

Abstract
Following the biosynthesis of polyketide backbones by polyketide synthases (PKSs), post-PKS modifications result in a significantly elevated level of structural complexity that renders the chemical synthesis of these natural products challenging. We report herein a total synthesis of the widely used polyketide insecticide spinosyn A by exploiting the prowess of both chemical and enzymatic methods. As more polyketide biosynthetic pathways are characterized, this chemoenzymatic approach is expected to become readily adaptable to streamlining the synthesis of other complex polyketides with more involved post-PKS modifications.
Keywords: polyketide macrolide, polyketide biosynthesis, post-polyketide synthase (PKS) modifications, chemoenzymatic synthesis, spinosyns
Natural products are rich sources of lead compounds for the development of new pharmaceuticals. Yet, their structural complexity renders their chemical synthesis challenging. There is a pressing need to develop efficient strategies for the synthesis of natural product-based or -like libraries for the drug discovery program.[1,2]
Macrolide polyketides are an important class of natural products having widespread clinical applications. Due to their complex structures, this class of secondary metabolites have often been targets for synthetic chemists to showcase newly developed methods. Over the years, chemical approaches to prepare macrolides have improved significantly to the point where Krische et al. reported a 14-step synthesis of deoxyerythronolide B in 2013.[3] Such remarkable innovations have greatly advanced the synthesis of linear polyketides and macrolactone backbones, which are biosynthetically assembled through the catalysis of polyketide synthases (PKSs).[4] However, many polyketides possess additional structural sophistication due to various post-PKS modifications of the initially formed linear and monocyclic structures.[5] Syntheses of polyketides whose structures are derived from a series of complicated post-PKS modifications are much more demanding.
Spinosyn A (1), a commercially important, polyketide-derived insecticide isolated from Saccharopolyspora spinosa,[6] consists of four fused rings, seventeen stereogenic centers, and multiple functional groups, including two unusual carbohydrate moieties, all of which are built through post-PKS modifications. Total syntheses of this complex framework have been reported by the Evans, Paquette, and Roush groups.[7-10] A Diels-Alder reaction (Evans, Roush) or an oxy-Cope rearrangement (Paquette) was employed to construct the octahydro-as-indacene core in these syntheses. Since, in all cases, the stereochemical control was directed by the conformation of the reactant, a major challenge of these early syntheses was the development of effective routes to assemble the stereocontrolling template motifs. To streamline the synthesis of more complex polyketides, We envisioned that the challenge of devising strategies for the stereo- and regiochemical control may better be met by using the corresponding biosynthetic enzymes.[11,12]
The entire biosynthetic pathway of spinosyn A has been elucidated as shown in Scheme 1.[13-19] Post-PKS modifications of the aglycone core commence following release of macrolactone 11 from the PKS acyl carrier protein. The monocyclic intermediate 11 is first primed via the SpnJ-catalyzed dehydrogenation at C-15 (11→12),[15] which facilitates the subsequent 1,4-dehydration catalyzed by SpnM.[19] The resulting intermediate 13 is susceptible to a transannular [4+2]-cycloaddition reaction (13 → 14) that can be accelerated by SpnF.[19] This cyclization step to form the tricyclic hexahydro-1H-indene intermediate 14 has attracted much attention, because SpnF may operate as a Diels-Alderase.[19-21]
Scheme 1.
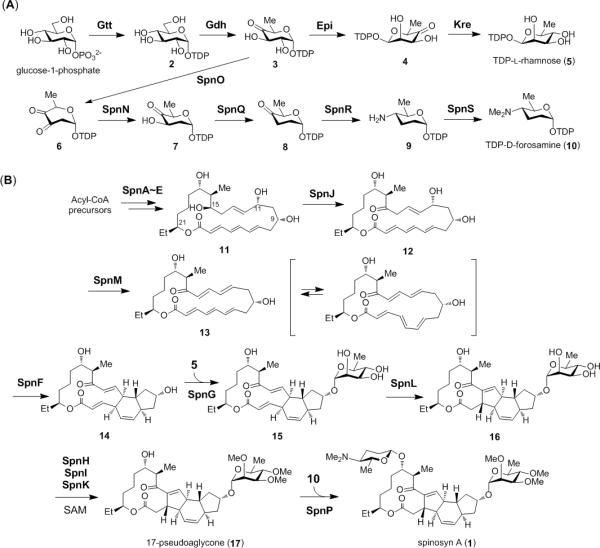
Established biosynthetic pathway for spinosyn A (1).
The C-C bond formation between C-2 and C-14, which is catalyzed by SpnL, occurs after the rhamnosylation of 14 via the action of SpnG.[17,18,22] This affords the tetracyclic core in 16, and the attached rhamnose moiety is subsequently permethylated in the presence of the SAM-dependent methyltransferases SpnH, SpnI, and SpnK to produce pseudoaglycone 17.[18] SpnP completes the biosynthesis of spinosyn A by catalyzing the final coupling of 17 with forosamine.[13,23] The two sugar appendages (rhamnose and forosamine) are both biosynthesized from TDP-D-glucose (2) through pathways that have been fully established (see Scheme 1A).[14,16,24]
Considering the wealth of information available in the chemical literature concerning the biosynthesis of polyketides, we envisioned that a chemoenzymatic synthesis, which exploits advantages offered by both chemical and biological approaches, would provide a practical alternative to conventional chemical synthesis for the assembly of complex polyketide structures. Herein, we report the first chemoenzymatic synthesis of spinosyn A encompassing chemical preparation of the spinosyn polyketide backbone and subsequent application of post-PKS modification enzymes to complete the final construction. Our results demonstrate that a “one pot, multi-step” chemoenzymatic synthesis is a viable and effective approach to prepare complex polyketide natural products.
The first phase of our synthesis is the preparation of macrolactone 11, which is biosynthesized via the actions of five polyketide synthases SpnA–E. Although 11 has never been isolated, its structure was deduced according to the established principles of linear polyketide chain assembly by type I PKSs[4] as well as the hydroxyl group stereochemistry prediction based on sequence analysis of the ketoreductase domains.[25] The retro-synthetic analysis for the preparation of 11 is depicted in Scheme 2. The linear polyketide chain 18 is assembled from three fragments via two C-C bond coupling reactions: a Julia-Kocieński olefination between sulfone 19 (fragment A) and aldehyde 20 (fragment B) to form the Δ12-(E)-olefin, and a palladium-catalyzed Stille coupling using stannyl dienoate 21 (fragment C) to generate the C-5/C-6 linkage. Subsequent cyclization of 18 by Yamaguchi macrolactonization completes the synthesis of 11 following global deprotection.
Scheme 2.

Retrosynthetic analysis.
Synthesis of fragment A (19) was initiated by applying Soai's asymmetric ethylation to Weinreb amide 22[26] using (−)-DBNP as the chiral catalyst.[27] Mosher ester analysis of product 23 confirmed the (S)-configuration at C-21. Aldehyde 24, prepared by O-silylation of 23 followed by DIBAL-H reduction, was utilized in an asymmetric aldol reaction with propionate 25, carrying a Crimmins' chiral auxiliary.[28] The anticipated syn-aldol adduct 26 was obtained in good yield with high diastereoselectivity (>19:1). The auxiliary group of 26 was reductively cleaved after O-silyl protection and the resulting hydroxyl group was oxidized using TPAP to afford aldehyde 27. Incorporation of a homoallyl extension was achieved by Brown's asymmetric allylation methodology.[29,30] The desired homoallylic alcohol 28 was produced as a single isomer at C-15.[31,32] The terminal olefin of 28 was transformed to a primary alcohol by Lemieux-Johnson oxidation under buffered conditions (pH 7) followed by NaBH4 reduction. Introduction of a 1H-phenyltetrazolyl-5-thioether moiety onto 29 to give 30 was accomplished under Mitsunobu conditions. Conversion of 30 to sulfone 31 was found problematic as common oxidants gave either a partially oxidized sulfoxide or no product at all. The desired sulfone was eventually obtained in 91% yield after overnight incubation at 4 °C with H2O2 in the presence of an ammonium molybdate catalyst.[33] Although cleavage of the TES group at C-21 was observed, it could be re-introduced quantitatively to complete the synthesis of fragment A (19).
Preparation of fragment B (20) started with regioselective epoxide opening of PMB-protected (S)-glycidol (32) using lithiated 1,3-dithiane to give 33. After O-silyl protection followed by hydrolysis of the 1,3-dithiane moiety, another application of Brown's allylation methodology afforded 35 with the desired stereochemistry at C-9 (d.r. > 20:1). Incorporation of the terminal vinyl iodide group began with TBS-protection of 35 followed by oxidative cleavage of the terminal olefin by Jin's procedure.[34] The resulting aldehyde 36 was subjected to Takai iodoolefination[35] providing 37 with >9:1 (E):(Z) stereoselectivity. Subsequent oxidative removal of the PMB ether followed by Dess-Martin oxidation completed the synthesis of fragment B (20).
Preparation of fragment C (21) was straightforward as shown in Scheme 4A. Regioselective addition of tributytin radical to propargyl alcohol (38) afforded (E)-vinylstannane 39. Oxidation of the hydroxyl group followed by Horner-Wadsworth-Emmons olefination of the resulting aldehyde furnished dienoate 21 in excellent overall yield. With all fragments in hand, a Julia-Kocieński olefination connecting 19 and 20 was achieved using KHMDS at –78 °C to give the desired product 40 with (E)-selectivity in 82% yield (Scheme 4B). Then, a palladium-catalyzed Stille coupling between 40 and fragment C (21) effectively generated the linear ketide 18. The final steps to complete the synthesis of 11 involved chemoselective deprotection of the TES group at C-21, hydrolysis of the ethyl ester moiety to afford seco-acid 41, and lactonization under Yamaguchi conditions to give macrolactone 42.[36] The finishing global deprotection of 42 proved to be more challenging encountering partial deprotection or decomposition under various conditions. We were pleased to find that treatment of 42 with HF•pyridine in ethanol at 4 °C for ca. 4 days eventually led to successful production of 11 in satisfactory yield (64%).
Scheme 4.

(A) Synthesis of fragment C (21) and (B) completion of preparation of 11. Reagents and conditions: a) Bu3SnH, AIBN, benzene, reflux, 50%; b) MnO2, acetone, rt, 85%; c) EtO2CCH2P(O)(OEt)2, NaH, THF, 0 °C to rt, 74%; d) KHMDS, THF, −78 °C, 82%; e) 21, Pd2(dba)3, Ph3As, DMF, rt, 70%; f) PPTS, EtOH, 0 °C; g) LiOH, THF, CH3OH, H2O, reflux, 2 steps 62%; h) 2,4,6-Cl3C6H3COCl, Et3N, THF; DMAP, toluene, rt, 75%; o) HF•pyr, EtOH, 4 °C, 4 days, 64%.
In the second phase of our synthesis, we opted to take advantage of our knowledge regarding spinosyn biosynthesis and the availability of all post-PKS tailoring enzymes, and apply a series of enzymatic transformations in one-pot to convert macrolactone 11 to 17-pseudoaglycone (17). As shown in Figure 1A, the genes responsible for post-PKS modifications in the spinosyn pathway are translationally controlled by at least four operons (operons I–IV).[13] In our previous study of O-methylation of the rhamnose moiety in spinosyn A, it was found that prudent coordination of the three rhamnose methyltransferases in vitro could generate distinct methylation patterns of rhamnose.[18] In fact, accumulation of mono- and di-methylated products could be avoided when 10 μM SpnI, 5 μM SpnK, and 1 μM SpnH were used to permethylate rhamnose. These results suggested that the in vivo control of rhamnose methylation is likely achieved via differential expression of these methyltransferase genes, spnH, spnI and spnK, which are located on three different operons (III, II, and I, respectively, see Figure 1A).[18]
Figure 1.

(A) The organization of putative operons encoding enzymes responsible for the post-PKS modifications in the spinosyn biosynthetic gene cluster. (B) HPLC analysis of the enzymatic tandem reactions to convert 11 to 17; 11 alone (a), enzyme reaction mixture (b), and 17
Hence, proper adjustment of the relative concentrations of enzymes catalyzing the tailoring reactions of spinosyn A biosynthesis may also be crucial to minimizing the possible accumulation of dead-end intermediates in a one-pot conversion of 11 to 17. While the actual mechanism for in vivo metabolic flux control is likely to be more complicated, we proceeded on the assumption that the expression levels of the genes (i.e., the concentration of the encoded enzymes) from the same operon would be similar. Thus, guided by results from the rhamnose permethylation work, the concentrations of enzymes from operon I (SpnM, SpnL, SpnK, and SpnJ) were set at 5 μM, those from operon II (SpnI) at 10 μM, and those from operon III (SpnH, SpnG) at 3 μM.[32]
SpnF catalyzes the [4+2] cycloaddition of 13 to yield 14 (Scheme 1).[19] The cyclization could also occur in the absence of SpnF, albeit at a reduced rate. Since 13 is susceptible to Michael addition by cellular nucleophiles and/or radicals, the physiological function of SpnF may be to prevent the formation of byproducts from such off-path reactions by accelerating the cycloaddition step. Since SpnF is the sole gene encoded in operon IV, the proper concentration of SpnF used in the incubation must be determined separately. Accordingly, a model system was devised in which the product profiles and yields of a series of incubations containing 12 and TDP-L-rhamnose with SpnM, SpnG, SpnL and varied concentrations of SpnF (0 to 20 μM) were analyzed. Our results showed that addition of 20 μM of SpnF clearly suppressed the formation of minor byproducts and elevated the yield of the tetracyclic octahydro-as-indacene product (Figure S1 and S2).[32] Hence, the concentration of SpnF in the in vitro experiment was set at 20 μM.
Having the concentrations of all enzymes required for post-PKS modifications adjusted, the one-pot reaction was conducted by incubation of 1 mM 11, excess SAM, and TDP-L-rhamnose (5) with the aforementioned enzymes in 50 mM Tris•HCl buffer (pH 8) at 30 °C. As shown in Figure 1B, transformation of 11 to product 17 was achieved with an overall conversion yield estimated to be 19.6% (average yield per step = 81.6%) based on HPLC analysis.[32]
To complete the synthesis of spionsyn A (1), the attachment of forosamine at C-17 of 17 was attempted using SpnP, which is the glycosyltransferase assigned for this transformation.[13] Specifically, SpnP was incubated with all enzymes involved in TDP-D-forosamine biosynthesis (SpnO, SpnN, SpnQ, SpnR, and SpnS),[14,16] TDP-4-keto-6-deoxyglucose (3), and 17 in one-pot. Unfortunately, production of 1 was not observed. Further sequence alignment and crystal structural analysis suggested that SpnP belongs to a group of glycosyltransferases requiring an auxiliary protein for activation.[23] However, no putative auxiliary protein gene could be found in the spinosyn biosynthetic gene cluster. Thus, the failure of SpnP to forosamylate 17 might be due to the absence of the cognate auxiliary protein to reconstitute its activity in vitro. Consequently, chemical glycosylation was adopted instead, and treatment of 17 and D-forosamine with BF3•OEt2 successfully led to spinosyn A (1).[32]
In summary, a chemoenzymatic strategy was effectively applied in our synthesis of spinosyn A. Construction of monocyclic precursor 11 was achieved chemically by assembling three synthesized fragments in a linear form followed by a controlled macrolactonization. The more challenging post-PKS modifications converting 11 to 17 were accomplished using a total of eight enzymes all in one-pot. While the overall enzymatic transformations were highly effective, it was rather unfortunate that direct transformation of 11 to spinosyn A (1) was hampered by missing the putative auxiliary protein for SpnP. This prevented the exploitation of the full capacity of the spinosyn biosynthetic machinery. Nevertheless, the feasibility of a chemoenzymatic synthesis of a complex polyketide-derived natural product has been demonstrated. When compared to the reported chemical syntheses of spinosyn A, our chemoenzymatic approach is not without its shortcomings. In particular, an in-depth understanding of the biosynthetic pathway is an essential prerequisite with its own set of challenges. Thus, it may be premature at the present time to claim a general applicability of the chemoenzymatic approach for the synthesis of complex natural products. However, continuing innovation in the fields of synthetic chemistry and natural product biosynthesis indicate that the current technical impediments to chemoenzymatic synthesis will be overcome before long. This approach is thus expected to become more readily adaptable in the future, and offer a valuable alternative for streamlining the synthesis of polyketides with more involved post-PKS modifications as well as the preparation of modified polyketide targets for mechanistic and pharmaceutical research.
Supplementary Material
Scheme 3.

Synthesis of (A) fragment A (19) and (B) fragment B (20). Reagents and conditions: a) (−)-DBNP, Et2Zn, hexanes, 0 °C, 66%, 92% ee; b) TESOTf, 2,6-lutidine, CH2Cl2, −78 °C; c) i-Bu2AlH, toluene, −78 °C, 2 steps 78%; d) TiCl4, (−)-sparteine, CH2Cl2, 0 °C, 84%; e) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C; f) LiBH4, Et2O, CH3OH, 0 °C, 2 steps 75%; g) TPAP, NMO, 4Å MS, CH2Cl2, rt; h) (+)-Ipc2B(allyl), Et2O, −78 °C, 2 steps 60%; i) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 87%; j) OsO4, NMO, THF, acetone, pH 7 buffer, rt; k) NaIO4, THF, pH 7 buffer, rt; l) NaBH4, EtOH, rt, 3 steps 67%; m) PTSH, PPh3, DIAD, THF, rt, 86%; n) (NH4)6Mo7O24, H2O2, EtOH, H2O, 4 °C; o) TESOTf, 2,6-lutidine, CH2Cl2, −78 °C, 2 steps 91%; p) 1,3-dithiane, n-BuLi, THF, 0 °C, 94%; q) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 95%; r) MeI, CaCO3, CH3CN, H2O, reflux, quantitative; s) (+)-Ipc2B(allyl), THF, −78 °C; t) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 2 steps 67%; u) OsO4, NaIO4, 2,6-lutidine, dioxane, H2O, rt, 83%; v) CrCl2, HCI3, dioxane, THF, rt, 87%; w) DDQ, aq CH2Cl2, rt; x) Dess-Martin periodinane, CH2Cl2, rt, 2 steps 83%.
Acknowledgments
** This work was supported by grants from the National Institutes of Health (GM035906 and GM040541) and the Welch Foundation (F-1511).
References
- [1].Bauer RA, Wurst JM, Tan DS. Curr. Opin. Chem. Biol. 2010;14:308–314. doi: 10.1016/j.cbpa.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Dandapani S, Marcaurelle LA. Nat. Chem. Biol. 2010;6:861–863. doi: 10.1038/nchembio.479. [DOI] [PubMed] [Google Scholar]
- [3].Gao X, Woo SK, Krische MJ. J. Am. Chem. Soc. 2013;135:4223–4226. doi: 10.1021/ja4008722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hertweck C. Angew. Chem. Int. Ed. 2009;48:4688–4716. doi: 10.1002/anie.200806121. [DOI] [PubMed] [Google Scholar]
- [5].Olano C, Méndez C, Salas JA. Nat. Prod. Rep. 2010;27:571–616. doi: 10.1039/b911956f. [DOI] [PubMed] [Google Scholar]
- [6].Kirst HA. J. Antibiot. 2010;63:101–111. doi: 10.1038/ja.2010.5. [DOI] [PubMed] [Google Scholar]
- [7].Evans D, Black W. J. Am. Chem. Soc. 1993;115:4497–4513. [Google Scholar]
- [8].Paquette L, Gao Z, Ni Z, Smith G. J. Am. Chem. Soc. 1998;120:2543–2552. [Google Scholar]
- [9].Paquette L, Collado I, Purdie M. J. Am. Chem. Soc. 1998;120:2553–2562. [Google Scholar]
- [10].Mergott DJ, Frank SA, Roush WR. Proc. Natl. Acad. Sci. USA. 2004;101:11955–11959. doi: 10.1073/pnas.0401247101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mortison JD, Sherman DH. J. Org. Chem. 2010;75:7041–7051. doi: 10.1021/jo101124n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kirschning A, Hahn F. Angew. Chem. Int. Ed. 2012;51:4012–4022. doi: 10.1002/anie.201107386. [DOI] [PubMed] [Google Scholar]
- [13].Waldron C, Matsushima P, Rosteck PR, Broughton MC, Turner J, Madduri K, Crawford KP, Merlo DJ, Baltz RH. Chem. Biol. 2001;8:487–499. doi: 10.1016/s1074-5521(01)00029-1. [DOI] [PubMed] [Google Scholar]
- [14].Hong L, Zhao Z, Liu H.-w. J. Am. Chem. Soc. 2006;128:14262–14263. doi: 10.1021/ja0649670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kim HJ, Pongdee R, Wu Q, Hong L, Liu H.-w. J. Am. Chem. Soc. 2007;129:14582–14584. doi: 10.1021/ja076580i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hong L, Zhao Z, Melançon CE, Zhang H, Liu H.-w. J. Am. Chem. Soc. 2008;130:4954–4967. doi: 10.1021/ja0771383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen Y-L, Chen Y-H, Lin Y-C, Tsai K-C, Chiu H-T. J. Biol. Chem. 2009;284:7352–7363. doi: 10.1074/jbc.M808441200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kim HJ, White-Phillip JA, Ogasawara Y, Shin N, Isiorho EA, Liu H.-w. J. Am. Chem. Soc. 2010;132:2901–2903. doi: 10.1021/ja910223x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kim HJ, Ruszczycky MW, Choi S-H, Liu Y-N, Liu H.-w. Nature. 2011;473:109–112. doi: 10.1038/nature09981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Oikawa H. B. Chem. Soc. Jpn. 2005;78:537–554. [Google Scholar]
- [21].Kim HJ, Ruszczycky MW, Liu H.-w. Cur. Opin. Chem. Biol. 2012;16:124–131. doi: 10.1016/j.cbpa.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Isiorho EA, Liu H.-w., Keatinge-Clay AT. Biochemistry. 2012;51:1213–1222. doi: 10.1021/bi201860q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Isiorho EA, Jeon B-S, Kim NH, Liu H.-w., Keatinge-Clay AT. Biochemistry. 2014;53:4292–4301. doi: 10.1021/bi5003629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Madduri K, Waldron C, Matsushima P, Broughton MC, Crawford K, Merlo DJ, Baltz RH. J. Ind. Microbiol. Biotechnol. 2001;27:399–402. doi: 10.1038/sj.jim.7000180. [DOI] [PubMed] [Google Scholar]
- [25].Siskos AP, et al. Chem. Biol. 2005;12:1145–1153. doi: 10.1016/j.chembiol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- [26].Molander GA, McWilliams JC, Noll BC. J. Am. Chem. Soc. 1997;119:1265–1276. [Google Scholar]
- [27].Soai K, Yokoyama S, Hayasaka T, Ebihara K. Chem. Lett. 1988:843–846. [Google Scholar]
- [28].Crimmins MT, King BW, Tabet EA, Chaudhary K. J. Org. Chem. 2001;66:894–902. doi: 10.1021/jo001387r. [DOI] [PubMed] [Google Scholar]
- [29].Brown HC, Randad RS, Bhat KS, Zaidlewicz M, Racherla US. J. Am. Chem. Soc. 1990;112:2389–2392. [Google Scholar]
- [30].Racherla US, Brown HC. J. Org. Chem. 1991;56:401–404. [Google Scholar]
- [31].Rychnovsky SD, Rogers BN, Richardson TI. Acc. Chem. Res. 1998;31:9–17. [Google Scholar]
- [32]. See supporting information.
- [33].Paquette LA, Chang S-K. Org. Lett. 2005;7:3111–3114. doi: 10.1021/ol0511833. [DOI] [PubMed] [Google Scholar]
- [34].Yu W, Mei Y, Kang Y, Hua Z, Jin Z. Org. Lett. 2004;6:3217–3219. doi: 10.1021/ol0400342. [DOI] [PubMed] [Google Scholar]
- [35].Takai K, Nitta K, Utimoto K. J. Am. Chem. Soc. 1986;108:7408–7410. doi: 10.1021/ja00279a068. [DOI] [PubMed] [Google Scholar]
- [36].Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. B. Chem. Soc. Jpn. 1979;52:1989–1993. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.