution at all three positions resulted in reduction of JNK3 inhibition activity, although these substitutions could improve the isoform selectivity (compounds 8f, 8g, 8h). While the meta-F compound 8g and the para-F compound 8h showed a slight decrease of JNK3 potency, a substitution at the ortho-position resulted in around 3-fold loss of potency as compared to the unsubstituted analog 8b (Table 1).
Table 1. Biochemical IC50 Values for JNK3 and JNK1 for SAR Studies of the Urea Moietya.
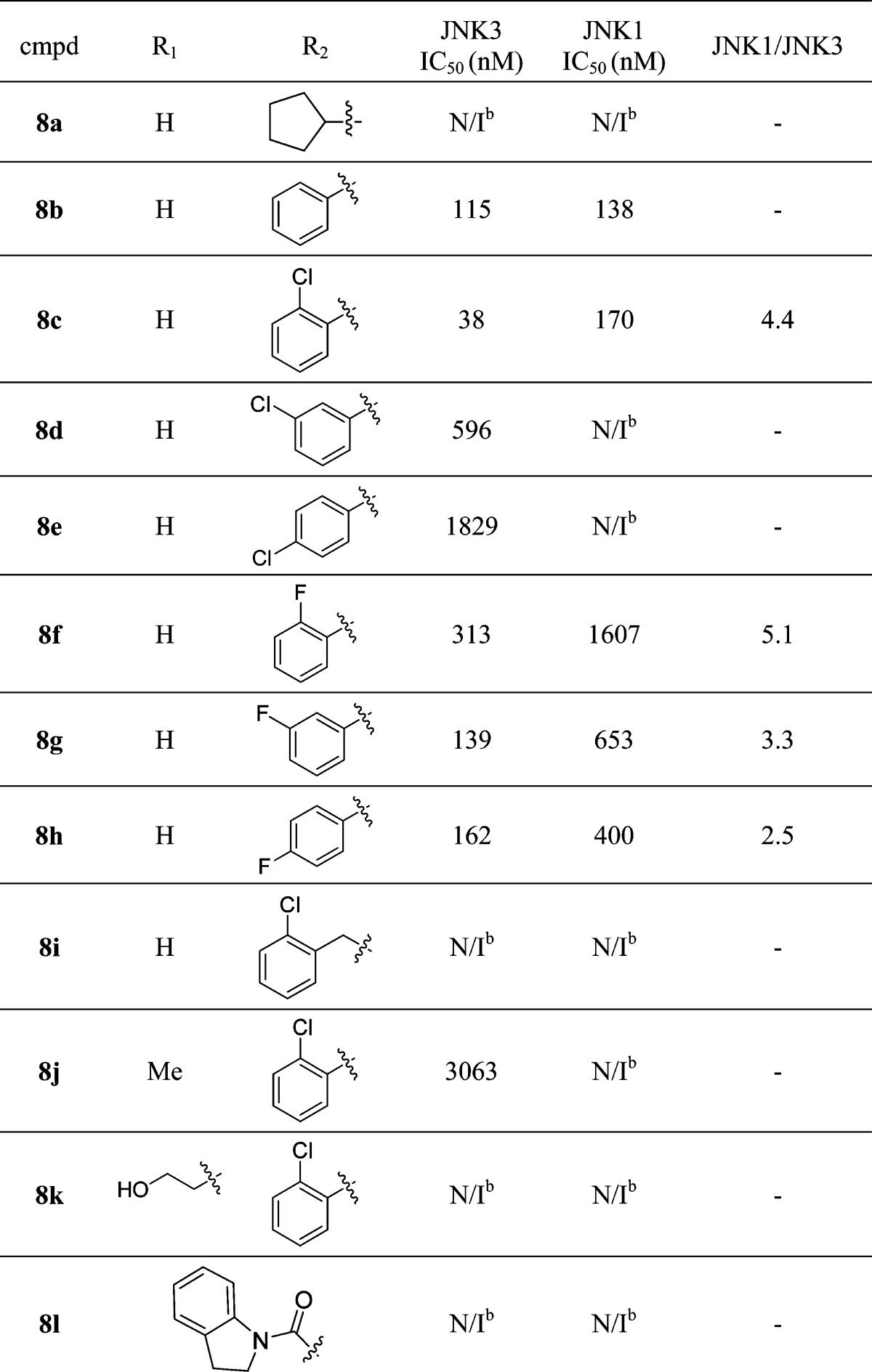
IC50 values are the mean of two or more experiments (with triplicate replicates for each experiment) with errors within 80% of the mean.
No inhibition up to 10 μM.
The unusual SAR observed for F-substituted compounds in Table 1 might be due to the F-bonding/interactions.46,47 Different types of interactions between fluorine atoms with protein functional groups have been reported (not found in other halogens), and in some protein environments, compounds with a fluorine substitution behave very differently from other halogen-substituted compounds.47 Such fluorine interactions include F–H hydrogen bonds, π-interactions with aromatic or guanidine groups, multipolar interactions, and/or F-nitrogen interactions (halogen bonding).47 For the aminopyrazole based JNK inhibitors, these special F-interactions could result in a change of inhibitor conformations in the pocket or a twist of the orientation of the phenyl ring inside hydrophobic pocket I. These changes in conformation or orientation could produce unfavorable binding of the inhibitor in the ATP pocket, and therefore cause a decrease in inhibition of JNK activity for compounds 8f–h.
Significant loss of potency also occurred with replacement of the o-Cl-phenyl moiety by an o-Cl-benzyl group (8i, Table 1). This result indicated that the hydrophobic selectivity pocket might not have enough room to tolerate a larger group or that the phenyl ring of the benzyl group could not assume a conformation/orientation that could give maximal hydrophobic interactions. The effect of substitutions on the phenyl urea NH was also studied. Unfortunately, any substitution to this NH group greatly reduced the JNK3 potency (Table 1). For example, a simple methyl substitution resulted in compound 8j which exhibited a JNK3 IC50 of 3063 nM. Larger substituents yielded even lower JNK3 inhibitory activity (compounds 8k and 8l, IC50 > 10 μM). As is demonstrated in the crystal structure (see Figure 4), this urea NH is involved in H-bonding interactions with protein residues, and its alkylation is predicted to reduce the JNK3 inhibitory activity.
Figure 4.
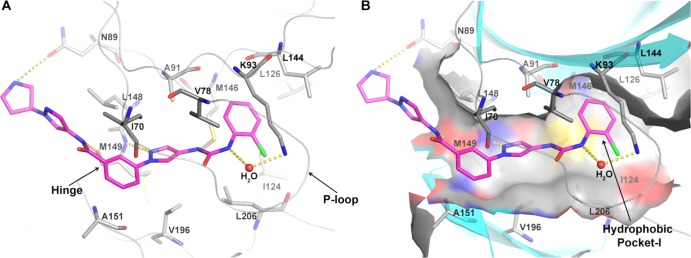
X-ray crystal structure of 26k in JNK3.
The feasibility of applying some minor structural modifications to the central phenyl group attached to the pyrazole N1 (region B, Figure 1) was explored next. Our preliminary SAR showed that a methyl substitution was tolerated here.36 We envisioned that introduction of a fluoro substitution on the benzene ring might introduce additional interactions that could perturb the JNK3 activity and selectivity, as well as improve DMPK properties. Therefore, compounds with a simple fluoro substitution at different positions of this benzene ring were investigated (Table 2). Although improvement in isoform selectivity was achieved (compared to SR-4326 and 8c) for the 2-fluoro substitution (16a) and the 6-fluoro substitution (16d), loss of JNK3 inhibition potency was observed for all fluoro-substituted compounds 16a–d, especially for the 3-fluoro-derivative 16b, which showed an IC50 value against JNK3 in the micromolar range (4588 nM). The reduction in JNK inhibitory potency could be due to the F-bondings/interactions46,47 previously mentioned and as observed in compounds 8f–h (Table 1). In addition, the 2-F analog 16a exhibited potent inhibition of cytochrome P450 isoforms CYP2C9 and CYP2D6 (see Table 5). Because of these detrimental features, F-substitutions on this benzene ring will not be applied in future optimizations.
Table 2. Biochemical IC50 Values for JNK3 and JNK1 for SAR Studies for the Middle Phenyl Moietya.
| cmpd | R | JNK3 IC50 (nM) | JNK1 IC50 (nM) | JNK1/JNK3 |
|---|---|---|---|---|
| 16a | 2-F | 80 | 2369 | 29.6 |
| 16b | 3-F | 4588 | N/Ib | − |
| 16c | 4-F | 71 | 180 | 2.5 |
| 16d | 6-F | 230 | 3691 | 16.1 |
IC50 values are the mean of two or more experiments (with triplicate replicates for each experiment) with errors within 80% of the mean.
No inhibition up to 10 μM.
Table 5. Inhibitor Selectivity and in Vitro DMPK Dataa.
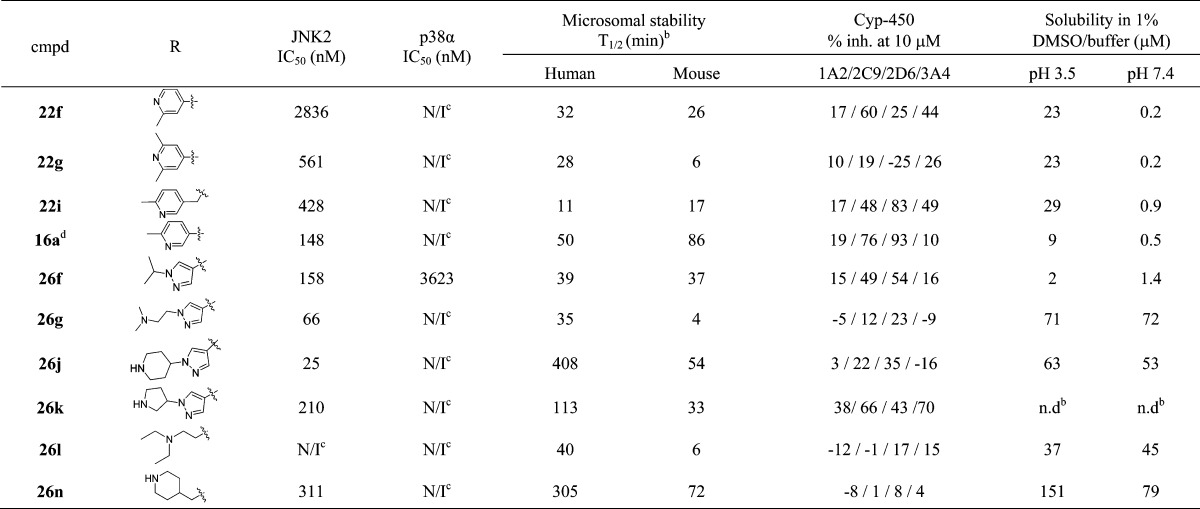
IC50 values are the mean of two or more experiments (with triplicate replicates for each experiment) with errors within 80% of the mean.
Not determined.
No inhibition up to 10 μM.
With fluoro-substitution on the middle phenyl ring at Region B.
The crystal structure published in our previous work indicated that these compounds were type I kinase inhibitors and showed that the amide carbonyl and the aromatic ring (in the amide moiety) did not have any significant interactions with the protein (only an H-bond interaction was observed between the amide NH and the protein backbone).36 Thus, structural modifications in this part will likely not affect the binding mode significantly. To test this observation, a series of replacements for the amide moiety in region C (Figure 1) with some amide bioisosters were investigated. As shown in Figure 3, the pyridine and pyrimidine based structures (20a–b), which have heteroatoms on the aromatic ring to form H-bonds, showed no JNK inhibitory activity. The triazole (21a) and the oxodiazole (21b) based amide isosters, which mimic the binding of the anilinoamide in region C, also gave no inhibition for JNK3 and JNK1. Moreover, alkylation of the amide NH (22a) led to complete loss of JNK3 inhibitory activity. These results indicated that the amide structure was critical for efficient binding and the amide moiety −CONH– was indispensable for this scaffold to achieve a high JNK3 affinity.
Figure 3.
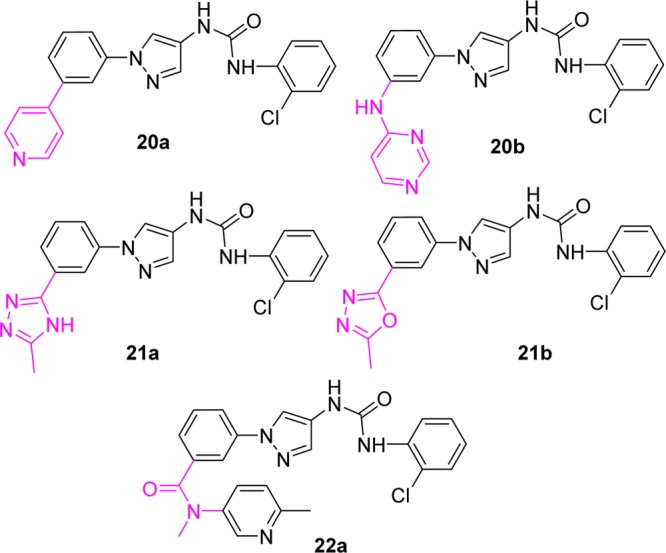
Compounds with low JNK3 inhibition activity with IC50 > 10 μM.
SAR studies of the amide moiety are a very important part of our efforts to improve both JNK3 inhibitory potency and isoform selectivity (against JNK1) for this aminopyrazole-based scaffold. The effect of substitution patterns on the pyridine ring for lead compound 8c was first explored. As shown in Table 3, moving the methyl group from 4′-position to 2′-position reduced JNK3 inhibitory potency by 3.7-fold but it reduced the JNK1 potency as much as 15.8-fold providing a benefit for the overall JNK3/JNK1 isoform selectivity (22b vs 8c). The dimethylpyridine compound 22c showed a JNK3 potency similar to that of the monosubstituted analogue 22b but a reduced isoform selectivity. Nevertheless, lead compound 8c still had the best JNK3 potency (38 nM). While the 2′-pyridine analogs 22d and 22e exhibited a significant loss of JNK3 inhibition potency, the 4′-pyridine compound, 22f, presented just a slightly lower JNK3 potency (98 nM vs 38 nM) but a better isoform selectivity (28-fold vs 4.4-fold, 22f vs 8c). The dimethyl-4′-pyridine compound 22g exhibited an even higher isoform selectivity (35-fold), although with a sacrifice in JNK3 potency. Interestingly, the pyrimidine amide 22h showed a similar JNK3 potency but had isoform selectivity slightly better than compound 8c. It is also important to note that the benzyl type amide 22i gave a weaker JNK3 inhibition but improved isoform selectivity as compared to the corresponding aniline type amide 8c and SR-4326. In summary, for pyridine and/or pyrimidine based amides, 3′-N derivatives gave better JNK3 potency but not satisfactory isoform selectivity (8c and 22h). On the other hand, 4′-N analogs exhibited good isoform selectivity but lower JNK3 inhibition activity (22f and 22g). Nevertheless, no compounds from this class could present both good JNK3 inhibition potency (IC50 < 50 nM) and excellent isoform selectivity (>50-fold).
Table 3. Biochemical IC50 Values for JNK3 and JNK1 for SAR Studies for the Amide Moietya.

IC50 values are the mean of two or more experiments (with triplicate replicates for each experiment) with errors within 80% of the mean.
No inhibition up to 10 μM.
Although the benzamide moiety (region C, Figure 1) extends away from hydrophobic pocket II and is exposed to solvents, results in Table 3 indicated that structures in region C can still significantly affect both JNK3 inhibitory activity and isoform selectivity. Considering that pyridine/pyrimidine amide compounds might also have cytotoxicity and poor microsomal stabilities (see Table 5 and Table 6), we expanded SAR studies for this part to other amide structures. Thus, amides based on five-membered heterocyclic ring systems were investigated. As shown in Table 4, the N-methylpyrazole compounds 26a–d showed a similar potency and isoform selectivity compared to lead compound SR-4326. Application of an N-methyl-triazole (26e) resulted in a 5-fold reduction in JNK3 inhibitory activity. Compounds having larger substituents on the NH of pyrazole groups gave better properties. For example, an N-isopropyl substitution on the pyrazole ring yielded a JNK3 inhibitor (26f) having better isoform selectivity compared to the N-methyl analog (26a).
Table 6. Cytotoxicity and Cell Based Potency Data for Selected Compounds.
| cytotoxicity in SHSY5Y cells after 48 h |
|||||
|---|---|---|---|---|---|
| cmpd | % cell viability at 10 μM | % cell viability at 30 μM | in-cell Western,a SHSY5Y IC50 (nM) | inhibition of 6-OHDA induced cell death, SHSY5Y IC50 (nM) | inhibition of 6-OHDA induced mitochondrial membrane depolarization,SHSY5Y IC50 (nM) |
| 22f | 7 | 5 | 866 | 2976 | 40 |
| 22g | 24 | 5 | 2331 | ndb | ndb |
| 22i | 80 | 56 | 905 | 13 | 17 |
| 26f | 84 | 16 | 3250 | ndb | 130 |
| 26j | 91 | 98 | 1436 | 568 | 25 |
| 26g | 105 | 97 | N/Ic | N/Ic | N/Ic |
| 26k | 112 | 97 | N/Ic | N/Ic | 31 |
| 26n | 92 | 98 | 1895 | 281 | 4 |
IC50 values are the mean of two or more experiments (with triplicate replicates for each experiment) with errors within 80% of the mean.
Not determined.
No inhibition up to 10 μM.
Table 4. Biochemical IC50 Values for JNK3 and JNK1 for SAR Studies for the Amide Moietya.

IC50 values are the mean of two or more experiments (with triplicate replicates for each experiment) with errors within 80% of the mean.
No inhibition at 10 μM.
Remarkably, those analogs with a substitution containing a dimethylamino, diethylamino, piperidine, or pyrrolidine group (26g–k) exhibited high JNK3 potency (IC50 of 21–34 nM) and good to excellent isoform selectivity (22- to 60-fold over JNK1). One compound, 26k, which had a substituent containing a pyrrolidine moiety, stood out and showed the highest JNK3 potency (IC50 < 1 nM) and isoform selectivity (>500-fold over JNK1 and >210-fold over JNK2; see Table 5). Careful inspection of the crystal structure of 26k indicated the potential for a hydrogen bond between the carbonyl of N89 in JNK3 with the pyrrolidinyl NH of 26k with an H-bond distance of 2.9 Å (Figure 4). This potential for tight interaction with optimal hydrogen bond distance and optimal H-bond angle was likely the reason for the potent inhibition of 26k (the energy minimized state of a computational model showed the N-pyrrolidinyl of 26k could rotate ∼90° to make a H-bond with N89). In contrast, 26j, which contained the six-member pipyridinyl ring and was not optimized for H-bond distance and angle, had weaker inhibition to JNK3 than 26k. This observation is likely true for all of the other analogs presented in Table 4. Excellent JNK3 isoform selective inhibitors were also obtained from amides containing no aromatic groups (alkylamides, 26l–q). As shown in Table 4, all the inhibitors prepared gave excellent isoform selectivity and fair to excellent JNK3 inhibitory activity. The much better JNK3 potency and the higher isoform selectivity (JNK3 over JNK1) for these compounds containing a side chain amino group could be due to extra H-bond interactions from the amino group, and a combination of these interactions and the hydrophobic interaction inside hydrophobic pocket I might be more favorable for JNK3 than for JNK1. These interactions are indicated in the crystal structure of 26k with JNK3 (Figure 4).
To better understand the observed SAR for these JNK3 isoform selective inhibitors, the crystal structure for 26k with JNK3 was solved at 1.8 Å (Figure 4). This crystal structure showed that 26k (the R-enantiomer) interacted with JNK3 through H-bonding with the hinge residue M149 with a bond distance of 2.7–3.2 Å. The hydrogen bond between the benzamide NH of 26k and the main-chain atoms of M149 in JNK3 would explain why substitution on the amide NH (22a) resulted in no JNK3 affinity (shown as yellow dotted lines). An extra H-bond also occurred between the urea NH (on the aniline side) and the side chain of residue K93 bridged by a water molecule. The existence of this interaction explains why N-alkylation of the urea NH led to loss of JNK3 inhibitory activity (compounds 8j, 8k, and 8l, Table 1). As mentioned, the N-pyrrolidinylpyrazole group made optimal H-bond interactions based on length and angle with residue N89, and the rest of the ring structure of 26k made tight hydrophobic interactions with residues in ATP binding pocket. The center phenyl ring made hydrophobic interactions with side chain of I70 and backbone of A151. The central pyrazole ring also made hydrophobic interactions with the side chains of A91 and V196.
The most significant hydrophobic interactions occurred at the terminal o-Cl-phenyl ring (attached to the urea moiety) and were tightly sandwiched between the side chains of K93 and M146. Nearby hydrophobic residues V78, M115, L126, L144, and L206 construct a hydrophobic pocket for the o-Cl-phenyl ring. These structural observations are consistent with the observed SAR results indicating that there was almost no JNK3 affinity if the urea moiety was replaced or the urea NH was alkylated. This structure also explains why there was a reduction of JNK3 inhibitory activity when the phenyl ring was substituted with a Cl on its meta- or para-position (8d and 8e), since there was not enough space inside this hydrophobic pocket. The isoform selectivity of JNK3 over JNK1 for aminopyrazole-based JNK3 inhibitors was also mainly due to interactions in this pocket, which has been documented in our previous publications.36
To evaluate the selectivity of these aminopyrazole based JNK3 inhibitors, several selected compounds were subjected to additional counter screening against JNK2 and p38. As shown in Table 5, almost all aminopyrazole compounds had no inhibition on the most closely related kinase p38 at a concentration of 10 μM with the exception of the N-alkyl substituted pyrazole amide 26f (3623 nM for p38). Moreover, these compounds had inhibitory activity for JNK2 with potency similar to that for JNK3 with only a few exceptions (22f, 22g, 22i, 26k, and 26l). To further evaluate the general kinase selectivity of these aminopyrazole based JNK3 inhibitors, compound 26n was subjected to a profiling study at a concentration of 10 μM in the full panel KINOMEscan (Ambit, San Diego, CA),48−50 a high throughput method for screening kinase inhibitors against a panel of 464 kinases. The TREEspot maps in Figure 5 revealed that 26n possessed an extremely high selectivity with significant inhibitions (>80% at 10 μM) observed only for JNK1/2/3, Clk2, haspin, Mek3, and Ysk4 (see Supporting Information for detailed profiling data).
Figure 5.

TREEspot interaction maps for 26n (also coded as SR-11935) in the panel profiling of 464 kinases.
Inhibition of four selected cytochrome P450 isoforms (1A2, 2C9, 2D6, and 3A4) was also tested at 10 μM, and results demonstrated that most of these aminopyrazole-based JNK3 inhibitors had generally low inhibition against all four enzymes. Generally, the pyrazole-amide compounds 26f, 26j, 26g, 26k presented lower inhibition for P450 isoforms compared to those pyridineamide based compounds (22e, 22f, 22i, 16a). The alkylamide based compounds gave even better P450 inhibition profiles. Compounds 26l and 26n exhibited almost no inhibition for all four selected isoforms even when tested at 10 μM. It is noteworthy that the F-substituted compound 16a exhibited potent inhibitions for isoforms CYP2C9 and CYP2D6.
The microsomal stability of these compounds in both human and mouse liver microsomes were evaluated. Briefly, the microsomal stabilities of most inhibitors based on the pyridine amide (16a, 22f, 22g, 22i) or the N-alkyl substituted pyrazoleamide (26f) were low with the exception of the F-substituted compound 16a (half-life was 50 min in human and 86 min in mouse). The presence of a secondary amine pyrrolidine ring or piperidine ring at the amide part was clearly a favorable factor for metabolic stability. For example, inhibitors 26g and 26l, which had no secondary amine substitutions, exhibited a stability of t1/2 = 35–40 min in human microsomes and only 4–6 min in mouse microsomes, while the corresponding inhibitors containing a secondary amine moiety (26j, 26k, 26n) had a much greater microsomal stability (t1/2 = 113–408 min in human and t1/2 = 33–72 min in mouse). The aqueous solubility was also tested for these compounds at both pH 3.5 and pH 7.4. As shown in Table 5, while the solubility for compounds containing the pyridineamide (22f, 22g, 22i) and N-alkyl substituted pyrazoleamide 26f were low, inhibitors with substituents containing a secondary or tertiary amine exhibited excellent solubility at both pH 3.5 and pH 7.4. Indeed, 26n had excellent solubility at both pH 3.5 and pH 7.4 (Table 5).
The cytotoxicity in SHSY5Y cells was evaluated for some selected lead inhibitors. As shown in Table 6, pyridineamide based inhibitors 22f and 22g showed significant cytotoxicity at both 10 and 30 μM, while the benzylamide based inhibitor 22i and the simple alkyl substituted pyrazoleamide 26f exhibited significant toxicity only at 30 μM. Remarkably, other pyrazoleamide and alkylamide based compounds were all nontoxic even at a high concentration of 30 μM in SHSY5Y cells, indicating that they were relatively safe JNK3 inhibitors. This was especially true for 26n which had >90% cell viability at both 10 and 30 μM.
Some compounds were also evaluated in vitro for their ability to inhibit the phosphorylation of c-Jun in neuronal SHSY5Y cells. In-cell Western blot analysis is a direct measure of JNK activity and was employed to investigate the ability of these JNK3 isoform selective inhibitors to inhibit c-Jun phosphorylation.20,36,41,51,52 As shown in Table 6, most of the compounds had an IC50 of around 1 μM or higher in this assay, indicating that they were reasonable JNK inhibitors. It is noteworthy that there was a large right shift between JNK3 biochemical potency and cell-based potency for these isoform selective JNK3 inhibitors (Table 6). The lack of JNK1 inhibition and the cell types might be part of the reasons.27,53 Indeed, because c-Jun phosphorylation is dependent on all three isoforms and SHSY5Y cells contain all three isoforms, the uninhibited JNK1 activity in these cells was the likely reason for the high cell-based IC50 values. In contrast, these same compounds showed good cell potency in other types of cell assays (Table 6) which were not as dependent on JNK1 nuclear activity for substrate phosphorylation but rather more centered on the role of JNK3 in mitochondrial dysfunction.3,4,51,52
These compounds showed protection from 6-OHDA-induced mitochondrial membrane depolarization and cell death in SHSY5Y cells, both of which are regulated and promoted by JNK activation.54 Improved cell potency was obtained in these two assays as compared to that of the in-cell Western assay. Interestingly, very good cell potency was seen in the mitochondrial membrane potential assays (IC50 < 50 nM) for most compounds (Table 6). The lower IC50 value in mitochondrial membrane potential assays compared to that in in-cell Western blot assays for these compounds (Table 6) could be due to the fact that JNK2/3 has a greater contribution to mitochondrial dysfunction than JNK1. This interpretation is consistent with the in vivo observation of Zhao and Herdegen where minimal to no JNK1 activity is seen on the mitochondria in neurodegenerative models.54 Thus, a small dose of these JNK2/3 isoform selective inhibitors is sufficient to inhibit JNK2/3 activity and to rescue mitochondrial membrane potential.
The high cell potency for isoform selective JNK2/3 inhibitors was further confirmed in our mitochondrial ROS inhibition assays in SHSY5Y cells. As shown in Figure 6, compound 26n significantly reduced the ROS generated by 6-OHDA (35 μM) at a concentration as low as 40 nM (IC50 < 40 nM). Similar cell potency in this specific assay was also observed for compound 26j (IC50 < 40 nM), showing this phenomenon is shared among the compounds in this class. Indeed, in contrast to biochemical assays, IC50 determinations of functional end points in cell-based assays often have steep slopes indicative of the functional readout that is being measured as opposed to the direct substrate competitive inhibition seen in a biochemical assay, and this could be why the cell-based IC50 is at the biochemical IC50 for 26k and lower than that for 26n. We have seen similar behavior in our aminopyrimidine class37 where the inhibition of ROS generation (∼1 nM) was lower than that of the biochemical IC50 value (50 nM). Thus, it may be that only a small amount of JNK3 and JNK2 inhibition goes a long way toward shutting down ROS generation and JNK mitochondrial function. Like 26n, the highlighted compound in the Kamenecka et al. study (9l, SR-3562) had exquisite selectivity over a panel of kinases. Thus, while we cannot rule out off-target kinase activity as a possible explanation for the lower IC50 in the cell-based assays, it is unlikely given the high selectivity of these compounds. Finally, we cannot rule out alternative interpretations such as other signaling pathways that do not have a kinase component contributing to ROS or MMP changes in the mitochondria, so this remains a possibility for the greater potency in ROS generation and MMP dissipation for these compounds.
Figure 6.

Inhibition of mitochondrial ROS generation by inhibitor 26n in SHSY5Y cells. SHSY5Y cells were treated with 35 μM 6-OHDA for 5 h, and mitochondrial ROS was measured by normalized MitoSOX fluorescence in the presence or absence of 40 nM, 370 nM, and 3 μM inhibitor. Statistical significance (p < 0.05) between control, untreated group, and 6-OHDA treated group is shown by ∗. Significance (p < 0.05) between 6-OHDA-treated groups and different concentrations of the inhibitor and 6-OHDA-treated groups is shown by ∗∗.
In order to evaluate the feasibility of using these JNK3 isoform selective inhibitors for in vivo applications, in vivo pharmacokinetic (PK) properties for selected compounds were studied in mice. As demonstrated in Table 7, these select lead compounds showed good PK properties in iv dosing. They had good Cmax and AUC (area under curve) values and fair half-lives (∼1–1.5 h). The clearance (Cl) and the volume of distribution (Vd) properties were also good for 22i, 26j, and 26k but were only fair for compound 26n. On the other hand, these compounds showed poor PK properties in oral dosing. For example, all of them exhibited almost no oral bioavailability (F values are all ≤1%). Therefore, the focus of future optimizations will be to improve oral PK properties if oral administration is preferred. However, these compounds, as JNK2/3 inhibitors for topical applications and/or in iv dosing, may be very useful to protect against retinal degeneration in glaucoma55 and could conceivably be used in this indication. Finally, since JNK3 selective inhibitors have the potential to be utilized in CNS disorders, we measured the brain penetration properties of 26n. After ip dosing (10 mg/kg in mice) brain levels at 2 h after dosing was approximately 55% of that found in the plasma. In addition, we also measured the brain concentration of 26n at 2 and 6 h after po and iv dosing. Because of the poor oral bioavailability of 26n (see Table 7) the brain levels at 2 and 6 h were below the lower limit of quantitation (∼5 nM) after oral dosing but were found to be 60 and 40 nM at 2 and 6 h, respectively, after iv dosing of 2 mg/kg (Table 8).
Table 7. In Vivo PK Data on Mice for Selected Lead JNK3 Inhibitorsa.
| cmpd | Cmax a (μM), iv | AUC a (μM·h), iv | T1/2 a (h), iv | Cl a ((mL/min)/kg), iv | Vd a (L/kg), iv | F a (%) po |
|---|---|---|---|---|---|---|
| 22i | 2.4 | 1.8 | 1.0 | 10 | 0.6 | 1.0 |
| 26j | 3.2 | 1.5 | 1.6 | 12 | 0.7 | 0.4 |
| 26k | 1.9 | 1.1 | 1.1 | 17 | 0.9 | 0.6 |
| 26n | 1.7 | 1.0 | 1.5 | 21 | 1.2 | 0.5 |
Dosed at 0.5 mg/kg for iv and at 3 mg/kg for po. All data were based on three determinations.
Table 8. Brain Concentration of 26n in Micea.
| dosing | 2 h | 6 h |
|---|---|---|
| iv (2 mg/kg) | 60 nM | 40 nM |
| po (5 mg/kg) | <5 nM | <5 nM |
Data were generated from three determinations.
Conclusion
We have developed a series of aminopyrazoleamide based JNK3 isoform selective inhibitors starting from the initial lead compound SR-4326. SAR investigation and optimization successfully yielded potent JNK3 inhibitors with high selectivity over p38 kinase, as well as high isoform selectivity against JNK1 and in some cases over JNK2. For example, compound 26n had an isoform selectivity of >50-fold for JNK3 against JNK1 and were potent in all three types of cell-based assays in SHSY5Y cells (in-cell Western target modulation, inhibition of 6-OHDA induced cell death, and inhibition of 6-OHDA induced mitochondrial membrane depolarization). Another lead inhibitor 26k was highly potent and selective with an IC50 value against JNK3 in the subnanomolar range and possessed an isoform selectivity of >500-fold over JNK1 and >20000-fold against p38α. In addition, 26n and 26k were noncytotoxic, potently inhibited mitochondrial dysfunction, and possessed good PK properties in iv dosing. In addition, 26n showed a plasma/brain ratio of ∼2:1. The X-ray crystal structure of 26k in JNK3 revealed a binding mode that can very well support the observed SAR, and this structure was also helpful for designing potent and selective JNK3 inhibitors. Future optimizations for these aminopyrazole-based compounds will be mainly focused on improving the pharmacokinetic properties in order to obtain better JNK3 isoform selective inhibitors for oral dosing. These studies will be reported in due course.
Experimental Section
Commercially available reagents and anhydrous solvents were used without further purification unless otherwise specified. Thin layer chromatography (TLC) analyses were performed with precoated silica gel 60 F254. The mass spectra were recorded by LC/MS with Finnigan LCQ Advantage MAX spectrometer of Thermo Electron. Flash chromatography was performed on prepacked columns of silica gel (230–400 mesh, 40–63 μm) by CombiFlash with EtOAc/hexane or MeOH/DCM as eluents. The preparative HPLC was performed on SunFire C18 OBD 10 μm (30 mm × 250 mm) with CH3CN + 50% MeOH/H2O + 0.1% TFA as eluents to purify the targeted compounds. Analytic HPLC was performed on Agilent Technologies 1200 series with CH3CN (solvent B)/H2O + 0.9% CH3CN + 0.1% TFA (solvent A) as eluents, and the targeted products were detected by UV in the detection range of 215–310 nm. All compounds were determined to be >95% pure by this method. NMR spectra were recorded with a Bruker 400 MHz spectrometer at ambient temperature with the residual solvent peaks as internal standards. The line positions of multiplets were given in ppm (δ), and the coupling constants (J) were given in hertz. The high-resolution mass spectra (HRMS, electrospray ionization) experiments were performed with Thermo Finnigan Orbitrap mass analyzer. Data were acquired in the positive ion mode at resolving power of 100 000 at m/z 400. Calibration was performed with an external calibration mixture immediately prior to analysis.
General Synthetic Procedures
A mixture of 4-nitro-1H-pyrazole 1 (10 mmol), 3-bromobenzoic acid 2 (20 mmol), CuI (2.0 mmol), trans-N,N-dimethylcyclohexane-1,2-diamine (4.0 mmol), and Cs2CO3 (30 mmol) in DMF (20 mL) was purged with argon and stirred for 12 h at 100 °C in a sealed tube. The reaction mixture was cooled, and filtered through a pad of silica gel, and rinsed with EtOAc. The resulting solution was concentrated in vacuo to yield a crude residue which was purified by chromatography on silica gel (EtOAc/hexane) to provide 3-(4-nitro-1H-pyrazol-1-yl)benzoic acid 3.
To a solution of 3-(4-nitro-1H-pyrazol-1-yl)benzoic acid 3 (5.0 mmol) in CH2Cl2 (20 mL) were added EDC (10 mmol), HOBt (10 mmol), and diisopropylethylamine (15 mmol), and the mixture was stirred for 30 min. Then the 6-methylpyridin-3-amine (5.5 mmol) was added, and the resulting mixture was stirred overnight. Water (50 mL) was added to the reaction mixture and extracted with EtOAc (2 × 100 mL). The resulting solution was concentrated in vacuo to yield a crude product (4).
This intermediate was hydrogenated in anhydrous methanol (100 mL) in the presence of 10% Pd/C (1.0 g) under a balloon of hydrogen for 3 h. The mixture was filtered through a Celite pad and evaporated. The residue was purified by chromatography on silica gel (dichloromethane/methanol) to give the 3-(4-amino-1H-pyrazol-1-yl)-N-(6-methylpyridin-3-yl)benzamide 5.
1-Chloro-2-isocyanatobenzene (0.12 mmol) was added to 3-(4-amino-1H-pyrazol-1-yl)-N-(6-methylpyridin-3-yl)benzamide (0.1 mmol) in CH2Cl2 (1.0 mL) at room temperature and stirred for 1 h. The solvent was evaporated and the residue was purified by reverse-phase preparative HPLC to give 3-(4-(3-(2-chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(6-methylpyridin-3-yl)benzamide 8c as a white powder.
3-(4-(2-(2-Chlorophenyl)acetamido)-1H-pyrazol-1-yl)-N-(6-methylpyridin-3-yl)benzamide (6a)
66% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.93 (s, 1H), 10.51 (s, 1H), 9.08 (d, J = 2.0 Hz, 1H), 8.70 (s, 1H), 8.42 (dd, J = 8.4, 2.0 Hz, 1H), 8.38 (s, 1H), 8.10–8.03 (m, 1H), 7.89 (d, J = 8.0 Hz, 1H), 7.82 (s, 1H), 7.68 (t, J = 8.0 Hz, 2H), 7.42–7.47 (m, 2H), 7.37–7.28 (m, 2H), 3.85 (s, 2H), 2.61 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C24H20ClN5O2, 446.1; found, 446.4
2-Chlorophenyl (1-(3-((6-Methylpyridin-3-yl)carbamoyl)phenyl)-1H-pyrazol-4-yl)carbamate (6b)
58% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.60–10.54 (m, 2H), 8.82 (d, J = 2.4 Hz, 1H), 8.52 (s, 1H), 8.38–8.33 (m, 1H), 8.11 (dd, J = 8.4, 2.6 Hz, 1H), 8.07–7.98 (m, 1H), 7.90–7.83 (m, 1H), 7.79 (s, 1H), 7.65 (t, J = 80 Hz, 1H), 7.60 (d, J = 7.6 Hz, 1H), 7.43 (m, 2H), 7.38–7.28 (m, 2H), 2.47 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H18ClN5O3, 448.1; found, 448.4
N-(1-(3-((6-Methylpyridin-3-yl)carbamoyl)phenyl)-1H-pyrazol-4-yl)-1H-indole-2-carboxamide (6c)
62% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ11.00 (s, 1H), 10.76 (s, 1H), 10.18 (s, 1H), 9.12 (s, 1H), 8.88 (s, 1H), 8.53–8.37 (m, 2H), 8.23 (t, J = 4.9 Hz, 2H), 8.12 (dd, J = 8.0, 1.2 Hz, 1H), 7.90 (d, J = 7.8 Hz, 2H), 7.71 (dd, J = 16.4, 8.4 Hz, 2H), 7.53–7.40 (m, 1H), 7.23–7.12 (m, 2H), 2.63 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C25H20N6O2, 437.2; found, 437.4.
N-(1-(3-((6-Methylpyridin-3-yl)carbamoyl)phenyl)-1H-pyrazol-4-yl)-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (6d)
35% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 11.10 (s, 1H), 10.89 (s, 1H), 9.80 (s, 1H), 9.69 (s, 1H), 9.04 (s, 1H), 8.83 (s, 1H), 8.48 (s, 1H), 8.37 (s, 1H), 8.17 (dd, J = 8.4, 1.6 Hz, 1H), 8.00 (d, J = 9.8 Hz, 2H), 7.84–7.71 (m, 1H), 7.59 (s, 1H), 7.37 (m, 3H), 4.49 (s, 2H), 3.56–3.52 (m, 1H), 3.49 (dd, J = 9.2, 4.4 Hz, 1H), 3.20 (dd, J = 16.8, 12.2 Hz, 1H), 2.62 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C26H24N6O2, 453.2; found, 453.3.
3-(4-((6-Bromo-1H-benzo[d]imidazol-2-yl)amino)-1H-pyrazol-1-yl)-N-(6-methylpyridin-3-yl)benzamide (7)
48% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 12.98 (s, 1H), 10.90 (s, 1H), 10.80 (s, 1H), 10.58 (s, 1H), 9.01 (s, 1H), 8.96 (s, 1H), 8.50 (s, 1H), 8.28 (s, 1H), 8.13 (d, J = 8.0 Hz, 1H), 8.04 (s, 1H), 8.00 (d, J = 8.0 Hz, 1H), 7.76 (t, J = 8.0 Hz, 1H), 7.36 (ddd, J = 56.6, 6.0, 3.2 Hz, 4H), 2.53 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H18BrN7O, 517.4; found, 517.6.
3-(4-(3-Cyclopentylureido)-1H-pyrazol-1-yl)-N-(6-methylpyridin-3-yl)benzamide (8a)
45% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.78 (s, 1H), 8.96 (s, 1H), 8.41 (s, 1H), 8.28 (s, 2H), 8.17 (s, 1H), 7.95 (dd, J = 8.1, 1.3 Hz, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.66 (s, 1H), 7.63–7.49 (m, 2H), 6.22 (s, 1H), 3.87 (dd, J = 13.4, 6.7 Hz, 1H), 2.51 (s, 3H), 1.76 (dt, J = 11.6, 5.8 Hz, 2H), 1.61–1.51 (m, 2H), 1.43–1.48 (m, 2H), 1.36–1.24 (m, 2H). LC/MS (ESI) m/z: [M + H]+ calcd for C22H24N6O2, 405.2; found, 405.3.
N-(6-Methylpyridin-3-yl)-3-(4-(3-phenylureido)-1H-pyrazol-1-yl)benzamide (8b)
58% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.82 (s, 1H), 8.99 (s, 1H), 8.81 (s, 1H), 8.69 (s, 1H), 8.52 (s, 1H), 8.32 (s, 2H), 8.07–7.96 (m, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.78 (d, J = 14.4 Hz, 1H), 7.61 (t, J = 8.0 Hz, 2H), 7.41 (d, J = 7.6 Hz, 2H), 7.26–7.17 (m, 2H), 6.90 (t, J = 7.2 Hz, 1H), 2.52 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H20N6O2, 413.2; found, 413.3.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(6-methylpyridin-3-yl)benzamide (8c)
63% yield in four steps (TFA salt, white solid); 1H NMR (DMSO-d6, 400 MHz) δ 10.88 (s, 1H), 9.45 (s, 1H), 9.04 (s, 1H), 8.62 (s, 1H), 8.40 (m, 3H), 8.20 (dd, J = 8.2, 1.6 Hz, 1H), 8.09 (d, J = 8.2 Hz, 1H), 7.94–7.81 (m, 2H), 7.69 (t, J = 8.0 Hz, 1H), 7.63 (s, 1H), 7.47 (dd, J = 8.0, 1.4 Hz, 1H), 7.38–7.25 (m, 1H), 7.10–6.96 (m, 1H), 2.59 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H19FN6O2, 447.1; found, 447.4.
3-(4-(3-(3-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(6-methylpyridin-3-yl)benzamide (8d)
64% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.89 (s, 1H), 9.21 (s, 1H), 9.05 (s, 1H), 8.99 (s, 1H), 8.61 (s, 1H), 8.39 (d, J = 8.6 Hz, 2H), 8.08 (dd, J = 8.1, 1.3 Hz, 1H), 7.88 (d, J = 8.2 Hz, 1H), 7.84 (s, 1H), 7.78 (s, 1H), 7.68 (t, J = 8.0 Hz, 1H), 7.64 (d, J = 7.9 Hz, 1H), 7.34–7.27 (m, 2H), 7.06–6.92 (m, 1H), 2.58 (d, J = 10.6 Hz, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H19FN6O2, 447.1; found, 447.4.
3-(4-(3-(4-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(6-methylpyridin-3-yl)benzamide (8e)
58% yield in four steps (TFA salt, white solid). H NMR (400 MHz, DMSO) δ 10.87 (s, 1H), 9.04 (s, 2H), 8.82 (s, 1H), 8.59 (s, 1H), 8.38 (s, 2H), 8.07 (d, J = 7.8 Hz, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.83 (s, 1H), 7.67 (t, J = 8.0 Hz, 2H), 7.52 (d, J = 8.8 Hz, 2H), 7.37–7.22 (m, 2H), 2.58 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H19FN6O2, 447.1; found, 447.3.
3-(4-(3-(2-Fluorophenyl)ureido)-1H-pyrazol-1-yl)-N-(6-methylpyridin-3-yl)benzamide (8f)
56% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.92 (s, 1H), 9.21 (s, 1H), 9.07 (s, 1H), 8.93 (s, 1H), 8.61 (s, 1H), 8.40 (s, 2H), 8.08 (dd, J = 8.1, 1.4 Hz, 1H), 7.89 (d, J = 8.1 Hz, 1H), 7.84 (s, 1H), 7.67 (dd, J = 15.0, 7.0 Hz, 2H), 7.54 (dt, J = 12.0, 2.4 Hz, 1H), 7.38–7.22 (m, 1H), 7.15 (dd, J = 8.2, 1.1 Hz, 1H), 6.78 (td, J = 8.4, 2.4 Hz, 1H), 2.58 (d, J = 13.2 Hz, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H19FN6O2, 431.2; found, 431.3.
3-(4-(3-(3-Fluorophenyl)ureido)-1H-pyrazol-1-yl)-N-(6-methylpyridin-3-yl)benzamide (8g)
55% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.98 (s, 1H), 9.14 (d, J = 7.2 Hz, 2H), 8.74 (d, J = 2.3 Hz, 1H), 8.68 (s, 1H), 8.46 (dd, J = 5.2, 3.6 Hz, 2H), 8.22 (td, J = 8.3, 1.6 Hz, 1H), 8.19–8.11 (m, 1H), 7.99–7.90 (m, 2H), 7.73 (dd, J = 16.4, 8.8 Hz, 2H), 7.30–7.34 (m, 1H), 7.27–7.14 (m, 1H), 7.14–7.01 (m, 1H), 2.66 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H19FN6O2, 431.2; found, 431.4.
3-(4-(3-(4-Fluorophenyl)ureido)-1H-pyrazol-1-yl)-N-(6-methylpyridin-3-yl)benzamide (8h)
58% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.94 (s, 1H), 9.08 (s, 1H), 9.03 (s, 1H), 8.87 (s, 1H), 8.59 (s, 1H), 8.46–8.36 (m, 2H), 8.07 (dd, J = 8.1, 1.3 Hz, 1H), 7.89 (d, J = 8.2 Hz, 1H), 7.83 (s, 1H), 7.68 (t, J = 8.0 Hz, 2H), 7.56–7.45 (m, 2H), 7.19–7.08 (m, 2H), 2.61 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H19FN6O2, 431.2; found, 431.3.
3-(4-(3-(3-Chlorobenzyl)ureido)-1H-pyrazol-1-yl)-N-(6-methylpyridin-3-yl)benzamide (8i)
32% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.97 (s, 1H), 9.45 (s, 1H), 9.12 (d, J = 2.0 Hz, 1H), 8.74 (s, 1H), 8.59 (s, 1H), 8.46 (dd, J = 8.6, 2.0 Hz, 1H), 8.42 (s, 1H), 8.14–8.07 (m, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.87 (s, 1H), 7.72 (t, J = 8.0 Hz, 2H), 7.50 (ddd, J = 8.0, 4.8, 2.0 Hz, 2H), 7.42–7.32 (m, 2H), 3.89 (s, 2H), 2.65 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C24H21N6O2, 461.1; found, 461.4.
3-(4-(3-(2-Chlorophenyl)-3-methylureido)-1H-pyrazol-1-yl)-N-(6-methylpyridin-3-yl)benzamide (8j)
21% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.96 (s, 1H), 9.10 (s, 1H), 8.51 (s, 1H), 8.48–8.37 (m, 2H), 8.34 (t, J = 1.8 Hz, 1H), 8.05–7.97 (m, 1H), 7.86 (dd, J = 6.8, 1.5 Hz, 1H), 7.75–7.70 (m, 2H), 7.63 (m, 2H), 7.55–7.34 (m, 3H), 3.18 (s, 3H), 2.62 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C24H21N6O2, 461.1; found, 461.2.
3-(4-(3-(2-Chlorophenyl)-3-(2-hydroxyethyl)ureido)-1H-pyrazol-1-yl)-N-(6-methylpyridin-3-yl)benzamide (8k)
23% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.59 (s, 1H), 8.84 (s, 1H), 8.52 (s, 1H), 8.31 (s, 1H), 8.30–8.23 (m, 1H), 8.16–8.08 (m, 1H), 8.02–7.94 (m, 1H), 7.84 (s, 1H), 7.73 (s, 1H), 7.63 (d, J = 4.0 Hz, 2H), 7.54 (s, 1H), 7.46 (s, 2H), 7.37–7.29 (m, 1H), 3.51–3.47 (m, 2H), 3.40–3.44 (m, 2H), 3.17 (s, 1H), 2.48 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C25H23N6O3, 491.2; found, 491.4.
N-(1-(3-((6-Methylpyridin-3-yl)carbamoyl)phenyl)-1H-pyrazol-4-yl)indoline-1-carboxamide (8l)
28% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 11.46 (s, 1H), 8.98 (s, 1H), 8.66 (s, 1H), 8.40 (s, 1H), 8.12 (dd, J = 8.0, 1.6 Hz, 1H), 8.00–7.91 (m, 3H), 7.91–7.82 (m, 2H), 7.71 (t, J = 8.0 Hz, 1H), 7.21 (d, J = 6.8 Hz, 1H), 7.13 (t, J = 7.6 Hz, 1H), 6.91 (dd, J = 11.6, 4.2 Hz, 1H), 6.57 (s, 1H), 4.10 (t, J = 8.8 Hz, 2H), 3.21 (t, J = 8.6 Hz, 2H), 2.66 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C25H22N6O2, 439.1; found, 439.4.
N-(2-Chlorobenzyl)-1-(3-((6-methylpyridin-3-yl)carbamoyl)phenyl)-1H-pyrazole-4-carboxamide (12a)
71% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.93 (s, 1H), 9.12 (s, 1H), 9.08 (s, 1H), 8.83 (t, J = 5.8 Hz, 1H), 8.48 (t, J = 1.8 Hz, 1H), 8.42 (d, J = 9.2 Hz, 2H), 8.30 (s, 1H), 8.15 (dd, J = 8.0, 1.6 Hz, 1H), 7.98 (d, J = 6.8 Hz, 1H), 7.75 (t, J = 8.0 Hz, 1H), 7.70 (d, J = 6.2 Hz, 1H), 7.48 (dd, J = 7.6, 1.6 Hz, 1H), 7.42 (dd, J = 7.4, 2.0 Hz, 1H), 7.34 (m, 2H), 4.55 (d, J = 5.7 Hz, 2H), 2.61 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C24H20ClN5O2, 446.1; found, 446.3.
3-(5-(2-Chlorobenzyl)-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridin-1-yl)-N-(6-methylpyridin-3-yl)benzamide (12b)
45% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.88 (s, 1H), 9.02 (s, 1H), 8.54 (s, 1H), 8.40 (t, J = 1.8 Hz, 1H), 8.34 (d, J = 8.4 Hz, 1H), 8.02 (dd, J = 8.1, 1.4 Hz, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.86–7.77 (m, 1H), 7.69 (t, J = 8.0 Hz, 1H), 7.64 (dd, J = 7.6, 1.6 Hz, 1H), 7.50–7.56 (m, 3H), 4.61 (s, 2H), 4.40–4.45 (m, 2H), 3.10–3.14 (m, 2H), 2.56 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C26H22ClN5O2, 472.2; found, 472.5.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-4-fluoro-N-(6-methylpyridin-3-yl)benzamide (16a)
47% yield in five steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.92 (s, 1H), 9.46 (s, 1H), 9.05 (s, 1H), 8.50 (dd, J = 7.6, 2.4 Hz, 1H), 8.46–8.31 (m, 3H), 8.18 (dd, J = 8.4, 1.4 Hz, 1H), 8.02 (ddd, J = 8.6, 4.4, 2.4 Hz, 1H), 7.91 (s, 1H), 7.74–7.69 (m, 1H), 7.69–7.60 (m, 1H), 7.47 (dd, J = 8.0, 1.4 Hz, 1H), 7.35–7.26 (m, 1H), 7.11–6.93 (m, 1H), 2.60 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H18ClFN6O2, 465.1; found, 465.3.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-5-fluoro-N-(6-methylpyridin-3-yl)benzamide (16b)
53% yield in five steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.58 (s, 1H), 9.45 (s, 1H), 8.80 (d, J = 2.4 Hz, 1H), 8.65 (s, 1H), 8.39 (s, 1H), 8.27 (s, 1H), 8.21 (dd, J = 8.4, 1.2 Hz, 1H), 8.07 (dd, J = 8.4, 2.4 Hz, 1H), 7.94 (dt, J = 10.0, 2.0 Hz, 1H), 7.90 (s, 1H), 7.66 (d, J = 9.0 Hz, 1H), 7.45 (dd, J = 8.0, 1.2 Hz, 1H), 7.36–7.20 (m, 2H), 7.00–7.04 (m, 1H), 2.45 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H18ClFN6O2, 465.1; found, 465.2.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-6-fluoro-N-(6-methylpyridin-3-yl)benzamide (16c)
22% yield in five steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.91 (s, 1H), 9.44 (s, 1H), 8.54 (d, J = 6.4 Hz, 1H), 8.48 (s, 1H), 8.39 (s, 1H), 8.24–8.14 (m, 2H), 8.14–8.07 (m, 1H), 8.04 (s, 1H), 7.94 (d, J = 6.4 Hz, 1H), 7.85 (s, 1H), 7.57 (t, J = 9.2 Hz, 1H), 7.47 (dd, J = 8.0, 1.6 Hz, 1H), 7.36–7.23 (m, 1H), 7.09–6.97 (m, 1H), 2.67 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H18ClFN6O2, 465.1; found, 465.4.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-2-fluoro-N-(6-methylpyridin-3-yl)benzamide (16d)
15% yield in five steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.79 (s, 1H), 9.53 (s, 1H), 8.93 (s, 1H), 8.74 (s, 1H), 8.48 (s, 1H), 8.37 (s, 1H), 8.27 (s, 2H), 8.05 (s, 1H), 7.94–7.99 (m, 1H), 7.56 (d, J = 8.0 Hz, 1H), 7.39 (d, J = 8.0, 1H), 7.29 (s, 1H), 7.14–7.18 (m, 1H), 7.10–7.13 (m, 1H), 7.00–7.06 (m, 1H), 2.60 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H18ClFN6O2, 465.1; found, 465.4.
1-(2-Chlorophenyl)-3-(1-(3-(pyridin-4-yl)phenyl)-1H-pyrazol-4-yl)urea (20a)
38% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 9.43 (s, 1H), 8.84 (d, J = 4.8 Hz, 2H), 8.69 (s, 1H), 8.39 (s, 1H), 8.28 (s, 1H), 8.24–8.09 (m, 3H), 8.02 (d, J = 8.1 Hz, 1H), 7.87–7.76 (m, 2H), 7.68 (t, J = 8.0 Hz, 1H), 7.46 (dd, J = 8.0, 1.2 Hz, 1H), 7.36–7.24 (m, 1H), 7.03 (td, J = 8.0, 1.6 Hz, 1H). LC/MS (ESI) m/z: [M + H]+ calcd for C21H16ClN5O, 390.1; found, 390.2.
1-(2-Chlorophenyl)-3-(1-(3-(pyrimidin-4-ylamino)phenyl)-1H-pyrazol-4-yl)urea (20b)
25% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.72 (s, 1H), 9.41 (s, 1H), 8.87 (s, 1H), 8.44 (s, 1H), 8.37 (s, 2H), 8.18 (d, J = 6.8 Hz, 2H), 7.81 (s, 1H), 7.58 (d, J = 7.6 Hz, 2H), 7.51 (d, J = 8.0 Hz, 1H), 7.46 (d, J = 7.0 Hz, 1H), 7.30 (s, 1H), 7.03 (s, 1H), 6.98 (d, J = 6.6 Hz, 1H). LC/MS (ESI) m/z: [M + H]+ calcd for C21H16ClN7O, 406.1; found, 406.3.
1-(2-Chlorophenyl)-3-(1-(3-(5-methyl-4H-1,2,4-triazol-3-yl)phenyl)-1H-pyrazol-4-yl)urea (21a)
28% yield in five steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 9.40 (s, 1H), 8.52 (s, 1H), 8.39 (s, 2H), 8.20 (d, J = 8.0, 1H), 7.87 (d, J = 8.0, 1H), 7.83 (m, 2H), 7.58 (t, J = 7.8 Hz, 1H), 7.47 (d, J = 8.0, 1H), 7.31 (t, J = 7.2 Hz, 1H), 7.16 (s, 1H), 7.04 (t, J = 7.6 Hz, 1H), 2.42 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C19H16ClN7O, 394.1; found, 394.3.
1-(2-Chlorophenyl)-3-(1-(3-(5-methyl-1,3,4-oxadiazol-2-yl)phenyl)-1H-pyrazol-4-yl)urea (21b)
37% yield in five steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 9.50 (s, 1H), 8.59 (s, 1H), 8.42 (s, 1H), 8.29 (s, 1H), 8.20 (d, J = 7.1 Hz, 1H), 8.04 (d, J = 8.0 Hz, 1H), 7.83 (s, 1H), 7.78 (d, J = 7.4 Hz, 1H), 7.63 (t, J = 7.8 Hz, 1H), 7.47 (d, J = 8.0 Hz, 1H), 7.31 (t, J = 7.8 Hz, 1H), 7.04 (t, J = 7.8 Hz, 1H), 2.45 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C19H15ClN6O2, 395.1; found, 395.2.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-methyl-N-(6-methylpyridin-3-yl)benzamide (22a)
48% yield in four steps (TFA salt, white solid); 1H NMR (DMSO-d6, 400 MHz) δ 9.88 (s, 1H), 9.42 (s, 1H), 8.59 (s, 1H), 8.36–8.39 (m, 1H), 8.32 (s, 1H), 8.21 (dd, J = 8.4, 1.6 Hz, 1H), 8.01 (d, J = 9.6 Hz, 1H), 7.90 (s, 1H), 7.86–7.78 (m, 2H), 7.62 (t, J = 8.0 Hz, 1H), 7.47 (dd, J = 8.0, 1.4 Hz, 1H), 7.31 (t, J = 7.2 Hz, 1H), 7.03 (dd, J = 10.8, 4.4 Hz, 1H), 3.77 (s, 3H), 2.17 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C24H21ClN6O2, 461.2; found, 461.5.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(2-methylpyridin-3-yl)benzamide (22b)
55% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.80 (s, 1H), 9.63 (s, 1H), 8.77–8.64 (m, 2H), 8.52 (dd, J = 4.8, 3.0 Hz, 2H), 8.44 (d, J = 8.0 Hz, 1H), 8.26 (dd, J = 8.3, 1.5 Hz, 1H), 8.17 (dd, J = 8.4, 1.2 Hz, 1H), 7.98 (d, J = 8.0 Hz, 1H), 7.93 (s, 1H), 7.88–7.80 (m, 1H), 7.75 (t, J = 8.0 Hz, 1H), 7.53 (dd, J = 8.0, 1.6 Hz, 1H), 7.42–7.32 (m, 1H), 7.14–7.06 (m, 1H), 2.73 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H19ClN6O2, 447.1; found, 447.4.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(2,6-dimethylpyridin-3-yl)benzamide (22c)
48% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.55 (s, 1H), 9.45 (s, 1H), 8.62 (s, 1H), 8.41 (d, J = 7.6 Hz, 2H), 8.19 (d, J = 8.3 Hz, 2H), 8.09 (d, J = 8.4 Hz, 1H), 7.89 (d, J = 7.6 Hz, 1H), 7.85 (s, 1H), 7.68 (t, J = 8.0 Hz, 1H), 7.57 (s, 1H), 7.47 (dd, J = 8.0, 1.6 Hz, 1H), 7.37–7.26 (m, 1H), 7.08–7.00 (m, 1H), 2.63 (s, 3H), 2.57 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C24H21ClN6O2, 461.1; found, 461.4.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(6-methylpyridin-2-yl)benzamide (22d)
32% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 11.10 (s, 1H), 9.60 (s, 1H), 8.72 (s, 1H), 8.43 (d, J = 14.2 Hz, 2H), 8.24–8.18 (m, 1H), 8.11–8.01 (m, 2H), 7.90 (d, J = 7.8 Hz, 1H), 7.84–7.74 (m, 2H), 7.62 (t, J = 8.0 Hz, 1H), 7.46 (dd, J = 8.0, 1.4 Hz, 1H), 7.30 (d, J = 7.2 Hz, 1H), 7.14–7.05 (m, 1H), 7.04 (d, J = 7.8 Hz, 1H), 2.53 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H19ClN6O2, 447.1; found, 447.4.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(5-methylpyridin-2-yl)benzamide (22e)
43% yield in five steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) 10.74 (s, 1H), 9.44 (s, 1H), 9.08 (s, 2H), 8.61 (s, 1H), 8.40 (d, J = 5.2 Hz, 2H), 8.21 (d, J = 8.2 Hz, 1H), 8.08 (d, J = 7.6 Hz, 1H), 7.88 (d, J = 12.4 Hz, 2H), 7.68 (t, J = 8.0 Hz, 1H), 7.47 (d, J = 7.8 Hz, 1H), 7.31 (t, J = 7.6 Hz, 1H), 7.04 (t, J = 7.2 Hz, 1H), 2.62 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H19ClN6O2, 447.1; found, 447.3.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(2-methylpyridin-4-yl)benzamide (22f)
22% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 11.42 (s, 1H), 9.45 (s, 1H), 8.63 (s, 2H), 8.40 (s, 2H), 8.20 (d, J = 6.8 Hz, 1H), 8.12 (s, 2H), 8.06–7.99 (m, 1H), 7.87 (s, 2H), 7.73 (d, J = 8.0 Hz, 1H), 7.48 (d, J = 9.4 Hz, 1H), 7.31 (s, 1H), 7.06 (d, J = 6.0 Hz, 1H), 2.66 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H19ClN6O2, 447.1; found, 447.3.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(2,6-dimethylpyridin-4-yl)benzamide (22g)
20% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 11.10 (s, 1H), δ 9.58 (s, 1H), 8.72 (s, 1H), 8.47–8.37 (m, 2H), 8.25–8.18 (m, 1H), 8.12–8.02 (m, 2H), 7.96–7.89 (m, 1H), 7.84–7.74 (m, 2H), 7.63 (t, J = 7.6 Hz, 1H), 7.51–7.42 (m, 1H), 7.31 (t, J = 7.8 Hz, 1H), 7.10 (d, J = 8.0 Hz, 1H), 7.05 (t, J = 7.6 Hz, 1H), 2.53–2.52 (m, 6H). LC/MS (ESI) m/z: [M + H]+ calcd for C24H21ClN6O2, 461.1; found, 461.4.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(2-methylpyrimidin-5-yl)benzamide (22h)
46% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.74 (s, 1H), 9.44 (s, 1H), 9.08 (s, 2H), 8.61 (s, 1H), 8.40 (d, J = 5.2 Hz, 2H), 8.21 (d, J = 8.2 Hz, 1H), 8.08 (d, J = 7.6 Hz, 1H), 7.88 (d, J = 12.4 Hz, 2H), 7.68 (t, J = 8.0 Hz, 1H), 7.47 (d, J = 7.8 Hz, 1H), 7.31 (t, J = 7.6 Hz, 1H), 7.04 (t, J = 7.2 Hz, 1H), 2.62 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H19ClN6O2, 448.1; found, 448.4.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-((6-methylpyridin-3-yl)methyl)benzamide (22i)
66% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 9.46 (s, 1H), 9.42 (t, J = 5.6 Hz, 1H), 8.76 (s, 1H), 8.58 (s, 1H), 8.41 (s, 1H), 8.34 (d, J = 8.2 Hz, 1H), 8.30 (s, 1H), 8.19 (dd, J = 8.4, 1.4 Hz, 1H), 8.01 (dd, J = 8.1, 1.3 Hz, 1H), 7.85–7.77 (m, 3H), 7.61 (t, J = 8.0 Hz, 1H), 7.47 (dd, J = 8.0, 1.4 Hz, 1H), 7.35–7.27 (m, 1H), 7.04 (td, J = 8.0, 1.6 Hz, 1H), 4.63 (d, J = 5.6 Hz, 2H), 2.67 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C24H21ClN6O2, 461.1; found, 461.2.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(1-methyl-1H-pyrazol-4-yl)benzamide (26a)
72% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.61 (s, 1H), 9.43 (s, 1H), 8.59 (s, 1H), 8.39 (s, 1H), 8.34 (s, 1H), 8.20 (dd, J = 8.3, 1.4 Hz, 1H), 8.06 (s, 1H), 8.06–7.98 (m, 1H), 7.84 (t, J = 3.6 Hz, 2H), 7.69–7.58 (m, 2H), 7.46 (dd, J = 8.0, 1.4 Hz, 1H), 7.36–7.24 (m, 1H), 7.04 (td, J = 8.0, 1.6 Hz, 1H), 3.84 (s, 3H). LC/MS (ESI) m/z: [M + H]+ calcd for C21H18ClN7O2, 436.1; found, 436.2.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(1-methyl-1H-pyrazol-3-yl)benzamide (26b)
68% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 11.07 (s, 1H), 9.42 (s, 1H), 8.66 (s, 1H), 8.38 (s, 2H), 8.23 (dd, J = 8.4, 1.6 Hz, 1H), 8.02 (dd, J = 8.1, 1.3 Hz, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.82 (s, 1H), 7.67–7.57 (m, 2H), 7.47 (dd, J = 8.0, 1.5 Hz, 1H), 7.38–7.26 (m, 1H), 7.08–6.97 (m, 1H), 6.63 (d, J = 2.4 Hz, 1H), 3.81 (s, 3H). LC–MS: 436 (M + H). LC/MS (ESI) m/z: [M + H]+ calcd for C21H18ClN7O2, 436.1; found, 436.4.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(1-methyl-1H-pyrazol-5-yl)benzamide (26c)
65% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.58 (s, 1H), 9.50 (s, 1H), 8.68 (s, 1H), 8.45 (s, 2H), 8.26 (dd, J = 8.4, 1.6 Hz, 1H), 8.14 (dd, J = 8.0, 1.4 Hz, 1H), 7.99–7.88 (m, 2H), 7.72 (t, J = 8.0 Hz, 1H), 7.56–7.45 (m, 2H), 7.41–7.32 (m, 1H), 7.14–7.05 (m, 1H), 6.33 (d, J = 1.9 Hz, 1H), 3.79 (s, 3H). LC–MS: 436 (M + H). LC/MS (ESI) m/z: [M + H]+ calcd for C21H18ClN7O2, 436.1; found, 436.2.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(1,3-dimethyl-1H-pyrazol-4-yl)benzamide (26d)
75% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, MeOD) δ 8.36 (s, 1H), 8.19 (s, 1H), 8.06–8.00 (m, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.79–7.72 (m, 2H), 7.65 (s, 1H), 7.53 (t, J = 8.0 Hz, 1H), 7.31 (dd, J = 8.0, 1.2 Hz, 1H), 7.21–7.17 (m, 1H), 6.99–6.92 (m, 1H), 3.74 (s, 3H), 2.16 (s, 3H). LC–MS: 436 (M + H). LC/MS (ESI) m/z: [M + H]+ calcd for C22H20ClN7O2, 450.1; found, 450.4.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(1-methyl-1H-1,2,4-triazol-3-yl)benzamide (26e)
66% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.91 (s, 1H), 9.43 (s, 1H), 8.63 (s, 1H), 8.39 (t, J = 13.4 Hz, 3H), 8.21 (dd, J = 8.4, 1.6 Hz, 1H), 8.05 (dd, J = 8.0, 1.4 Hz, 1H), 7.92–7.81 (m, 2H), 7.63 (t, J = 7.9 Hz, 1H), 7.47 (dd, J = 8.0, 1.4 Hz, 1H), 7.36–7.26 (m, 1H), 7.08–6.99 (m, 1H), 3.87 (s, 3H). LC–MS: 436 (M + H). LC/MS (ESI) m/z: [M + H]+ calcd for C20H17ClN8O2, 437.1; found, 437.3.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(1-isopropyl-1H-pyrazol-4-yl)benzamide (26f)
44% yield in five steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.60 (d, J = 7.2 Hz, 1H), 9.44 (s, 1H), 8.59 (s, 1H), 8.44–8.32 (m, 2H), 8.21 (dd, J = 8.3, 1.5 Hz, 1H), 8.09 (d, J = 3.6 Hz, 1H), 8.06–7.97 (m, 1H), 7.84 (d, J = 6.8 Hz, 2H), 7.70–7.58 (m, 2H), 7.47 (dd, J = 8.0, 1.5 Hz, 1H), 7.36–7.26 (m, 1H), 7.09–7.00 (m, 1H), 4.55–4.47 (m, 1H), 1.42 (dd, J = 6.8, 2.0 Hz, 7H). LC–MS: 436 (M + H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H22ClN8O2, 464.2; found, 464,4.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(1-(2-(dimethylamino)ethyl)-1H-pyrazol-4-yl)benzamide (26g)
54% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, MeOD) δ 8.36 (s, 1H), 8.20 (t, J = 1.8 Hz, 1H), 8.13 (s, 1H), 8.02 (dd, J = 8.4, 1.6 Hz, 1H), 7.87–7.82 (m, 1H), 7.74 (d, J = 8.4 Hz, 1H), 7.68 (s, 1H), 7.64 (s, 1H), 7.52 (t, J = 8.0 Hz, 1H), 7.31 (dd, J = 8.0, 1.4 Hz, 1H), 7.18 (d, J = 1.6 Hz, 1H), 6.95 (dd, J = 7.8, 1.6 Hz, 1H), 4.57–4.32 (m, 2H), 3.59 (t, J = 5.7 Hz, 2H), 2.87 (s, 6H). LC/MS (ESI) m/z: [M + H]+ calcd for C24H25ClN8O2, 493.2; found, 493,5.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(1-(2-(diethylamino)ethyl)-1H-pyrazol-4-yl)benzamide (26h)
54% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, MeOD) δ 8.48 (s, 1H), 8.32 (t, J = 2.0 Hz, 1H), 8.28 (s, 1H), 8.14 (dd, J = 8.4, 1.6 Hz, 1H), 8.01–7.93 (m, 1H), 7.86 (dd, J = 5.4, 4.0 Hz, 1H), 7.78 (d, J = 14.8 Hz, 2H), 7.65 (t, J = 8.0 Hz, 1H), 7.43 (dd, J = 8.0, 1.4 Hz, 1H), 7.33–7.24 (m, 1H), 7.10–6.98 (m, 1H), 4.61 (t, J = 5.9 Hz, 2H), 3.72 (t, J = 6.0 Hz, 2H), 3.32–3.28 (m, 4H), 1.34 (t, J = 7.2 Hz, 6H). LC/MS (ESI) m/z: [M + H]+ calcd for C26H29ClN8O2, 521.2; found, 521.3.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(1-(2-(pyrrolidin-1-yl)ethyl)-1H-pyrazol-4-yl)benzamide (26i)
55% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.74 (s, 1H), 9.49 (s, 1H), 8.59 (s, 1H), 8.42 (s, 1H), 8.36 (s, 1H), 8.28 (s, 1H), 8.20 (dd, J = 8.4, 1.6 Hz, 1H), 8.03 (dd, J = 8.0, 1.4 Hz, 1H), 7.86 (d, J = 7.0 Hz, 2H), 7.74 (s, 1H), 7.65 (t, J = 7.9 Hz, 1H), 7.47 (dd, J = 8.0, 1.4 Hz, 1H), 7.36–7.27 (m, 1H), 7.09–7.00 (m, 1H), 4.52 (t, J = 6.0 Hz, 2H), 3.64 (s, 2H), 3.49 (s, 2H), 3.01 (s, 2H), 1.90–1.94 (m, 4H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H22ClN8O2, 519.2; found, 519.5.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(1-(piperidin-4-yl)-1H-pyrazol-4-yl)benzamide (26j)
62% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, MeOD) δ 8.50 (s, 1H), 8.33 (s, 1H), 8.25 (s, 1H), 8.15 (dd, J = 8.4, 1.6 Hz, 1H), 7.99 (d, J = 6.0 Hz, 1H), 7.87 (d, J = 8.4 Hz, 1H), 7.76 (s, 1H), 7.73 (s, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.46–7.39 (m, 1H), 7.30 (d, J = 6.9 Hz, 1H), 7.11–7.01 (m, 1H), 3.59 (d, J = 13.6 Hz, 2H), 3.51–3.46 (m, 1H), 3.29–3.19 (m, 2H), 2.40–2.27 (m, 4H). LC/MS (ESI) m/z: [M + H]+ calcd for C25H25ClN8O2, 505.2; found, 505.4.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(1-(pyrrolidin-3-yl)-1H-pyrazol-4-yl)benzamide (26k)
53% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 10.72 (s, 1H), 9.48 (s, 1H), 9.10 (s, 1H), 8.59 (d, J = 7.7 Hz, 1H), 8.42 (s, 1H), 8.36 (s, 1H), 8.28 (s, 1H), 8.20 (dd, J = 8.4, 1.6 Hz, 1H), 8.02 (dt, J = 11.4, 5.6 Hz, 1H), 7.86 (d, J = 5.2 Hz, 1H), 7.75 (s, 1H), 7.62 (dt, J = 19.8, 8.0 Hz, 1H), 7.47 (dt, J = 8.0, 2.0 Hz, 1H), 7.36–7.22 (m, 1H), 7.04 (ddd, J = 11.0, 4.6, 2.3 Hz, 1H), 5.33–5.12 (m, 1H), 3.77–3.27 (m, 5H), 2.42 (m, 1H), 2.26 (m, 1H). LC/MS (ESI) m/z: [M + H]+ calcd for C24H23ClN8O2, 496.2; found, 496.4.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(2-(diethylamino)ethyl)benzamide (26l)
73% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, CH3OD) δ 8.44 (s, 1H), 8.23 (d, J = 1.7 Hz, 1H), 8.12 (dd, J = 8.2, 1.4 Hz, 1H), 7.97–7.91 (m, 1H), 7.81–7.72 (m, 2H), 7.60 (t, J = 7.9 Hz, 1H), 7.41 (dd, J = 8.0, 1.4 Hz, 1H), 7.33–7.25 (m, 1H), 7.09–7.02 (m, 1H), 3.53 (t, J = 6.4 Hz, 2H), 3.27–3.20 (m, 4H), 2.05 (dt, J = 19.2, 5.6 Hz, 2H), 1.33 (t, J = 7.2 Hz, 6H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H27ClN6O2, 455.2; found, 455.4.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(3-(pyrrolidin-1-yl)propyl)benzamide (26m)
69% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, MeOD) δ 8.45 (s, 1H), 8.23 (t, J = 1.8 Hz, 1H), 8.13 (dd, J = 8.4, 1.5 Hz, 1H), 8.00–7.89 (m, 1H), 7.84–7.71 (m, 2H), 7.60 (t, J = 8.0 Hz, 1H), 7.43 (dd, J = 8.0, 1.4 Hz, 1H), 7.30 (ddd, J = 8.0, 7.6, 1.5 Hz, 1H), 7.12–6.92 (m, 1H), 3.78–3.62 (m, 2H), 3.55 (t, J = 6.5 Hz, 2H), 3.29 (d, J = 8.0 Hz, 2H), 3.11 (dd, J = 10.8, 8.0 Hz, 2H), 2.23–2.13 (m, 2H), 2.13–1.90 (m, 4H). LC–MS: 467 (M + H). LC/MS (ESI) m/z: [M + H]+ calcd for C24H27ClN6O2, 467.2; found, 467.5.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(piperidin-4-ylmethyl)benzamide (26n)
67% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 9.45 (s, 1H), 8.79 (t, J = 6.0 Hz, 1H), 8.56 (s, 1H), 8.40 (s, 1H), 8.24 (t, J = 1.7 Hz, 1H), 8.19 (dd, J = 8.3, 1.5 Hz, 1H), 7.98 (dd, J = 8.0, 1.2 Hz, 1H), 7.82 (s, 1H), 7.75 (d, J = 8.0 Hz, 1H), 7.58 (t, J = 8.0 Hz, 1H), 7.47 (dd, J = 8.0, 1.6 Hz, 1H), 7.34–7.27 (m, 1H), 7.07–7.00 (m, 1H), 3.43 (d, J = 11.3 Hz, 2H), 3.22 (t, J = 6.0 Hz, 2H), 2.90 (q, J = 10.4 Hz, 2H), 2.75 (s, 1H), 1.89 (d, J = 14.2 Hz, 2H), 1.80–1.84 (m, 1H), 1.38 (dd, J = 24.8, 10.8 Hz, 2H). LC/MS (ESI) m/z: [M + H]+ calcd for C23H25ClN6O2, 453.2; found, 453.9.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-((1-methylpiperidin-4-yl)methyl)benzamide (26o)
72% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 9.47 (s, 1H), 8.80 (t, J = 6.0 Hz, 1H), 8.56 (d, J = 6.4 Hz, 2H), 8.41 (s, 1H), 8.24 (d, J = 1.6 Hz, 2H), 8.19 (dd, J = 8.3, 1.5 Hz, 1H), 7.97 (dd, J = 8.0, 1.2 Hz, 1H), 7.81 (s, 1H), 7.75 (d, J = 8.0 Hz, 1H), 7.58 (t, J = 8.0 Hz, 1H), 7.46 (dd, J = 8.0, 1.6 Hz, 1H), 7.33–7.27 (m, 1H), 7.11–6.98 (m, 1H), 3.35 (s, 3H), 3.28 (d, J = 12.6 Hz, 2H), 3.23 (t, J = 6.1 Hz, 2H), 2.94–2.77 (m, 2H), 1.83 (d, J = 12.4 Hz, 2H), 1.35 (d, J = 12.4 Hz, 2H), 1.29–1.23 (m, 1H). LC/MS (ESI) m/z: [M + H]+ calcd for C24H27ClN6O2, 467.2; found, 467.6.
N-(Azetidin-3-yl)-3-(4-(3-(2-chlorophenyl)ureido)-1H-pyrazol-1-yl)benzamide (26p)
58% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 9.51 (s, 1H), 9.32 (d, J = 6.4 Hz, 1H), 8.56 (s, 1H), 8.44 (s, 1H), 8.28 (t, J = 2.0 Hz, 1H), 8.23–8.15 (m, 1H), 8.05–7.98 (m, 1H), 7.84 (s, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.66–7.59 (m, 1H), 7.47 (dd, J = 8.0, 1.4 Hz, 1H), 7.36–7.27 (m, 1H), 7.08–6.99 (m, 1H), 4.85 (m, 1H), 4.18 (m, 4H), 3.26 (m, 1H). LC/MS (ESI) m/z: [M + H]+ calcd for C20H19ClN6O2, 411.1; found, 411.4.
3-(4-(3-(2-Chlorophenyl)ureido)-1H-pyrazol-1-yl)-N-(pyrrolidin-3-yl)benzamide (26q)
65% yield in four steps (TFA salt, white solid); 1H NMR (400 MHz, DMSO) δ 9.45 (s, 1H), 8.80 (s, 1H), 8.56 (s, 1H), 8.41 (s, 1H), 8.25 (s, 1H), 8.19 (d, J = 8.4 Hz, 1H), 8.00 (d, J = 8.1 Hz, 1H), 7.84 (s, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.61 (t, J = 8.0 Hz, 1H), 7.52–7.40 (m, 1H), 7.30 (dd, J = 11.6, 4.4 Hz, 1H), 7.10–6.95 (m, 1H), 4.54 (d, J = 6.1 Hz, 1H), 3.47 (d, J = 6.3 Hz, 3H), 3.30 (s, 1H), 3.25–3.16 (m, 1H), 2.23 (dd, J = 13.2, 7.6 Hz, 1H), 2.12–2.00 (m, 1H). LC/MS (ESI) m/z: [M + H]+ calcd for C21H21ClN6O2, 425.1; found, 425.4.
JNK-1/2/3 and p38 Assays
Homogeneous time-resolved fluorescence assay–enzyme inhibition studies were performed in 384-well polystyrene homogeneous time-resolved fluorescence plates (Grainier) for 1 h at ambient temperature (∼22 °C) with 0.5 μM biotinylated FL-ATF-2, 1.25 μM ATP, 0.75 nM activated JNK3α1 or JNK2 or JNK1 (with a control in the absence of kinase for determining the basal signal) in 10 μL volumes containing the final concentrations of the following: 50 mM Hepes, pH 7.0, 2.5 mM MgCl2, 0.1 mg/mL bovine serum albumin, 1 mM dl-dithiothreitol, 0.01% Triton X-100 (all from Sigma-Aldrich), and 5% DMSO (with or without compound). A 10-point titration of all compounds was carried out in 3-fold dilutions from 10 μM to 0.5 nM. After 22 min, the kinase reaction was terminated by addition of 10 μL of quenching solution (1× Lance buffer, detection reagents, streptavidin-xlAPC (200 nM), and europium cryptate-labeled rabbit polyclonal anti-phospho-ATF-2 (1 nM) were from Cis-Bio). The homogeneous time-resolved fluorescence signal was detected using an EnVision plate reader 1 h after quenching. The data from four different experiments were averaged and presented as the mean ± SD. IC50 values were determined by fitting the data to the equation for a four-parameter logistic.3,4,37,51
Cell Culture
SHSY5Y cells (ATCC) were grown at 37 °C and 5% CO2 in DMEM/F:12 (Invitrogen) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin. To ensure that the cells were actively growing, only cells at ∼80% confluency and between passages five and 15 were used in the experiments. The cells were serum starved for 24 h in DMEM/F:12 medium containing 2% FBS before any treatment.
Mitochondrial Membrane Depolarization
Mitochondrial membrane depolarization was monitored by MitoTracker Orange CMTRos (Invitrogen) fluorescence. SHSY5Y cells (60 000 cells/well) were seeded in black walled, clear bottomed 96-well plates. Cells were incubated with compounds for 30 min before the addition of 35 μM 6-hydroxydopamine (6-OHDA) for 4 h. After the incubation, cells were stained with 500 nM MitoTracker Orange CMTRos for 30 min under growth conditions. Cells were washed twice in Hank’s buffer salt solution (HBSS), and placed in prewarmed HBSS for fluorescent recordings. Fluorescence was detected at 576 nm (exciting at 554 nm) on a SpectraMax e5 plate reader (Molecular Devices). Mitochondrial membrane depolarization was normalized to cell abundance by staining the cells with Hoechst 33342 (excitation, 350 nm; emission, 450 nm).
Cell Viability
Cell viability of SHSY5Y cells was monitored by MTT assay (Cayman Chemical). Cells (60 000 cells/well) were seeded in a 96-well plate (clear bottom) and treated as described in the text. At the culmination of each treatment the cells were treated with the MTT reagent. Absorbance was monitored in a SpectraMax e5 plate reader (Molecular Devices).
Measurement of Compound Cytotoxicity
Cytotoxicity of the compounds was monitored by MTT assay (Cayman Chemical). SHSY5Y cells (60 000 cells/well) were seeded in a 96-well plate (clear bottom) and incubated with different concentrations (0–30 μM) of compounds for 48 h. At the culmination of the treatment the cells were treated with the MTT reagent. Absorbance was monitored in a SpectraMax e5 plate reader (Molecular Devices).
In Cell Western Assay
SHSY5Y cells (60 000 cells/well) were plated in a clear-bottomed Packard View black 96-well plate in 100 μL of 10% FBS DMEM:F12 medium and were allowed to attach overnight. Next day, the cells were treated with the compounds for 1 h prior to induction of the JNK pathway activation. The cells were treated with 35 μM 6-OHDA for 4–5 h. Cells were then fixed in 4% paraformaldehyde in PBS for 20 min at room temperature with no shaking. They were then washed once with 0.1 M glycine to neutralize paraformaldehyde for 5 min. Cells were permeabilized with 0.2% Triton X-100 in PBS for 20 min at room temperature on orbital shaker after which they were washed once with PBS for 5 min. They were then incubated with Licor blocking buffer in PBS (1:1 dilution in PBS) for 1–1.5 h rocking at room temperature. Cells were incubated with primary antibody p-c-Jun S63 Ab (Cell Signaling no. 9261) 1:100 dilution in Licor blocking buffer overnight at 4 °C. Next day, they were washed twice with PBS–0.1% Tween 20 (PBST) washing solution for 5 min each at room temperature on the orbital shaker, followed by one wash with Licor blocking buffer containing 0.05% Tween-20 for 5 min on the shaker at room temperature. The cells were then incubated with secondary antibody goat anti-rabbit IR800 (1:500 dilution) for 1 h at room temperature in the dark (covered the plate with foil) in Licor blocking buffer-containing Tween-20. Following this, cells were washed twice with PBST for 5 min each at room temperature and then once with Licor blocking buffer-containing 0.05% Tween-20. The wells were then incubated with ToPro 3 stain (nucleic acid staining), diluted 1:4000 in Licor blocking buffer or Licor blocking buffer with 0.05% Tween-20 for 30 min at room temperature in the dark. Finally the plates were washed twice with PBS and analyzed using the Odyssey LICOR infrared scanner.3,4,20,36,37,51,52,56
Crystallization
Purification of JNK3 39-402 and its crystallization with AMPPNP was done following previously published procedure.36,37,57 Compound 26k was soaked into the crystal by adding 2 mM compound into the crystallization drop and incubating for 24 h. The crystal was then transferred to a mounting loop, and excess soaking solution was removed and flash frozen by plunging into liquid nitrogen.
Data Collection and Refinement
A diffraction data set was collected at LS-CAT beamline 21-ID-G (APS) using Marmosiac 300 CCD detector (Mar Research). The data set was processed with autoProc with XDS as the data reduction engine. The PDB code 1JNK was used as the molecular replacement model. Quick molecular replacement using Phaser in Phenix suite properly positoned JNK3, 26k was identified as positive densities in ATP binding pocket. Restraints and coordintates for the compound was generated using eLBOW in Phenix suite and incorporated into the JNK3 coordinate using the graphics program Coot. The model was then refined using autoBuster with TLS (translation, libration, and screw-motion), water update, and unknown ligand search options turned on. The model was manually inspected and adjusted after each refinement cycle using Coot. The refinement was completed after the free and crystallographic R-factors stabilized. Data processing and refinement statistics are given in Supporting Information. Structural analysis and figure preparations were done using PyMol. The coordinate and the structure factor are deposited to wwPDB with the PDB code 4WH2.
Acknowledgments
This work was supported by DOD Grant W81XWH-12-1-0431 and NIH Grant GM103825 (P.V.L.).
Glossary
Abbreviations Used
- JNK
c-jun NH2-terminal kinase
- MAPK
mitogen-activated protein kinase
- ATF2
activating transcription factor 2
- ATP
adenosine triphosphate
- 6-OHDA
6-hydroxydopamine
- MMP
mitochondrial membrane potential
- AD
Alzheimer’s disease
- PD
Parkinson’s disease
- ALS
amyotrophic lateral sclerosis
- AUC
pharmacokinetic area under curve
- Cl
pharmacokinetic clearance
- Cmax
pharmacokinetic maximum concentration
- F
percent oral bioavailability
- HLMS
human liver microsomal stability
- Vd
volume of distribution
- SAR
structure–activity relationship
- DMPK
drug metabolism and pharmacokinetics
Supporting Information Available
Details for the synthesis of 12, 16, 22, and 26; LC purity data; protocols for JNKs and p38 enzyme assays; protocols of DMPK studies. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Gupta S.; Barrett T.; Whitmarsh A. J.; Cavanagh J.; Sluss H. K.; Derijard B.; Davis R. J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. [PMC free article] [PubMed] [Google Scholar]
- Bogoyevitch M. A.; Ngoei K. R.; Zhao T. T.; Yeap Y. Y.; Ng D. C. c-Jun N-terminal kinase (JNK) signaling: recent advances and challenges. Biochim. Biophys. Acta 2010, 1804, 463–475. [DOI] [PubMed] [Google Scholar]
- Chambers J. W.; Howard S.; LoGrasso P. V. Blocking c-Jun N-terminal kinase (JNK) translocation to the mitochondria prevents 6-hydroxydopamine-induced toxicity in vitro and in vivo. J. Biol. Chem. 2013, 288, 1079–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers J. W.; Pachori A.; Howard S.; Iqbal S.; LoGrasso P. V. Inhibition of JNK mitochondrial localization and signaling is protective against ischemia/reperfusion injury in rats. J. Biol. Chem. 2013, 288, 4000–4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusgen W.; Boehm R.; Zhao Y.; Herdegen T.; Waetzig V. Specific activities of individual c-Jun N-terminal kinases in the brain. Neurosci. 2009, 161, 951–959. [DOI] [PubMed] [Google Scholar]
- LoGrasso P.; Kamenecka T. Inhibitors of c-jun-N-terminal kinase (JNK). Mini-Rev. Med. Chem. 2008, 8, 755–766. [DOI] [PubMed] [Google Scholar]
- Siddiqui M. A.; Reddy P. A. Small molecule JNK (c-Jun N-terminal kinase) inhibitors. J. Med. Chem. 2010, 53, 3005–3012. [DOI] [PubMed] [Google Scholar]
- Weston C. R.; Davis R. J. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 2007, 19, 142–149. [DOI] [PubMed] [Google Scholar]
- Mohit A. A.; Martin J. H.; Miller C. A. P49(3f12) kinase—a novel map kinase expressed in a subset of neurons in the human nervous-system. Neuron 1995, 14, 67–78. [DOI] [PubMed] [Google Scholar]
- Derijard B.; Hibi M.; Wu I. H.; Barrett T.; Su B.; Deng T.; Karin M.; Davis R. J. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 1994, 76, 1025–1037. [DOI] [PubMed] [Google Scholar]
- Hibi M.; Lin A.; Smeal T.; Minden A.; Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993, 7, 2135–2148. [DOI] [PubMed] [Google Scholar]
- Kyriakis J. M.; Banerjee P.; Nikolakaki E.; Dai T.; Rubie E. A.; Ahmad M. F.; Avruch J.; Woodgett J. R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 1994, 369, 156–160. [DOI] [PubMed] [Google Scholar]
- Chang L. F.; Jones Y.; Ellisman M. H.; Goldstein L. S. B.; Karin M. JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev. Cell 2003, 4, 521–533. [DOI] [PubMed] [Google Scholar]
- Kim E. K.; Choi E. J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta, Mol. Basis Dis, 2010, 1802, 396–405. [DOI] [PubMed] [Google Scholar]
- Salh B. c-Jun N-terminal kinases as potential therapeutic targets. Expert Opin. Ther. Targets 2007, 11, 1339–1353. [DOI] [PubMed] [Google Scholar]
- Yang D. D.; Kuan C. Y.; Whitmarsh A. J.; Rincon M.; Zheng T. S.; Davis R. J.; Rakic P.; Flavell R. A. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 1997, 389, 865–870. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Shi X. Q.; Zhang Q. J.; Hampong M.; Paddon H.; Wahyuningsih D.; Pelech S. Nocodazole-induced p53-dependent c-Jun N-terminal kinase activation reduces apoptosis in human colon carcinoma HCT116 cells. J. Biol. Chem. 2002, 277, 43648–43658. [DOI] [PubMed] [Google Scholar]
- Yoon S. O.; Park D. J.; Ryu J. C.; Ozer H. G.; Tep C.; Shin Y. J.; Lim T. H.; Pastorino L.; Kunwar A. J.; Walton J. C.; Nagahara A. H.; Lu K. P.; Nelson R. J.; Tuszynski M. H.; Huang K. JNK3 perpetuates metabolic stress induced by Abeta peptides. Neuron 2012, 75, 824–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsello T.; Forloni G. JNK signalling: a possible target to prevent neurodegeneration. Cur. Pharm. Des. 2007, 13, 1875–1886. [DOI] [PubMed] [Google Scholar]
- Chambers J. W.; Pachori A.; Howard S.; Ganno M.; Hansen D. Jr.; Kamenecka T.; Song X.; Duckett D.; Chen W.; Ling Y. Y.; Cherry L.; Cameron M. D.; Lin L.; Ruiz C. H.; Lograsso P. Small molecule c-jun-N-terminal kinase (JNK) inhibitors protect dopaminergic neurons in a model of Parkinson’s disease. ACS Chem. Neurosci. 2011, 2, 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker C. E.; Khan S.; Cameron M. D.; Robertson H. A.; Robertson G. S.; LoGrasso P. JNK inhibition protects dopamine neurons and provides behavioral improvement in a rat 6-hydroxydopamine model of Parkinson’s disease. ACS Chem. Neurosci. 2011, 2, 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan C. Y.; Burke R. E. Targeting the JNK signaling pathway for stroke and Parkinson’s diseases therapy. Curr. Drug Targets: CNS Neurol. Disord. 2005, 4, 63–67. [DOI] [PubMed] [Google Scholar]
- Hunot S.; Vila M.; Teismann P.; Davis R. J.; Hirsch E. C.; Przedborski S.; Rakic P.; Flavell R. A. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 665–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G. Y.; Zhang Q. G. Agents targeting c-Jun N-terminal kinase pathway as potential neuroprotectants. Expert Opin. Invest. Drugs 2005, 14, 1373–1383. [DOI] [PubMed] [Google Scholar]
- Bennett B. L.; Sasaki D. T.; Murray B. W.; O’Leary E. C.; Sakata S. T.; Xu W. M.; Leisten J. C.; Motiwala A.; Pierce S.; Satoh Y.; Bhagwat S. S.; Manning A. M.; Anderson D. W. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 13681–13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez C.; Powers D. A.; Tamayo N. p38 MAP kinase inhibitors: many are made, but few are chosen. Curr. Opin. Drug Discovery Dev. 2005, 8, 421–430. [PubMed] [Google Scholar]
- Alam M.; Beevers R. E.; Ceska T.; Davenport R. J.; Dickson K. M.; Fortunato M.; Gowers L.; Haughan A. F.; James L. A.; Jones M. W.; Kinsella N.; Lowe C.; Meissner J. W.; Nicolas A. L.; Perry B. G.; Phillips D. J.; Pitt W. R.; Platt A.; Ratcliffe A. J.; Sharpe A.; Tait L. J. Synthesis and SAR of aminopyrimidines as novel c-Jun N-terminal kinase (JNK) inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 3463–3467. [DOI] [PubMed] [Google Scholar]
- Angell R. M.; Atkinson F. L.; Brown M. J.; Chuang T. T.; Christopher J. A.; Cichy-Knight M.; Dunn A. K.; Hightower K. E.; Malkakorpi S.; Musgrave J. R.; Neu M.; Rowland P.; Shea R. L.; Smith J. L.; Somers D. O.; Thomas S. A.; Thompson G.; Wang R. N-(3-Cyano-4,5,6,7-tetrahydro-1-benzothien-2-yl)amides as potent, selective, inhibitors of JNK2 and JNK3. Bioorg. Med. Chem. Lett. 2007, 17, 1296–1301. [DOI] [PubMed] [Google Scholar]
- Bowers S.; Truong A. P.; Neitz R. J.; Neitzel M.; Probst G. D.; Hom R. K.; Peterson B.; Galemmo R. A.; Konradi A. W.; Sham H. L.; Toth G.; Pan H.; Yao N. H.; Artis D. R.; Brigham E. F.; Quinn K. P.; Sauer J. M.; Powell K.; Ruslim L.; Ren Z.; Bard F.; Yednock T. A.; Griswold-Prenner I. Design and synthesis of a novel, orally active, brain penetrant, tri-substituted thiophene based JNK inhibitor. Bioorg. Med. Chem. Lett. 2011, 21, 1838–1843. [DOI] [PubMed] [Google Scholar]
- Cao J. R.; Gao H.; Bemis G.; Salituro F.; Ledeboer M.; Harrington E.; Wilke S.; Taslimi P.; Pazhanisamy S.; Xie X. L.; Jacobs M.; Green J. Structure-based design and parallel synthesis of N-benzyl isatin oximes as JNK3 MAP kinase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 2891–2895. [DOI] [PubMed] [Google Scholar]
- Christopher J. A.; Atkinson F. L.; Bax B. D.; Brown M. J. B.; Champigny A. C.; Chuang T. T.; Jones E. J.; Mosley J. E.; Musgrave J. R. 1-Aryl-3,4-dihydroisoquinoline inhibitors of JNK3. Bioorg. Med. Chem. Lett. 2009, 19, 2230–2234. [DOI] [PubMed] [Google Scholar]
- Gaillard P.; Jeanclaude-Etter I.; Ardissone V.; Arkinstall S.; Cambet Y.; Camps M.; Chabert C.; Church D.; Cirillo R.; Gretener D.; Halazy S.; Nichols A.; Szyndralewiez C.; Vitte P. A.; Gotteland J. P. Design and synthesis of the first generation of novel potent, selective, and in vivo active (benzothiazol-2-yl)acetonitrile inhibitors of the c-Jun N-terminal kinase. J. Med. Chem. 2005, 48, 4596–4607. [DOI] [PubMed] [Google Scholar]
- Graczyk P. P.; Khan A.; Bhatia G. S.; Palmer V.; Medland D.; Numata H.; Oinuma H.; Catchick J.; Dunne A.; Ellis M.; Smales C.; Whitfield J.; Neame S. J.; Shah B.; Wilton D.; Morgan L.; Patel T.; Chung R.; Desmond H.; Staddon J. M.; Sato N.; Inoue A. The neuroprotective action of JNK3 inhibitors based on the 6,7-dihydro-5H-pyrrolo[1,2-a]imidazole scaffold. Bioorg. Med. Chem. Lett. 2005, 15, 4666–4670. [DOI] [PubMed] [Google Scholar]
- He Y. J.; Kamenecka T. M.; Shin Y. S.; Song X. Y.; Jiang R.; Noel R.; Duckett D.; Chen W. M.; Ling Y. Y.; Cameron M. D.; Lin L.; Khan S.; Koenig M.; LoGrasso P. V. Synthesis and SAR of novel quinazolines as potent and brain-penetrant c-jun N-terminal kinase (JNK) Inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 1719–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R.; Duckett D.; Chen W.; Habel J.; Ling Y. Y.; LoGrasso P.; Kamenecka T. M. 3,5-Disubstituted quinolines as novel c-Jun N-terminal kinase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 6378–6382. [DOI] [PubMed] [Google Scholar]
- Kamenecka T.; Habel J.; Duckett D.; Chen W. M.; Ling Y. Y.; Frackowiak B.; Jiang R.; Shin Y. S.; Song X. Y.; LoGrasso P. Structure-activity relationships and X-ray structures describing the selectivity of aminopyrazole inhibitors for c-Jun N-terminal kinase 3 (JNK3) over p38. J. Biol. Chem. 2009, 284, 12853–12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenecka T.; Jiang R.; Song X. Y.; Duckett D.; Chen W. M.; Ling Y. Y.; Habel J.; Laughlin J. D.; Chambers J.; Figuera-Losada M.; Cameron M. D.; Lin L.; Ruiz C. H.; LoGrasso P. V. Synthesis, biological evaluation, X-ray structure, and pharmacokinetics of aminopyrimidine c-jun-N-terminal kinase (JNK) inhibitors. J. Med. Chem. 2010, 53, 419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenitsky V. P.; Delgado M.; Nadolny L.; Sahasrabudhe K.; Ayala L.; Clareen S. S.; Hilgraf R.; Albers R.; Kois A.; Hughes K.; Wright J.; Nowakowski J.; Sudbeck E.; Ghosh S.; Bahmanyar S.; Chamberlain P.; Muir J.; Cathers B. E.; Giegel D.; Xu L.; Celeridad M.; Moghaddam M.; Khatsenko O.; Omholt P.; Katz J.; Pai S.; Fan R.; Tang Y.; Shirley M. A.; Benish B.; Blease K.; Raymon H.; Bhagwat S.; Henderson I.; Cole A. G.; Bennett B.; Satoh Y. Aminopurine based JNK inhibitors for the prevention of ischemia reperfusion injury. Bioorg. Med. Chem. Lett. 2012, 22, 1427–1432. [DOI] [PubMed] [Google Scholar]
- Noel R.; Shin Y.; Song X. Y.; He Y. J.; Koenig M.; Chen W. M.; Ling Y. Y.; Lin L.; Ruiz C. H.; LoGrasso P.; Cameron M. D.; Duckett D. R.; Kamenecka T. M. Synthesis and SAR of 4-(pyrazol-3-yl)-pyridines as novel c-jun N-terminal kinase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2732–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckle T.; Biamonte M.; Grippi-Vallotton T.; Arkinstall S.; Cambet Y.; Camps M.; Chabert C.; Church D. J.; Halazy S.; Jiang X.; Martinou I.; Nichols A.; Sauer W.; Gotteland J. P. Design, synthesis, and biological activity of novel, potent, and selective (benzoylaminomethyl)thiophene sulfonamide inhibitors of c-Jun-N-terminal kinase. J. Med. Chem. 2004, 47, 6921–6934. [DOI] [PubMed] [Google Scholar]
- Shin Y. S.; Chen W. M.; Habel J.; Duckett D.; Ling Y. Y.; Koenig M.; He Y. J.; Vojkovsky T.; LoGrasso P.; Kamenecka T. M. Synthesis and SAR of piperazine amides as novel c-jun N-terminal kinase (JNK) inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 3344–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocks M. J.; Barber S.; Ford R.; Leroux F.; St-Gallay S.; Teague S.; Xue Y. Structure-driven HtL: design and synthesis of novel aminoindazole inhibitors of c-Jun N-terminal kinase activity. Bioorg. Med. Chem. Lett. 2005, 15, 3459–3462. [DOI] [PubMed] [Google Scholar]
- Swahn B. M.; Huerta F.; Kallin E.; Malmstrom J.; Weigelt T.; Viklund J.; Womack P.; Xue Y.; Ohberg L. Design and synthesis of 6-anilinoindazoles as selective inhibitors of c-Jun N-terminal kinase-3. Bioorg. Med. Chem. Lett. 2005, 15, 5095–5099. [DOI] [PubMed] [Google Scholar]
- Swahn B. M.; Xue Y.; Arzel E.; Kallin E.; Magnus A.; Plobeck N.; Viklund J. Design and synthesis of 2′-anilino-4,4′-bipyridines as selective inhibitors of c-Jun N-terminal kinase-3. Bioorg. Med. Chem. Lett. 2006, 16, 1397–1401. [DOI] [PubMed] [Google Scholar]
- Neitz R. J.; Konradi A. W.; Sham H. L.; Zmolek W.; Wong K.; Qin A.; Lorentzen C.; Nakamura D.; Quinn K. P.; Sauer J. M.; Powell K.; Ruslim L.; Chereau D.; Ren Z.; Anderson J.; Bard F.; Yednock T. A.; Griswold-Prenner I. Highly selective c-Jun N-terminal kinase (JNK) 3 inhibitors with in vitro CNS-like pharmacokinetic properties II. Central core replacement. Bioorg. Med. Chem. Lett. 2011, 21, 3726–3729. [DOI] [PubMed] [Google Scholar]
- Fang X.; Yin Y.; Wang B.; Yao L.; Chen Y. T.; Schröter T.; Weiser A.; Pocas J.; Wayne G.; Cameron M. D.; Lin L.; Ruiz C.; Khan S.; Schürer S. C.; Pachori A.; LoGrasso P.; Feng Y. Tetrahydroisoquinoline derivatives as potent and selective Rho kinase inhibitors. J. Med. Chem. 2010, 53, 5727–5737. [DOI] [PubMed] [Google Scholar]
- Zhou P.; Zou J. W.; Tian F. F.; Shang Z. C. Fluorine bonding; How does it work in protein–ligand interactions?. J. Chem. Inf. Model. 2009, 49, 2344–2355. [DOI] [PubMed] [Google Scholar]
- Fabian M. A.; Biggs W. H. 3rd; Treiber D. K.; Atteridge C. E.; Azimioara M. D.; Benedetti M. G.; Carter T. A.; Ciceri P.; Edeen P. T.; Floyd M.; Ford J. M.; Galvin M.; Gerlach J. L.; Grotzfeld R. M.; Herrgard S.; Insko D. E.; Insko M. A.; Lai A. G.; Lelias J. M.; Mehta S. A.; Milanov Z. V.; Velasco A. M.; Wodicka L. M.; Patel H. K.; Zarrinkar P. P.; Lockhart D. J. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [DOI] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [DOI] [PubMed] [Google Scholar]
- Chambers J. W.; Cherry L.; Laughlin J. D.; Figuera-Losada M.; Lograsso P. V. Selective inhibition of mitochondrial JNK signaling achieved using peptide mimicry of the Sab kinase interacting motif-1 (KIM1). ACS Chem. Biol. 2011, 6, 808–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers J. W.; LoGrasso P. V. Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J. Biol. Chem. 2011, 286, 16052–16062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.; Wang S. Y.; Clampit J. E.; Gum R. J.; Haasch D. L.; Rondinone C. M.; Trevillyan J. M.; Abad-Zapatero C.; Fry E. H.; Sham H. L.; Liu G. Discovery of a new class of 4-anilinopyrimidines as potent c-Jun N-terminal kinase inhibitors: synthesis and SAR studies. Bioorg. Med. Chem. Lett. 2007, 17, 668–672. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Herdegen T. Cerebral ischemia provokes a profound exchange of activated JNK isoforms in brain mitochondria. Mol. Cell. Neurosci. 2009, 41, 186–195. [DOI] [PubMed] [Google Scholar]
- Fernandes K. A.; Harder J. M.; Fornarola L. B.; Freeman R. S.; Clark A. F.; Pang I. H.; John S. W.; Libby R. T. JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol. Dis. 2012, 46, 393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y.; Chambers J. W.; Iqbal S.; Koenig M.; Park H.; Cherry L.; Hernandez P.; Figuera-Losada M.; Lograsso P. V. A small molecule bidentate-binding dual inhibitor probe of the LRRK2 and JNK kinases. ACS Chem. Biol. 2013, 8, 1747–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X.; Gu Y.; Fox T.; Coll J. T.; Fleming M. A.; Markland W.; Caron P. R.; Wilson K. P.; Su M. S. Crystal structure of JNK3: a kinase implicated in neuronal apoptosis. Structure 1998, 6, 983–991. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.