

Abstract
The inflammasome is a highly regulated protein complex that triggers caspase-1 activation and subsequent secretion of IL-1β and IL-18. Recognition of microbial components and danger signals by NOD-like receptor (NLR) family members in the cytosol promotes inflammasome activation and downstream inflammatory cytokine production. Pathogen recognition by NLRs and downstream release of inflammasome-derived cytokines are important in host defense against numerous infections. Recent studies have also identified a unique role for inflammasome regulation in the induction and pathogenesis of multiple autoimmune and inflammatory disorders. We now know that obesity-related factors and endogenous markers of cellular stress can lead to unchecked activation of the inflammasome and provoke inflammation and subsequent destruction of vital organs. This review will highlight recent findings that link inflammasome signaling to the progression of autoinflammatory and autoimmune diseases. We will focus on the contribution of inflammasome activation to the pathogenesis of autoinflammatory and autoimmune diseases that are of major significance to human health including type 2 diabetes, atherosclerosis, multiple sclerosis and type 1 diabetes.
Keywords: Obesity, atherosclerosis, multiple sclerosis, type 2 diabetes, type 1 diabetes, NLR, Nlrp3 inflammasome, caspase-1
Introduction
Over the past 10 years we have witnessed the exciting identification of a new class of pattern-recognition molecules known as the Nod-like receptor (NLR) family of molecules. Structurally NLR proteins contain a protein-protein binding domain (either a CARD or pyrin domain), a centrally located NACHT effector domain, and a C-terminal domain containing leucine-rich repeats. To date, 22 NLR family genes have been identified in the human genome and at least 34 NLR related genes have been discovered in mice (Ting et al., 2008). NLRs recognize both pathogen- and danger-associated molecular patterns and are thus important sensors of cellular stress that result from infection and cellular instability (Franchi et al., 2006; Kanneganti, 2010; Kanneganti et al., 2007; Lamkanfi & Kanneganti, 2010; Lamkanfi et al., 2007; Shaw et al., 2010; Shaw et al., 2011; Zaki et al., 2011). Activation of the NLR proteins NLRP3, NLRP1B, and NLRC4 as well as a recently identified HIN-200 protein absent in melanoma 2 (AIM2) results in the recruitment of the inflammasome-adaptor protein, ASC (also known as PYCARD), and pro-caspase-1 into a molecular platform known as the inflammasome (Figure 1). These multi protein complexes mediate the proximity-induced autoactivation of caspase-1. Active caspase-1 subsequently cleaves pro-IL-1β and pro-IL-18, which is required for their secretion and inflammatory properties. In addition to tightly controlling the activation of IL-1β and IL-18, inflammasome signaling can also influence other important biological processes including autophagy and cell death.
Figure 1. Activation of the inflammasome.
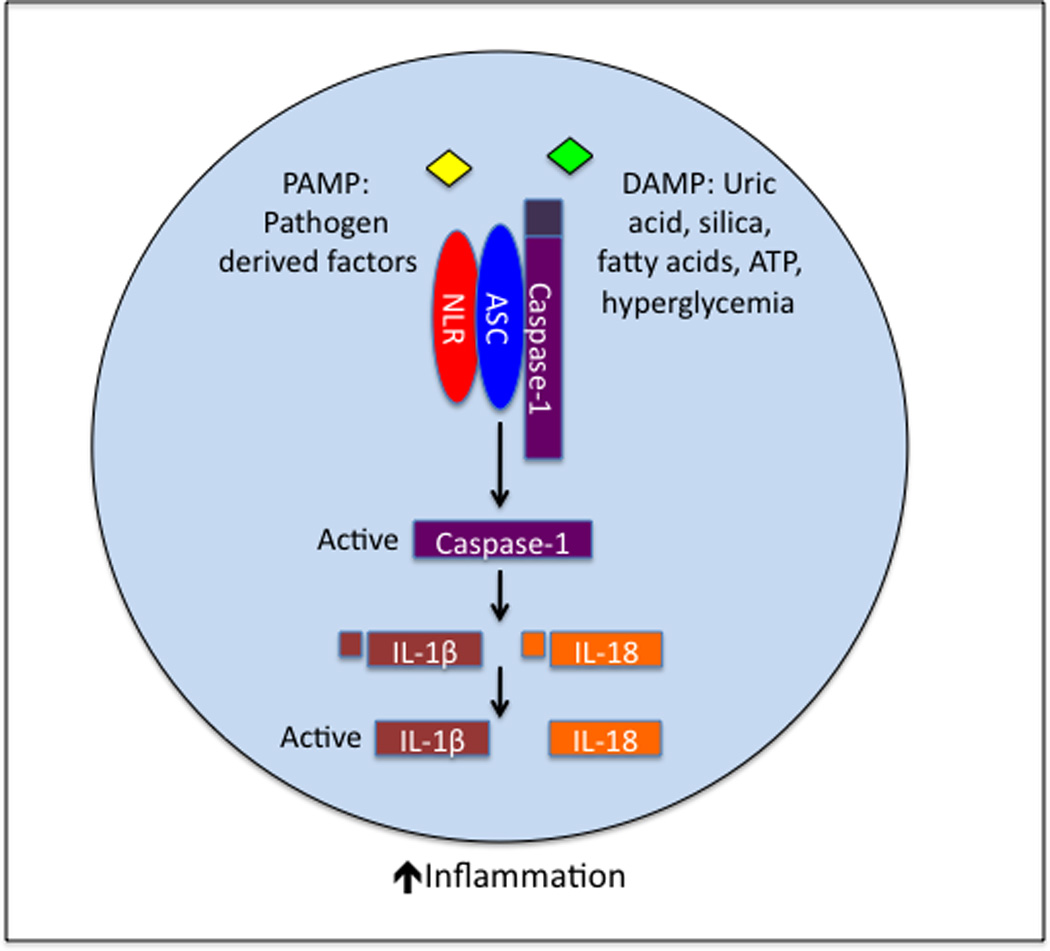
Pathogen- and danger-associated molecular patterns (PAMPs and DAMPs, respectively) are detected by NLRs in the cytosol. Once activated, the NLR undergoes a conformational change, resulting in the recruitment of ASC and caspase-1 into a protein complex known as the inflammasome. Formation of the inflammasome molecular platform triggers self-cleavage and activation of caspase-1. Active caspase-1 subsequently cleaves the pro-forms of IL-1β and IL-18, which is required for their secretion and biological activity. Inflammasome-derived cytokines (IL-1β and IL-18) are potent mediators of inflammation and immune cell activation. Their immunogenic activity plays a central role in host defense against numerous pathogens. However, unchecked regulation of IL-1β and IL-18 activation in responses to DAMPs (e.g., cellular and metabolic distress, environmental insults, metabolites, etc.) results in chronic inflammation and contributes to the induction and pathogenesis of numerous inflammatory and autoimmune diseases.
Both IL-1β and IL-18 are potent mediators of inflammation and immune responses. IL-1β secretion triggers production of IL-6 and TNF-α, which in turn elicits cell migration and immune cell infiltration into tissue (Sims & Smith, 2010). Furthermore IL-1β also promotes the generation and maintenance of IFN-γ and IL-17 producing T cells, which are required for the pathogenesis of numerous autoimmune diseases including multiple sclerosis and type 1 diabetes (Ben-Sasson et al., 2009). IL-18 is also a proinflammatory cytokine that incites immune cell recruitment and activation (Dinarello, 2007). Specifically, IL-18 can directly influence natural killer (NK) cell and T cell effector responses (Dinarello, 1994; Nakanishi et al., 2001).
Similar to Toll-like receptors (TLRs), NLRs also recognize pathogen-derived molecules and are involved in the first line of defense during infection (Kanneganti, 2010; Lamkanfi et al., 2007). In contrast to TLRs that bind pathogen ligands on the cell surface and in vesicles, NLRs respond to molecules and stress signals in the cytosol. The importance of NLR-mediated recognition of pathogens to the generation of protective host responses has been reviewed extensively in the literature. This article will instead focus on NLR mediated activation of the inflammasome in response to specific danger-associated molecular patterns (DAMPs) and in the induction and progression of autoinflammatory, autoimmune and obesity-associated diseases (Shaw et al., 2011). DAMPs that trigger inflammasome activation include exogenous stress-inducing agents (asbestos, silica, and alum), endogenous instigators of cellular and metabolic distress (ATP, uric acid, and mitochondrial dysfunction) and obesity-related factors (fatty acids, ceramides, ROS and hyperglycemia).
Mutations in NLR-inflammasome proteins are associated with both monogenic and polygenic human inflammatory disorders. Most of these rare genetic disorders are caused by mutations that result in enhanced inflammasome activation. For instance, mutations that cause abnormal activation of the NLRP3 inflammasome are collectively known as the cryopyrin-associated periodic syndromes (CAPS). This family of diseases clinically manifests as episodes of fever, rash, joint pain, systemic amyloidosis, central nervous system impairments, and joint and bone deformations. Many of these genetic inflammasome disorders that result from uncontrolled production of proinflammatory cytokines are responsive to IL-1R antagonist treatment (Church & McDermott, 2009; Hoffman et al., 2004). The involvement of inflammasome signaling in these rare disorders has been extensively described elsewhere (Aksentijevich et al., 2007; Davis et al., 2011). Rather, the goal of this review is to highlight the emerging involvement of the NLR-inflammasome axis in major inflammatory and autoimmune diseases that are of significant threat to human health. We will focus on recent findings in the field that demonstrate a pivotal role for inflammasome regulation in obesity driven inflammatory disorders (insulin-resistance, type 2 diabetes and atherosclerosis) and autoimmune diseases (multiple sclerosis and type 1 diabetes).
Obesity
The ability of adipose tissue to expand in response to chronic caloric excess is a critical adaptive response. In obese individuals, adipose tissue can constitute up to 50% of total body mass. In addition to its function as an energy storage site, adipose tissue also releases hormone-like mediators known as adipokines that are involved in the regulation of energy balance and insulin-sensitization. Obesity is associated with self-directed tissue inflammation where local or systemic factors other than infectious agents activate the cells of innate immune system. During the development of obesity, adipocytes undergo considerable differentiation and expansion to store lipids (Shoelson et al., 2006). However, immune cell infiltration and activation within the adipose tissue is a major source of pro-inflammatory cytokines that impair adipocyte function in chronic obesity. Inflammation is also known to induce fibrosis. In obesity, development of adipose tissue fibrosis is thought to participate in inability of adipocytes to expand and store lipid (Divoux et al., 2010; Khan et al., 2009). The resultant “lipid spill over” from dysfunctional or necrotic adipocytes participates in dyslipidemia, lipotoxicity and serves as a source of endogenous DAMPs that can be sensed by specific PRR mediated activation of the cells of innate immune system. Thus, alterations in adipose tissue and development of chronic inflammation are the hallmarks of obesity and are at least partially responsible for the induction of insulin resistance. In addition to its function as an energy storage site, adipose tissue also releases hormone-like mediators known as adipokines that are involved in the regulation of insulin-sensitization.
Several recent studies demonstrated that the NLRP3 Inflammasome senses obesity-associated DAMPs and is an important mechanism that participates in the development of insulin-resistance (Stienstra et al., 2010; Vandanmagsar et al., 2011; Wen et al., 2011; Zhou et al., 2010). The influx of macrophages, T and B cells in adipose tissue in obesity and release of pro-inflammatory mediators by these cells cause insulin-resistance. The mechanisms that regulate the activation of these immune cells in adipose tissue are still largely unclear; however, NLRP3 inflammasome activation in adipose tissue macrophages may be one of regulators of immune activation in obesity. Importantly, inflammasome-mediated caspase-1 activation in adipose tissue and liver has been shown to impair insulin-signaling and glucose homeostasis (Stienstra et al., 2010). Studies using knock-in reporter mice suggest that Nlrp3 is largely expressed in myeloid cells (Guarda et al., 2011) and it appears that hematopoietic compartment may play a predominant role in sensing of obesity-related DAMPs and subsequent production of IL-1β and IL-18 (Vandanmagsar et al., 2011; Wen et al., 2011). Obesity is associated with increase in ceramides, saturated fatty acids, ROS, mitochondrial dysfunction and ATP release from necrotic adipocytes. All these factors have been shown to activate the NLRP3 inflammasome in macrophages (Lamkanfi et al., 2009; Stienstra et al., 2010; Vandanmagsar et al, 2011.; Wen et al., 2011; Zhou et al., 2010). Of note, the Caspase-1 and IL-1β activity was found to be enhanced in both diet-induced and genetically prone obese mouse models (Stienstra et al., 2010; Vandanmagsar et al., 2011). Furthermore, IL-1β directs adipocytes to a more insulin-resistant phenotype during differentiation. Importantly, caspase-1 inhibitors were effective in improving insulin sensitivity and metabolic functions of adipocytes in diseased mice. In addition, elimination of NLRP3 inflammasome activation in obesity, reduced IL-18 and the number of effector-memory T cells within adipose tissue together with lower IFNγ production. Together, inhibition of NLRP3 inflammasome lowers obesity-associated inflammation and improves insulin-sensitivity.
Type 2 Diabetes
Type 2 diabetes (T2D) is an inflammatory disorder characterized by insulin resistance and uncontrolled glucose levels. Ultimately, chronic inflammation in both the pancreas and adipose tissue causes impaired responsiveness to insulin and results in the development of disease. Deleterious complications that arise as a result of elevated blood sugar levels include renal failure, coronary artery disease, blindness, and stroke. Both genetic and lifestyle factors contribute to the development of this disease. Moreover, metabolic and dietary factors that are associated with increased weight gain and inflammation have been proposed to link obesity to T2D (Kolb & Mandrup-Poulsen, 2005; Pradhan, 2007). As a result of the sharp rise in obesity rates, T2D has become one of the foremost threats to global health.
It is well accepted that inflammatory cytokines secreted by immune cells and activated macrophages are centrally involved in the induction and maintenance of pancreatic and adipose inflammation (Donath & Shoelson, 2011). Early work in the field identified IL-6 and TNF-α as important effectors in disease progression and severity (Gregor & Hotamisligil, 2011). Although it is widely agreed that both of these cytokines regulate macrophage infiltration and tissue damage during disease (Nikolajczyk et al., 2011), T2D therapeutics that target these inflammatory cytokines have been disappointing in clinical trials (Alexandraki et al., 2006; Nikolajczyk et al., 2011; Ofei et al., 1996; Paquot et al., 2000). Inflammasome derived cytokines, on the other hand, have emerged as central regulators in the inflammatory response that impairs pancreatic and adipose tissue function and imparts insulin resistance. For instance, IL-1β can induce insulin resistance in adipocytes and promote apoptosis of β-cells in the pancreas (Bendtzen et al., 1986; Lagathu et al., 2006). IL-18 is also upregulated in T2D patients and has been associated with increased secondary renal failure and atherosclerosis (Blankenberg et al., 2002; Thorand et al., 2005). Finally, IL-1R antagonists have shown great promise in T2D clinical trials (Larsen et al., 2007). In these studies, IL-1 signaling blockade was found to stabilize blood glucose levels and improve β-cell function.
The factors that trigger the induction of IL-1β and subsequent progression of T2D have remained enigmatic for many years. Saturated fatty acids and other obesity-related metabolites are elevated in T2D patients and have been suspected to contribute to inflammation and disease progression. However, the mechanistic link between metabolites that are associated with high fat diets and chronic inflammatory conditions has remained elusive. Recent discoveries indicate that obesity-related factors (i.e., metabolites and changes to adipose tissue) can trigger the activation of the inflammasome and that this potentiates the secretion of IL-1β and subsequent disease pathogenesis. It was recently shown that palmitate, one of the most abundant saturated fatty acids in the plasma of T2D patients, can induce the activation of the NLRP3-ASC inflammasome and trigger the secretion of IL-1β and IL-18 (Wen et al., 2011) (Figure 2). Palmitate induced secretion of inflammasome cytokines impinges on insulin signaling in numerous target organs and, as a result, promotes hyperglycemia, insulin insensitivity, and steatohepatitis. In presence of serine palmitoyl transferase and ceramides synthase, palmitate is further converted into sphinganine, dihydroceramide and ceramide. Interestingly, ceramide, which is also elevated within pancreas and is known to impair pancreatic function was also found to activate the NLRP3-inflammasome and may contribute to obesity-induced T2D in a similar fashion (Vandanmagsar et al., 2011).
Figure 2. Inflammasome activation in atherosclerosis and type 2 diabetes.
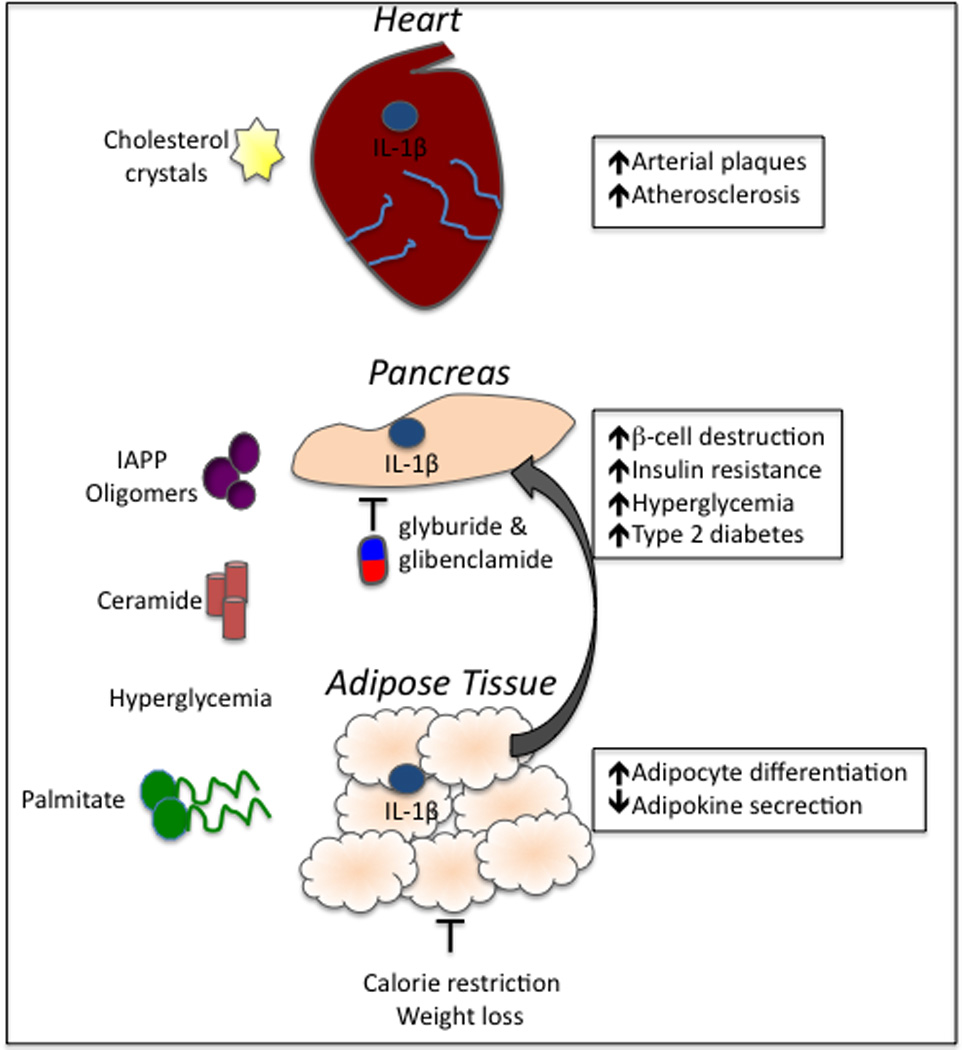
Obesity-related factors have emerged as important activators of inflammasome-derived cytokines. Cholesterol crystals that develop as a result of high-fat diets and obesity deposit in the arterial walls and incite inflammation via the activation of the NLRP3 inflammasome. Unrestrained production of IL-1β and IL-18 induces the production of additional inflammatory cytokines and provokes the recruitment of immune cells, including macrophages. Inflammasome activation in the cardiovascular system can ultimately result in plaque rupture. Obesity-derived metabolites, including IAAP oligomers and fatty acids (palmitate and ceramide), can induce inflammasome activation, which contributes to the induction and progression of T2D. Furthermore, hyperglycemia and alteration of adipose tissue causes metabolic distress and direct downstream activation of inflammasome signaling. Secretion of IL-1β and IL-18 in the pancreas causes the destruction of insulin producing cells (β-cells) and hyperglycemia. Inflammasome-mediated inflammation in the adipose tissue impairs adipokine production, which is an important regulator of insulin-sensitization. Two commonly prescribed T2D drugs, glyburide and glibenclamide, limit disease partially through their inhibition of inflammasome activation. Weight loss can also limit the secretion of inflammasome-induced cytokines.
Another obesity-related factor, islet amyloid polypeptide (IAPP), was also discovered to trigger inflammasome activation and potentiate T2D (Masters et al., 2010). IAPP is a metabolite secreted at the same time as insulin by β-cells in the pancreas. During the progression of T2D, IAPP forms amyloid deposits in the pancreas (Clark et al., 1988; Wei et al., 2011). This formation of amyloid plaques is a major hallmark of T2D and has been speculated to exacerbate disease severity. We now know that IAPP contributes to T2D by triggering NLRP3 inflammasome mediated secretion of IL-1β. Furthermore, recent studies also found that glyburide, a drug used to treat T2D, can suppress IAPP-mediated IL-1β production (Lamkanfi et al., 2009). Deposition of IAPP in pancreatic grafts has also been indicated as a negative predictor of transplantation to treat severe T2D (Westermark et al., 2008; Westermark et al., 1995). Thus, IAPP induced activation of the inflammasome may also play a separate role in transplant rejection. Additional studies are needed to delineate the role of inflammasome-derived cytokines in the destruction of transplanted islets. These recent discoveries in the inflammasome field have helped to resolve longstanding questions regarding the link between obesity derived metabolites and T2D progression and identify inflammasome-induced production of IL-1β as a central mediator of disease. A recent study extended these findings by making the interesting observation that calorie restriction and exercise-mediated weight loss in obese individuals with T2D results in reduced expression of NLRP3 (Vandanmagsar et al., 2011). Furthermore, improvement of T2D in morbidly obese patients that undergo weight loss through bariatric surgery is associated with significant reduction in IL-1β and NLRP3 inflammasome expression (Moschen et al., 2011) suggesting that this pathway is of high clinical relevance in the management of obesity and diabetes.
Collectively, inflammasome research has greatly improved our understanding of the relationship that links inflammation and diet to the induction and progression of T2D. Importantly, recent work has demonstrated that inflammasome-induced cytokines promote disease following stimulation by high fat diet metabolites and adipose tissue changes that occur during weight gain. These studies have helped to elucidate additional molecular pathways that can be specifically targeted to provide novel therapeutics for T2D. It is possible that other metabolic changes that result from obesity will also play an important part in the pathogenesis of disease via regulation of the inflammasome. Furthermore, there are numerous other unique NLR molecules that we are just beginning to understand in the context of inflammation and disease. These NLRs may also play central roles in T2D and other obesity driven diseases. Studies that focus on these molecules will undoubtedly provide novel insight into regulation of inflammation during disease and offer new molecular interactions to target in the design of improved treatment strategies for T2D.
Atherosclerosis
Atherosclerosis is an inflammatory disorder in which the artery wall thickens as a result of the accumulation of fats and cholesterol. Immune cells, including macrophages and T cells, are prominent mediators of the inflammation that promotes disease progression. Much like T2D, atherosclerosis pathogenesis is believed to be greatly affected by obesity-related factors. Numerous pieces of clinical data suggest that inflammasome-derived cytokines play a pivotal role in disease. For one, IL-1β and IL-1R levels are elevated in arterial plaques and expression levels are linked to disease severity (Galea et al., 1996; Moyer et al., 1991). Likewise, the prevalence of an IL1RN polymorphism that confers enhanced IL-1 production is negatively correlated with arterial plaque size (Francis et al., 1999; Olofsson et al., 2009). Finally, IL-18 concentrations in the blood are a predictor of atherosclerosis related death in patients (Blankenberg et al., 2002; Mallat et al., 2001). Even though the importance of immune cell derived inflammation in atherosclerotic vessel walls is well established, the factors that incite and prolong the inflammation have remained elusive. Within the last year, we have learned that cholesterol crystals are an important early contributor to inflammation and tissue damage during atherosclerosis pathogenesis. Importantly, cholesterol crystals were discovered to induce disease by triggering NLRP3-mediated activation of the inflammasome and promoting the release of both IL-1β and IL-18 (Duewell et al., 2010) (Figure 2).
One of the most touted treatments for atherosclerosis in recent years has been a class of acylcoenzyme A: cholesterol acyltransferase (ACAT) inhibitors. These drugs were expected to reduce cholesterol levels and plaque formation. However, in many cases treatment with ACAT inhibitors actually promoted the formation of coronary plaques (Meuwese et al., 2009; Nissen et al., 2006). Our new understanding of the role of cholesterol crystals in the promotion of inflammasome activation may help to explain these unexpected and discouraging ACAT inhibitor clinical trial results. The inhibition of cholesterol droplet storage by this line of drugs actually promotes crystal formation (Chang et al., 2006). Thus, one could speculate that it is the deposition of crystals that exacerbates disease with this treatment via inflammasome-mediated induction of inflammatory cytokines. In the future, it will be important to determine whether addition of inflammasome inhibitors or IL-1 antagonists with ACAT inhibitors prove beneficial in the treatment of atherosclerosis.
Multiple sclerosis
Multiple sclerosis (MS) is a debilitating neuroinflammatory disease that occurs when autoreactive T cells gain entry into the central nervous system (CNS) and destroy myelin-producing oligodendrocytes. T cell-derived cytokines, including IL-17, IFN-γ, and GM-CSF, are primarily responsible for the disease symptoms that occur when the myelin sheath that insulates the neurons is damaged during MS (Codarri et al., 2011; El-Behi et al., 2011; Goverman, 2009). Inflammatory dendritic cells (DCs) and macrophages also contribute to disease induction and progression by activating these autoreactive T cells and secreting inflammatory cytokines in the CNS.
Clinical studies have identified an important role for inflammasome-derived cytokines in MS disease pathogenesis. For instance, IL-1β and naturally occurring IL-1R antagonist gene polymorphisms were shown to be associated with MS disease severity (de Jong et al., 2002; Schrijver et al., 1999). Furthermore, individuals with a relatively high ratio of IL-1β relative to the naturally occurring IL-1R antagonist are genetically predisposed to MS (Arend, 2002; de Jong et al., 2002). Elevated expression of caspase-1 has also been observed in MS lesions (Huang et al., 2004; Ming et al., 2002). The central role of IL-1 in neurodegenerative disease has been further substantiated in the experimental autoimmune encephalomyelitis (EAE) mouse model of MS. Caspase-1 and IL-1R were found to be required for the development of EAE (Furlan et al., 1999; Jacobs et al., 1991). Mechanistically IL-1 was shown to potentiate neuroinflammation by promoting the differentiation and maintenance of autoreactive IL-17 producing T cells and causing cell death in the CNS (Chung et al., 2009; Sutton et al., 2006; Sutton et al., 2009). Although caspase-1 mediated activation of IL-1β is known to be an important contributor to EAE, the exact inflammasome platform that is required to induce caspase-1 mediated progression of EAE still remains to be formally determined. There have been conflicting data on the importance of the NLRP3 inflammasome in EAE pathology (Gris et al., 2010; Shaw et al., 2010). Thus, future work is needed to clarify these disparate findings and to also identify the specific inflammasome-platform that promotes neuroinflammation in MS. It is possible that multiple redundant inflammasome complexes are involved in caspase-1 activation during MS.
One of the most successful therapies in the treatment of MS symptoms to date is IFN-β administration (Comi et al., 2001). This strategy has been especially effective in the treatment of patients in the relapsing-remitting phase of disease progression. Although this treatment has been effective in the management of disease symptoms for many MS patients, the mode of action of this therapy has remained a subject of great debate. A recent report has suggested that IFN-β attenuates the course and severity of MS by regulating inflammasome activation and subsequent IL-1 production (Guarda et al., 2011). These studies found that type I interferon potently repressed the activity of the NLRP1 and NLRP3 inflammasomes, thereby suppressing caspase-1 dependent IL-1β secretion in mice. Importantly, they were able to clinically translate these observations and found that IFN-β treatment does in fact markedly diminish inflammasome activation and downstream IL-1β maturation in MS patients. These findings shed light onto the mechanism of type 1 IFN-mediated protection in MS and also highlight the importance of the inflammasome-IL-1 axis in this debilitating disease. Although IFN-β therapies are effective in alleviating MS symptoms for many patients, they can also cause numerous adverse side effects including flu-like symptoms, liver damage, and increased susceptibility to certain infections. Thus, targeted approaches to specifically disrupt inflammasome activation may provide improved therapies for MS patients.
Type 1 Diabetes
In type 1 diabetes (T1D), autoreactive CD4+ T cells cause β-cell destruction in the pancreas (Lehuen et al., 2010). Similar to T2D, disease is characterized by uncontrolled glucose levels and insulin resistance. Unlike T2D, where obesity related inflammation is a major driver of disease, T1D development is associated with dysregulated T cell responses. Insulin replacement therapy is used to manage T1D; however, fluctuations in blood sugar levels still occur and result in deleterious damage to numerous organs. We have only recently begun to unravel the role of inflammasome-dependent cytokines in T1D pathogenesis. As mentioned above, IL-1β has been conclusively shown to promote β cell apoptosis and is thus also believed to contribute to pancreatic damage and insulin resistance during T1D progression. Recently, a coding polymorphism in NLRP1 was demonstrated to confer susceptibility to T1D (Magitta et al., 2009). Furthermore, two single-nucleotide polymorphisms in NLRP3 were also identified in a separate study as a predisposing factor for T1D (Pontillo et al., 2010). Because of these promising findings and others, a multicenter randomized trial was recently launched to investigate the effectiveness of IL-1R antagonists in the treatment of T1D in patients (Mandrup-Poulsen et al., 2010) (www.clinicaltrials.gov).
Additional in vivo studies that investigate the ability of inflammasome activation to influence T1D development and progression are needed going forward. Analysis of the role that the inflammasome plays in disease induction and priming of diabetogenic T cells in the non-obese diabetic (NOD) mouse model should help to shed light on this important topic (Anderson & Bluestone, 2005). The factors that unleash autoreactive T cells on the pancreas have not been formally identified to date. This gap in T1D understanding has hindered the development of specific therapeutics. Regulation of inflammasome-derived cytokines may prove to be especially important in the priming of autoreactive T cells that destroy β-cells.
Future considerations and closing remarks
Recent discoveries have identified the NLR-inflammasome axis as a pivotal regulator of numerous diverse autoinflammatory and autoimmune disorders. The effectiveness of IL-1R blockade in the treatment of many of these diseases, including T2D, suggests that additional therapeutics that target inflammasome activation may provide novel strategies to treat these devastating disorders. The identification of the central role of inflammasome-derived cytokines in disease progression and the discovery of stress/danger associated signals that trigger this activation have provided an important foundation in our understanding of the etiology of many human diseases. Despite these recent advancements, numerous important questions remain to be addressed in order to gain a more complete understanding of inflammasome-mediated control of inflammatory and autoimmune diseases.
For instance, extensive research has characterized the regulation of the inflammasome in innate cells, especially macrophages. However, NLRs and inflammasome-associated proteins are also expressed in a wide range of cells including epithelial cells and adaptive immune cells (Guarda et al., 2011; Kufer & Sansonetti, 2011). The biological relevance of inflammasome signaling pathways in non-innate cells has only recently been appreciated (Yazdi et al., 2010). Foam cells and endothelial cells are major producers of IL-1β in atherosclerotic plaques (Galea et al., 1996; Moyer et al., 1991); however, the inflammasome platforms that mediate this production and how this contribution to the inflammatory milieu affects disease still needs to be formally defined. Future studies that probe the function of the NLR-inflammasome axis in cells other than macrophages and DCs will facilitate in the development of new therapeutics and enhance our knowledge of basic inflammasome biology.
Recent studies have clearly delineated a role for IL-1β in numerous human diseases. However, the role of IL-18 in autoimmune and autoinflammatory disease has only been defined for a few disorders. It is possible that IL-1β is the major driver of inflammasome-mediated inflammatory disease. However, this is unlikely considering the potent ability of IL-18 to promote IFN-γ production by T cells and innate cells. Furthermore, IL-18 also promotes the migration and infiltration of inflammatory cells into organs. Future studies using IL-18R deficient mice are needed to elucidate the influence of IL-18 on inflammatory disease pathogenesis. A possible explanation for the failure of IL-1R therapies to reduce disease severity in particular clinical trials may be potentially due to the ability of IL-18 to compensate for IL-1β as an inflammatory mediator in these diseases. Combined IL-1 and IL-18 blockade may be required to achieve efficacy in the treatment of certain diseases such as rheumatoid arthritis where IL-1R antagonism alone was not successful.
Inflammasome signaling can also affect biological process other than inflammatory cytokine production. For instance, the NLR-inflammasome axis directly influences cell death and autophagy in infectious models (Harris et al., 2011). Importantly, regulation of cell turnover and autophagy both play prominent roles in inflammation and autoimmunity (Levine & Deretic, 2007; Lleo et al., 2007; Rock & Kono, 2008; Rudin & Thompson, 1997). The regulation of cell death and autophagy by inflammasome activation in the context of autoimmune disease and danger/stress-induced inflammation have not been extensively studied to date and is thus an exciting area of future research.
Recent advancements in the field of inflammasome biology have greatly enhanced our understanding of the etiology of numerous inflammatory and autoimmune diseases. These studies have positioned inflammasomes as central regulators that link cellular stress that results from obesity-induced inflammation, metabolic distress, and other danger/stress signals to the induction and progression of autoinflammatory disease. It has become clear that inflammasome activation can also significantly influence T cell mediated autoimmune diseases including MS and type 1 diabetes. These findings and future discoveries in the inflammasome field should provide novel strategies to treat numerous debilitating and chronic inflammatory and autoimmune diseases.
Acknowledgements
We apologize to authors whose work could not be referenced in this review due to space limitations. This work was supported by National Institute of Health Grants AR056296, AI088177, and the American Lebanese Syrian Associated Charities (ALSAC).
Footnotes
Competing interest statement
The authors declare no competing financial interests.
References
- Aksentijevich I, Putnam C, Remmers EF, Mueller JL, Le J, Kolodner RD, Moak Z, Chuang M, Austin F, Goldbach-Mansky R, Hoffman HM, Kastner DL. The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007;56(4):1273–1285. doi: 10.1002/art.22491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandraki K, Piperi C, Kalofoutis C, Singh J, Alaveras A, Kalofoutis A. Inflammatory process in type 2 diabetes: The role of cytokines. Ann N Y Acad Sci. 2006;1084:89–117. doi: 10.1196/annals.1372.039. [DOI] [PubMed] [Google Scholar]
- Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- Arend WP. The balance between IL-1 and IL-1Ra in disease. Cytokine Growth Factor Rev. 2002;13(4–5):323–340. doi: 10.1016/s1359-6101(02)00020-5. [DOI] [PubMed] [Google Scholar]
- Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, Dinarello CA, Paul WE. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci U S A. 2009;106(17):7119–7124. doi: 10.1073/pnas.0902745106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtzen K, Mandrup-Poulsen T, Nerup J, Nielsen JH, Dinarello CA, Svenson M. Cytotoxicity of human pI 7 interleukin-1 for pancreatic islets of Langerhans. Science. 1986;232(4757):1545–1547. doi: 10.1126/science.3086977. [DOI] [PubMed] [Google Scholar]
- Blankenberg S, Tiret L, Bickel C, Peetz D, Cambien F, Meyer J, Rupprecht HJ. Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation. 2002;106(1):24–30. doi: 10.1161/01.cir.0000020546.30940.92. [DOI] [PubMed] [Google Scholar]
- Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006;22:129–157. doi: 10.1146/annurev.cellbio.22.010305.104656. [DOI] [PubMed] [Google Scholar]
- Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30(4):576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church LD, Mcdermott MF. Rilonacept in cryopyrin-associated periodic syndromes: the beginning of longer-acting interleukin-1 antagonism. Nat Clin Pract Rheumatol. 2009;5(1):14–15. doi: 10.1038/ncprheum0959. [DOI] [PubMed] [Google Scholar]
- Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, Cooper GJ, Holman RR, Turner RC. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 1988;9(4):151–159. [PubMed] [Google Scholar]
- Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12(6):560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- Comi G, Filippi M, Barkhof F, Durelli L, Edan G, Fernandez O, Hartung H, Seeldrayers P, Sorensen PS, Rovaris M, Martinelli V, Hommes OR. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet. 2001;357(9268):1576–1582. doi: 10.1016/s0140-6736(00)04725-5. [DOI] [PubMed] [Google Scholar]
- Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jong BA, Huizinga TW, Bollen EL, Uitdehaag BM, Bosma GP, Van Buchem MA, Remarque EJ, Burgmans AC, Kalkers NF, Polman CH, Westendorp RG. Production of IL-1beta and IL-1Ra as risk factors for susceptibility and progression of relapse-onset multiple sclerosis. J Neuroimmunol. 2002;126(1–2):172–179. doi: 10.1016/s0165-5728(02)00056-5. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. The interleukin-1 family: 10 years of discovery. FASEB J. 1994;8(15):1314–1325. [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-18 and the pathogenesis of inflammatory diseases. Semin Nephrol. 2007;27(1):98–114. doi: 10.1016/j.semnephrol.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Divoux A, Tordjman J, Lacasa D, Veyrie N, Hugol D, Aissat A, Basdevant A, Guerre-Millo M, Poitou C, Zucker Jd, Bedossa P, Clement K. Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes. 2010;59(11):2817–2825. doi: 10.2337/db10-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11(2):98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12(6):568–575. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L, Mcdonald C, Kanneganti TD, Amer A, Nunez G. Nucleotide-binding oligomerization domain-like receptors: intracellular pattern recognition molecules for pathogen detection and host defense. J Immunol. 2006;177(6):3507–3513. doi: 10.4049/jimmunol.177.6.3507. [DOI] [PubMed] [Google Scholar]
- Francis SE, Camp NJ, Dewberry RM, Gunn J, Syrris P, Carter ND, Jeffery S, Kaski JC, Cumberland DC, Duff GW, Crossman DC. Interleukin-1 receptor antagonist gene polymorphism and coronary artery disease. Circulation. 1999;99(7):861–866. doi: 10.1161/01.cir.99.7.861. [DOI] [PubMed] [Google Scholar]
- Furlan R, Martino G, Galbiati F, Poliani PL, Smiroldo S, Bergami A, Desina G, Comi G, Flavell R, Su MS, Adorini L. Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J Immunol. 1999;163(5):2403–2409. [PubMed] [Google Scholar]
- Galea J, Armstrong J, Gadsdon P, Holden H, Francis SE, Holt CM. Interleukin-1 beta in coronary arteries of patients with ischemic heart disease. Arterioscler Thromb Vasc Biol. 1996;16(8):1000–1006. doi: 10.1161/01.atv.16.8.1000. [DOI] [PubMed] [Google Scholar]
- Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. 2009;9(6):393–407. doi: 10.1038/nri2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, Huang M, Schneider M, Miller SD, Ting JP. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol. 2010;185(2):974–981. doi: 10.4049/jimmunol.0904145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, Tschopp J. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34(2):213–223. doi: 10.1016/j.immuni.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Guarda G, Zenger M, Yazdi AS, Schroder K, Ferrero I, Menu P, Tardivel A, Mattmann C, Tschopp J. Differential expression of NLRP3 among hematopoietic cells. J Immunol. 2011;186(4):2529–2534. doi: 10.4049/jimmunol.1002720. [DOI] [PubMed] [Google Scholar]
- Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, Kornfeld H, Fitzgerald KA, Lavelle EC. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286(11):9587–9597. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman HM, Rosengren S, Boyle DL, Cho JY, Nayar J, Mueller JL, Anderson JP, Wanderer AA, Firestein GS. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 2004;364(9447):1779–1785. doi: 10.1016/S0140-6736(04)17401-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WX, Huang P, Hillert J. Increased expression of caspase-1 and interleukin-18 in peripheral blood mononuclear cells in patients with multiple sclerosis. Mult Scler. 2004;10(5):482–487. doi: 10.1191/1352458504ms1071oa. [DOI] [PubMed] [Google Scholar]
- Jacobs CA, Baker PE, Roux ER, Picha KS, Toivola B, Waugh S, Kennedy MK. Experimental autoimmune encephalomyelitis is exacerbated by IL-1 alpha and suppressed by soluble IL-1 receptor. J Immunol. 1991;146(9):2983–2989. [PubMed] [Google Scholar]
- Kanneganti TD. Central roles of NLRs and inflammasomes in viral infection. Nat Rev Immunol. 2010;10(10):688–698. doi: 10.1038/nri2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27(4):549–559. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, Zhang BB, Bonaldo P, Chua S, Scherer PE. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol. 2009;29(6):1575–1591. doi: 10.1128/MCB.01300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb H, Mandrup-Poulsen T. An immune origin of type 2 diabetes? Diabetologia. 2005;48(6):1038–1050. doi: 10.1007/s00125-005-1764-9. [DOI] [PubMed] [Google Scholar]
- Kufer TA, Sansonetti PJ. NLR functions beyond pathogen recognition. Nat Immunol. 2011;12(2):121–128. doi: 10.1038/ni.1985. [DOI] [PubMed] [Google Scholar]
- Lagathu C, Yvan-Charvet L, Bastard JP, Maachi M, Quignard-Boulange A, Capeau J, Caron M. Long-term treatment with interleukin-1beta induces insulin resistance in murine and human adipocytes. Diabetologia. 2006;49(9):2162–2173. doi: 10.1007/s00125-006-0335-z. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Kanneganti TD. Nlrp3: an immune sensor of cellular stress and infection. Int J Biochem Cell Biol. 2010;42(6):792–795. doi: 10.1016/j.biocel.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Kanneganti TD, Franchi L, Nunez G. Caspase-1 inflammasomes in infection and inflammation. J Leukoc Biol. 2007;82(2):220–225. doi: 10.1189/jlb.1206756. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, Lee WP, Hoffman HM, Dixit VM. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009;187(1):61–70. doi: 10.1083/jcb.200903124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356(15):1517–1526. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- Lehuen A, Diana J, Zaccone P, Cooke A. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol. 2010;10(7):501–513. doi: 10.1038/nri2787. [DOI] [PubMed] [Google Scholar]
- Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7(10):767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lleo A, Invernizzi P, Selmi C, Coppel RL, Alpini G, Podda M, Mackay IR, Gershwin ME. Autophagy: highlighting a novel player in the autoimmunity scenario. J Autoimmun. 2007;29(2–3):61–68. doi: 10.1016/j.jaut.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magitta NF, Boe Wolff AS, Johansson S, Skinningsrud B, Lie BA, Myhr KM, Undlien DE, Joner G, Njolstad PR, Kvien TK, Forre O, Knappskog PM, Husebye ES. A coding polymorphism in NALP1 confers risk for autoimmune Addison's disease and type 1 diabetes. Genes Immun. 2009;10(2):120–124. doi: 10.1038/gene.2008.85. [DOI] [PubMed] [Google Scholar]
- Mallat Z, Corbaz A, Scoazec A, Besnard S, Leseche G, Chvatchko Y, Tedgui A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation. 2001;104(14):1598–1603. doi: 10.1161/hc3901.096721. [DOI] [PubMed] [Google Scholar]
- Mandrup-Poulsen T, Pickersgill L, Donath MY. Blockade of interleukin 1 in type 1 diabetes mellitus. Nat Rev Endocrinol. 2010;6(3):158–166. doi: 10.1038/nrendo.2009.271. [DOI] [PubMed] [Google Scholar]
- Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z, Mullooly N, Mielke LA, Harris J, Coll RC, Mills KH, Mok KH, Newsholme P, Nunez G, Yodoi J, Kahn SE, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol. 2010;11(10):897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwese MC, De Groot E, Duivenvoorden R, Trip MD, Ose L, Maritz FJ, Basart DC, Kastelein JJ, Habib R, Davidson MH, Zwinderman AH, Schwocho LR, Stein EA. ACAT inhibition and progression of carotid atherosclerosis in patients with familial hypercholesterolemia: the CAPTIVATE randomized trial. JAMA. 2009;301(11):1131–1139. doi: 10.1001/jama.301.11.1131. [DOI] [PubMed] [Google Scholar]
- Ming X, Li W, Maeda Y, Blumberg B, Raval S, Cook SD, Dowling PC. Caspase-1 expression in multiple sclerosis plaques and cultured glial cells. J Neurol Sci. 2002;197(1–2):9–18. doi: 10.1016/s0022-510x(02)00030-8. [DOI] [PubMed] [Google Scholar]
- Moschen AR, Molnar C, Enrich B, Geiger S, Ebenbichler CF, Tilg H. Adipose and Liver Expression of IL-1 Family Members in Morbid Obesity and Effects of Weight Loss. Mol Med. 2011 doi: 10.2119/molmed.2010.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer CF, Sajuthi D, Tulli H, Williams JK. Synthesis of IL-1 alpha and IL-1 beta by arterial cells in atherosclerosis. Am J Pathol. 1991;138(4):951–960. [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol. 2001;19:423–474. doi: 10.1146/annurev.immunol.19.1.423. [DOI] [PubMed] [Google Scholar]
- Nikolajczyk BS, Jagannathan-Bogdan M, Shin H, Gyurko R. State of the union between metabolism and the immune system in type 2 diabetes. Genes Immun. 2011 doi: 10.1038/gene.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen SE, Tuzcu EM, Brewer HB, Sipahi I, Nicholls SJ, Ganz P, Schoenhagen P, Waters DD, Pepine CJ, Crowe TD, Davidson MH, Deanfield JE, Wisniewski LM, Hanyok JJ, Kassalow LM. Effect of ACAT inhibition on the progression of coronary atherosclerosis. N Engl J Med. 2006;354(12):1253–1263. doi: 10.1056/NEJMoa054699. [DOI] [PubMed] [Google Scholar]
- Ofei F, Hurel S, Newkirk J, Sopwith M, Taylor R. Effects of an engineered human anti-TNF-alpha antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes. 1996;45(7):881–885. doi: 10.2337/diab.45.7.881. [DOI] [PubMed] [Google Scholar]
- Olofsson PS, Sheikine Y, Jatta K, Ghaderi M, Samnegard A, Eriksson P, Sirsjo A. A functional interleukin-1 receptor antagonist polymorphism influences atherosclerosis development The interleukin-1beta:interleukin-1 receptor antagonist balance in atherosclerosis. Circ J. 2009;73(8):1531–1536. doi: 10.1253/circj.cj-08-1150. [DOI] [PubMed] [Google Scholar]
- Paquot N, Castillo MJ, Lefebvre PJ, Scheen AJ. No increased insulin sensitivity after a single intravenous administration of a recombinant human tumor necrosis factor receptor: Fc fusion protein in obese insulin-resistant patients. J Clin Endocrinol Metab. 2000;85(3):1316–1319. doi: 10.1210/jcem.85.3.6417. [DOI] [PubMed] [Google Scholar]
- Pontillo A, Brandao L, Guimaraes R, Segat L, Araujo J, Crovella S. Two SNPs in NLRP3 gene are involved in the predisposition to type-1 diabetes and celiac disease in a pediatric population from northeast Brazil. Autoimmunity. 2010;43(8):583–589. doi: 10.3109/08916930903540432. [DOI] [PubMed] [Google Scholar]
- Pradhan A. Obesity, metabolic syndrome, and type 2 diabetes: inflammatory basis of glucose metabolic disorders. Nutr Rev. 2007;65(12 Pt 2):S152–S156. doi: 10.1111/j.1753-4887.2007.tb00354.x. [DOI] [PubMed] [Google Scholar]
- Rock KL, Kono H. The inflammatory response to cell death. Annu Rev Pathol. 2008;3:99–126. doi: 10.1146/annurev.pathmechdis.3.121806.151456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Thompson CB. Apoptosis and disease: regulation and clinical relevance of programmed cell death. Annu Rev Med. 1997;48:267–281. doi: 10.1146/annurev.med.48.1.267. [DOI] [PubMed] [Google Scholar]
- Schrijver HM, Crusius JB, Uitdehaag BM, Garcia Gonzalez MA, Kostense PJ, Polman CH, Pena AS. Association of interleukin-1beta and interleukin-1 receptor antagonist genes with disease severity in MS. Neurology. 1999;52(3):595–599. doi: 10.1212/wnl.52.3.595. [DOI] [PubMed] [Google Scholar]
- Shaw PJ, Lamkanfi M, Kanneganti TD. NOD-like receptor (NLR) signaling beyond the inflammasome. Eur J Immunol. 2010;40(3):624–627. doi: 10.1002/eji.200940211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PJ, Lukens JR, Burns S, Chi H, Mcgargill MA, Kanneganti TD. Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J Immunol. 2010;184(9):4610–4614. doi: 10.4049/jimmunol.1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PJ, Mcdermott MF, Kanneganti TD. Inflammasomes and autoimmunity. Trends Mol Med. 2011;17(2):57–64. doi: 10.1016/j.molmed.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116(7):1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol. 2010;10(2):89–102. doi: 10.1038/nri2691. [DOI] [PubMed] [Google Scholar]
- Stienstra R, Joosten LA, Koenen T, Van Tits B, Van Diepen JA, Van Den Berg SA, Rensen PC, Voshol PJ, Fantuzzi G, Hijmans A, Kersten S, Muller M, Van Den Berg WB, Van Rooijen N, Wabitsch M, Kullberg BJ, Van Der Meer JW, Kanneganti T, Tack CJ, Netea MG. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010;12(6):593–605. doi: 10.1016/j.cmet.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203(7):1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31(2):331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Thorand B, Kolb H, Baumert J, Koenig W, Chambless L, Meisinger C, Illig T, Martin S, Herder C. Elevated levels of interleukin-18 predict the development of type 2 diabetes: results from the MONICA/KORA Augsburg Study, 1984–2002. Diabetes. 2005;54(10):2932–2938. doi: 10.2337/diabetes.54.10.2932. [DOI] [PubMed] [Google Scholar]
- Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, Hoffman HM, Hugot JP, Inohara N, Mackenzie A, Maltais LJ, Nunez G, Ogura Y, Otten LA, Philpott D, Reed JC, et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28(3):285–287. doi: 10.1016/j.immuni.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17(2):179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Jiang P, Xu W, Li H, Zhang H, Yan L, Chan-Park MB, Liu XW, Tang K, Mu Y, Pervushin K. The molecular basis of distinct aggregation pathways of islet amyloid polypeptide. J Biol Chem. 2011;286(8):6291–6300. doi: 10.1074/jbc.M110.166678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12(5):408–415. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark GT, Westermark P, Berne C, Korsgren O. Widespread amyloid deposition in transplanted human pancreatic islets. N Engl J Med. 2008;359(9):977–979. doi: 10.1056/NEJMc0802893. [DOI] [PubMed] [Google Scholar]
- Westermark P, Eizirik DJ, Pipeleers DG, Hellerstrom C, Andersson A. Rapid deposition of amyloid in human islets transplanted into nude mice. Diabetologia. 1995;38(5):543–549. doi: 10.1007/BF00400722. [DOI] [PubMed] [Google Scholar]
- Yazdi AS, Drexler SK, Tschopp J. The role of the inflammasome in nonmyeloid cells. J Clin Immunol. 2010;30(5):623–627. doi: 10.1007/s10875-010-9437-y. [DOI] [PubMed] [Google Scholar]
- Zaki MH, Lamkanfi M, Kanneganti TD. The Nlrp3 inflammasome: contributions to intestinal homeostasis. Trends Immunol. 2011;32(4):171–179. doi: 10.1016/j.it.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunolrr. 2010;11(2):136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]