

Abstract
The interferon consensus sequence binding protein (Icsbp) is a transcription factor that influences multiple aspects of myelopoiesis. Expression of Icsbp is decreased in the bone marrow of human subjects with chronic myeloid leukemia (CML), and studies in murine models suggest that Icsbp functions as an anti-oncogene for CML. We previously identified a set of Icsbp target genes that may contribute to this anti-oncogene effect. The set includes PTPN13, the gene encoding Fas-associated phosphatase 1 (Fap1, a Fas antagonist). We previously demonstrated that myeloid progenitor cells from Icsbp-knockout mice exhibit Fap1-dependent Fas resistance. In the present study, we determined that the Fas resistance of Bcr−abl+ cells is Icsbp- and Fap1-dependent. We also found that treatment of Bcr−abl+ bone marrow cells with a Fap1-blocking peptide prevents in vitro selection of a tyrosine kinase inhibitor (TKI)-resistant population. Therefore, these results have implications for therapeutic targeting of the Fas-resistant leukemia stem cell population and addressing TKI resistance in CML.
Keywords: Leukemia, gene regulation, apoptosis
Introduction
The interferon consensus sequence binding protein is an interferon regulatory transcription factor with leukemia suppressor activity (referred to as Icsbp or Irf8) [1-3]. Expression of Icsbp is restricted to hematopoietic stem cells (HSCs), myeloid and lymphoid progenitors, and maturing phagocytes and B-cells. Icsbp functions as a transcriptional activator or repressor, depending upon identity of the cis element and the cellular context [1,4-7]. Gene expression profiling of bone marrow cells from subjects with chronic myeloid leukemia (CML) determined that Icsbp expression is decreased in comparison to normal bone marrow [2,8,9]. These studies additionally determined that expression of Icsbp increases during remission with tyrosine kinase inhibitors (TKIs), but decreases with development of TKI resistance, and is lowest during progression to blast crisis (BC) [8,9].
Several murine models support a leukemia suppressor role for Icsbp. For example, mice with IRF8 gene disruption develop a myeloproliferative neoplasm (MPN) that is characterized by increased circulating granulocytes [3]. In IRF8-/- mice, this MPN progresses to acute myeloid leukemia (AML) by ~12 months [3,10]. In a more CML-specific model, murine bone marrow was transduced with a p210 Bcr–abl-expression vector and transplanted into syngeneic mice [11]. These mice develop an MPN that evolves to myeloid BC over time [11]. Further studies determined that Icsbp expression is decreased in the bone marrow of these mice, and that engineered re-expression of Icsbp decreases myeloproliferation and delays BC [12].
To identify molecular mechanisms involved in leukemia suppression, we performed chromatin immunoprecipitation-based high-throughput screening for Icsbp target genes. The identified set was enriched for genes encoding phagocyte effector proteins, consistent with previously identified Icsbp target genes. However, target genes of potential significance to leukemogenesis were also identified, including genes that regulate cytokine-induced proliferation and programmed cell death [6,7,13]. The gene encoding Fas-associated phosphatase 1 (Fap1; the PTPN13 gene) was in the latter cluster [6]. This is of interest, because the expression profile of Fap1 is the reciprocal of the Icsbp expression profile at all clinical stages of CML [9].
Fap1 interacts with the Fas C-terminus, resulting in Fas dephosphorylation and inhibition of Fas-induced apoptosis [14,15]. Other investigators determined that a tripeptide with Fas C-terminal amino acids (serine–leucine–valine; SLV) specifically blocks the interaction of Fap1 with Fas. These investigators found that treatment of epithelial cell lines with SLV peptide increases sensitivity to Fas-induced apoptosis [15].
We found that Icsbp interacts with a specific cis element and represses PTPN13 gene transcription. Icsbp-induced PTPN13 repression increases during myelopoiesis, suggesting that Icsbp mediates sensitivity to Fas-induced apoptosis as differentiation proceeds [6]. We found that knockdown of Icsbp in myeloid cell lines increases Fap1 expression and confers resistance to Fas-induced apoptosis [6]. Fap1 expression and Fas resistance are also increased in primary myeloid progenitor cells from IRF8-/- mice in comparison to wild-type (WT) mice. In those prior studies, Fas resistance was reversed by treatment of Icsbp knockdown cells or IRF8-/- murine bone marrow cells with SLV peptide, thereby implicating Fap1 in this process [6].
Expression of p210 Bcr–abl in myeloid cell lines or primary murine bone marrow progenitor cells decreases Icsbp mRNA and protein in a TK-dependent manner [7]. Investigating the functional association between Icsbp, Fap1 and Fas resistance in CML was the goal of the present study. A Fas-resistant subset of CML leukemia stem cells (LSCs) has been identified that persists during TKI treatment, even in subjects with complete cytogenetic remission [16,17]. These cells are hypothesized to expand over time, predisposing to development of overt TKI resistance or BC [16]. Therefore, targeting the Fap1/Fas interaction in CML might complement the action of TKI agents.
Materials and methods
Plasmids
Icsbp cDNA was obtained from Ben Zion-Levi (Technion, Haifa, Israel) and subcloned into the pcDNA expression vector. p210 Bcr–Abl cDNA in the MIGR1 vector was obtained from R. Bhatia (City of Hope). shRNAs and scrambled control sequences for Icsbp were designed using the Promega website and subcloned into the pLKO.1-puro vector (from Kathy Rundell, Northwestern University, Chicago, IL). Several were tested and the most efficient for suppression used (see [6,7]).
A double-stranded, synthetic oligonucleotide with three copies of the −587 to −627 bp sequence from the PTPN13 promoter (a previously described Icsbp-binding cis element) was subcloned into a minimal promoter/luciferase reporter vector (pGL3-promoter; Stratagene) [6]. Another construct was generated with 670 bp of the PTPN13 5’ flank (starting at the transcription start site) subcloned into a luciferase reporter vector (p-GL3-basic).
Oligonucleotides
Oligonucleotides were synthesized by MWG Biotech (Piedmont, NC). Double-stranded oligonucleotides used in DNA-affinity purification represent the −587 to −627 bp PTPN13 promoter sequence (5′-CTCCCGGAGTCTGTTTCTAATTTCTGCAAATGATTGTGG-3′), or a non-Icsbp-binding, mutant form of the PRDI-consensus (5′-tgtctttgtctttgtctt-3′) [6].
Myeloid cell line culture
The human myelomonocytic leukemia line U937 [18] was obtained from Andrew Kraft (Hollings Cancer Center, Medical University of South Carolina, Charleston, SC). The human myeloblastic cell line Kg1 was obtained from Leonidas Platanias (Northwestern University, Chicago). Cells were maintained as described [6,7].
Murine bone marrow culture
Animal studies were approved by the Animal Care and Use Committees of Northwestern University and Jesse Brown VA. Bone marrow mononuclear cells were obtained from the femurs of WT C57/BL6 mice. Cells were transduced with a retroviral vector to express p210 Bcr–abl or empty control vector [7]. Some cells were cultured under HSC conditions in Dulbecco’s modified Eagle’s (DME) medium supplemented with 10% fetal calf serum, 1% penicillin–streptomycin (pen-strep), 10 ng/mL murine interleukin 6 (IL6; R&D Systems Inc., Minneapolis, MN), 10 ng/mL murine recombinant IL3 (R&D Systems) and 100 ng/mL stem cell factor (SCF; R & D Systems). Sca1+ cells were separated using the Miltenyi magnetic bead system, according to the manufacturer’s instructions (Miltenyi Biotec, Auburn, CA). Granulocyte monocyte progenitors (GMPs) were cultured in DME medium supplemented with 10% fetal calf serum, 1% pen-strep, 20 ng/mL murine granulocyte macrophage-colony stimulating factor (GM-CSF; R & D Systems), 100 ng/mL SCF and 10 ng/mL murine recombinant IL3. CD34+ cells were obtained using a magnetic bead purification technique, as above. Some cultured GMPs were differentiated over 48 h in 10 ng/mL of G-CSF or M-CSF to granulocytes or monocytes, respectively [6,7,19]. These cultured cells were used in studies of mRNA and protein expression, and apoptosis, as described below.
For drug-resistance studies, transduced cells were cultured under GMP conditions with the addition of imatinib (IM) at an initial dose of 0.2 μg/mL (versus sham treatment with buffer). The dose of IM was gradually increased over 6 weeks to 2.0 μg/mL. In some experiments, cells were also incubated with VLS or SLV peptides (10 μM). For some of these studies, Fas-sensitive cells were depleted prior to culture in IM or SLV/VLS peptide by incubation with Fas-agonist antibody (CH11) or control antibody (24 h, 37 °C, 5% CO2), followed by removal of apoptotic cells using the Miltenyi magnetic bead system (Dead Cell Removal Kit; Miltenyi Biotec).
Chromatin co-immunoprecipitation
Kg1 cells were incubated briefly in formaldehyde-supplemented medium and cell lysates sonicated to generate chromatin fragments of < 200 bp [6,7]. Lysates were immunoprecipitated with Icsbp or irrelevant control antibody, as in [6,7].
Quantitative real time PCR
Primers were designed with Applied Biosystems software and real time polymerase chain reaction (PCR) was performed using SYBR Green and the “standard curve” method. For some studies, RNA was isolated using TRIzol reagent (Gibco-BRL, Gaithersburg, MD). Result were normalized to 18S. The same set of control cDNA was used to generate the standard curve for analysis of various samples, thereby permitting comparison between samples and between experiments. For cell line transfection experiments, the standard curve was generated with cDNA from non-transfected cells. For murine bone marrow experiments, the standard curve was generated with cDNA from WT murine bone marrow growing in GM-CSF, IL3 and SCF. In some experiments, comparison of expression levels for different messages was performed by normalizing to Ct numbers (i.e. for comparison of relative Sca1, CD34 and CD38 expression).
For other studies, chromatin that co-precipitated with Icsbp or control antibody was amplified with primers flanking the Icsbp-binding PTPN13 promoter cis element (−669 to −646: 5′-GTCGTGCTTGCACAGCTCCGCTCT-3′ and −512 to −489: 5′-ACCTTGCATCAGACAGTGTCTCTC-3′). Results were normalized to total chromatin.
Myeloid cell line transfections and reporter gene assays
Kg1 cells were transfected by electroporation with vectors to express p210 Bcr–abl, or empty MIGR1 vector plus a vector with a neomycin phosphotransferase cassette (pSRα) (30 μg each). Stable pools were selected in G418 (0.5 mg/mL).
U937 cells were transfected with vectors to express various combinations of Icsbp, p210 Bcr–abl or control, and minimal promoter/firefly luciferase reporter constructs with three copies of the −587 to −627 bp PTPN13 sequence (ptpn13GL3-p) or control (pGL3-p) (70 μg), and a cytomegalovirus (CMV)/Renilla luciferase vector (for transfection efficiency). Transfectants were assayed for firefly and Renilla luciferase after 48 h [6,7].
Apoptosis assays
Cells were incubated with Fas-agonist antibody CH11 or control. In some experiments, cells were incubated for the last 12 h with SLV peptide (or control VLS peptide) [15]. Apoptosis assays were performed using annexin V/propidium iodide double staining. The cells were analyzed on a Becton-Dickinson FACScan flow cytometer (Cambridge, MA).
Western blots
Cells were lysed by boiling in 2 × sodium dodecyl sulfate (SDS) sample buffer. Lysate proteins (50 μg) were separated by SDS-polyacrylamide gel electrophoresis (PAGE) (8% acrylamide) and transferred to nitrocellulose. Western blots were serially probed with various antibodies plus tubulin (loading control). Each experiment was repeated at least three times with different batches of lysate proteins. To quantify band density, blots were electronically scanned and evaluated using Un-Scan-It software (Silk Scientific, Orem, UT).
DNA-affinity purification assays
Nuclear proteins (300 μg) were incubated with biotin-labeled ds oligonucleotide probe representing the −587 to −627 bp PTPN13 promoter or a non-Icsbp-binding PRDI-mutant sequence in DNA-affinity purification assay (DAPA) buffer (25 mM HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid], pH 7.6, 60 mM KCl, 5 mM MgCl2, 7.5% glycerol, 0.1 mM ethylenediaminetetraacetic acid [EDTA], 1 mM dithiothreitol [DTT] and 0.25% Triton X-100). DNA–protein complexes were precipitated with 50 μL of a 50% slurry of neutravidin-coated agarose beads (Pierce, Rockford, IL). Proteins bound to the beads were separated by SDS-PAGE and transferred to nitrocellulose. Western blots were probed with an antibody to Icsbp.
Statistical analysis
Statistical significance was determined by paired Student’s t-test or analysis of variance (ANOVA) methods (for analysis of > 2 groups) using SigmaPlot and SigmaStat software. Statistical significance is reported as standard error. For transfection, apoptosis and real time PCR experiments, n values represent the number of independent experiments, each of which was performed in triplicate. Differences are considered statistically significant if p < 0.05 (95% confidence interval).
Results
Bcr–abl increases Fap1 expression in myeloid cells
We first investigated Fap1 expression in Bcr−abl+ cells. For these studies, we generated stable transfectants of the Kg1 myeloid leukemia cell line using a p210 Bcr–abl expression vector or empty control vector [7]. We used the p210 form in preference to the p190 form because p210 is more common in adult CML and is most frequently associated with myeloid BC. In previous studies, we found that knockdown of Icsbp in Kg1 cells increases Fap1 expression and Fap1-dependent Fas resistance [6]. Kg1 cells are relatively sensitive to Fas-induced apoptosis in comparison to other myeloid leukemia cell lines (i.e. U937 cells, employed in studies below) [6]. This relative Fas sensitivity of Kg1 cells correlates with relatively increased expression of Icsbp and decreased Fap1 expression in comparison to U937 cells [6].
Bcr−abl+ Kg1 cells and control cells were analyzed for Fap1 and Icsbp mRNA expression by real time PCR, and protein expression by Western blot. We found significantly more Fap1 mRNA [Figure 1(A)] in Bcr−abl+ Kg1 cells in comparison to control cells (p < 0.0001, n = 3). This correlated with increased Fap1 protein in Bcr−abl+ cells [Figure 1(B)]. As in previous studies, we found significantly less Icsbp mRNA (p < 0.0001, n = 3) [Figure 1(A)] and protein [Figure 1(B)] in Bcr−abl+ Kg1 cells in comparison to control cells [7].
Figure 1.
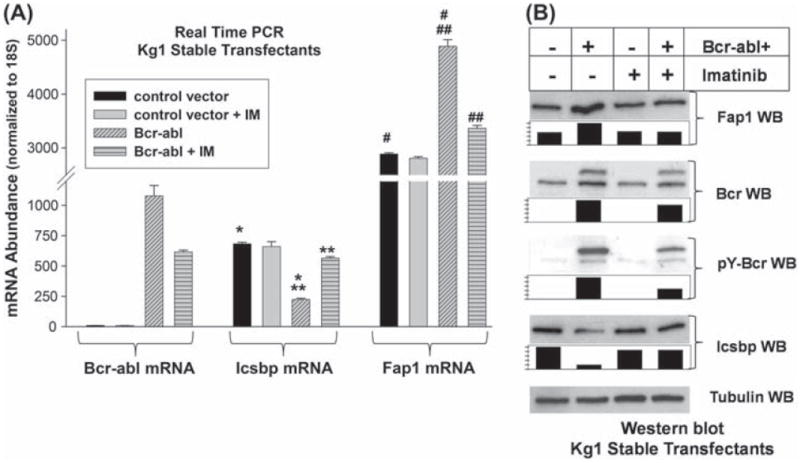
Bcr–abl increases Fap1 expression in a tyrosine kinase dependent manner in Kg1 myeloid cells. (A) Bcr–abl increases Fap1 mRNA expression in Kg1 myeloid cells. Kg1 cells were stably transfected with a vector to express Bcr–abl (p210) or empty control vector. Expression of Fap1, Icsbp and Bcr–abl mRNA was determined by real time PCR. Transfectants were analyzed with or without treatment with the tyrosine kinase inhibitor imatinib (IM). Statistically significant differences in expression are indicated by *, **, # or ## (p<0.001, n = 3). (B) Bcr–abl increases Fap1 protein expression in Kg1 myeloid cells. Kg1 stable transfectants, described above, were also analyzed for expression of Fap1, Icsbp and Bcr–abl protein. Cells were analyzed with versus without IM treatment, and cell lysates were separated by SDS-PAGE. Western blots were probed with antibodies to Fap1, Icsbp, Bcr, phospho-Bcr (as an indication of Bcr–abl activity) or tubulin (as a loading control). Blots were scanned, band density was quantified, normalized to loading control, and graphed relative to samples from non-IM treated Bcr−abl+ cells (set at a value of 1).
To determine whether inhibition of Bcr–abl TK activity alters Fap1 expression, some stable transfectants were treated with imatinib (2.0 μM). We found that IM treatment of Bcr−abl+ Kg1 transfectants significantly decreased expression of Fap1 mRNA (p = 0.0003, n = 3) [Figure 1(A)] and protein [Figure 1(B)] and increased expression of Icsbp mRNA (p < 0.0001, n = 3) and protein. The dose of IM used in this study consistently inhibits Bcr–abl tyrosine kinase activity in Kg1 stable transfectants, as determined by Bcr–abl auto-phosphorylation [Figure 1(B)] [7,20].
However, these studies were performed in a leukemia cell line that exhibits abnormal cell survival prior to introduction of the Bcr–abl oncogene. Therefore, we also investigated the effect of Bcr–abl on Fap1 expression in primary bone marrow cells. For these studies, bone marrow mononuclear cells were isolated from C57 Black 6 mice and transduced with a retroviral vector to express p210 Bcr–abl, or with empty control vector (MIGR1) (as in our previous studies) [7,20]. This retroviral vector also expresses green fluorescent protein (GFP), and ~80% of cells were GFP+ with our transduction technique. Some transduced cells were cultured in IL3, IL6 and SCF for 48 h, followed by isolation of Sca1+ cells. This process enriches the HSC population [19]. Other cells were cultured in GM-CSF, IL3 and SCF followed by isolation of CD34+ cells. This population is enriched for bi-potential GMPs, the population that is most similar to the LSC in chronic phase CML [19,21-23]. Some of the latter cells were cultured for an additional 24 h in G-CSF or M-CSF to induce granulocyte or monocyte differentiation, respectively. Such ex vivo differentiated cells are -80% CD34−CD38+ and positive for Gr1 or Mac1 (indicating granulocyte or monocyte differentiation) [19].
Transduced murine bone marrow cells were analyzed for expression of Fap1, Icsbp and Bcr–abl mRNA by real time PCR. In studies of control cells, we found that Fap1 expression is not significantly different in HSCs versus GMPs (p = 0.6, n = 4), but decreases significantly with granulocyte or monocyte differentiation (p < 0.0001, n = 4) [Figure 2(A)]. In Bcr−abl+ GMPs, expression of Fap1 mRNA is significantly greater than in control GMPs (p < 0.00001, n = 4) [Figure 2(A)]. This difference is also reflected at the protein expression level [Figure 2(B)]. Fap1 expression does not decrease with differentiation of Bcr–abl+ cells [Figure 2(A)]. In contrast, there is no significant difference in Fap1 expression in control HSCs versus Bcr−abl+ HSCs (p = 0.4, n = 4). Significantly less Icsbp is expressed in Bcr−abl+ cells in comparison to control cells (p < 0.002, n = 4), consistent with our previous results [Figures 2(A) and 2(B)].
Figure 2.
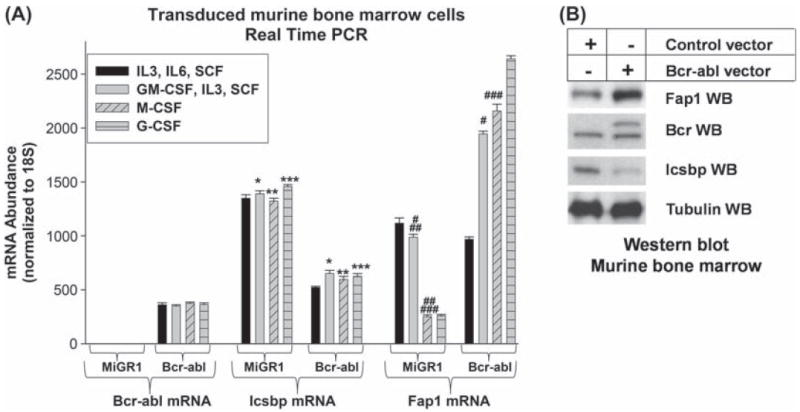
Bcr–abl increases Fap1 expression in a tyrosine kinase dependent manner in transduced primary murine bone marrow cells. (A) Bcr–abl increases Fap1 mRNA expression in primary murine bone marrow cells. Murine bone marrow mononuclear cells were transduced with a retroviral vector to express Bcr–abl (p210) or with empty control vector (MIGR1). Cells were cultured under hematopoietic stem cell (HSC: IL3/IL6/SCF) or granulocyte/monocyte progenitor (GMP: IL3/GM-CSF/SCF) conditions, or ex vivo differentiated to granulocytes (with G-CSF) or monocytes (with M-CSF). Cells were analyzed for Fap1, Icsbp or Bcr–abl expression by real time PCR. Statistically significant differences are indicated by *, **, ***, #, ## or ### (p<0.01, n = 4). (B) Bcr–abl increases Fap1 protein expression in primary murine bone marrow cells. Primary transduced cells, described above, were also analyzed for expression of Fap1, Icsbp and Bcr–abl protein. Cells were cultured under GMP conditions, and cell lysates were separated by SDS-PAGE. Western blots were probed with antibodies to Fap1, Icsbp, Bcr or tubulin (as a loading control).
Resistance to IM and other TKIs develops in 50% of human subjects treated with these agents, and is associated with increased probability of progression to blast crisis (BC) [24-27]. We used an in vitro model to investigate the impact of IM resistance on expression of Fap1 and Icsbp in Bcr−abl+ cells. For these studies, primary murine bone marrow cells were transduced with a Bcr–abl expression vector and cultured under GMP conditions, as above. Bcr−abl+ cells were cultured with or without IM (initial dose of 0.2 μM increasing to 2.0 μM over 6 weeks versus sham treatment) [28]. Culture in IM under these conditions results in selection of a resistant population. IM-resistant and IM-naive Bcr−abl+ cells used in these studies were cultured for a total of 3 months. Empty vector transduced cells were cultured for 48 h under GMP conditions as a control for these studies. Fap1 and Icsbp mRNA was quantified by real time PCR.
We found increased Fap1 mRNA and decreased Icsbp mRNA in IM-resistant Bcr−abl+ cells in comparison to IM-naive Bcr−abl+ cells (p < 0.0001, n = 6) [Figure 3(A)]. These differences in Icsbp and Fap1 mRNA correlate with protein expression in these cells [Figure 3(A)]. Bcr–abl expression is less in IM-resistant Bcr−abl+ cells in comparison to IM-naive cells (mRNA; p = 0.002, n = 6) [Figures 3(A) and 3(B)]. This suggests that IM resistance develops due to point mutation of the BCR–ABL transgene, rather than increased transgene copy number/expression. Cultured cells were also analyzed to determine whether development of IM resistance alters the differentiation stage of long-term cultured Bcr−abl+ cells. For these studies, the cultured cells were analyzed by real time PCR for relative expression of Sca1, CD34 and CD38. We found that both types of long-term Bcr−abl+ cultures were enriched for CD34+ cells with relatively less CD38 expression [Figure 3(C)]. We found that CD34 expression was slightly but significantly increased and CD38 expression was decreased in IM-resistant cells in comparison to IM-naive Bcr−abl+ cells (p < 0.006, n = 3).
Figure 3.
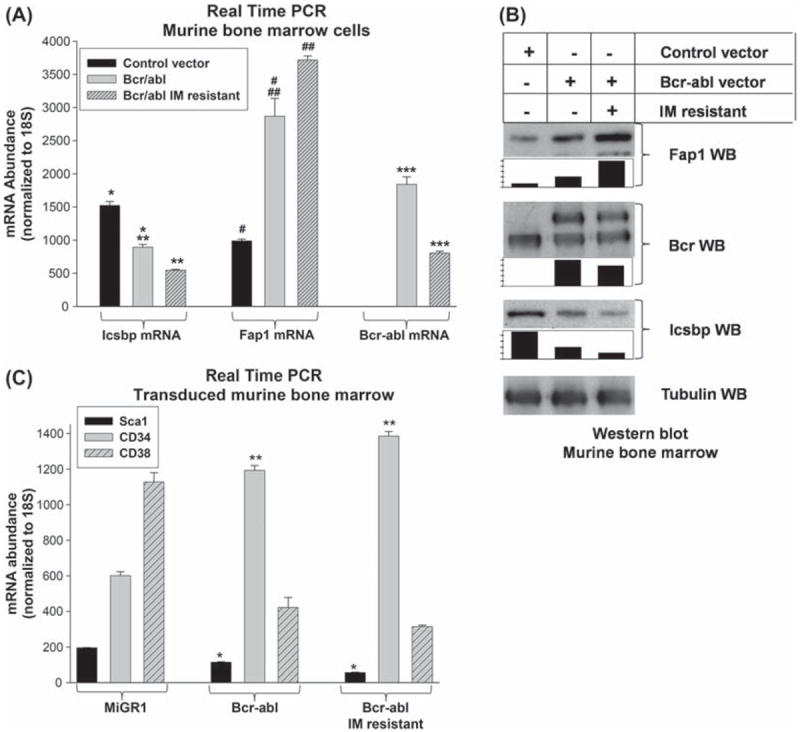
Tyrosine kinase resistance influences Fap1 expression in Bcr–abl+ cells. (A) Expression of Fap1 mRNA is increased in imatinib-resistant Bcr–abl+ primary murine bone marrow cells. Murine bone marrow cells were transduced with a retroviral vector to express Bcr–abl or with empty MIGR1 control vector. Cells were cultured under GMP conditions, and some Bcr–abl+ cells were cultured with increasing doses of IM to induce in vitro TKI resistance. Cells were analyzed for expression of Fap1, Icsbp and Bcr–abl by real time PCR. Statistically significant differences are indicated by *, **, ***, # or ## (p ≤0.002, n = 6). (B) Expression of Fap1 protein in Bcr–abl+ primary murine bone marrow cells is increased by ex vivo resistance to imatinib (IM). Transduced murine bone marrow cells, described above, were analyzed for expression of Fap1, Icsbp and Bcr–abl protein. Cell lysates were separated by SDS-PAGE and Western blots were serially probed with antibodies to Fap1, Icsbp, Bcr and tubulin (as a loading control). Blots were scanned, band density was quantified, normalized to loading control, and graphed relative to samples from IM-naive Bcr–abl+ cells (set at a value of 1). (C) IM-resistant and IM-naive Bcr–abl+ primary murine bone marrow cells are enriched for CD34 expression. Transduced bone marrow cells, described above, were also analyzed by real time PCR for expression of Sca1, CD34 and CD38. Standard curve method was used to permit comparison between samples, and results were normalized to Ct numbers to compare expression of different genes. Statistically significant differences are indicated by * and ** (p< 0.006, n = 3).
Bcr–abl increases PTPN13 promoter activity in an Icsbp-dependent manner
We next determined whether increased PTPN13 promoter activity results in increased Fap1 expression in Bcr−abl+ cells. For these studies, we used a reporter construct that includes 670 bp of the PTPN13 promoter (the minimal promoter sequence that is repressed by Icsbp) [6]. This PTPN13-promoter/reporter construct (or empty vector control) was co-transfected into a myeloid cell line with a vector to express Bcr–abl (or empty expression vector). We used U937 myeloid leukemia cells for these studies, since this line expresses abundant Fap1 [6].
We found that PTPN13 promoter activity is significantly greater in Bcr−abl+ transfectants in comparison to control cells (p < 0.0001, n = 4) [Figure 4(A)]. Therefore, we investigated the role of TK activity in Bcr–abl-induced PTPN13 promoter activity. For these studies, U937 cells were co-transfected as above, and reporter activity was determined with versus without IM treatment. We found that IM significantly decreases Bcr–abl-induced PTPN13 reporter activity in a dose dependent manner [Figure 4(A)] (p < 0.001, n = 4 for all comparisons). We found a comparable effect of Bcr–-abl expression and IM treatment (2.0 μM) on the expression of endogenous Fap1 mRNA in U937 cells [Figure 4(B)] (p < 0.01, n = 3 for control cells versus Bcr−abl+ cells, and for Bcr−abl+ cells with versus without IM treatment).
Figure 4.
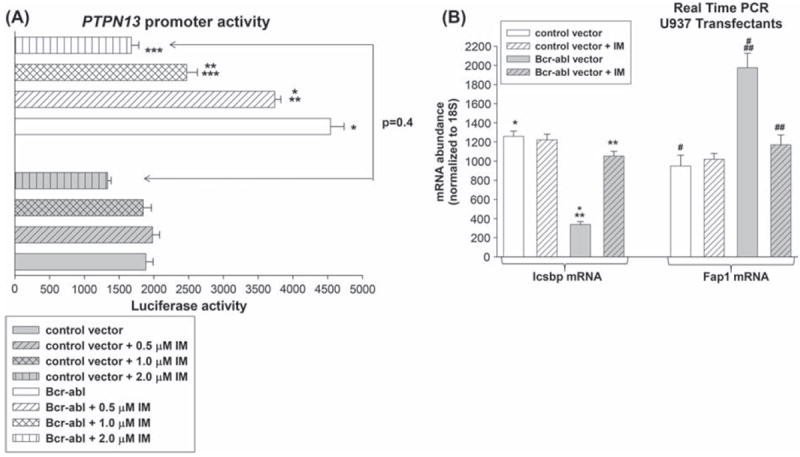
Bcr–abl activates the PTPN13 promoter in a tyrosine kinase dependent manner. (A) Bcr–abl induces activation of the PTPN13 promoter in a tyrosine kinase dependent manner. U937 cells were co-transfected with a reporter vector with 670 bp of the PTPN13 promoter (or empty control vector), and a vector to express Bcr–abl (or empty expression vector). Transfectants were analyzed for luciferase reporter expression with versus without various amounts of the tyrosine kinase inhibitor, imatinib (IM). Statistically significant differences in reporter expression are indicated by *, ** or *** (p<0.001, n = 4). (B) Bcr–abl increases Fap1 mRNA expression in U937 transfectants. U937 cells were transfected with a vector to express Bcr–abl (or empty vector control). Cells were analyzed with or without treatment with IM (2.0 μM) for expression of Icsbp and Fap1 by real time PCR. Statistically significant differences are indicated by *, **, ***, #, ## or ### (p< 0.001, n = 3).
We also investigated the contribution of decreased Icsbp to PTPN13 promoter activity in Bcr−abl+ cells. Our previous studies identified a specific PTPN13 cis element that is repressed by Icsbp [6]. Therefore, we determined whether Bcr–abl influences activity of this cis element in an Icsbp-dependent manner. For these studies, U937 cells were co-transfected with a reporter construct with three copies of the Icsbp-binding PTPN13 cis element linked to a minimal promoter (or minimal promoter control vector), and a vector to express p210 Bcr–abl (or empty vector). We found that Bcr–abl significantly increases activity of this PTPN13 cis element (p < 0.0001, n = 6) [Figure 5(A)].
Figure 5.
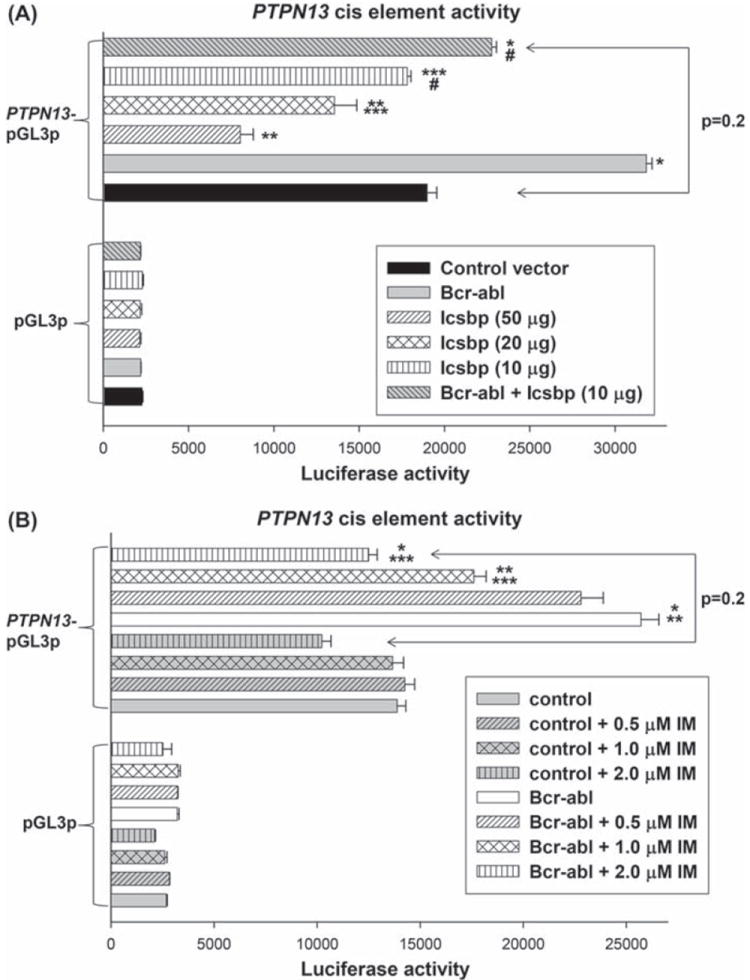
Bcr–abl activates the Icsbp-binding PTPN13 cis element in an Icsbp dependent manner. (A) Bcr–abl induces activation of the Icsbp-binding PTPN13 cis element in an Icsbp dependent manner. U937 cells were co-transfected with an artificial promoter construct with multiple copies of the Icsbp-binding PTPN13 cis element (or minimal promoter/reporter control vector) and vectors to express various combinations of Icsbp and Bcr–abl (or relevant control vectors). Luciferase reporter activity was determined. Statistically significant differences in reporter activity are indicated by *, **, *** or # (p<0.01, n =6). (B) Bcr–abl induces activation of an Icsbp-binding PTPN13 cis element in a tyrosine kinase dependent manner. U937 cells were co-transfected with an artificial promoter construct with multiple copies of the Icsbp-binding PTPN13 cis element (or minimal promoter/reporter control vector) and vectors to express Bcr–abl or control expression vector. Transfectants were analyzed for luciferase reporter activity after treatment with various amounts of imatinib (IM). Statistically significant differences in reporter activity are indicated by *, ** or ***(p<0.001, n = 6).
Next, U937 cells were co-transfected with the PTPN13 cis element-containing construct (or control vector) and a dose titration of Icsbp expression vector. The goal of these studies was to identify the maximum level of Icsbp expression that does not alone influence PTPN13 cis element activity (10 μg of plasmid; p = 0.7, n = 6). Based on these results, U937 cells were co-transfected with the PTPN13 cis element-containing reporter vector, Bcr–abl expression vector and Icsbp expression vector (10 μg). We found that re-expression of Icsbp reverses the effect of Bcr–abl on this PTPN13 cis element [Figure 5(A)]. To determine whether TK activity mediates increased PTPN13 cis element activity in Bcr−abl+ cells, some transfectants were treated with IM. We found that IM decreases the effect of Bcr–abl on PTPN13 cis element activity in a dose-dependent manner [Figure 5(B)].
We also investigated whether Icsbp-knockdown rescues PTPN13 cis element activity in IM treated Bcr−abl+ transfectants. In initial experiments, U937 cells were co-transfected with the PTPN13-cis element containing reporter construct and a dose titration of plasmid vector to express Icsbp-specific shRNAs. The goal of these studies was to identify the maximal amount of plasmid that did not alter PTPN13 cis element activity [Figure 6(A)]. For these studies, we used Icsbp shRNA vectors (and scrambled control vectors) that were validated in our previous investigations [6,7]. U937 cells were also co-transfected with the PTPN13 cis element-containing construct, a vector to express Bcr–abl and a vector to express Icsbp-specific shRNAs (10 μg). We found that Icsbp knockdown blocks the effect of IM on PTPN13 cis element activity in Bcr−abl+ transfectants (p = 0.6, n = 6 for comparison of IM-treated Bcr−abl+ transfectants with versus without Icsbp-specific shRNA) [Figure 6(B)].
Figure 6.
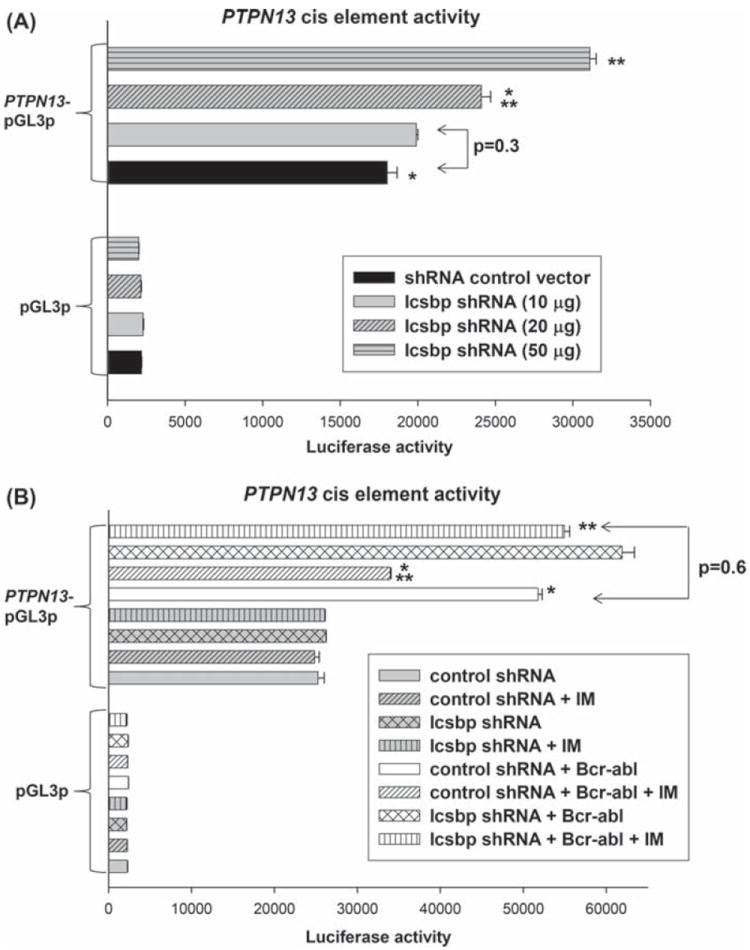
Bcr–abl activates the Icsbp-binding PTPN13 cis element in a tyrosine kinase dependent manner. (A) Knockdown of endogenous Icsbp increases activity of an Icsbp-binding PTPN13 cis element in a dose dependent manner. U937 cells were co-transfected with an artificial promoter construct with multiple copies of the Icsbp-binding PTPN13 cis element (or minimal promoter/reporter control vector) and vectors to express various amounts of an Icsbp-specific shRNA (or scrambled shRNA control vector). Transfectants were analyzed for luciferase reporter activity. Statistically significant differences in reporter activity are indicated by * or **(p<0.001, n = 6). (B) Icsbp-knockdown rescues Bcr–abl induced activation of the PTPN13 cis element in IM treated transfectants. U937 cells were co-transfected with an artificial promoter construct with multiple copies of the Icsbp-binding PTPN13 cis element (or minimal promoter/reporter control vector) and vectors to express various combinations of Bcr–abl, Icsbp-specific shRNA or relevant control vectors. Some transfectants were treated with IM to inhibit Bcr–abl-TK activity, and transfectants were analyzed for luciferase reporter activity. Statistically significant differences in reporter activity are indicated by * or **(p< 0.001, n = 4).
Bcr–abl decreases Icsbp binding to the PTPN13 cis element
To clarify the mechanism for Icsbp-dependent PTPN13 regulation by Bcr–abl, we determined the effect of Bcr–abl on interaction of Icsbp with the PTPN13 cis element. We first investigated this question using an in vitro DNA affinity purification assay. For these studies, cell lysate proteins from Bcr−abl+ or control vector transfected Kg1 cells were incubated with biotin labeled, double stranded oligonucleotide probes representing the PTPN13 cis element or with a non-Icsbp-binding oligonucleotide sequence. Oligonucleotides and interacting proteins were purified by affinity of the biotin label for an avidin matrix, and identified by Western blot. We found a decrease in total and PTPN13-bound Icsbp in Bcr−abl+ cells [Figure 7(A)].
Figure 7.
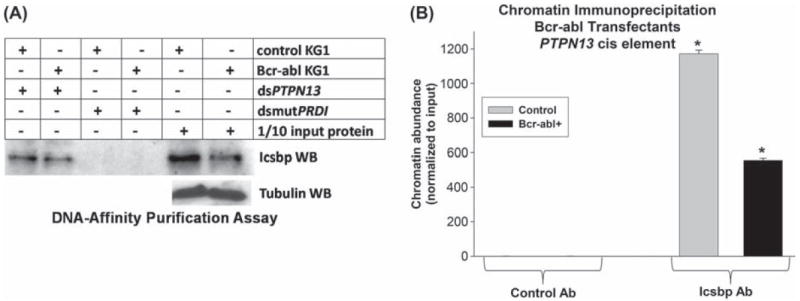
Bcr–abl expression decreases Icsbp expression and binding to the PTPN13 cis element in myeloid cells. (A) Bcr–abl expression in Kg1 myeloid cells decreases in vitro Icsbp-binding to the PTPN13 cis element. Kg1 cells were stably transfected with a vector to express Bcr–abl or empty control vector, as described above. Cell lysates were incubated with a biotin-labeled probe with the Icsbp-binding PTPN13 cis element versus a mutant form of the Icsbp-binding PRDI sequence. DNA-bound proteins were isolated by affinity of the probe to neutravidin matrix. Affinity purified proteins were separated by SDS-PAGE and identified by probing Western blots with an antibody to Icsbp. Protein concentrations were normalized by probing “total input ” protein lanes with an antibody to tubulin. (B) Bcr–abl expression in Kg1 myeloid cells decreases in vivo Icsbp binding to the PTPN13 cis element. Bcr–abl+ and control Kg1 stable transfectants, described above, were analyzed for in vivo Icsbp binding by chromatin immunoprecipitation. Cell lysates were subjected to immunoprecipitation with an antibody to Icsbp or control antibody. Co-precipitating chromatin was amplified by real time PCR with primers that flank the Icsbp-binding PTPN13 cis element. Statistically significant difference is indicated by * (p<0.0001, n = 3).
We also investigated Icsbp binding to the PTPN13 cis element in vivo. For these studies, chromatin was immuno-precipitated from Bcr−abl+ or control Kg1 cells with an antibody to Icsbp. Recovered chromatin was amplified by real time PCR using primers flanking the Icsbp-binding PTPN13 cis element [6,20]. Chromatin that co-precipitated with an irrelevant, control antibody was a negative control for these studies, and results were normalized to non-precipitated (total input) chromatin. We found a significant decrease in occupancy of the PTPN13 cis element by Icsbp in Bcr−abl+ Kg1 cells in comparison to control cells (p < 0.0001, n = 3) [Figure 7(B)].
Bcr–abl induces Fap1-dependent Fas resistance in myeloid cells
We investigated the role of increased Fap1 in Fas resistance of Bcr−abl+ cells using Kg1 stable transfectants, described above. Bcr−abl+ or control Kg1 cells were treated with a Fas-agonist antibody (Ab) and assayed for apoptosis by annexin V staining and flow cytometry [6]. Fas-Ab fails to induce significant apoptosis in Bcr−abl+ Kg1 cells, although apoptosis is induced in control cells under these conditions [Figure 8(A)]. To determine the role of Fap1 on Fas resistance, cells were treated with the SLV peptide (to block Fap1/Fas interaction) or VLS control peptide. We found that treatment of Bcr−abl+ Kg1 cells with SLV peptide rescued Fas-Ab induced apoptosis; apoptosis in these cells is not significantly different from that in control cells treated with Fas-Ab (p = 0.4, n = 4) [Figure 8(A)]. In control experiments, there was no significant difference in apoptosis in Bcr−abl+ versus control Kg1 cells without Fas-Ab (± SLV peptide). We found that the addition of VLS control peptide did not alter apoptosis under any of the assay conditions, so this peptide was used as a negative control (i.e. results were not different from results without any peptide treatment).
Figure 8.
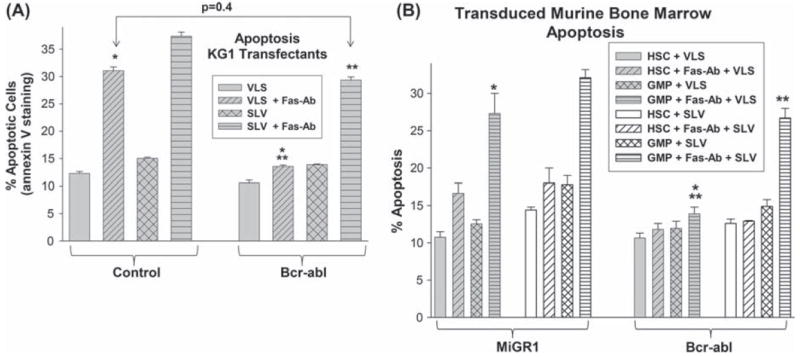
Bcr–abl induces Fap1-dependent Fas resistance in myeloid progenitor cells. (A) Bcr–abl+ Kg1 cells exhibit Fap1-dependent Fas resistance. Kg1 cells were stably transfected with a vector to express Bcr–abl or with empty control vector, as described above. Cells were pretreated with SLV peptide (Fap1-blocking) or VLS control peptide followed by treatment with Fas-agonist antibody (or control antibody). Cells were analyzed for apoptosis by staining for annexin V and flow cytometry. Statistically significant differences in apoptosis are indicated by * or ** (p<0.001, n = 6). (B) Bcr–abl+ primary murine myeloid progenitor cells exhibit Fap1-dependent Fas resistance. Primary murine bone marrow cells were transduced with a vector to express Bcr–abl or with empty vector control, as described above. Cells were cultured under HSC or GMP conditions, or differentiated with G-CSF or M-CSF, and pretreated with SLV or VLS control peptide, followed by treatment with Fas-agonist antibody (or control antibody). Cells were analyzed for apoptosis by staining for annexin V and flow cytometry. Statistically significant differences in apoptosis are indicated by * or ** (p<0.0001, n = 4).
We also investigated the impact of Bcr–abl on Fas-induced apoptosis using transduced primary murine bone marrow cells, as described above. We found that Bcr−abl+ HSCs and GMPs were significantly less sensitive to Fas-Ab induced apoptosis in comparison to control cells (p < 0.001, n = 6) [Figure 8(B)]. We also found that treatment with Fap1-blocking SLV peptide reverses Fas resistance of Bcr−abl+ GMPs (p < 0.0001, n = 6 for comparison between Bcr−abl+ GMPs with SLV versus VLS). In contrast, treatment with SLV peptide does not significantly alter Fas resistance of the Bcr−abl+ HSC population (p = 0.5, n = 6 for experiments with Bcr−abl+ HSCs with SLV versus VLS). These results suggest that Fas resistance in Bcr−abl+ GMPs is Fap1-dependent, but resistance in Bcr−abl+ HSC is not, consistent with Fap1 expression levels in these cells. This is relevant, because the LSC in CML chronic phase is the Sca1 −CD34+ CD38− population (GMP-like) in murine models, rather than the Sca1+ CD34− population (HSC-like) [19,21-23]. In control experiments, there was no difference in apoptosis in Bcr−abl+ versus control cells in the absence of Fas-Ab (± SLV peptide; not shown). VLS control peptide treatment was not significantly different from no peptide for any condition (not shown).
Fap1 inhibition prevents development of TKI resistance in Bcr−abl+ myeloid progenitor cells
We next determined the effect of IM resistance on Fas-induced apoptosis using the in vitro IM resistance model, described above. For these studies, IM-naive Bcr−abl+, IM-resistant Bcr−abl+ and control cells were assayed for Fas-Ab induced apoptosis after treatment with SLV peptide (or VLS control), high-dose IM (5 μM) or both. We found that both IM-resistant and IM-naive Bcr−abl+ cells exhibit significantly less Fas-induced apoptosis in comparison to control cells (p < 0.0001, n = 4) [Figure 9(A)]. Fas-induced apoptosis of IM-naive Bcr−abl+ cells is increased significantly by treatment with Fap1-blocking, SLV peptide (p < 0.0002, n = 4) [similar to Figure 5(B)]. In contrast, treatment with SLV peptide does not alter Fas-induced apoptosis in IM-resistant Bcr−abl+ cells (p = 0.6, n = 6) [Figure 9(A)].
Figure 9.
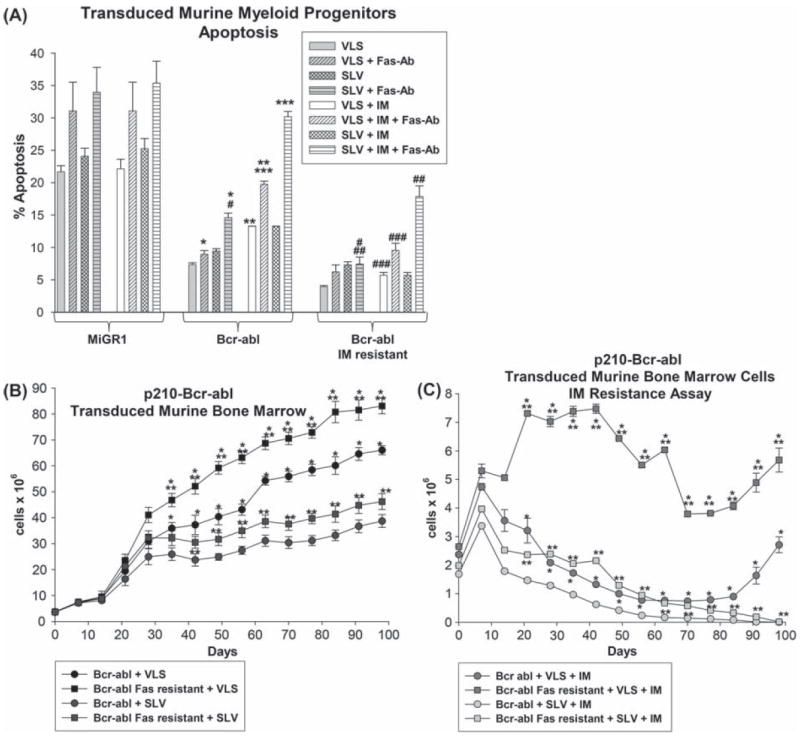
Fap1-dependent Fas insensitivity influences development of IM resistance in Bcr–abl+ myeloid progenitor cells. (A) IM resistance decreases Fas sensitivity in Bcr–abl+ primary murine myeloid progenitor cells. Primary murine bone marrow cells were transduced with a vector to express Bcr–abl or with vector control and cultured under GMP conditions. Some Bcr–abl+ cells were treated with increasing doses of IM to induce in vitro TKI resistance, as described above. Cells were pretreated with SLV versus VLS, and high-dose IM (5 μM) versus sham treatment, followed by treatment with Fas-agonist Ab (or control). Apoptosis was determined by flow cytometry for annexin V staining. Statistically significant differences in apoptosis are indicated by *, **, ***, #, ## or ### (p<0.001, n = 4). (B) Fas-resistant Bcr–abl+ primary murine myeloid progenitor cells expand more rapidly in vitro in comparison to the total Bcr–abl+ population. Primary murine bone marrow cells were transduced with a vector to express Bcr–abl and cultured under GMP conditions with SLV or VLS peptide. Prior to culture, some cells were treated with a Fas-agonist antibody, and apoptotic cells were removed (indicated as the Fas-resistant population). Viable cells were counted each week. Statistically significant differences in cell numbers at a given time point are indicated by *, ** or *** (p<0.01, n = 3). (C) Fap1-blocking SLV peptide impairs development of IM resistance by Bcr–abl+ cells in vitro. Primary bone marrow cells were transduced with a Bcr–abl expression vector and some cells were depleted of the Fas-sensitive subpopulation, as above. Cells were cultured under GMP conditions with or without an increasing dose of IM (0.2 μg/mL to 2.0 μg/mL over 6 weeks), with SLV or VLS (control) peptide in the culture. Viable cells were counted each week. Statistically significant differences in cell numbers at a given time point are indicated by *, ** or *** (p<0.01, n = 3).
Treatment of IM-naive Bcr−abl+ cells with high-dose IM increases both baseline and Fas-induced apoptosis. However, this higher IM dose has little effect on apoptosis in IM-resistant Bcr−abl+ cells (p = 0.2, n = 4) [Figure 9(A)]. Treatment with SLV peptide plus IM increases Fas-induced apoptosis in both IM-resistant and IM-naive Bcr−abl+ cells; however, the effect is significantly greater in IM-naive cells (p < 0.0001, n = 4) [Figure 9(A)].
We also used this model to investigate the role of Fap1 in development of IM resistance. As in the experiments above, murine bone marrow cells were transduced with Bcr–abl expression vector (or control vector). To determine whether the Fas-resistant subpopulation of cells is more susceptible to development of IM resistance, some Bcr−abl-transduced cells were incubated with Fas-agonist antibody and annexin V positive cells were removed prior to culture (referred to as the Fas-resistant population). Cells cultured in an increasing dose of IM were compared to cells cultured without IM (IM-naive cells). SLV or VLS peptides were added to the cultures undergoing selection and to IM-naive cultures. Cells were counted each week.
In the absence of IM, we found that incubation of Bcr−abl+ cells with SLV peptide impairs growth in comparison to VLS control peptide [Figure 9(B)]. Depletion of Fas-sensitive cells increased cell growth in Bcr−abl+ cultures treated with SLV or VLS peptide [Figure 9(B)]. Cells transduced with control vector survived in culture for ~2 weeks under all conditions (not shown).
IM treatment of Bcr−abl+ cultures significantly decreased the number of cells during initial weeks of culture under all conditions [Figure 9(C)]. Cultures with IM plus SLV peptide grew more slowly in comparison to IM plus VLS control peptide. In comparison, growth in Bcr−abl+ IM-treated cultures that were depleted of Fas-sensitive cells was greater than growth in total cell cultures (in with SLV or VLS). After several months of IM selection, control VLS peptide-treated, Bcr−abl+ cells began to increase in number [Figure 9(C)]. In contrast, IM-treated Bcr−abl+ cultures that were incubated with SLV peptide did not undergo this period of expansion [Figure 9(C)]. There were no viable cells in the IM plus SLV treated Bcr−abl+ cultures at 91 days, and no viable cells in the Fas-resistant IM plus SLV treated Bcr−abl+ at 98 days [Figure 9(C)].
Discussion
In previous studies, we determined that decreased Icsbp expression results in Fap1-dependent resistance to Fas-induced apoptosis [6]. In the present study, we show that impaired Icsbp expression in Bcr−abl+ myeloid progenitor cells contributes to Fap1-dependent Fas resistance. We demonstrate the involvement of Bcr–abl TK activity in this process, and that Fap1-dependent insensitivity to Fas-induced apoptosis increases with development of IM resistance. CML cells are recognized as being relatively resistant to Fas-induced apoptosis, but mechanisms for this were not well defined. Fas expression is not consistently decreased in CML, and expression of FasL in CML bone marrow actually increases with progressive disease [29,30]. Since Fas resistance worsens with drug resistance and BC, the latter is thought to be an unsuccessful attempt at compensation [31]. Fas resistance is hypothesized to be a source of persistence and/or expansion of CML-LSCs, even in complete cytogenetic remission [17].
Our previous studies suggest that Fap1 protects progenitor cells from Fas-induced apoptosis during normal myelopoiesis. This would be especially important in the presence of metabolic stress or increased reactive oxygen species, both of which increase FasL [32]. Metabolic stress and increased reactive oxygen species are also found during the emergency granulopoiesis response to infectious challenge. Under these conditions, Fap1 expression in GMP would permit survival and expansion of these cells as a part of the innate immune response. During myelopoiesis, decreasing Fap1 expression would permit scheduled death of circulating granulocytes and monocytes, either after activation, or diapedesis into the tissues.
In contrast, aberrantly increased Fap1 expression in CML would adversely influence myelopoiesis. The resultant Fas resistance would result in sustained survival of Bcr−abl+ GMPs/LSCs. Such cells provide a potential reservoir for the development of additional mutations that lead to overt TKI resistance or BC. In addition, increased Fap1 expression confers relative Fas resistance to circulating, post-mitotic CML cells. This would facilitate accumulation of such cells in the circulation and tissues. These results are consistent with clinical observations that the apoptosis pathway is generally “primed” in CML, but defective.
Our findings suggest that development of IM resistance in Bcr−abl+ GMPs results in decreased Icsbp expression, increased Fap1 expression and increasing Fas resistance. In previous studies, we showed that treating myeloid cells with SLV peptide decreases Fap1/Fas interaction, and increases Fas phosphorylation and apoptosis in response to Fas agonists [6]. In the present work, we find that treatment with SLV peptide abrogates Fas resistance in Bcr−abl+ cells, and this effect is enhanced by treatment with IM. In contrast, we found that Fas insensitivity of IM-resistant Bcr–abl+ cells is not efficiently reversed by treatment with Fap1-blocking peptide, with or without the addition of high-dose IM. This suggests interventions to increase Fas sensitivity via Fap1 would be most effective early in disease, as a TKI adjuvant.
We hypothesize that Fap1-dependent Fas insensitivity influences the development of TKI resistance in CML. Consistent with this, we found that incubation in Fap1-blocking SLV peptide prevents emergence of an IM-resistant, Bcr−abl+ population in vitro. Also consistent with our hypothesis, we found that depletion of Fas-sensitive cells from a Bcr−abl+ population accelerates the development of IM resistance in vitro. However, the presence of SLV peptide prevents development of IM resistance even in this Fas-resistant population, also suggesting a role for Fap1 in Fas resistance.
These studies use a peptide with the three C-terminal amino acids from Fas to block Fap1. The immediate translational applications of this work for treatment of CML are limited, because peptide therapeutics are complicated by technical concerns regarding solubility and stability. However, the fact that a tripeptide is sufficient to block the Fas/Fap1 interaction suggests that a small molecule might be developed with the same effect. Additionally, increased Fap1 has also been associated with Fas resistance in colon cancer, suggesting wider applications for targeting the Fap1/Fas interaction [33,34]. These are areas of interest for future investigations.
Acknowledgments
These studies were supported by a Merit Review Grant from the Veteran’s Administration (E.A.E.); and NIH R01 HL088747 (to E.A.E.).
Footnotes
Potential conflict of interest: Disclosure forms provided by the authors are available with the full text of this article at www.informahealthcare.com/lal.
References
- 1.Driggers PH, Ennist DL, Gleason SL, et al. An interferon gamma-regulated protein that binds the interferon-inducible enhancer element of major histocompatibility complex class I genes. Proc Natl Acad Sci USA. 1990;87:3743–3747. doi: 10.1073/pnas.87.10.3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt M, Nagel S, Proba J, et al. Lack of interferon consensus sequence binding protein transcripts in human myeloid leukemias. Blood. 1998;91:22–29. [PubMed] [Google Scholar]
- 3.Holtschke T, Lohler J, Kanno J, et al. Immuno-deficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 1996;87:307–315. doi: 10.1016/s0092-8674(00)81348-3. [DOI] [PubMed] [Google Scholar]
- 4.Eklund EA, Jalava A, Kakar R. The transcription factors PU.1, IRF-1 and ICSBP cooperate to increase gp91-phox expression. J Biol Chem. 1998;273:13957–13965. doi: 10.1074/jbc.273.22.13957. [DOI] [PubMed] [Google Scholar]
- 5.Eklund EA, Kakar R. Recruitment of CREB-binding protein by PU.1, IFN-regulatory factor 1, and the IFN consensus sequence-binding protein is necessary for IFNγ induced p67phox and gp91phox expression. J Immunol. 1999;163:6095–6105. [PubMed] [Google Scholar]
- 6.Huang W, Zhu C, Wang H, et al. The interferon consensus sequence binding protein (ICSBP/IRF8) represses PTPN13 gene transcription in differentiating myeloid cells. J Biol Chem. 2008;283:7921–7935. doi: 10.1074/jbc.M706710200. [DOI] [PubMed] [Google Scholar]
- 7.Huang W, Zhou W, Saberwal G, et al. Interferon consensus sequence binding protein (ICSBP) decreases beta-catenin activity in myeloid cells by repressing GAS2 transcription. Mol Cell Biol. 2010;30:4575–4594. doi: 10.1128/MCB.01595-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmidt M, Hochhaus A, Nitsche A, et al. Expression of ICSBP in chronic myeloid leukemia correlates with pretreatment risk features and cytogenetic response. Blood. 2001;97:3648–3650. doi: 10.1182/blood.v97.11.3648. [DOI] [PubMed] [Google Scholar]
- 9.Oncomine research edition. Ann Arbor, MA: University of Michigan; Available from: www.oncomine.org. [Google Scholar]
- 10.Konieczna I, Horvath E, Wang H, et al. Constitutive activation of SHP2 cooperates with ICSBP-deficiency to accelerate progression to acute myeloid leukemia. J Clin Invest. 2008;118:853–867. doi: 10.1172/JCI33742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X, Ren R. Bcr-Abl efficiently induces a myeloproliferative disease and production of excess interleukin-3 and granulocyte-macrophage colony-stimulating factor in mice: a novel model for chronic myelogenous leukemia. Blood. 1998;92:3829–3840. [PubMed] [Google Scholar]
- 12.Hao SX, Ren R. Expression of ICSBP is down-regulated in bcr-abl-induced murine CML-like disease and forced co-expression of ICSBP inhibits bcr-abl-induced MPD. Mol Cell Biol. 2000;20:1149–1161. doi: 10.1128/mcb.20.4.1149-1161.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu CL, Saberwal G, Lu YF, et al. The interferon consensus sequence binding protein (ICSBP) activates transcription of the gene encoding Neurofibromin 1 (NF1) J Biol Chem. 2004;279:50874–50885. doi: 10.1074/jbc.M405736200. [DOI] [PubMed] [Google Scholar]
- 14.Saras J, Claesson-Welsh L, Heldin CH, et al. Cloning and characterization of PTPL1, a PTP with similarities to cytoskeletal-associated proteins. J Biol Chem. 1994;269:24082–24089. [PubMed] [Google Scholar]
- 15.Yanagisawa J, Takahashi M, Kanki H, et al. The molecular interaction of Fas and Fap1. J Biol Chem. 1997;272:8539–8545. doi: 10.1074/jbc.272.13.8539. [DOI] [PubMed] [Google Scholar]
- 16.Roeder I, Horn M, Glauche I, et al. Dynamic modeling of imatinib-treated CML: functional insights & clinical implications. Nat Med. 2006;12:1181–1184. doi: 10.1038/nm1487. [DOI] [PubMed] [Google Scholar]
- 17.Michor F, Hughes TP, Iwasa Y, et al. Dynamics of CML. Nature. 2005;435:1267–1270. doi: 10.1038/nature03669. [DOI] [PubMed] [Google Scholar]
- 18.Larrick JW, Fischer DG, Anderson SJ, et al. Characterization of a human macrophage-like cell line stimulated to differentiate in vitro: a model of macrophage functions. J Immunol. 1980;125:6–12. [PubMed] [Google Scholar]
- 19.Wang H, Lindsey S, Konieczna I, et al. Constitutively active SHP2 cooperates with HoxA10-overexpression to induce AML. J Biol Chem. 2009;284:2549–2567. doi: 10.1074/jbc.M804704200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang W, Hu L, Bei L, et al. The leukemia-associated fusion protein Tel-platelet-derived growth factor receptor β (Tel-PdgfRβ) inhibits transcriptional repression of PTPN13 gene by interferon consensus sequence binding protein (Icsbp) J Biol Chem. 2012;287:8110–8125. doi: 10.1074/jbc.M111.294884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisterer W, Jiang X, Christ O, et al. Different subsets of primary CML stem cells engraft immuno-de3 cient mice and produce a model of the human disease. Leukemia. 2005;19:435–441. doi: 10.1038/sj.leu.2403649. [DOI] [PubMed] [Google Scholar]
- 22.Jamieson CHM, Ailles LE, Dylla SJ, et al. Granulocyte-macrophage progenitors as candidate leukemia stem cells in blast crisis CML. N Engl J Med. 2004;351:657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 23.Minami Y, Stuart SA, Ikawa T, et al. Bcr/abl-transformed GMP as myeloid leukemia stem cells. Proc Natl Acad Sci USA. 2008;105:17967–17972. doi: 10.1073/pnas.0808303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drucker BJ, Guilhot F, O’Brien SG, et al. Five-year follow up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 25.Okimoto RA, Van Etten R. Road toward optimal initial therapy for chronic myeloid leukemia. Cur Opin Hematol. 2011;18:89–97. doi: 10.1097/MOH.0b013e32834399a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cortes J, Hochhaus A, Hughes T, et al. Front-line and salvage therapies with tyrosine kinase inhibitors and other treatments in chronic myeloid leukemia. J Clin Oncol. 2011;29:524–531. doi: 10.1200/JCO.2010.31.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitchell B, Deninger M. Risk stratification in newly diagnosed CML. Leuk Lymphoma. 2011;52:4–11. doi: 10.3109/10428194.2010.546916. [DOI] [PubMed] [Google Scholar]
- 28.Miething C, Grundler R, Mugler C, et al. Retroviral insertional mutagenesis identifies RUNX genes involved in chronic myeloid leukemia disease persistence under imatinib treatment. Proc Natl Acad Sci USA. 2007;104:4594–4599. doi: 10.1073/pnas.0604716104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turkina AG, Logacheva NP, Stromskaya TP, et al. Studies of some mechanisms of drug resistance in CML. Adv Exp Med Biol. 1999;457:477–488. doi: 10.1007/978-1-4615-4811-9_52. [DOI] [PubMed] [Google Scholar]
- 30.Kim KM, Lee K, Hong YS, et al. Fas-mediated apoptosis and expression of related genes in human malignant hematopoietic cells. Exp Mol Med. 2000;32:246–254. doi: 10.1038/emm.2000.41. [DOI] [PubMed] [Google Scholar]
- 31.Lickliter JD, Kratzke RA, Nguyen PL, et al. Fas ligand is highly expressed in acute leukemia and during the transformation of chronic myeloid leukemia to blast crisis. Exp Hematol. 1999;27:1519–1527. doi: 10.1016/s0301-472x(99)00091-0. [DOI] [PubMed] [Google Scholar]
- 32.Liu WH, Cheng YC, Chang LS. ROS-mediated p38alpha MAPK activation and ERK inactivation responsible for upregulation of Fas and FasL and autocrine Fas-mediated cell death in phospholipase A(2)-treated U937 cells. J Cell Physiol. 2009;219:642–651. doi: 10.1002/jcp.21713. [DOI] [PubMed] [Google Scholar]
- 33.Xiao ZY, Wu W, Eagleton N, et al. Silencing Fas-associated phosphatase 1 expression enhances efficiency of chemotherapy for colon carcinoma with oxaliplatin. World J Gastroenterol. 2010;16:112–118. doi: 10.3748/wjg.v16.i1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao H, Song E, Chen J, et al. Expression of Fap1 by human colon adenocarcinoma: implication for resistance against Fas-mediated apoptosis in cancer. Br J Cancer. 2004;91:1718–1725. doi: 10.1038/sj.bjc.6602136. [DOI] [PMC free article] [PubMed] [Google Scholar]