

Abstract
A duplexed, functional multiaddition high throughput screen and subsequent iterative parallel synthesis effort identified the first highly selective and CNS penetrant glucagon-like peptide-1R (GLP-1R) positive allosteric modulator (PAM). PAM (S)-9b potentiated low-dose exenatide to augment insulin secretion in primary mouse pancreatic islets, and (S)-9b alone was effective in potentiating endogenous GLP-1R to reverse haloperidol-induced catalepsy.
Introduction
Glucagon-like peptide-1 (GLP-1) is a key incretin hormone with diverse physiological functions in both the periphery and central nervous system (CNS) that are mediated by the GLP-1 receptor (GLP-1R), a family B G protein-coupled receptor (GPCR).1−3 GLP-1R is activated by four endogenous GLP-1 peptides (GLP-1 (1–37), GLP-1 (7–37), GLP(1–36)NH2, and GLP-1 (7–36)NH2) and a fifth structurally analogous peptide oxyntomodulin.4,5 Due to its key role in the potentiation of insulin secretion and suppression of glucagon scretion, GLP-1R is a major focus of therapeutic discovery for type II diabetes, and several peptide-based drugs have been developed, including exenatide, 1, and liragulitide, 2, administered by subcutaneous injection.6−10 In the CNS, GLP-1R is important for neuroprotection, learning, and memory as well as neurogenesis.11,12 While these synthetic peptides overcome the short half-life of endogenous GLP-1, adverse events (e.g., nausea and GI distress) contribute to relatively poor long-term adherence.4−10 Recently, efforts have been directed toward the development of orally bioavailable, nonpeptide approaches, particularly small-molecule positive allosteric modulators (PAMs), for which several chemotypes 3–7 have been reported (Figure 1).13−18 While allosteric modulation19,20 is an attractive approach for GLP-1R modulation, these early PAM tool compounds suffer from weak potency and/or efficacy, ligand and/or stimulus bias13−20 as well as innate electrophilicity (2 covalently modifies GLP-1R).21,22 Thus, we decided to pursue the discovery of novel GLP-1R PAMs to enable the in vitro and in vivo assessment of the therapeutic potential of GLP-1 potentiation in the periphery, and importantly, the CNS, for which no CNS penetrant GLP-1R PAMs have been reported.
Figure 1.

Structures 3–7 of reported GLP-1 PAMs and ago-PAMs. All known GLP-1 PAMs have noatble limitations in assessing the GLP-1 potentiation mechanism in the periphery and CNS.
Results and Discussion
High-Throughput Screen
We performed a duplexed (GLP-1R and glucagon receptor (GR)) triple-add, functional high-throughput screen to identify GLP-1R PAM and glucagon PAM leads.18 For this effort, we screened our internal collection (175478 compounds) in human GLP-1R 9-3-H cells measuring intracellular calcium mobilization as well as a secondary GloSensor cAMP assay because both read-outs are GLP-1R primary coupling pathways which identified 98 primary hits.18 PAMs and ago-PAMs active in both calcium and cAMP assays, that were devoid of activity at the GR, as well as in a counter-screen against the melanocortin 4 receptor (MC4R), were further profiled as putative GLP-1R PAM leads.18
Chemistry
Of the confirmed hits (Figure 2), our attention focused on 8, an intriguing racemic 1-oxo-2,9-dihydro-1H-pyrido[3,4-b]indole-4-amide scaffold, which was active in both primary assays (GLP-1R EC50 = 4.1 μM, pEC50 = 5.38 ± 0.13, GLP-1R% max = 58.9 ± 2.3) and inactive at GR and MC4R with good physiochemical properties (MW = 434, clogP = 3.7). Following the route shown in Scheme 1, we synthesized the single enantiomers (S)-8 (GLP-1R EC50 = 2.4 μM, pEC50 = 5.69 ± 0.12, GLP-1R% max = 53.4 ± 2.03) and (R)-8 (GLP-1R EC50 >10 μM) and found that all the activity resided in the (S)-8 enantiomer.23 Commercial acid 9 was coupled to either (S)- or (R)-tert-butyl-2-(aminomethyl)pyrrolidine-1 carboxylate to provide 10. Deprotection of the Boc moiety under acidic conditions delivered 11, which underwent a subsequent reductive amination reaction with acetaldehyde to give (S)- and (R)-8 in 62–68% overall yield for the three step sequence. (S)-8 afforded no calcium response in the absence of GLP-1 but potentiated an EC20 concentration of GLP-1 and had no effect on GR (GLP-1R EC50 = 2.4 μM, pEC50 = 5.69 ± 0.12, GLP-1R% max = 53.4 ± 2.0) and (R)-8 (GLP-1R EC50 >10 μM) (Figure 2B). In a standard fold-shift experiment, (S)-8 induced a modest ∼2-fold shift of the GLP-1 concentration–response curve (CRC), and increased the GLP-1%max (from 100.3 to 129.6).19,20,23
Figure 2.

Structures and pharmacology of GLP-1R PAMs. (A) Structures of 8 and (S)-8. (B) PAM CRCs for (S)-8 in the presence of an EC20 concentration of either GLP-1 or glucagon.
Scheme 1. Synthesis of (S)- and (R)-8 and Route for Analogue Synthesis.

Reagents and conditions: (a) HATU, DIEA, DMF, (S)- or (R)-tert-butyl-2-(aminomethyl)pyrrolidine-1 carboxylate, 82–85%; (b) 4 M HCl in dioxane, DCM, 99%; (c) acetaldehyde, NaBH(OAc)3, DCM, 81–83%.
Preliminary DMPK evaluation of (S)-8 demonstrated a clean P450 inhibition profile (1A2, 2C9, 2D6 IC50s >20 μM; 3A4 IC50 = 3.2 μM), good plasma free fraction (fu >10% in rat and human), but high liver microsome-predicted clearance (rat CLhep = 69 mL/min/kg) and in vivo clearance (92 mL/min/kg) due to N-dealkylation of the N-ethyl pyrrolidine and oxidation of the N-cylopentyl moiety.23 Nevertheless, (S)-8 afforded an ∼1.5-fold potentiation of glucose-stimulated insulin secretion in primary mouse islets in the presence of 2.23,24 Encouraged by these early data, we initiated an analogue library effort to evaluate SAR following the route described in Scheme 1. SAR proved to be steep. Replacement of the N-cylcopentyl moiety of (S)-8 with N-Me, N-isobutyl led to inactive compounds (GLP-1 EC50s >10 μM), as did deletion of the indole N-Me. Due to the rapid N-dealkylation of the N-ethyl pyrrolidine, we surveyed a broader diversity of amide congeners, but all proved to be devoid of GLP-1R PAM activity. Finally, we elected to survey alternative N-alkyl pyrrolidines 9 (Table 1) following the route depicted in Scheme 1. This effort identified pure PAMs ((S)-9c-e) as well as ago-PAMs ((S)-9b), but once again, little structural diversity was tolerated. In general, these were low efficacy PAMs (48–68% GLP-1 max), but (S)-9b, (S)-2-cyclopentyl-N-((1-isopropylpyrrolidin2-yl)-9-methyl-1-oxo-2,9-dihydro-1H-pyrrido[3,4-b]indole-4-carboxamide, appeared worthy of further inspection (GLP-1 EC50 = 1.3 μM, 59.2% GLP-1 max).
Table 1. Structure and Activities of Analogues 9.
| entry | R | GLP-1R EC50 (μM) | GLP-1R pEC50a | GLP-1R maxa (%) |
|---|---|---|---|---|
| (S)-8 | Et | 2.4 | 5.69 ± 0.12 | 53.4 ± 2.03 |
| (S)-9a | n-Pr | 6.5 | 5.18 ± 0.13 | 48.4 ± 2.77 |
| (S)-9b | i-Pr | 1.3 | 5.88 ± 0.15 | 59.2 ± 2.53 |
| (S)-9c | 2-OMeBn | 12.1 | 4.92 ± 0.09 | 68.3 ± 3.31 |
| (S)-9d | n-hexyl | 10.3 | 4.99 ± 0.09 | 55.7 ± 2.63 |
| (S)-9e | CH2CH2Ph | 14.1 | 4.74 ± 0.19 | 68.5 ± 7.96 |
GLP-1 pEC50 and GLP-1 max data reported as averages + SEM from our calcium mobilization assay; n = 3.
Molecular Pharmacology
Again, SAR was driven on human GLP-1R 9-3-H cells measuring intracellular calcium mobilization in response to an EC20 concentration of GLP-1.18,23 This proved challenging due to the relative instability and short half-life of endogenous GLP-1 in cells and in vivo. Still, it was important to identify PAMs that could potentiate the endogenous peptide, as other reported PAMs displayed ligand bias and diminished activity at native GLP-1. To expand the utility of (S)-9b, we evaluated its propensity for ligand bias by assessing its ability to potentiate subthreshold concentrations (∼EC20s) of native GLP-1 and the synthetic peptide agonists exenatide, 1, and liragulitide, 2 (Figure 3). Here, (S)-9b, an ago-PAM (direct activation of GLP-1 at higher concentrations, e.g., ∼20% max efficacy at 30 μM) robustly potentiates both GLP-1 and synthetic peptide 1. In fact, (S)-9b was more efficacious with 1 than GLP-1, while liragulitide was less efficacious and the CRC did not plateau at 30 μM. Overall, these findings reveal potentiation across three ligands, suggesting a lack of substantial ligand bias. As with (S)-8, the maximum fold-shift of the GLP-1 CRC was 1.6-fold at 30 μM, but the efficacy increased from 100% to 140%.
Figure 3.

Molecular pharmacology of (S)-9b. (A) CRC of (S)-9b in the presence and absence of an EC20 of GLP-1 (EC50 = 1.8 μM, pEC50 = 5.74 ± 0.2, 59.2 ± 2% GLP-1 max). (B) CRC of (S)-9b in the presence and absence of an EC20 of 1 (EC50 = 8.4 μM, pEC50 = 5.07 ± 0.3, 71.5 ± 5% 1 max). (C) CRC of (S)-9b in the presence and absence of an EC20 of 2 (EC50 = 30 μM, pEC50 = 4.5 ± 0.2, 59.2 ± 1% 1 max). (D) Fold-shift experiment of GLP-1 in the presence of increasing concentrations of (S)-9b, with a maximum fold-shift of 1.6-fold at 20 μM.
On the basis of observations with previously reported GLP-1R PAMs 3–5,14,15 we also evaluated the pharmacological response of (S)-9b on β-arrestin recruitment and receptor internalization (Figure 4) by potentiation of liragulitide, 2. Very weak impact on β-arrestin recruitment was noted, along with a more significant effect on GLP-1 receptor internalization. These findings were in agreement with those reported with 3–6.14,15
Figure 4.
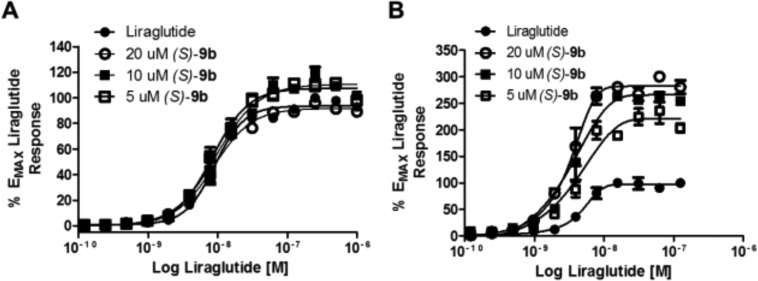
β-Arrestin recruitment and receptor internalization molecular pharmacology of (S)-9b. Fold shift experiment of liraglutide in the presence of 5, 10, and 20 μM (S)-9b on the recruitment of β-arrestin (A) and GLP-1 receptor internalization (B) using the PathHunter Express DiscoveRx assay platform. Efficacy of receptor internalization was increased by 2.26-fold at 20 μM, while there was little to no effect on β-arrestin recruitment. Data are normalized to the maximal response of liraglutide alone and fit to a four-parameter logistic equation with variable slope. Values are expressed as mean ± SEM, n = 3.23
Peptide agonist 1 has been shown to potentiate glucose-induced insulin secretion in primary mouse pancreatic islets, and, based on the ability of (S)-9b to potentiate 1, we wanted to determine if (S)-9b could potentiate the effects of low dose 1 under low and high glucose conditions. As shown in Figure 5, in the presence of 1, (S)-9b significantly increased insulin secretion beyond that of either glucose alone or glucose and 1.23,24
Figure 5.
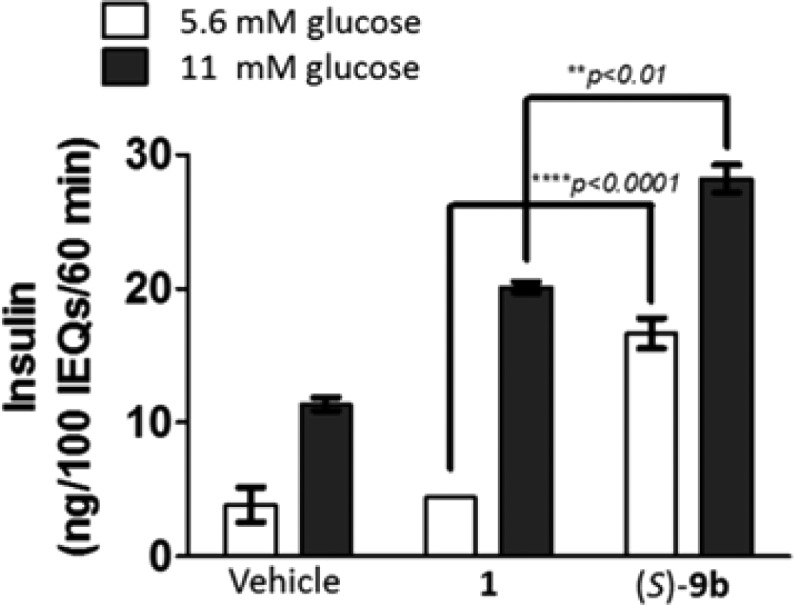
Measurement of the effect of 30 μM (S)-9b on potentiation of glucose-stimulated insulin secretion in primary mouse islets in the presence of 10 nM 1.23,24 Islets were isolated from C57BL/6 male mice, size-matched into wells of a 12-well plate, and treated with vehicle, exendin-4, or exendin-4 + (S)-9b at both low (5.6 mM) and high (11 mM) glucose for 60 min. n = 3. Data are represented as mean insulin response ± SEM per 100 islet equivalents and analyzed using 2-way ANOVA followed by Sidak’s multiple comparisons test. **p < 0.01, ****p < 0.0001.
Drug Metabolism and Disposition
Having achieved efficacy in primary tissue, we next evaluated the DMPK profile of (S)-9b to assess its potential as an in vivo tool (Table 2).23 (S)-9b, like (S)-8, displays an elevated plasma clearance (CLp, 72 mL/min/kg) that approaches hepatic blood flow in male Sprague–Dawley rats. Coupled with a high volume of distribution predicted at steady-state (Vss, 4.0 L/kg), (S)-9b produced a half-life (t1/2) of approximately 1 h in vivo (Table 2). Importantly, (S)-9b was found to possess CNS permeability (Kp, 2.7, Kp,uu, 0.25), a first among reported GLP-1 PAMs, although a Kp,uu < 1 did indicate a lack of true equilibrium at the Tmax of 0.5 h. The elevated clearance and poor associated oral bioavailability (F, <1%, observed for (S)-9b in rat was consistent with the predicted hepatic clearance (Table 2). Rat and human liver microsome incubations revealed the principal routes of biotransformation for (S)-9b to be cyclopentyl oxidation and oxidative N-dealkylation of the pyrrolidine moiety, respectively. The propensity of (S)-9b to mediate a P450 drug–drug interaction (DDI) was assessed in an in vitro cassette microsome inhibition assay of 1A2, 2C9, 2D6, and 3A4. The results of the inhibition screen indicated (S)-9b to possess low risk of mediating a DDI, displaying micromolar IC50 values against these P450 enzymes (Table 2). While oral delivery of this agent is limiting, intraperitoneal dosing readily enables in vivo work to be performed. To assess ancillary pharmacology, and to ensure that in vivo activity was due to potentiation of GLP-1, (S)-9b was profiled in a Eurofin radioligand binding panel of 68 GPCRs, ion channels, and transporters at a concentration of 10 μM, which revealed no significant off-target activity (no inhibition >50%@10 μM, i.e, Kis >10 μM).23
Table 2. DMPK Profile of (S)-9b.
| parameter | (S)-9b |
|---|---|
| MW | 434.28 |
| TPSA | 59.7 |
| cLogP | 3.51 |
| P450 inhibition | IC50 (μM) |
|---|---|
| P450 (1A2, 2C9, 3A4, 2D6) | >30, >30, 3.89, 10.1 |
| in vitro PKa | |
|---|---|
| rat CLHEP (mL/min/kg) | 69.9 |
| human CLHEP (mL/min/kg) | 19.9 |
| rat plasma fu | 0.158 |
| human plasma fu | 0.146 |
| rat brain homogenate fu | 0.015 |
| in vivo rat distribution (IP, 10 mg/kg, 0.5 h) | |
|---|---|
| plasma (nM) | 181 |
| brain (nM) | 481 |
| brain:plasma (Kp, Kp,uu) | 2.7, 0.27 |
| in vivo rat PK (IV, 1 mg/kg; PO, 1 mg/mL) | |
|---|---|
| CLobs (mL/min/kg) | 72 |
| t1/2 (min) | 58 |
| Vss (L/kg) | 4.0 |
| %F | <1 |
Liver microsome clearance was predicted using the well-stirred model with 20 and 45 g liver per kg body weight and 21 and 70 mL/kg hepatic blood flow for human and rat, respectively.
Behavioral Pharmacology
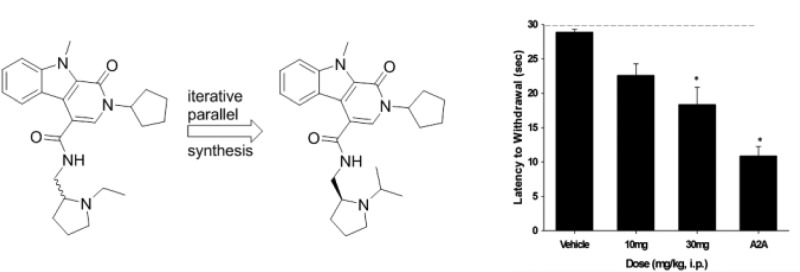
In numerous preclinical studies models of Parkinson’s disease, 1 reverses key deficits and arrests progression;9,11,25,26 additionally, the peptide agonist is neuroprotective and neurorestorative in 6-OHDA rats.25 In a recent clinical trial with 1 (despite very low and variable CNS exposure), 1 improved both cognition and motor symptoms.26 We and others routinely employ haloperidol-induced catalepsy (HIC) as a first tier pharmacodynamic assay to assess potential symptomatic benefit for novel anti-Parkinsonian mechanisms.27 Interestingly, neither GLP-1 agonists, such as 1 or 2, or PAMs have been evaluated in this preclinical model of the motor symptoms of PD. We determined the ability of (S)-9b to reverse the cataleptic state induced by haloperidol; these studies were performed in comparison to a positive control, the A2A antagonist preladenant.27 We tested both 10 and 30 mg/kg doses, administered ip, for their ability to reverse the catalepsy induced by two doses of haloperidol, a screening dose of 0.75 mg/kg, and a more robust challenge at 1.5 mg/kg (Figure 6). Excitingly, at the lower challenge of 0.75 mg/kg haloperidol, statistically significant reversal in catalepsy was noted at both the 10 mg/kg (36.3% reversal) and 30 mg/kg (50.4% reversal) doses. For the 1.5 mg/kg dose of haloperidol, the same trend is noted, but significance is only achieved at the 30 mg/kg dose (36.6% reversal); data for the A2A antagonist preladenant is shown for comparison (62% reversal). In satellite animals, the 10 mg/kg dose afforded brain levels of 481 nM, below the GLP-1 PAM EC50 value of (S)-9b; however, the EC50 is based on an EC20 concentration of GLP-1. We do not known the GLP-1 tone in the CNS or the central concentration of the other three peptidic forms of GLP-1 or oxyntomodulin; thus, the potency and efficacy of GLP-1 potentiation by (S)-9b could be significantly higher in vivo. Moreover, the absolute CNS concentration 1 is low in both preclinical species as well as humans, despite displaying robust efficacy, perhaps speaking to high receptor reserve of GLP-1 in the CNS. To further eliminate nonmechanism based efficacy in this model, we performed a spontaneous locomotor activity assay and noted no effect on locomotion or sedative effects; coupled with the clean ancillary pharmacology in the Eurofins panel, it is reasonable to assume the efficacy is due to potentiation of GLP-1.23 Thus, the GLP-1R ago-PAM (S)-9b is the first example of GLP-1R activation displaying efficacy in a haloperidol-induced catalepsy model, and importantly, (S)-9b is efficacious by potentiation of endogenous GLP-1 as opposed to potentiation of exogenously administered 1 or 2.
Figure 6.

GLP-1R ago-PAM (S)-9b produces a dose-dependent reversal of haloperidol induced catalepsy in rats. (A) (S)-9b at doses of 10 and 30 mg/kg ip (10% Tween 80) significantly reverse a 0.75 mg/kg ip dose of haloperdiol. (B) (S)-9b at doses of 30 mg/kg ip significantly reverse a 1.5 mg/kg ip dose of haloperidol and the clinically validated A2A antagonist preladenant is shown for comparison. Catalepsy was measured as the latency to withdraw the forepaws from a horizontal bar with a cutoff of 30 s. Vertical bars represent the means ± SEM of 10–12 rats/treatment group. *p < 0.0001 vs vehicle by Dunnett’s test.
Conclusion
In summary, we have developed a novel, CNS-penetrant GLP-1 ago-PAM (S)-9b, VU0453379, wherein enantiospecific GLP-1 PAM activity was noted, with all activity residing in the (S)-enantiomer. This new chemotype was devoid of ligand bias, potentiating endogenous GLP-1 as well as synthetic peptide agonists 1 and 2. Favorable physiochemical properties and a good disposition profile enabled efficacy in native tissues, potentiating low dose 1 insulin secretion in primary mouse islets and recapitulating, via the PAM mechanism, efficacy in a preclinical PD model and highlighting PD as an exciting therapeutic target for GLP-1R PAMs. While data with (S)-9b is very encouraging, current efforts are focused on further improving both GLP-1R PAM potency and DMPK profile, while further dissecting the molecular pharmacology of GLP-1 PAMs and better understanding PK/PD relationships. These studies are in progress and will be reported in due course.
Experimental Section
Chemistry. All compounds were purified to ≥95% as determined by analytical LCMS (214 nm, 254 nm and ELSD) as well as 1H NMR. The general chemistry, experimental information, and syntheses of all other compounds are supplied in the Supporting Information.
(S)-2-Cyclopentyl-N-((1-isopropylpyrrolidin-2-yl)methyl)-9-methyl-1-oxo-2,9-dihydro-1H-pyrido[3,4-b]indole-4-carboxamide, (S)-9b
To a round-bottom flask was added at room temperature (S)-2-cyclopentyl-9-methyl-1-oxo-N-(pyrrolidin-2-ylmethyl)-2,9-dihydro-1H-pyrido[3,4-b]indole-4-carboxamide (200 mg, 0.51 mmol) dissolved in CH2Cl2 (4 mL) and dry acetone (100 μL). The resultant mixture was stirred at room temperature for 5 min before adding sodium triacetoxyborohydride (150 mg, 0.71 mmol), at which point the mixture was stirred an additional 4 h. Upon completion by LC/MS, the reaction was quenched with sodium bicarbonate (5 mL) and extracted with CH2Cl2. The combined organic layers were dried by passage through a phase separator and concentrated in vacuo. The orange residue was taken up in dimethyl sulfoxide and purified via reverse-phase preparative HPLC using acetonitrile in water with 0.5% NH4OH added to elute. Pure fractions were pooled and concentrated to dryness in vacuo to afford desired product as a foamy yellow solid in 73% yield. Specific rotation [α](23)/D = −20.6° (c = 1.0, MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.32 (d, J = 8.2 Hz, 1H), 7.49–7.45 (m, 2H), 7.39–7.37 (m, 1H), 7.24–7.20 (m, 1H), 7.08 (m, 1H), 5.47–5.43 (m, 1H), 4.24 (s, 3H), 3.74–3.68 (m, 1H), 3.38–3.32 (m, 1H), 3.12–3.07 (m, 1H), 3.02–2.95 (m, 1H), 2.94–2.89 (m, 1H), 2.57–2.51 (m, 1H), 2.25–2.22 (m, 2H), 1.96–1.81 (m, 3H) 1.81–1.67 (m, 7H), 1.12 (d, J = 6.6 Hz, 3H), 1.02 (d, J = 6.4 Hz, 3H). 13C NMR (100.6 MHz, CDCl3) δ (ppm): 166.93, 156.23, 141.05, 126.54, 126.47, 124.77, 124.06, 120.09, 119.92, 113.34, 109.64, 58.17, 55.52, 49.70, 47.36, 42.47, 32.35, 32.32, 31.05, 29.06, 24.41, 23.48, 22.23, 16.24. HRMS (TOF, ES+) calcd for C26H35N4O2 (M + 1), 435.5915; found, 435.5917.
Acknowledgments
This work was generously supported by resources of the Tennessee Valley Healthcare System, the NIH GM06232 (C.W.L., K.D.N., and J.S.D.), the Warren Family and Foundation for establishing the William K. Warren, Jr. Chair in Medicine (C.W.L.), a Culpepper Medical Scholarship (K.D.N), American Diabetes Association research award (K.D.N), Vanderbilt Diabetes and Research Training Center Pilot and Feasibility award (KDN), and the Vanderbilt Diabetes and Research Training Center (DK020593).
Glossary
Abbreviations Used
- GLP-1
glucagon-like peptide 1
- CRC
concentration–response curve
- PAM
positive allosteric modulator
- PBL
plasma:brain level
- PD
Parkinson’s disease
- HIC
haloperidol-induced catalepsy
Supporting Information Available
Experimental procedures and spectroscopic data for selected compounds, detailed pharmacology and DMPK methods. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∇ L.C.M. and K.D.N. contributed equally
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Koole C.; Pabreja K.; Savage E. E.; Wootten D.; Furness S. G.; Miller L. J.; Christopoulos A.; Sexton P. M. Recent advances in understanding GLP-1R (glucagon-like peptide-1 receptor) function. Biochem. Soc. Trans. 2013, 41, 172–179. [DOI] [PubMed] [Google Scholar]
- Dillon J. S.; Tanizawa Y.; Wheeler M. B.; Leng X. H.; Ligon B. B.; Rabin D. U.; Yoo-Warren H.; Permutt M. A.; Boyd A. III. Cloning and functional expression of the human glucagon-like peptide-1 (GLP-1) receptor. Endocrinology 1993, 133, 1907–1910. [DOI] [PubMed] [Google Scholar]
- Hayes M. R. Neuronal and intracellular signaling pathways mediating GLP-1 energy balance and glycemic effects. Physiol. Behav. 2012, 106, 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estall J. L.; Druker D. J. Glucagon and glucagon-like peptide receptors as drug targets. Curr. Pharm. Des. 2006, 12, 1731–1750. [DOI] [PubMed] [Google Scholar]
- Baggio L. L.; Druker D. J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [DOI] [PubMed] [Google Scholar]
- Calanna S.; Christensen M.; Holst J. J.; Laferrere B.; Visboll T.; Knop F. K. Secretion of glucagon-like peptide-1 with type 2 diabetes mellitus: systematic review and meta-analyses of clinical studies. Diabetologia 2013, 56, 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier J. J. GLP-1 receptor agonists for individual treatment of type 2 diabetes mellitus. Nature Rev. Endocrinol. 2012, 8, 728–742. [DOI] [PubMed] [Google Scholar]
- Edwards C. M.; Stanley S. A.; Davis R.; Bynes A. E.; Frost G. S.; Seal L. J.; Ghatei M. A.; Bloom S. R. Exendin-4 reduces fasting and postprandial glucose and decreases energy intake in healthy volunteers. Am. J. Physiol.: Endocrinol. Metab. 2001, 281, E151–E161. [DOI] [PubMed] [Google Scholar]
- Knudsen L. B.; Nielsen P. F.; Huusfeldt P. O.; Johansen N. L.; Madsen K.; Pedersen F. Z.; Thorgersen H.; Wilken M.; Agerso H. Potent derivative of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily dosing adminstration. J. Med. Chem. 2000, 43, 1664–1669. [DOI] [PubMed] [Google Scholar]
- Derosa G.; Maffioloi P. GLP-1 agonists exenatide and liraglutide: a review about their safety and efficacy. Curr. Clin. Pharmacol. 2012, 7, 214–228. [DOI] [PubMed] [Google Scholar]
- Perry T.; Haughey N. J.; Mattson M. P.; Egan J. M.; Greig N. H. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J. Pharmacol. Exp. Ther. 2002, 302, 881–888. [DOI] [PubMed] [Google Scholar]
- During M. J.; Cao L.; Zuzga D. S.; Francis J. S.; Fitzsimons H. L.; Jiao X.; Bland R. J.; Klugmann M.; Bank W. A.; Drucker D. J.; Haile C. N. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nature Med. 2003, 9, 1173–1179. [DOI] [PubMed] [Google Scholar]
- Willard F. S.; Bueno A. B.; Sloop K. W. Small molecule discovery at the glucagon-like peptide-1 receptor. Exp. Diabetes Res. 2012, 2012, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koole C.; Savage E. E.; Christopoulos A.; Miller L. J.; Sexton P. M.; Wootten D. Minireview: Signal bias, allosterism, and polymorphic variation at the GLP-1R: Implications for drug discovery. Mol. Endocrinol. 2013, 27, 1234–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koole C.; Wootten D.; Simms J.; Valant C.; Sridhar R.; Woodman O. L.; Miller L. J.; Summers R. J.; Christopoulos A.; Sexton P. M. Allosteric ligands of the glucagon-like peptide-1 receptor (GLP-1R) differentially modulate endogenous and exogenous peptide responses in a pathway-selective manner: implications for drug screening. Mol. Pharmacol. 2010, 78, 456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willard F. S.; Wootten D.; Showalter A. D.; Savage E. E.; Ficorilli J.; Farb T. B.; Bokvist K.; Alsina-Fernandez J.; Furness S. G. B.; Christopoulos A.; Sexton P. M.; Sloop K. W. Small molecule allosteric modulation of the glucagon-like peptide-1 receptor enhances the insulinotropic effect of oxyntomodulin. Mol. Pharmacol. 2012, 82, 1066–1073. [DOI] [PubMed] [Google Scholar]
- De Graff C.; Rein C.; Piwinica D.; Giordanetto F.; Rognan D. Structure-based discovery of allosteric modualtors of two related class B G-protein-coupled receptors. ChemMedChem 2011, 6, 2159–2169. [DOI] [PubMed] [Google Scholar]
- Morris L. C.; Days E. L.; Turney M.; Mi D.; Lindsley C. W.; Weaver C. D.; Niswender K. D. A duplexed high-throughput screen to identify allosteric modulators of the glucagon-like peptide 1 and glucagon receptor. J. Biomol. Screening 2014, 19, 839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. J.; Lindsley C. W.; Meiler J.; Niswender C. M. Opportunities and challenges in the discovery of allosteric modulators of GPCRs for the treatment of CNS disorders. Nature Rev. Drug Discovery 2014, 13, 692–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melancon B. J.; Hopkins C. R.; Wood M. R.; Emmitte K. A.; Niswender C. M.; Christopoulos A.; Conn P. J.; Lindsley C. W. Allosteric modulation of 7 transmembrane spanning receptors: theory, practice and opportunities for CNS drug discovery. J. Med. Chem. 2012, 55, 1445–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng H.; Sharma R.; McDonald T. S.; Edmonds D. J.; Fortin J.-P.; Li X.; Stevens B. D.; Griffith D. A.; Limberakis C.; Nolte W. M.; Price D. A.; Jackson M.; Kalgutkar A. S. Demonstration of the innate electrophilicity of 4-(3-(benzyloxy)phenyl)-2-(ethylsulfinyl)-6-trifluoromethyl)pyrimidine (BETP), a small molecule positive allosteric modulator of the glucagon-like peptide-1. Drug Metab. Dispos. 2013, 41, 1470–1479. [DOI] [PubMed] [Google Scholar]
- Nolte W. M.; Fortin J.-P.; Stevens B. D.; Aspnes G. E.; Griffith D. A.; Hoth L. R.; Ruggeri R. B.; Mathiowetz A. M.; Limberakis C.; Hepworth D.; Carpino P. A. A potentiator of the orthosteric ligand activity at GLP-1R acts via covalent modification. Nature Chem. Biol. 2014, 10, 629–633. [DOI] [PubMed] [Google Scholar]
- For full experimental details (chemistry and pharmacology), please see the Supporting Information.
- Brissova M.; Shiota M.; Nicholson W. E.; Gannon M.; Knobe S. M.; Piston D. W.; Wright C. V.; Powers A. C. Reduction in pancreatic transcription factor PDX-1 impairs glucose-stimulated insulin secretion. J. Biol. Chem. 2002, 277, 11225–11232. [DOI] [PubMed] [Google Scholar]
- Harkavyi A.; Abuirmeuileh A.; Lever R.; Kingsbury A. E.; Biggs C. S.; Whitton P. S. Glucagon-like peptide 1 receptor stimulation reverse key deficits in distinct rodent models of Parkinson’s disease. J. Neuroinflammation 2008, 5, 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avile-Olmos I.; Disckson J.; Kefalopoulou Z.; Djamshidian A.; Ell P.; Soderlund T.; Whitton P.; Wyse R.; Isaacs T.; Lees A.; Limousin P.; Foltynie T. Exenatide and the treatment of patients with Parkinson’s disease. J. Clin. Med. 2013, 123, 2730–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C. K.; Bubser M.; Thompson A. D.; Dickerson J. W.; Blobaum A. L.; Bridges T. M.; Morrsion R. D.; Daniels S. J.; Jadhav S.; Engers D. W.; Italiano K.; Bode J.; Daniels J. S.; Lindsley C. W.; Hopkins C. R.; Conn P. J.; Niswender C. M. The mGlu4 positive allosteric modulator VU0364770 produces efficacy alone and in combination with L-DOPA or an adenosine A2A antagonist in preclinical rodent models of Parkinson’s disease. J. Pharmacol. Exp. Ther. 2012, 340, 404–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.